

Abstract
Alcohol dehydrogenases can act as powerful catalysts in the preparation of optically pure γ‐hydroxy‐δ‐lactones by means of an enantioconvergent dynamic redox isomerization of readily available Achmatowicz‐type pyranones. Imitating the traditionally metal‐mediated “borrowing hydrogen” approach to shuffle hydrides across molecular architectures and interconvert functional groups, this chemoinspired and purely biocatalytic interpretation effectively expands the enzymatic toolbox and provides new opportunities in the assembly of multienzyme cascades and tailor‐made cellular factories.
Keywords: asymmetric synthesis, biocatalysis, enantioconvergent processes, heterocycles, redox isomerization
With regard to novel strategies en route to more step‐economic, waste conscious, and environmentally benign, as well as elegant and selective synthesis protocols, the development of redox‐neutral activation scenarios utilizing redox‐active catalysts for the traceless generation of reactive intermediates from readily available building blocks represents a breakthrough in modern catalysis.1 Over the years, the “borrowing hydrogen” methodology pioneered by Williams, exploiting the reversible dehydrogenation of alcohols in order to promote both carbon–carbon and carbon–heteroatom coupling reactions by generating carbonyl intermediates has become a prime example for the synthetic utility of dehydrogenation catalysts.2 In addition to bimolecular coupling reactions, the “borrowing hydrogen” approach also proved equally powerful for the redox isomerization of unsaturated alcohols to saturated aldehydes and ketones,3 where particularly chiral iridium‐based systems provide a straightforward access to stereochemically defined complex carbonyl products.4 Traditionally, transition‐metal‐based complexes act as an intermediary storage system for the hydride equivalents extracted in the initial dehydrogenation that become the reducing agent for intermediates formed during the coupling process or isomerization, for example, iminium species or enones. In principle, similar hydride storage can also be achieved in a biocatalytic fashion as illustrated in the pioneering example by Hollmann et al., where the interplay of nicotinamide‐dependent enzymes and the reversible formation and consumption of NADH as a biological hydride equivalent was successfully utilized in the redox isomerization of cyclohexenol to cyclohexanone.5, 6, 7
In a recent addition to the ever growing toolbox of “borrowing hydrogen” catalysis, Guo, Tang et al. presented in 2015 an iridium‐based procedure for the redox isomerization of Achmatowicz pyranones (6‐hydroxy‐2H‐pyran‐3(6H)‐ones) to yield hydroxylated δ‐lactones in a substrate‐controlled diastereoselective fashion (Scheme 1, top).8 Considering the easy access to the substrates by Achmatowicz oxidation of furfuryl alcohols,9, 10 this dynamic stereoconvergent transformation thus offers a streamlined route to synthetically valuable lactones from simple biogenic building blocks such as furfural. Inspired by this work, and due to our personal scientific interest in the use of enzymes for the Achmatowicz rearrangement11 and related heterocyclic transformations12 for synthetic and biosynthetic purposes, we started a campaign to evaluate the potential of nicotinamide‐dependent alcohol dehydrogenases as biological mimics of the abovementioned metal complexes in stereoselective redox isomerizations, to be used as stand‐alone biocatalytic tools as well as to serve as powerful modules in more complex multienzymatic cascades (Scheme 1, bottom).
Scheme 1.
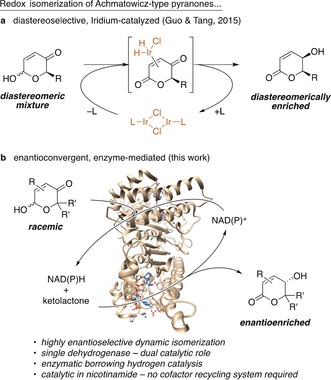
a) Classical “borrowing hydrogen” in an iridium‐mediated redox isomerization of Achmatowicz‐type pyranones. b) Nicotinamide‐dependent dehydrogenases as bifunctional, enantioselective isomerization catalysts.
