

Abstract
The global configuration of individual, surface-adsorbed molecules of the giant muscle protein titin, labeled with rhodamine conjugates, was followed with confocal microscopy. Fluorescence-emission intensity was reduced because of self-quenching caused by the close spacing between rhodamine dye molecules that formed dimers. In the presence of chemical denaturants, fluorescence intensity increased, reversibly, up to 5-fold in a fast reaction; the kinetics were followed at the single-molecule level. We show that dimers formed in a concentrated rhodamine solution dissociate when exposed to chemical denaturants. Furthermore, titin denaturation, followed by means of tryptophan fluorescence, is dominated by a slow reaction. Therefore, the rapid fluorescence change of the single molecules reflects the direct action of the denaturants on rhodamine dimers rather than the unfolding/refolding of the protein. Upon acidic denaturation, which we have shown not to dissociate rhodamine dimers, fluorescence intensity change was minimal, suggesting that dimers persist because the unfolded molecule has contracted into a small volume. The highly contractile nature of the acid-unfolded protein molecule derives from a significant increase in chain flexibility. We discuss the potential implications this finding could have for the passive mechanical behavior of striated muscle.
Titins are 3.0–3.7 MDa filamentous proteins extending between the Z- and M-lines of the sarcomere (1–4). Titin is a tandem array of Ig type C2 and fibronectin (FN) type III domains (≈300 per molecule) interspersed with unique sequences, most notably the Pro-, Glu-, Val-, and Lys-rich PEVK segment, and the N2A and N2B segments (5). The I-band segment of titin acts as a molecular spring, whose elastic properties define the passive or restoring mechanical properties of striated muscle (6–9). By contrast, titin's A-band segment is thought to function as a scaffold that defines structural regularity within the A band (10). Recent single-molecule experiments often have used titin as a model system because of a large size that makes it relatively easily accessible to such investigations. Single-molecule-mechanics experiments using either optical tweezers (11–13) or atomic force microscopy (AFM; ref. 14) described titin as an entropic chain in which domain unfolding occurs at high forces and refolding occurs at low forces. These experiments formed the basis of many theoretical and modeling works that addressed the mechanisms of mechanically driven protein folding (15–20). Individual titin molecules can be made visible under aqueous conditions after labeling with fluorophores. Fluorescently labeled titin molecules have recently been studied under various conditions: equilibration to a surface (21–23), stretching with meniscus force (22, 23) and direct manipulation (24), and chemical denaturation (25, 26). Chemical denaturation has been shown to cause an abrupt and significant rise in the fluorescence intensity of Oregon Green 488 (Molecular Probes) maleimide-labeled titin molecules, which has been interpreted to derive from abolishing the fluorescence self-quenching present in the native state of the protein (26). An understanding of the exact mechanisms of fluorescence self-quenching and its abolition during protein unfolding, however, have remained elusive.
Here, we followed the global configuration of fluorescently labeled individual titin molecules during chemically driven unfolding. The configuration of titin is complex because, on the one hand, it is determined by the conformation of the local protein matrix (β-barrel structure of the Ig and FN domains), and, on the other hand, it is influenced by the overall shape of the chain of tandemly arranged domains, which behaves as an entropic, random coil (27). Thus, interactions between fluorophores bound along the primary structure of titin are affected by the native protein structure, the shape of the chain, and by fluctuations in both of these structural parameters.
Titin prepared from rabbit back muscle (28, 29) was labeled overnight on ice in the presence of ≈1650× molar excess of tetramethylrhodamine-5-(and 6)-iodoacetamide (TMRIA) in AB buffer (25 mM imidazole-HCl, pH 8/0.2 M KCl/4 mM MgCl2/1 mM EGTA/0.01% NaN3/1 mM DTT/0.2% Tween 20/40 μg/ml leupeptin/20 μM E-64). Protein and dye aggregates and the unbound dye fraction were removed by chromatography (Sepharose CL-2B, 0.8 × 120 cm, equilibrated with AB buffer). According to electrophoretic analysis (data not shown), the titin sample contained a mixture of T1 (mother molecule) and T2 (proteolytic fragment lacking a 1,200-kDa segment from the Z-line end; ref. 30). The labeling ratio, determined as TMRIA concentration per titin concentration, varied between 80–140 TMRIA molecules per titin molecule. Titin concentration was determined by measuring OD280 (ɛ280 = 1.43 ml/mg) and by protein microassay (31) using BSA as standard. TMRIA concentration was determined by measuring OD555 in the presence of 6 M guanidine hydrochloride (GdnHCl) to diminish the absorption spectral effects of self-quenching (see below). ɛ555 = 59,000 cm−1M−1 was used, as determined by us for TMRIA dissolved in water.