Whereas the formation of transient metal hydride species can potentially lead to selectivity issues such as competition between C=C and C=O hydrogenation13 or acceptorless dehydrogenation, [14] we envisaged that a “borrowing hydrogen” approach by means of a nicotinamide‐coupled storage of reduction equivalents would make it possible to take advantage of the exquisite chemoselectivity of biological catalysts while at the same time rendering the reaction redox neutral without the requirement of cofactor regeneration systems. Moreover, the strict irreversibility of the biocatalytic lactol dehydrogenation would provide the necessary driving force to enable smooth conversion towards the desired γ‐hydroxy‐δ‐lactones. Most importantly, the combination of a rapid mutarotation of six‐membered lactols in the aqueous biocatalysis medium and the inherent enantioselectivity of dehydrogenases in carbonyl reductions was believed to create a dynamic scenario in which even racemic, symmetrically substituted hydroxypyranones could be isomerized into one stereochemically defined product in an enantioconvergent manner with a theoretical yield of 100 %.15
The spirocyclic pyranone 1 a, originally derived from oxidative ring expansion of 1‐furylcyclohexanol, was employed as model substrate to elucidate the general applicability of our design to replace metal‐based “borrowing hydrogen” catalysts by nicotinamide‐dependent alcohol dehydrogenases. To our positive surprise, out of a set of 26 commercial biocatalysts, taken from the ketoreductase screening kits of Codexis Inc. and evoCatal GmbH, in total 16 enzymes exhibited significant activity and led to the formation of the desired γ‐hydroxy‐δ‐lactone 2 a in moderate to mostly excellent yield (Scheme 2). Worth noting, the biocatalysts engage in the transformation of two distinctly different functional group couples (lactol/lactone and alcohol/ketone) by taking advantage of their ability to catalyze both dehydrogenations and reductions, obviating the more tedious search for enzyme pairs to address the specific tasks. In many cases, however, the observed enantioselectivity remained mediocre at best. Yet, five of the tested biocatalysts exhibited synthetically useful stereoinduction in the terminating ketone reduction giving rise to the highly optically enriched spirolactone 2 a. Gratifyingly, in addition to the four S‐selective ADHs (P1‐B05: 89 % ee,P2‐D11: 98 % ee, P2‐G03: 98 % ee, and evo 1.1.200: 91 % ee) also a complementary enzyme with decent R‐selectivity (P2‐D12: 86 % ee) could be identified, thereby meeting a crucial requirement of any stereoselective method to be considered as synthetically useful tool.
Scheme 2.
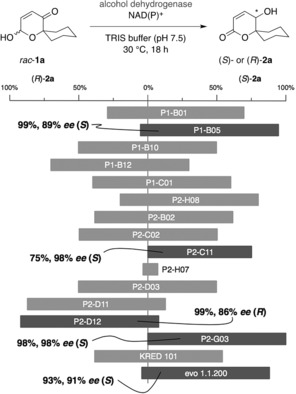
Reactivity of biocatalysts from commercial ketoreductase kits for the redox isomerization of pyranone 1 a.
Thanks to the rather general reactivity of multiple alcohol dehydrogenases in the redox isomerization to γ‐hydroxy‐δ‐lactones, the method proved to be generally applicable on a series of symmetrically substituted Achmatowicz pyranones, even though a secondary screening was required to identify most suitable biocatalysts for substrates structurally less related to model compound 1 a (Scheme 3). Both the S‐selective ketoreductase P2‐G03 as well as the enantiocomplementary P2‐D12 exhibited high activity on spirocyclic substrates bearing all‐carbon backbones and the products were isolated in good yield in most cases. Interestingly, the oxaspiro[4.5]decene scaffold 1 b showed slightly lower selectivity, compared to the oxaspiro[5.5]undecene 1 a and the oxaspiro[3.5]nonene 1 c, in the S‐series while the opposite effect was observed in the R‐series, where (R)‐2 b was obtained with an excellent optical purity of 96 % ee. Also incorporation of an oxetane moiety into the spirocyclic framework was well accepted, yielding hydroxylactone (S)‐2 e in 93 % ee, which, considering the bioisosteric relationship between carbonyls and oxetanes, resembles a hydrolytically fortified cyclic anhydride mimic. For the redox isomerization of substrates bearing simple alkyl groups instead of the cyclic α,α‐disubstitution pattern, another set of ketoreductases showed good activity and after treatment with catalytic amounts of evo 1.1.200 or P1‐B05, lactones (S)‐2 f and (S)‐2 g were isolated in 73 % yield and in optically pure form. Notably, even the unsubstituted pyranone 1 h was successfully isomerized to yield (S)‐2 h in 95 % ee indicating a pronounced face discrimination despite an apparently marginal steric distinction (sp2‐CH vs. sp3‐CH2).
Scheme 3.
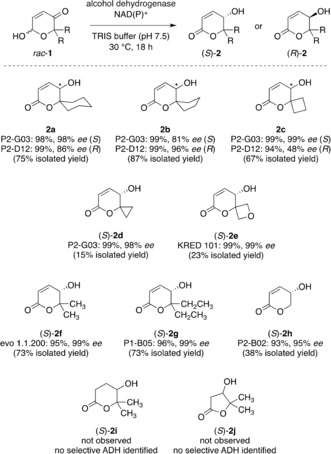
Hydroxyenone isomerase activity: scope and limitations.
Absolute configurations of the redox isomerization products were assigned by means of vibrational and electronic CD spectroscopy on representative hydroxylactones (Supporting Information, Figures 1–6), in addition to the comparison of optical rotations to previously reported compounds.16 To our surprise, saturated analogues such as 1 i or 1 j did not engage in a similar redox isomerization, which might reflect a certain geometrical or electronical prerequisite to support the biological “borrowing hydrogen” process.
The development of novel, enzyme‐based chemoinspired methodologies opens up great opportunities to implement biological tools in more complex multicatalytic strategies. One such tool is the application of oxidase/peroxidase couples as biocatalysts for the Achmatowicz rearrangement giving rise to the hydroxypyranones that was previously developed in our lab.11 Hence, we were pleased to find that furfuryl alcohol 3 undergoes smooth oxidative ring expansion in the presence of chloroperoxidase (CPO) and glucose/glucose oxidase (GOx) to the racemic 1 f, which could be subsequently subjected to the optimized formal hydroxyenone isomerase process to provide the enantiomerically pure lactone (S)‐2 f in 72 % overall yield through a purely biocatalytic reaction cascade (Scheme 4 a). Moreover, it turned out that reaction sequences were not limited to cascades based on oxidative furan ring expansions but that the enzymatic redox isomerization could be coupled to other transformations. Here, an organocatalytic crossed‐benzoin reaction between phthaldialdehyde and formaldehyde proved useful to deliver the aromatic non‐Achmatowicz hydroxypyranone rac‐5 (Scheme 4 b).17 An enzyme screening on this alternative substrate structure revealed several effective biocatalysts for the enantioconvergent transformation to the optically enriched lactone 6. Again, both S‐ and R‐configured 6 could be isolated in very high yield and good to excellent enantiopurity by either of two enantiocomplementary alcohol dehydrogenases. Furthermore, a base‐mediated ring contraction gave access to the corresponding γ‐lactone (S)‐7 with complete conservation of optical activity. Since both 6 and 7 represent core scaffolds in numerous complex natural products such as echinolactone B (8) and simplexene B (9),18, 19 a focus in our upcoming studies lies on the elucidation of the extended scope of the hydroxyenone isomerases on this particular substrate family.
Scheme 4.
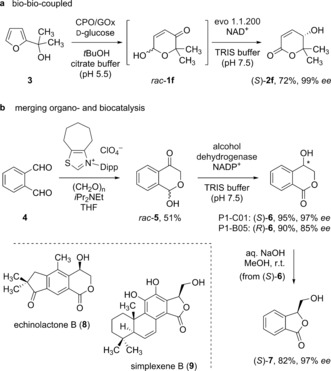
Design of catalytic cascades utilizing the biocatalytic hydroxyenone isomerase module. Dipp: 2,6‐diisopropylphenyl.
Especially the approach to assemble multienzyme cascades based on natural tools emulating classical synthetic transformations seems appealing as it will facilitate bringing traditional retrosynthetic thinking into biological settings.20 To illustrate the immediate synthetic impact of the developed hydroxyenone isomerase, the direct combination of peroxidase‐mediated furan ring rearrangement and ketoreductase‐catalyzed redox isomerization was utilized in the preparation of a series of δ‐valerolactone natural products (Scheme 5).