Fluorescently labeled titin molecules, at a concentration of 0.2–0.8 μg/ml, were allowed to bind to a glass microscope coverslip surface for 1 min; the surface then was washed with AB buffer containing 1.5% (vol/vol) 2-mercaptoethanol to reduce photobleaching. The sample was examined with laser scanning confocal microscopy (Bio-Rad) using 120–360 μW optical power, 514 nm excitation wavelength (λex), and a 550-nm longpass filter for detection of emission. Single titin molecules appeared as bright spots with a mean diameter (d) of 0.3 μm (Fig. 1a Upper), which is comparable to the resolution limit of the microscope used here. This result suggests that the dimensions of the surface area occupied by a molecule are less than or equal to the diffraction limit (21). For rapid exchange of solution, we used a flow-through microchamber. The chamber was a sandwich of a coverslip, a 100-μm-thick Parafilm spacer (American National Can, Neenah, WI) and a thick glass plate with two tubes inserted for inward and outward flow. Considering that the flow was laminar (Reynolds number = ≈10−3) at a mean rate (v) of 10−3 m/s, and assuming nonsignificant flow retardation at the chamber surface, the transition time (tt) of a single molecule from one solution to the other can be expressed as tt = d/v, which was ≈0.3 ms. The fluorescence intensity of the molecules was quantitated by several methods: (i) mean grayscale intensity of each particle, (ii) peak value of the Gaussian function fitted onto the spatial intensity profile (21), and (iii) particle area after binary transition using a given grayscale threshold value. Upon the addition of GdnHCl, the fluorescence intensity of the molecules increased abruptly and significantly, and the number of fluorescent particles visible increased (Fig. 1a Lower), similarly to the observations of Zhuang et al. (26). When GdnHCl was injected in the microchamber in steps of increasing concentration, the fluorescence of the same titin molecule increased in a stepwise fashion; at 7 M GdnHCl, its intensity was about four times that observed in the absence of the denaturant (Fig. 1b). The mean fluorescence intensity of an ensemble of many molecules in several different microscopic fields of view increased as a function of GdnHCl concentration. A concentration of 8 M GdnHCl caused an approximately 5-fold increase in the mean fluorescence intensity (Fig. 1c). Notably, a large variation was observed around the mean intensity, which is probably caused by variations in the response of the individual titin molecules to the denaturant. The kinetics of fluorescence change was measured by following the intensity of individual molecules as a function of time during the addition and removal of 5.5 M GdnHCl (Fig. 1d). The fluorescence increase in the presence of GdnHCl was reversible: after the removal of the denaturant, the fluorescence intensity returned to the initial level. The time-dependent intensity changes were fitted with single-exponential functions that gave mean time constants of 0.51 s (±0.09 SEM, n = 13) and 0.73 s (±0.12 SEM, n = 13) for the fluorescence rise and decay transitions, respectively. For comparison, these times are an order of magnitude longer than those recorded by Zhuang et al. (26) for single titin molecules labeled with Oregon Green 488. Thus, in native titin molecules, the fluorescence of chain-associated rhodamine molecules is reduced, most likely by quenching caused by the proximity of the dye molecules. This interaction, called self-quenching (26), then becomes reduced upon chemical denaturation.
Figure 1.

(a) Confocal microscopic image of surface-adsorbed, fluorescently labeled titin molecules before (Upper) and after (Lower) the addition of 5.5 M GdnHCl. (b) Effect of a stepwise increase of GdnHCl concentration on the fluorescence intensity of a single titin molecule. (c) Mean fluorescence intensity of an ensemble of surface-adsorbed titin molecules as a function of GdnHCl concentration. Data points represent mean ± SEM for an average 261 molecules per field of view. (d) Kinetics of fluorescence increase and decrease of a single titin molecule upon the addition (Left) and removal (Right), respectively, of 5.5 M GdnHCl. Fluorescence intensities were normalized to the intensity at 0 M GdnHCl.