Scheme 5.
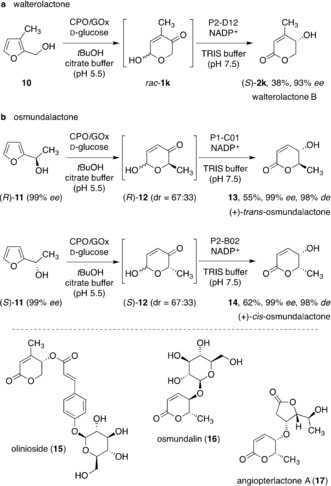
Application in the purely enzymatic total synthesis of walterolactone B and the osmundalactones, key building blocks en route to complex heterocyclic natural products.
Walterolactone B (2 k), found as a secondary metabolite in Cornus walteri and part of the aglycon of olinioside (15),21, 22 was thus obtained with good optical purity starting from 3‐methylfurfuryl alcohol (10) in a fully biocatalytic two‐step sequence (Scheme 5 a). Not limited to symmetrically disubstituted pyranones, the hydroxyenone isomerase could further be used in the synthesis of δ‐chiral δ‐valerolactones. Whilst the iridium‐based original template is currently still limited to the direct synthesis of cis‐configured γ‐hydroxy‐δ‐lactones due to its substrate‐controlled character, the biocatalytic interpretation thereof manages to substantially expand the target scope. As a catalyst‐controlled system, either cis‐ or trans‐configured products can be selectively addressed by the right choice of enantioenriched pyranone starting materials and complementary ketoreductase biocatalysts. Hence, both trans‐ (13) and cis‐osmundalactone (14) were successfully synthesized from optically pure 1‐furylethanol (11, obtained via bioreduction of acetylfuran)23 under the previously mentioned ring expansion/isomerization sequence without significant matched/mismatched effects and excellent enantio‐ and diastereopurity of the two epimeric natural products (Scheme 5 b),24 which themselves form an important platform for more complex target structures (such as 16 or 17).25
On the longer term, the discovery of novel artificial biocatalysis modules provides tremendous opportunities to create tailored cellular bioproduction factories with a much wider synthetic scope and more flexibility. In order to explore the possibilities to implement the non‐natural hydroxyenone isomerase tool into a truly biological environment, a set of different recombinant E. coli strains expressing ketoreductases26 were tested in the whole‐cell transformation of the pyranone rac‐1 f. Particularly, bacteria expressing alcohol dehydrogenase from Lactobacillus kefir 27 exhibited good turnover and offered perfect enantioselectivity in the in vivo production of the δ‐lactone (S)‐2 f (Scheme 6).
Scheme 6.
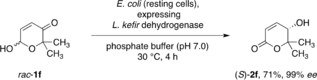
In vivo redox isomerization by recombinant E. coli expressing L. kefir alcohol dehydrogenase as formal hydroxyenone isomerase.
While the use of commercial enzyme preparations offers the advantage of a more general applicability of the method in synthesis, the black box character of these systems needs to be regarded with caution. Hence, to shed light on the underlying processes, numerous experiments targeting the nature of active species and the fundamental reaction steps have been conducted. Considering the structural similarity of the Achmatowicz lactols and naturally occurring sugars, contamination by, for example, glucose‐active proteins such as glucose dehydrogenase (GDH) and glucose oxidase could potentially account for the observed lactol dehydrogenation. However, spectrophotometric studies on the stability of rac‐1 f in presence of GDH or GOx clearly rule out any reactivity of either of the glucose‐oxidizing enzymes on these non‐natural hemiacetal substrates (Figure S7). In order to endorse our hypothesis of one single dehydrogenase acting first as a dehydrogenation catalyst to form a ketolactone and NAD(P)H followed by its role as a ketoreductase to furnish the formally redox isomerized products (see also Scheme 1 b), L. kefir ADH produced from E. coli was thoroughly purified28 and utilized as a biocatalyst (Figure S8). In the presence of NADP+, the purified LkADH did clearly exhibit good performance in the redox isomerization of 1 f, confirming the independence of the general process from other protein cocatalysts (Figure S9).29 From a mechanistic perspective, the redox isomerization can be initiated by the suggested lactol oxidation as well as by a reductive first step that would result in a hydroxylactol intermediate. Yet, when 1 f was treated with evo 1.1.200 in the presence of GDH/glucose or isopropanol, providing a more reducing environment and limiting the concentration of available NAD+, significantly reduced conversions and the formation of various side products was observed. In addition, isotopic labelling studies revealed a direct coupling of the dehydrogenation and the ketone reduction through the nicotinamide cofactor, as deuterated 1 f‐d1 was cleanly converted to its isomer (S)‐2 f‐d1 with high deuterium incorporation in the product (Scheme 7 a, Figures S10–S12). Based on crossover experiments utilizing equimolar mixtures of two 1 f isotopologues (Scheme 7 b, Figure S13), the extensive H/D scrambling between the CH3 and the CD3 derivatives suggests a pathway where dehydrogenation and ketoreduction occur as a distinct two‐step process with dissociation and reassociation of the reduced cofactor and the corresponding ketolactone (Figure S14); the latter step is also observed in reaction mixtures by GC and NMR.
Scheme 7.
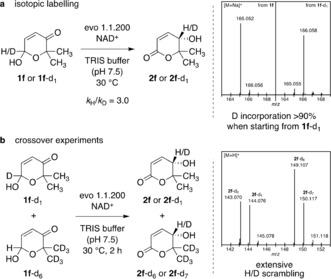
Labelling studies on the nature of free nicotinamides and their role as intermediary hydrogen storage systems.
In summary, with the identification of an artificial hydroxyenone isomerase activity of alcohol dehydrogenases we were able to develop a novel enantioconvergent protocol for the redox isomerization of readily available Achmatowicz pyranones to yield γ‐hydroxy‐δ‐lactones in a highly enantio‐ and diastereoselective fashion. In addition to the exploitation of this biocatalytic instrument as a stand‐alone tool, the formal hydroxyenone isomerase proved to be compatible with existing catalytic strategies to access the hydroxypyranone substrates and was successfully used in multicatalytic cascade reactions. Moreover, this novel enzymatic module could ultimately be encoded into a cellular system where the isomerization was successfully conducted in vivo.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We gratefully acknowledge support by the Suomen Akatemia (grant no 298250, JD), the Novo Nordisk Fonden (NNF17OC0025092, YCL), the COST action CA15106 (CHAOS), the Fonds der Chemischen Industrie (FCI; Liebig fellowship for C.M.), the Deutsche Forschungsgemeinschaft (DFG) through the Cluster of Excellence RESOLV (“Ruhr Explores SOLVation”, EXC 1069), and Carbolution Chemicals GmbH.
Y.-C. Liu, C. Merten, J. Deska, Angew. Chem. Int. Ed. 2018, 57, 12151.
Contributor Information
Dr. Yu‐Chang Liu, http://www.deskalab.com
Dr. Christian Merten, Email: christian.merten@ruhr-uni-bochum.de.
Prof. Dr. Jan Deska, Email: jan.deska@aalto.fi.
References
- 1. Watson A. J. A., Williams J. M. J., Science 2010, 329, 635–636. [DOI] [PubMed] [Google Scholar]
- 2.Recent reviews:
- 2a. Hamid M. H. S. A., Slatford P. A., Williams J. M. J., Adv. Synth. Catal. 2007, 349, 1555–1575; [Google Scholar]
- 2b. Guillena G., Ramón D. J., Yus M., Chem. Rev. 2010, 110, 1611–1641; [DOI] [PubMed] [Google Scholar]
- 2c. Dobereiner G. E., Crabtree R. H., Chem. Rev. 2010, 110, 681–703; [DOI] [PubMed] [Google Scholar]
- 2d. Ketcham J. M., Shin I., Montgomery T. P., Krische M. J., Angew. Chem. Int. Ed. 2014, 53, 9142–9150; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 9294–9302; [Google Scholar]
- 2e. Corma A., Navas J., Sabater M. J., Chem. Rev. 2018, 118, 1410–1459. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Ahlsten N., Lundberg H., Martín-Matute B., Green Chem. 2010, 12, 1628–1633; [Google Scholar]
- 3b. Slugovc C., Rüba E., Schmid R., Kirchner K., Organometallics 1999, 18, 4230–4233; [Google Scholar]
- 3c. García-Álvarez J., Gimeno J., Suárez F. J., Organometallics 2011, 30, 2893–2896. [Google Scholar]
- 4.