The molecular mechanisms behind self-quenching and its reduction in the presence of denaturant were explored by fluorescence and absorption spectroscopy of TMRIA-labeled titin in solution. The intensity in both the excitation and emission spectra increased significantly in the presence of GdnHCl (Fig. 2a). The emission intensity increased 4-fold (λem = 580 nm) in the presence of 5.4 M GdnHCl. In the absorption spectra, a significant peak at 518 nm—absent from the excitation spectra—appeared in addition to the one at 555 nm (Fig. 2b). Whereas the 555-nm peak corresponds to rhodamine monomers, the peak at 518 nm is caused by the presence of nonfluorescent rhodamine dimers (32–35). In the presence of increasing concentrations of GdnHCl, the dimer peak became reduced, whereas the monomer peak increased. Thus, self-quenching in rhodamine-labeled native titin molecules is caused by dimer formation. In the presence of increasing concentrations of GdnHCl, an increasing fraction of the nonfluorescent rhodamine dimer population is converted to monomers, thereby increasing the fluorescence intensity. This shift between the rhodamine dimer and monomer populations is indicated by the progressive change in the OD518/OD555 ratio (Fig. 2b Inset), which corresponds to the dimer/monomer ratio (36). For TMRIA, an OD518/OD555 range of 0.4–1.3 has been reported (36), with 1.3 and 0.4 corresponding to the purely dimeric and monomeric forms, respectively. Considering that we found an OD518/OD555 of 1.2 for native titin, the majority of the chain-associated rhodamine molecules are in the form of dimers. The OD518/OD555 value of 0.6 in the denatured state indicates that a fraction of rhodamine persists in the dimeric state, probably because it is unaffected by the action of the denaturant. Because the probability of dimer formation increases with rhodamine concentration (32, 35, 37), the presence of a large dimer population suggests that titin has collected the associated fluorophores into a small volume. As a result, the dye molecules are in close proximity, allowing self-quenching.
Figure 2.

Effect of GdnHCl on the spectral characteristics of TMRIA-labeled titin. Fluorescence excitation and emission (a) and absorption (b) spectra of TMRIA-labeled titin as a function of increasing GdnHCl concentration. (λex = 550; λem = 580 nm.) (b Inset) OD518/OD555 ratio, obtained from the absorption spectra of TMRIA-labeled titin, as a function of GdnHCl concentration.
To determine whether the fluorescence increase upon chemical denaturation is related to the conformational changes associated with the unfolding of titin or to a direct action of the denaturant on the fluorophores, we added increasing concentrations of GdnHCl to rhodamine dimers formed in a highly concentrated aqueous rhodamine B solution (Fig. 3). We used 5 mM rhodamine B, in which the rhodamine population is dominated by dimers, and measured the absorption spectra by using a home-built cuvette with an ≈100-μm path length. The addition of increasing concentrations of GdnHCl resulted in a progressive transition of the dimeric rhodamine population to the monomeric state. At 4.5 M GdnHCl, nearly all of the rhodamine were in the monomeric form. We found that denaturation by other methods (urea, SDS) also resulted in the conversion of dimers to monomers (data not shown). The dissociation of rhodamine dimers by urea has been observed before (38). Conceivably, chemical denaturants disrupt the weak interactions that hold the rhodamine dimer together. Such interactions, however, are unlikely to significantly contribute to stabilizing the three-dimensional structure of the titin molecule, as the unfolding force spectrum of TMRIA-labeled titin, recorded using AFM, was similar to that of the unlabeled molecule (M.S.Z. & H. L. Granzier, unpublished data). The AFM experiments did not reveal distinct rhodamine-dimer rupture events, indicating that the dimer bond energies are much smaller than the free energy of titin domain unfolding (17–28 kBT; refs. 16, 39). Thus, we assume that the dimer bonds do not significantly alter the conformation of titin. It also is unlikely that the rhodamine dimers in titin are formed by the binding of free rhodamine molecules to molecules that are attached to titin. If that were the case, the free rhodamine molecules would be washed away upon the addition of the denaturant, and fluorescence intensity would not reduce to the original level after denaturant removal. Our findings suggest that the abrupt and significant increase in the fluorescence intensity of single, rhodamine-labeled titin molecules upon GdnHCl treatment (Fig. 1) is due to, or dominated by, a direct effect of the denaturant on rhodamine dimers.
Figure 3.

(a) Absorption spectra of 5 mM aqueous rhodamine B solution as a function of GdnHCl concentration. The spectra were normalized to OD555. (a Inset) OD518/OD555 ratio, obtained from the absorption spectra of 5 mM rhodamine B, as a function of GdnHCl concentration. (b) Intrinsic tryptophan fluorescence-emission spectra of titin before (0 M GdnHCl) and after the addition of 4.5 M GdnHCl. (λex = 295 nm.) (b Inset) Kinetics of fluorescence decay coupled to titin denaturation. Buffer: 30 mM potassium phosphate, pH 7.0/0.6 M KCl. Arrow marks the addition of GdnHCl to a final concentration of 4.5 M. (λem = 340 nm.)