- 4a. Mantilli L., Gérard D., Torche S., Besnard C., Mazet C., Angew. Chem. Int. Ed. 2009, 48, 5143–5147; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 5245–5249; [Google Scholar]
- 4b. Li J.-Q., Peters B., Andersson P. G., Chem. Eur. J. 2011, 17, 11143–11145; [DOI] [PubMed] [Google Scholar]
- 4c. Li H., Mazet C., Acc. Chem. Res. 2016, 49, 1232–1241. [DOI] [PubMed] [Google Scholar]
- 5. Gargiulo S., Opperman D. J., Hanefeld U., Arends I. W. C. E., Hollmann F., Chem. Commun. 2012, 48, 6630–6632. [DOI] [PubMed] [Google Scholar]
- 6.For a nicotinamide-decoupled asymmetric redox isomerization, see: Martínez-Montero L., Gotor V., Gotor-Fernández V., Lavandera I., ACS Catal. 2018, 8, 2413–2419. [Google Scholar]
- 7.For nicotinamide-coupled “borrowing hydrogen” biocatalysis in reactions other than isomerizations, see:
- 7a. Wandrey C., Fiolitakis E., Wichmann U., Kula M.-R., Ann. N. Y. Acad. Sci. 1984, 434, 194–205; [DOI] [PubMed] [Google Scholar]
- 7b. Resch V., Fabian W. M. F., Kroutil W., Adv. Synth. Catal. 2010, 352, 993–997; [Google Scholar]
- 7c. Mutti F. G., Knaus T., Scrutton N. S., Breuer M., Turner N. J., Science 2015, 349, 1525–1529; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7d. Huang L., Romero E., Ressmann A. K., Rudroff F., Hollmann F., Fraaije M. W., Kara S., Adv. Synth. Catal. 2017, 359, 2142–2148. [Google Scholar]
- 8. Wang H.-Y., Yang K., Bennett S. R., Guo S.-R., Tang W., Angew. Chem. Int. Ed. 2015, 54, 8756–8759; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 8880–8883. [Google Scholar]
- 9. Achmatowicz O., Bukowski P., Szechner B., Zwierzchowska Z., Zamojski A., Tetrahedron 1971, 27, 1973–1996. [Google Scholar]
- 10.Recent reviews:
- 10a. Deska J., Thiel D., Gianolio E., Synthesis 2015, 47, 3435–3450; [Google Scholar]
- 10b. van der Pijl F., van Delft F. L., Rutjes F. J. P. T., Eur. J. Org. Chem. 2015, 4811–4829; [Google Scholar]
- 10c. Ghosh A. K., Brindisi M., RSC Adv. 2016, 6, 111564–111598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.
- 11a. Thiel D., Doknić D., Deska J., Nat. Commun. 2014, 5, 5278; [DOI] [PubMed] [Google Scholar]
- 11b. Thiel D., Blume F., Jäger C., Deska J., Eur. J. Org. Chem. 2018, 2717–2725; [Google Scholar]
- 11c. Blume F., Sprengart P., Deska J., Synlett 2018, 29, 1293–1296. [Google Scholar]
- 12.
- 12a. Manzuna Sapu C., Bäckvall J.-E., Deska J., Angew. Chem. Int. Ed. 2011, 50, 9731–9734; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 9905–9908; [Google Scholar]
- 12b. Hammel M., Deska J., Synthesis 2012, 44, 3789–3796; [Google Scholar]
- 12c. Skrobo B., Deska J., Org. Lett. 2013, 15, 5998–6001; [DOI] [PubMed] [Google Scholar]
- 12d. Naapuri J., Rolfes J. D., Keil J., Manzuna Sapu C., Deska J., Green Chem. 2017, 19, 447–452; [Google Scholar]
- 12e. Skrobo B., Schlörer N. E., Neudörfl J.-M., Deska J., Chem. Eur. J. 2018, 24, 3209–3217. [DOI] [PubMed] [Google Scholar]
- 13.