To test whether the kinetics of TMRIA fluorescence increase is comparable to the kinetics of titin unfolding, we monitored the intrinsic tryptophan fluorescence as a function of time after the addition of 4.5 M GdnHCl (39). Denaturation resulted in a 25% decrease in emission intensity at 340 nm. The steady-state fluorescence-emission spectra (Fig. 3b) display a decrease in fluorescence intensity and a 16-nm red shift, suggesting that upon denaturation, tryptophan fluorescence is quenched by exposure to the polar solvent. The tryptophan fluorescence decay data (Fig. 3b Inset) were best-fitted with a double-exponential function using time constants of 26.91 s (±0.71 SEM) and 234.37 s (±4.23 SEM), respectively. Both of these processes are significantly slower than the denaturant-induced increase in the fluorescence of Oregon Green 488 maleimide-labeled (26) or TMRIA-labeled titin (Fig. 1d). Although the unfolding of a single-Ig-domain protein has been reported to occur rapidly (40), the complexity of titin's global structure probably places constraints on the rate of complete denaturation. It is not inconceivable that the Oregon Green 488 fluorescence measured by Zhuang et al. (26) monitors a fast-unfolding intermediate related to that seen in single-Ig-domain proteins (40). However, the rapid kinetics of TMRIA fluorescence change seen here is most likely related to the denaturant-induced dimer dissociation reaction rather than the unfolding of titin.
Besides dissociating rhodamine dimers, the addition of GdnHCl results in the eventual unfolding of titin. Indeed, we have previously found that single, mechanically manipulated titin molecules become completely unfolded within 5 min in 4 M GdnHCl (41). To examine whether the changes in TMRIA fluorescence reflect structural changes associated with the unfolding of titin, the protein was denatured with a method that does not directly influence rhodamine dimers. We found that lowering pH with HCl did not result in a significant dissociation of rhodamine dimers (Fig. 4a) and a change in the spectral properties of TMRIA (Fig. 4a Inset). Therefore, acidic denaturation was used to unfold titin. Upon lowering the pH to 1.9 the intrinsic tryptophan fluorescence decreased by 16% in a rapid reaction that was complete within the time of manual mixing, suggesting that acid-induced titin denaturation may proceed via local kinetic intermediates that are different from those during chemical denaturation. To measure the ratio of rhodamine dimers and monomers in acid-denatured titin, the pH was lowered by adding HCl step by step, and the absorption spectrum, at equilibrium, was recorded at each stage (Fig. 4b). The OD518/OD555 value decreased moderately, from 1.10 at pH 6.8 to 1.02 at pH 2.5 (Fig. 4b Inset), indicating that the titin-bound rhodamine dimer population remained nearly unchanged. The effect of acidic denaturation was tested on single, surface-adsorbed titin molecules as well (Fig. 4c). Lowering pH from 7.7 (Fig. 4c Upper) to 2.0 (Lower) failed to cause an increase in TMRIA fluorescence intensity. The mean fluorescence intensity of the acid-denatured titin molecules did not change significantly during a time period of 30 min, but their fluorescence increased as soon as GdnHCl was added (Fig. 4d). Thus, although the GdnHCl-induced fluorescence increase is dominated by a direct action on the rhodamine dimers, the unfolding of TMRIA-labeled titin, induced by acidic denaturation, is not reflected in a significant change of fluorescence-emission intensity.
Figure 4.

(a) Effect of HCl (solid line, pH 7.7; dashed line, pH 2.0) on the absorption spectrum of 5 mM aqueous rhodamine B solution. (Inset) Fluorescence-emission spectra of 40 μM TMRIA solution at pH 7.0 and 1.0. (λex = 550 nm.) (b) Absorption spectrum of TMRIA-labeled titin as a function of pH. (b Inset) OD518/OD555 ratio, obtained from the absorption spectra of TMRIA-labeled titin, as a function of pH. (c) Confocal microscopic image of surface-adsorbed, fluorescently labeled titin molecules before (pH 7.7, Upper) and after (Lower) the addition of a pH 2.0 buffer solution. (d) Mean fluorescence intensity obtained for an ensemble of surface-adsorbed titin molecules as a function of time after acidic denaturation (at HCl, pH 2.0) and the addition of 5.5 M GdnHCl (at GdnHCl). The pH of the GdnHCl solution was 7.7. Data points represent mean ± SEM for an average of 187 molecules per field of view. Data were normalized to the average of the first five points that represent control settings.
Our results indicate that the fluorescence of TMRIA-labeled titin molecules is quenched because of the formation of dimers of rhodamine, and the unfolding of titin does not result in a significant dissociation of the chain-associated dimers. The increase in fluorescence intensity observed during chemical denaturation (with GdnHCl, urea, and SDS) is the result of direct interaction between the denaturant and the rhodamine dimers rather than a marker of protein unfolding. The mechanisms of concentration quenching in rhodamine dimers are not precisely known; collisional quenching, dark-complex formation, and energy transfer have been implicated (35). Dimer formation is distance-dependent (42); therefore, dimers appear only if the spatial proximity between rhodamine monomers is sufficiently high, as in a concentrated solution. By measuring the ratio of dimers and monomers, the average spatial proximity (which is analogous to the concentration) of the dye molecules may be estimated. Thus, considering that titin is studded with TMRIA whose ensemble behavior is monitored, we may derive information about titin's global structure if we make the simplifying assumptions that (i) the fluorophores are more or less evenly distributed along the titin chain, and (ii) they contribute equally to dimer formation. In native titin, the spatial arrangement of the rhodamine molecules is determined by the domain structure. The dimers are formed by the interaction of rhodamine molecules that are either on the same domain or on closely spaced cysteines of neighboring domains, because the distance between point dipoles in the dimer (R ≈ 0.6 nm; ref. 42) is smaller than the average interdomain spacing of Ig or FN domains (≈4.6 nm; ref. 43). In the completely unfolded titin, the native three-dimensional structure is collapsed. In the first approximation, titin is a linear chain with a nearly constant frequency of 13 ± 3 cysteines per 1000 residues along its contour, except in the PEVK segment that lacks cysteines (5). Considering the labeling ratio obtained in this work and assuming an equal accessibility of the cysteines to TMRIA, approximately every fourth cysteine residue was labeled. If the segments of the unfolded titin chain are allowed to diffuse and interact with each other within the space allotted, or if they can freely rearrange during denaturation, then the ratio of dimers and monomers describes the average concentration of rhodamine in the volume occupied by a single titin molecule. The volume of the single titin molecule then can be calculated as V = n/MA, where n is the average number of TMRIA molecules on titin, M is the molar concentration of TMRIA (mol/liter), and A is Avogadro's number (6.023 × 1023). According to TMRIA concentration calibration (data not shown, and ref. 44), the dye concentration in the volume occupied by a single, acid-denatured titin molecule is in the order of mM. Thus, at 136 TMRIA molecules per titin and an assumed TMRIA concentration of 1 mM, the mean volume of a single, acid-denatured titin molecule is 0.23 × 10−3 μm3, which gives a mean radius of 0.054 μm for a spherical particle. For comparison, the equilibrium dimensions of a denatured titin molecule may be calculated from its statistical chain parameters. The mean-square end-to-end distance is obtained from the contour (L) and persistence (P) lengths as follows (45):
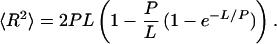 |
For a contour length of 10 μm (5) and a persistence length of 1.6 nm (46), the mean-square end-to-end distance of the unfolded titin molecule is ≈3 × 10−2 μm2, which gives a radius of gyration (47) of ≈0.07 μm, calculated by
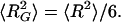 |
Although the radii obtained with the two different methods are in fair agreement, the discrepancy between the results may be explained by the following factors: (i) underestimation of the labeling ratio; (ii) axial inhomogeneities in the labeling ratio; (iii) uncertainty in determining the local rhodamine concentration; and (iv) the highly compact nature of the acid-denatured titin molecule. (i) The labeling ratio is obtained by measuring the OD555 of TMRIA-labeled titin in 6 M GdnHCl so that as much of the rhodamine dimer population as possible is converted to monomers. However, as the OD518/OD555 values indicate, a small dimer population persists even in the presence of the denaturant, which results in an underestimation of the number of TMRIA molecules bound along titin. (ii) It may be possible that the TMRIA molecules are not distributed evenly along the primary structure of titin but are located in clusters. In such a scenario, the local rhodamine concentration is overestimated, yielding an underestimated molecular volume. (iii) The local rhodamine concentration is obtained by comparing the OD518/OD555 ratio with that obtained for a series of different concentrations of rhodamine B and TMRIA. Besides the rhodamine concentration, the OD518/OD555 ratio is affected by the nature of the solvent, the local hydrophobicity (34), and by the ratio of 5- and 6-TMRIA isoforms that have different dimerization-binding constants (44). Overestimation of the local rhodamine concentration will yield an underestimated molecular volume. (iv) Finally, acid-denatured titin may assume a conformation that is more compact than the mechanically unfolded molecule. To calculate the molecular dimensions, we used a persistence length of 1.6 nm, which was obtained from the complete mechanical unfolding of single titin molecules (46). It is conceivable that the acid-denatured titin chain has a shorter persistence length, allowing the molecule to contract into a compact state and, thereby, into a small volume. Considering the relatively high ionic strength (0.2 M), it also may be possible that acid-denatured titin adopts a molten-globule configuration with a small relative volume (48). Predictions of chain statistics and our measurements on TMRIA-labeled titin suggest that the volumes occupied by native and unfolded titin are not significantly different. In support of this notion, we found that the light scattering (at λ = 400 nm) by titin was not increased upon denaturation (data not shown). Mean molecular volume is applicable only to titin in solution. When deposited onto a surface, titin occupies two-dimensional space in a configuration determined by its contour and persistence lengths, similarly to other flexible chain molecules such as DNA (45), and its global configuration becomes stabilized by interactions with the surface at several points along its contour. The lack of a fluorescence-intensity increase during acidic denaturation (Fig. 4 c and d) indicates that the apparent rhodamine dimer concentration, and, therefore, the dye-molecule proximity, is not reduced significantly in such surface-adsorbed titin molecules. Although the constraints placed on the unfolding reaction by surface adsorption remain to be established, it is possible that the high rhodamine dimer concentration is maintained either by kinetic traps resembling the native state or by the freedom for structural rearrangement and formation of highly compact structures between the surface attachment points.
The similarity of rhodamine dimer concentration in native and unfolded titin indicates that the spatial proximity of the dye molecules in these states are similar. In the native state, the spatial proximity is maintained by the three-dimensional structure of the protein matrix. In the unfolded state, by contrast, the forces holding the three-dimensional native structure together are absent. Therefore, the contraction of the 10-μm long (5) unfolded titin chain, without the aid of intramolecular interactions into a volume similar to that occupied by the native molecule, must involve a significantly increased chain flexibility (or reduced bending rigidity). Thus, unfolded titin has a shorter persistence length than the native molecule. Previous experiments using dynamic light scattering (27) and single-molecule mechanics (11, 13, 14) have indicated that the persistence length of the unfolded titin chain is at least 10× shorter than that of the native chain. The high flexibility of unfolded domains or quasi-unfolded protein elements (such as the PEVK; ref. 43) may have important implications for the passive mechanical behavior of striated muscle. The forces required to extend a flexible polymer chain are greater than those required to extend a rigid one. Whereas physiologically occurring, repeated folding–unfolding transitions in titin are debated (43, 49), it is conceivable that unfolding of some of titin's globular domains does take place during pathological overextension of the sarcomere. The increased flexibility of the denatured domain, along with the contour-length gain, may provide a safety mechanism that prevents further denaturation along the molecule and facilitates domain refolding by contraction into a small volume, once the molecule is allowed to relax.
In conclusion, we have shown that the global configuration of titin at the single molecule level may be examined by following the fluorescence of chain-associated rhodamine molecules. The fluorescence of TMRIA-labeled titin is quenched because of the formation of nonfluorescent rhodamine dimers. In the unfolded state of titin, the rhodamine dimers prevail, suggesting that the molecule has contracted, by means of a significantly increased chain flexibility, into a small volume that allows the formation of concentration-dependent dimers. Studying chain-associated rhodamine dimers may be particularly useful in the combination of the fluorescence microscopic method with single-molecule manipulation techniques. In such an arrangement, the rhodamine dimers are expected to dissociate upon domain unfolding (resulting in a fluorescence increase) because the molecule is extended and the dye molecules become axially diluted. Then, by following local fluctuations in fluorescence intensity, the dynamics of mechanically driven protein unfolding may be spatially resolved.
Acknowledgments
This work was supported by Hungarian Science Foundation Grants OTKA F025353, T032700, and T023209, Hungarian Ministry of Education Grant FKFP 0463/99, European Union Grant HPRN CT 2000 00091, and the Hungarian Academy of Sciences. M.S.Z.K. is a Howard Hughes Medical Institute International Research Scholar and is recipient of the Bolyai János Fellowship of the Hungarian Academy of Sciences.
Abbreviations
- TMRIA
tetramethylrhodamine-5-(and 6)-iodoacetamide
- GdnHCl
guanidine hydrochloride
References
- 1.Freiburg A, Trombitas K, Hell W, Cazorla O, Fougerousse F, Centner T, Kolmerer B, Witt C, Beckmann J S, Gregorio C C, et al. Circ Res. 2000;86:1114–1121. doi: 10.1161/01.res.86.11.1114. [DOI] [PubMed] [Google Scholar]
- 2.Maruyama K. FASEB J. 1997;11:341–345. doi: 10.1096/fasebj.11.5.9141500. [DOI] [PubMed] [Google Scholar]
- 3.Trinick J, Tskhovrebova L. Trends Cell Biol. 1999;9:377–380. doi: 10.1016/s0962-8924(99)01641-4. [DOI] [PubMed] [Google Scholar]
- 4.Wang K. Adv Biophys. 1996;33:123–134. [PubMed] [Google Scholar]
- 5.Labeit S, Kolmerer B. Science. 1995;270:293–296. doi: 10.1126/science.270.5234.293. [DOI] [PubMed] [Google Scholar]
- 6.Horowits R, Kempner E S, Bisher M E, Podolsky R J. Nature (London) 1986;323:160–164. doi: 10.1038/323160a0. [DOI] [PubMed] [Google Scholar]
- 7.Trombitás K, Jin J-P, Granzier H L. Circ Res. 1995;77:856–861. doi: 10.1161/01.res.77.4.856. [DOI] [PubMed] [Google Scholar]
- 8.Granzier H L M, Helmes M, Trombitás K. Biophys J. 1996;70:430–442. doi: 10.1016/S0006-3495(96)79586-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Linke W A, Ivemeyer M, Olivieri N, Kolmerer B, Ruegg J C, Labeit S. J Mol Biol. 1996;261:62–71. doi: 10.1006/jmbi.1996.0441. [DOI] [PubMed] [Google Scholar]
- 10.Trinick J. Curr Biol. 1996;6:258–260. doi: 10.1016/s0960-9822(02)00472-4. [DOI] [PubMed] [Google Scholar]
- 11.Kellermayer M S Z, Smith S B, Granzier H L, Bustamante C. Science. 1997;276:1112–1116. doi: 10.1126/science.276.5315.1112. [DOI] [PubMed] [Google Scholar]
- 12.Kellermayer M S Z, Smith S B, Bustamante C, Granzier H L. J Struct Biol. 1998;122:197–205. doi: 10.1006/jsbi.1998.3988. [DOI] [PubMed] [Google Scholar]
- 13.Tskhovrebova L, Trinick J, Sleep J A, Simmons R M. Nature (London) 1997;387:308–312. doi: 10.1038/387308a0. [DOI] [PubMed] [Google Scholar]
- 14.Rief M, Gautel M, Oesterhelt F, Fernandez J M, Gaub H E. Science. 1997;276:1109–1112. doi: 10.1126/science.276.5315.1109. [DOI] [PubMed] [Google Scholar]
- 15.Lu H, Isralewitz B, Krammer A, Vogel V, Schulten K. Biophys J. 1998;75:662–671. doi: 10.1016/S0006-3495(98)77556-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rief M, Fernandez J M, Gaub H E. Phys Rev Lett. 1998;81:4764–4767. [Google Scholar]
- 17.Paci E, Karplus M. J Mol Biol. 1999;288:441–459. doi: 10.1006/jmbi.1999.2670. [DOI] [PubMed] [Google Scholar]
- 18.Zhang B, Xu G, Evans J S. Biophys J. 1999;77:1306–1315. doi: 10.1016/S0006-3495(99)76980-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Klimov D K, Thirumalai D. Proc Natl Acad Sci USA. 1999;96:6166–6170. doi: 10.1073/pnas.96.11.6166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Leckband D. Annu Rev Biophys Biomol Struct. 2000;29:1–26. doi: 10.1146/annurev.biophys.29.1.1. [DOI] [PubMed] [Google Scholar]
- 21.Grama L, Somogyi B, Kellermayer M S Z. Single Mol. 2001;2:239–248. doi: 10.1073/pnas.191494098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tskhovrebova L, Trinick J. Biophys J. 1999;76:A9. [Google Scholar]
- 23.Tskhovrebova L, Trinick J. Adv Exp Med Biol. 2000;481:163–173. doi: 10.1007/978-1-4615-4267-4_10. [DOI] [PubMed] [Google Scholar]
- 24.Kellermayer M S Z, Grama L, Somogyi B. Biophys J. 2000;78:A392. [Google Scholar]
- 25.Zhuang X, Ha T, Kim H D, Chu S, Labeit S. Biophys J. 1999;76:A9. [Google Scholar]
- 26.Zhuang X, Ha T, Kim H D, Centner T, Labeit S, Chu S. Proc Natl Acad Sci USA. 2000;97:14241–14244. doi: 10.1073/pnas.97.26.14241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Higuchi H, Nakauchi Y, Maruyama K, Fujime S. Biophys J. 1993;65:1906–1915. doi: 10.1016/S0006-3495(93)81261-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Soteriou A, Gamage M, Trinick J. J Cell Sci. 1993;104:119–123. doi: 10.1242/jcs.104.1.119. [DOI] [PubMed] [Google Scholar]
- 29.Kellermayer M S Z, Granzier H L M. FEBS Lett. 1996;380:281–286. doi: 10.1016/0014-5793(96)00055-5. [DOI] [PubMed] [Google Scholar]
- 30.Wang K, Ramirez M R, Palter D. Proc Natl Acad Sci USA. 1984;81:3685–3689. doi: 10.1073/pnas.81.12.3685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bradford M M. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 32.Levshin L V, Bocharov V G. Op Spectrosc. 1961;10:330–333. [Google Scholar]
- 33.Rohatgi K K, Singhal G S. J Phys Chem. 1966;70:1695–1701. [Google Scholar]
- 34.Selwyn J E, Steinfeld J I. J Phys Chem. 1972;76:762–774. [Google Scholar]
- 35.Plant A L. Photochem Photobiol. 1986;44:453–459. doi: 10.1111/j.1751-1097.1986.tb04692.x. [DOI] [PubMed] [Google Scholar]
- 36.Hamman B D, Oleinikov A V, Jokhadze G G, Bochkariov D E, Traut R R, Jameson D M. J Biol Chem. 1996;271:7568–7573. doi: 10.1074/jbc.271.13.7568. [DOI] [PubMed] [Google Scholar]
- 37.Baranova E G. Op Spectrosc. 1965;18:230–234. [Google Scholar]
- 38.Rohatgi K K, Singhal G S. J Phys Chem. 1963;67:2844–2846. [Google Scholar]
- 39.Soteriou A, Clarke A, Martin S, Trinick J. Proc R Soc London Ser B. 1993;254:83–86. doi: 10.1098/rspb.1993.0130. [DOI] [PubMed] [Google Scholar]
- 40.Goto Y, Hamaguchi K. J Mol Biol. 1982;156:911–926. doi: 10.1016/0022-2836(82)90147-4. [DOI] [PubMed] [Google Scholar]
- 41.Kellermayer M S Z, Smith S B, Bustamante C, Granzier H L. Biophys J. 2001;80:852–863. doi: 10.1016/S0006-3495(01)76064-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Packard B Z, Toptygin D D, Komoriya A, Brand L. Proc Natl Acad Sci USA. 1996;93:11640–11645. doi: 10.1073/pnas.93.21.11640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Trombitás K, Greaser M, Labeit S, Jin J-P, Kellermayer M, Helmes M, Granzier H. J Cell Biol. 1998;140:1–7. doi: 10.1083/jcb.140.4.853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Burghardt T P, Lyke J E, Ajtai K. Biophys Chem. 1996;59:119–131. doi: 10.1016/0301-4622(95)00118-2. [DOI] [PubMed] [Google Scholar]
- 45.Rivetti C, Guthold M, Bustamante C. J Mol Biol. 1996;264:919–932. doi: 10.1006/jmbi.1996.0687. [DOI] [PubMed] [Google Scholar]
- 46.Kellermayer M S Z, Smith S, Bustamante C, Granzier H L. Adv Exp Med Biol. 2000;481:111–126. doi: 10.1007/978-1-4615-4267-4_7. [DOI] [PubMed] [Google Scholar]
- 47.Cantor C, Schimmel P R. Biophysical Chemistry III. The Behavior of Biological Macromolecules. San Francisco: Freeman; 1980. [Google Scholar]
- 48.Fink A L, Calciano L J, Goto Y, Kurotsu T, Palleros D. Biochemistry. 1994;33:12504–12511. doi: 10.1021/bi00207a018. [DOI] [PubMed] [Google Scholar]
- 49.Helmes M, Trombitás K, Centner T, Kellermayer M S Z, Labeit S, Linke W A, Granzier H. Circ Res. 1999;84:1339–1352. doi: 10.1161/01.res.84.11.1339. [DOI] [PubMed] [Google Scholar]