- 13a. Trost B. M., Kulawiec R. J., J. Am. Chem. Soc. 1993, 115, 2027–2036; [Google Scholar]
- 13b. Martín-Matute B., Bogár K., Edin M., Kaynak F. B., Bäckvall J.-E., Chem. Eur. J. 2005, 11, 5832–5842. [DOI] [PubMed] [Google Scholar]
- 14.
- 14a. Fujita K.-i., Tanino N., Yamaguchi R., Org. Lett. 2007, 9, 109–111; [DOI] [PubMed] [Google Scholar]
- 14b. Ngo A. H., Adams M. J., Do L. H., Organometallics 2014, 33, 6742–6745. [Google Scholar]
- 15. Mohr J. T., Moore J. T., Stoltz B. M., Beilstein J. Org. Chem. 2016, 12, 2038–2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Considering the close structural analogy, comparison of optical rotation values seems sufficient as the dominating chromophore (the α,β-unsaturated lactone) is identical for all structures and should therefore result in the same optical rotation signs. See also: Comprehensive Chiroptical Spectroscopy (Eds.: N. Berova, P. L. Polavarapu, K. Nakanishi, R. W. Woody), Wiley, Hoboken, 2013. [Google Scholar]
- 17. Kuhl N., Glorius F., Chem. Commun. 2011, 47, 573–575. [DOI] [PubMed] [Google Scholar]
- 18. Suzuki S., Murayama T., Shiono Y., Phytochemistry 2005, 66, 2329–2333. [DOI] [PubMed] [Google Scholar]
- 19. Favre-Godal Q., Dorsaz S., Queiroz E. F., Marcourt L., Ebrahimi S. N., Allard P.-M., Voinesco F., Hamburger M., Gupta M. P., Gindro K., Sanglard D., Wolfender J.-L., J. Nat. Prod. 2015, 78, 2994–3004. [DOI] [PubMed] [Google Scholar]
- 20.
- 20a. Turner N. J., O'Reilly E., Nat. Chem. Biol. 2013, 9, 285–288; [DOI] [PubMed] [Google Scholar]
- 20b. Green A. P., Turner N. J., Perspect. Sci. 2016, 9, 42–48. [Google Scholar]
- 21. Kim K. H., Shin Y. J., Choi S. U., Lee K. R., Bull. Korean Chem. Soc. 2011, 32, 2443–2445. [Google Scholar]
- 22. Nyandat E., Rwekiki E., Galeffi C., Palazzino G., Nicoletti M., Phytochemistry 1993, 33, 1493–1496. [Google Scholar]
- 23. Blume F., Liu Y.-C., Thiel D., Deska J., J. Mol. Catal. B Enzym. 2016, 134, 280–284. [Google Scholar]
- 24. Buchanan M. S., Hashimoto T., Takaoka S., Asakawa Y., Phytochemistry 1995, 40, 1251–1257. [Google Scholar]
- 25.
- 25a. Hollenbeak K. H., Kuehne M. E., Tetrahedron 1974, 30, 2307–2316 (osmundalin); [Google Scholar]
- 25b. Yu Y.-M., Yang J.-S., Peng C.-Z., Caer V., Cong P.-Z., Zou Z.-M., Lu Y., Yang S.-Y., Gu Y.-C., J. Nat. Prod. 2009, 72, 921–924 (angiopterlactones). [DOI] [PubMed] [Google Scholar]
- 26. Liu Y.-C., Guo C., Liu Y., Wang H.-B., Wu Z.-L., Org. Biomol. Chem. 2017, 15, 2562–2568. [DOI] [PubMed] [Google Scholar]
- 27. Yamamoto H., Kudoh M., Appl. Microbiol. Biotechnol. 2013, 97, 8087–8096. [DOI] [PubMed] [Google Scholar]
- 28. Weckbecker A., Hummel W., Biocatal. Biotransform. 2006, 24, 380–389. [Google Scholar]
- 29.Due to the heterogenicity of some of the commercial ADH samples (see also Figure S8), an involvement of other ADH-like impurities cannot be ruled out entirely in those cases. Yet, since the redox isomerization itself is achieved by a combination of the two native ADH functions, it seems well feasible that the assigned main proteins in these mixtures are capable of carrying the reaction forward.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary