

Abstract
Two symmetrically positioned redox active tyrosine residues are present in the photosystem II (PSII) reaction center. One of them, TyrZ, is oxidized in the ns–μs time scale by P680+ and reduced rapidly (μs to ms) by electrons from the Mn complex. The other one, TyrD, is stable in its oxidized form and seems to play no direct role in enzyme function. Here, we have studied electron donation from these tyrosines to the chlorophyll cation (P680+) in Mn-depleted PSII from plants and cyanobacteria. In particular, a mutant lacking TyrZ was used to investigate electron donation from TyrD. By using EPR and time-resolved absorption spectroscopy, we show that reduced TyrD is capable of donating an electron to P680+ with t1/2 ≈ 190 ns at pH 8.5 in approximately half of the centers. This rate is ≈105 times faster than was previously thought and similar to the TyrZ donation rate in Mn-depleted wild-type PSII (pH 8.5). Some earlier arguments put forward to rationalize the supposedly slow electron donation from TyrD (compared with that from TyrZ) can be reassessed. At pH 6.5, TyrZ (t1/2 = 2–10 μs) donates much faster to P680+ than does TyrD (t1/2 > 150 μs). These different rates may reflect the different fates of the proton released from the respective tyrosines upon oxidation. The rapid rate of electron donation from TyrD requires at least partial localization of P680+ on the chlorophyll (PD2) that is located on the D2 side of the reaction center.
Tyrosyl radicals play key roles in the mechanisms of a wide range of enzymes. Understanding these roles and how proteins are able to control these reactive species to carry out quite specific chemical reactions has been an important aim of researchers in this area (for review, see ref. 1). One of the earliest demonstrations of redox active tyrosines was in photosystem II (PSII), the water oxidizing enzyme, in which a tyrosyl radical, designated tyrosine Z⋅ (TyrZ⋅), is thought to play a key role in the active site, abstracting electrons and possibly protons from the substrate water that is bound to the highly oxidized Mn cluster (2–5).
PSII contains a second tyrosyl radical, TyrD⋅, which is stable during enzyme function and which is located in a 2-fold rotationally symmetrical position to TyrZ on a subunit (D2) adjacent to that (D1) in which water oxidation takes place. TyrZ and TyrD are equally situated relative to the central chlorophyll (Chl) pair (PD1 and PD2) with the center-to-center distances for TyrZ–PD1 and TyrD–PD2 being ≈12.4 Å (6). It is this pair of Chls that bears the photogenerated cation (P680+) that is considered to be the oxidant for the tyrosines (refs. 6–12; for recent reviews see refs. 2 and 3). The existing enzyme may have evolved from an ancestor in which the core was a homodimer with the two tyrosines having identical redox functions (13).
Despite homologous positions in subunits D1 and D2, TyrZ and TyrD exhibit extremely different kinetics, different redox potentials, and play completely different functional roles in the enzyme. Thus, they constitute an ideal system for understanding how the protein environment controls the reactivity of these species.
Several studies have focused on the differences in the kinetics of formation and decay of the two radicals. Most of these studies were conducted on Mn-depleted PSII because TyrZ⋅ is much longer lived in this material, decaying with t1/2 values of 20–600 ms, in contrast with oxygen-evolving PSII in which the t1/2 values are 0.03–1 ms (14–15). Compared with TyrZ⋅, TyrD⋅ is very stable; it decays with t1/2 in the minutes–hours time range (refs. 16–18; for reviews, see refs. 2 and 3).
TyrD⋅ is thought to be immobilized in a site with a well defined and ordered H-bond interaction between its phenol oxygen and a proton from D2 His-189 (in cyanobacteria; D2 His-190 in higher plants) (19). Moreover, the site is well shielded from the lumen (20–23) and is thought to be relatively hydrophobic. The TyrZ⋅ site is quite different. In the absence of Mn, TyrZ⋅ is readily accessible from the lumen (22–24) and is thought to be in a more hydrophilic environment. In addition, TyrZ⋅ exhibits a much more disordered H-bond, and the ring seems to be more mobile (25, 26). The hydrogen bond to TyrZ⋅ seems to involve D1 His-190 directly or indirectly (i.e., by means of one or more water molecules) (for discussion, see refs. 2 and 3).
In the reduced state, TyrD and TyrZ are likely to be protonated and to
form a hydrogen bond to D2 His-189 and D1 His-190, respectively (27,
28). For TyrZ, indications were obtained for a minor population in
which the hydrogen bonds to either water, a hydroxylated amino acid
side chain, or to the protonated imidazole side chain of His (i.e.,
HisH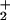 instead of HisH) (27).
instead of HisH) (27).
Because TyrZ and TyrD are protonated in the reduced state and deprotonated in the oxidized state (Tyr⋅), the oxidation involves both electron and proton transfer. In PSII lacking the Mn-cluster, the t1/2 (190 ns–30 μs) of the electron transfer from TyrZ to P680+ was reported to depend on the biological origin of PSII, the presence of Ca2+, and the pH (23, 24, 29–31). The rate increased with the pH from tens of μs at acidic pH to μs or sub-μs at basic pH, exhibiting pKa values of 8.3 (23), 7.0 (24), and 7.5 (31). This pKa was ascribed to the ionization of either TyrZ or D1-His-190 (23, 24, 31). In the latter case, the suggestion is that at low pH, the His is protonated and has to be deprotonated before it is able to accept a proton from TyrZ. In contrast, at high pH (>8–9), the His is thought to be deprotonated already and, therefore, the electron transfer can occur more rapidly (sub-μs or μs) (31, 32).
It is widely accepted that electron donation from TyrD to P680+ is relatively slow compared with TyrZ, although its redox potential is much lower (TyrD, ≈0.75 V; TyrZ, 0.95 V–1.1 V). This notion is largely based on the work of Buser et al. (33), who measured the yield of oxidation of all of the electron donors to P680+ at pH 5.0–7.5 in Mn-depleted PSII. A global fit gave a t1/2 = 10–20 ms for the electron transfer from TyrD to P680+ at pH values between 5.0 and 7.5. Thus, the oxidation yield appeared to be practically independent of pH over this range (33).
Intriguingly, indications for TyrD electron donation to P680+ with a rate similar to TyrZ exist in the older literature, but these have been largely overlooked by the field (34, 35). The present work was performed to readdress the electron donation rate from TyrD to P680+ by using kinetic absorption and EPR spectroscopies and a mutant lacking TyrZ.
Materials and Methods
Construction of D1-Y161F Mutants and Isolation of PSII Particles from Synechocystis sp.
The D1-Y161F mutation was constructed in the psbA-2 gene of the cyanobacterium Synechocystis sp. strain PCC 6803 (36). The plasmid bearing this mutation was transformed into a host strain of Synechocystis that lacks all three psbA genes (37) and contains a hexahistidine-tag (His-tag) fused to the C terminus of CP47 (38). Single colonies were selected for their ability to grow on solid media containing 5 μg/ml kanamycin monosulfate (37). The control wild-type′ strain was constructed in identical fashion as the D1-Y161F mutant except that the transforming plasmid carried no site-directed mutation. The designation wild-type′ differentiates this strain from the native wild-type strain that contains all three psbA genes and is sensitive to antibiotics. Cells were propagated and PSII particles were isolated as described (38). The PSII particles were isolated in a buffer containing 50 mM 2-(N-morpholino)ethanesulfonic acid (Mes)–NaOH (pH 6.0), 20 mM CaCl2, 5 mM MgCl2, 0.03% (wt/vol) N-dodecyl β-d-maltoside, 25% (vol/vol) glycerol, and concentrated to ≈1 mg of Chl per ml (38). The concentration of Mes was decreased to 5 mM by 10-fold dilution and was followed by reconcentration. The final solution contained 5 mM Mes–NaOH (pH 6.0), 20 mM CaCl2, 5 mM MgCl2, 0.03% (wt/vol) N-dodecyl β-d-maltoside, and 25% (vol/vol) glycerol. Mn-depleted wild-type′ PSII particles were treated with NH2OH and EDTA, as described (32). After concentration to ≈1 mg of Chl per ml, the concentration of Mes in the Mn-depleted PSII particles was decreased to 5 mM by dilution and reconcentration, as described above. The mutant and wild-type′ PSII cores were stored at 77 K at a concentration of about 1.5 mg Chl per ml.
After thawing, 20 mM Tris⋅HCl (pH 8.5) was added to the PSII. The PSII particles then were dark-adapted for 2 h at room temperature with 8 mM K3Fe3+(CN)6/8 mM K4Fe2+(CN)6. These conditions result in an almost complete reduction of TyrD. For time-resolved absorption measurements, the samples were diluted to 0.15 mg Chl per ml in 15 mM NaCl/0.3 M sucrose and 50 mM of one of the following buffers: CAPSO/NaOH (pH 9.5), Tris⋅HCl (pH 8.5 and pH 8.0), Hepes/NaOH (pH 7.5), or Mes/NaOH (pH 6.5). For EPR, 40 μl of concentrated PSII was adjusted to pH 8.5 (pH 6.5) by adding 12.5 μl of 0.4 M Tris⋅HCl at pH 8.5 (or Mes/NaOH pH 6.5) and then put into a small volume (≈50 μl) quartz flat cell.
EPR Spectroscopy.
X-band EPR spectra and kinetics were recorded with an ESP 300 spectrometer (Bruker, Karlsruhe, Germany) in a flat cell [small size for Synechocystis sp., normal size (≈400 μl) for spinach]. Excitation flashes were provided with a Spectra-Physics GCR-230–10 Nd-YAG laser (532 nm, 550 mJ, 8 ns).
Time-Resolved Absorption at Room Temperature.
Excitation flashes (300 ps, 532 nm) were provided with an Nd-YAG laser (Quantel, Les Ulis, France) with an energy of about 1.5 mJ/cm2. The measuring light was a continuous laser diode emitting at 820 nm (SDL-5411-G1, Spectra Diode Labs, San Jose, CA). A cut-off filter RG780 was placed before the cuvette and an 820-nm interference filter (10-nm bandwidth) after the cuvette (path length = 10 mm, 2 mm broad). The measuring-light intensity and the flash-induced changes were monitored behind the sample with a photodiode (FND 100, EG & G, Salem, MA) connected to an amplifier (HCA, 28dB, DC-325 MHz; FEMTO, Berlin,) and a digital storage oscilloscope DSA 602A with plug-in 11A52 (DC-20 MHz, Tektronix, Les Ulis, France). The kinetic data were fitted into a multiexponential decay with a Marquart least-square algorithm. The program was kindly provided by P. Setif (Saclay). The very-fast phase with a t1/2 = ≈3 ns was assigned to exited states of loosely bound Chls and were not considered in the fittings.
GEPASI 3 was downloaded from
http://gepasi.dbs.aber.ac.uk/softw/gepasi.html (for review, see
ref. 39). The simulations were based on Scheme
S1. The rate constant
kRec = 800 s−1
was taken from the literature (33). No direct measurement of
k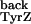 is available in the literature.
k
is available in the literature.
k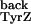 was calculated based on the
equilibrium between TyrZ+ and
P680+, which was estimated to be about
103 at pH 8.5 (23, 40, 41). This value
yields a k
was calculated based on the
equilibrium between TyrZ+ and
P680+, which was estimated to be about
103 at pH 8.5 (23, 40, 41). This value
yields a k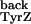 of about 3.4 ×
103 s−1 based on a
kTyrZ = 3.4 ×
106 s−1 at pH 8.5, as
measured in the present study.
of about 3.4 ×
103 s−1 based on a
kTyrZ = 3.4 ×
106 s−1 at pH 8.5, as
measured in the present study.
Scheme 1.

Results
Fig. 1 A, C, and E shows the EPR experiments at pH 8.5 performed with the D1-Y161F PS II mutant (i.e., the TyrZ-less mutant), in which the typical tyrosine radical spectrum arises solely from TyrD. Fig. 1 B, D, and F show the results obtained with wild-type′ PSII.
Figure 1.

Room temperature EPR studies of TyrZ⋅/TyrD⋅ kinetics on Mn-depleted PSII particles from Synechocystis sp. strain PCC 6803 at pH 8.5. The measurements were performed on the mutant D1-Y161F (A, C, E), where the electron donor TyrZ was replaced by a Phe, and on the wild-type′ PSII (B, D, F). (A and B) Traces “dark” show the spectrum of the dark-adapted samples. Trace “after 5 fl” is the spectrum measured after 5 saturating flashes (immediately after the experiment shown in C and D), whereas the trace “after 50 fl” shows the spectrum taken after 50 flashes (four scans per spectrum). (C and D) The time course of the EPR signal from (oxidized) Tyr⋅ measured at 3,464 G (arrows A and B). The five arrows indicate the saturating laser flashes given to the samples. (E and F) The time course of the EPR signal from (oxidized) Tyr⋅ measured at 3,464 G (arrows in A and B) after illumination with 50 flashes. The signal was averaged over 16 or 32 flashes (repetition rate = 8 s). EPR conditions: power, 20 mW; microwave frequency, 9.6 GHz; modulation amplitude, 4.5 G; modulation frequency, 100 kHz. Conversion time: 82 ms for C/D, 5.12 ms for E/F. Time constant: 41 ms for C/D, 2.56 ms for E/F.
Dark incubation of PSII from wild-type and D1-Y161F mutant from Synechocystis sp. for 2 h at pH 8.5 in a buffer containing K3Fe3+(CN)6 and K4Fe2+(CN)6 (see Materials and Methods) resulted in the reduction of TyrD. Fig. 1 A and B (traces labeled “dark”) shows that in both samples, the amplitude of the TyrD⋅ signal is less than 5% of that measured after 50 flashes (Fig. 1 A and B, traces labeled “after 50 fl”). A further series of 50 flashes did not result in an additional increase (data not shown), indicating that TyrD was fully oxidized after 50 flashes.
The kinetics of TyrD⋅ and/or TyrZ⋅ were measured by monitoring the EPR signal at 3,465 G (1 G = 0.1 mT; see Fig. 1 A and B arrow) in these samples. This magnetic field value is outside the magnetic-field range in which signals from Chl and carotenoid cations could contribute significantly (42, 43).
Fig. 1C shows the time course of TyrD⋅ formation and decay during a series of five flashes separated by 8 s each (flashes are indicated by arrows) in D1-Y161F PSII. The first flash induced a signal with an amplitude similar to that measured after 50 flashes (Fig. 1A). This observation indicates that TyrD is transiently fully oxidized after one flash. The slow time-resolution of the EPR spectrometer (i.e., 82 ms) used did not allow the kinetics of TyrD⋅ to be resolved. About 30% of the signal decayed within 1 s, whereas the other 70% was relatively stable. The second flash oxidized the re-reduced fraction of TyrD, and again, about one third of this fraction decayed within 1 s. With each flash, the stable signal approached the level of fully oxidized TyrD⋅ (Fig. 1A). After the fifth flash, the signal was monitored for 42 s (Fig. 1C shows only 22 s), showing the beginning of a very slow decay (t1/2 estimated to be 30 min). Immediately after the recording of this trace, the TyrD⋅ spectrum was measured (Fig. 1A, trace “after 5 fl”), confirming that almost all TyrD was oxidized after five flashes.
Fig. 1E corresponds to the light-induced signal (averaged over 32 flashes) measured immediately after the recording of the trace in C. The absence of a reversible Tyr⋅ signal is as expected, because TyrZ is absent in this mutant.
Fig. 1D shows the time course of Tyr⋅ formation (TyrD⋅ and/or TyrZ⋅) and decay after five flashes separated by 8 s each (flashes indicated by arrows) in wild-type′ PSII. The first flash oxidized one equivalent of Tyr. About 15% of Tyr⋅ decayed within a few seconds; the other 85% were relatively stable. On the following flashes, one equivalent of Tyr⋅ is formed,¶ but the fraction of the signal that was stable became smaller from flash to flash. The stable signal is attributed to TyrD⋅ because of the similar spectrum and decay rate as seen for TyrD⋅ in the Z-less mutant (Fig. 1 A and B; trace “after 5 fl”) (see also ref. 14). Thus, after the first flash, about 85% of the TyrD was oxidized‖, while the remainder was oxidized by the subsequent flashes.
Fig. 1F shows the flash-induced TyrZ⋅ signal of wild-type′ PSII, in which TyrD is fully oxidized after 50 preflashes (see also ref. 44). Because it could be averaged (16 flashes, spaced by 8s), this trace was measured with a higher time resolution and shows that all of the TyrZ was oxidized upon each flash (i.e., it has the same intensity as the fully oxidized TyrD in Fig. 1A).
Fig. 2 shows the flash-induced absorption changes at 820 nm of wild-type′ and D1-Y161F PSII mutant from Synechocystis sp. at pH 8.5. Chl cations absorb at this wavelength; thus, all of the kinetics show an immediate increase caused by the fast formation of P680+ followed by a decay that reflects the re-reduction of P680+ by electron donor(s).
Figure 2.

Flash-induced absorption transients at 820 nm in PSII particles from Synechocystis sp. strain PCC 6803 at pH 8.5. The traces from the wild-type′ PSII are denoted wt′; the traces from the TyrZ-less mutant are denoted D1-Y161F. The traces show the first and the tenth flash after dark adaptation of the samples with a mixture of 8 mM K4[Fe2+(CN−)6] and 8 mM K3[Fe3+(CN−)6] to reduce TyrD. The initial amplitudes of the traces from the wild-type′ were normalized to that of the D1-Y161F.
Before the first flash, TyrD was reduced (see above) and thus available as an electron donor to P680+. For the time window shown in Fig. 2, the kinetic traces could be satisfactorily fitted with three components. The initial spike present during the first 10 ns was omitted for the fits (see Material and Methods). For the first flash on the wild-type′, the fit yielded a fast phase with t1/2 = 169 ns (70%), a slower phase with t1/2 = 895 ns (21%), and a constant (9%). The constant reflects the slower decays, which were also measured here by using a longer time range (data not shown) and were fitted with t1/2 = 112 μs (4%), 820 μs (5%), and 4.3 ms (1%) (see also refs. 30 and 31). The kinetics after the tenth flash of the wild-type′ were not significantly different from the first flash [t1/2 = 186 ns (67%), 950 ns (22%), constant (11%)] (Fig. 2).
Surprisingly, the kinetic traces of the D1-Y161F mutant upon the first flash revealed about the same t1/2 values as the wild-type′, although the amplitudes were somewhat different [t1/2 = 183 ns (45%), 675 ns (22%), constant (33%)]. The trace after the tenth flash was very different; i.e., the constant (μs/ms phases) increased dramatically at the expense of the sub-μs phases [fit: t1/2 = 222 ns (7%), 1160 ns (13%), constant (80%)]. All of the fits of the wild-type′ and mutant samples at different pHs revealed similar rates, which were in the following range: fast phase, t1/2 = 190 ± 35 ns; slow phase, t1/2 = 900 ± 300 ns. EPR experiments showed that almost all TyrD is oxidized after the first flash (see above). Therefore, the fast phase that occurs upon the first flash, which is almost absent after the tenth flash, can be attributed to electron donation from TyrD to P680+, with a t1/2 = ≈190 ns. The phase with a t1/2 = ≈900 ns is also diminished after the tenth flash, suggesting its assignment to P680+ reduction by TyrD in a fraction of centers. The higher amplitude of the μs/ms phases (i.e., the constant in our fit) in the D1-Y161F mutant than in the wild-type′ that occurs upon the first flash is partially due to the presence of some residual oxidized TyrD (≈5% measured by EPR) before the flash experiment (see Fig. 1 A and B and Discussion).
Treatment of the D1-Y161F PSII mutant and wild-type′ PSII with
NH2OH [instead of
K3Fe3+(CN)6/K4Fe2+(CN)6]
to reduce TyrD⋅ did not significantly change the rates and
amplitudes of the traces, when taking into account the longer lifetime
of Q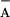 in the presence of NH2OH.
in the presence of NH2OH.
The P680+ reduction in the TyrZ-less mutant
(D1-Y161F) was also measured at different pH values (6.5–9.5; Fig.
3B). TyrD was prereduced by
dark-incubation in the presence of 1 mM NH2OH (as
verified by the lack of a TyrD⋅ signal in EPR). The amplitude of
the long-lived P680+ [i.e., the constant at this
time scale (0–4 μs); assigned to
P680+/Q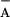 recombination]
increased at the expense of the sub-μs phases (assigned to electron
donation from TyrD) upon lowering the pH. At pH 6.5, fitting suggested
re-reduction of P680+ by TyrD in 10% and by
recombination with Q
recombination]
increased at the expense of the sub-μs phases (assigned to electron
donation from TyrD) upon lowering the pH. At pH 6.5, fitting suggested
re-reduction of P680+ by TyrD in 10% and by
recombination with Q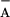 in 90% of the centers. This
estimate is in line with the corresponding EPR experiments,
which revealed that only about 16% of all TyrD is oxidized after one
flash at pH 6.5 (Fig. 3A).
in 90% of the centers. This
estimate is in line with the corresponding EPR experiments,
which revealed that only about 16% of all TyrD is oxidized after one
flash at pH 6.5 (Fig. 3A).
Figure 3.

(A) Room temperature EPR studies of TyrD⋅ kinetics on Mn-depleted PSII TyrZ-less mutant from Synechocystis sp. at pH 6.5 and pH 8.5. The kinetics were measured at 3,464 G, and a flash was given at time 0. Other parameters were as in Fig. 1D. (B) Flash-induced absorption transients at 820 nm in the Mn-depleted TyrZ-less mutant (D1-Y161F) from Synechocystis sp. at different values of pH as indicated in the figure. TyrD was prereduced by incubating for 90 min in 1 mM NH2OH. (C) The relative amplitude of the sum of the fast components (<1 μs, corresponding to TyrD electron donation) at the different pH values from Fig. 3B. The solid line shows a single proton titration fit of the data points.
Fig. 3C shows the sum of the amplitudes of the two sub-μs phases (corresponding to electron donation from TyrD to P680+) plotted against the pH. The experimental data show a strong pH dependence and were fitted to a single proton titration, which led to a pKa of 7.7 (Fig. 3C).
Discussion
The use of a D1-Y161F (i.e., TyrZ-less) mutant allowed a direct measurement of the re-reduction rate of P680+ by TyrD. Two rates with t1/2 = ≈190 ns and 900 ns could be attributed to this electron donation. These rates are four to five orders of magnitude faster than the generally accepted rate (reviewed in ref. 45).
Buser et al. (33) reported a TyrD donation rate to P680+ slower than that reported here (t1/2 = 10–20 ms at pH 5.0–7.5). The two measurements agree at low pH (≤6.5) but disagree at higher pH. Buser et al. (33) measured low yields of TyrD⋅ at pH 7.5 in the presence of 0.4 mM ascorbate and 20 s after the flash, and this led them to calculate a much slower rate. Experiments (data not shown) performed under these conditions with similar materials (PSII membranes from spinach) gave similar yields. We suggest that the low yield is due to the ascorbate that may reduce TyrD⋅ after the flash and QA before the flash, leading to an underestimation of TyrD⋅ yield.
The electron transfer rate from TyrD is very similar to that from
TyrZ to P680+ at higher pH [measured in the presence of
oxidized TyrD (Fig. 2 and ref. 30)]. The sub-μs electron donation
from TyrD to P680+ was measured in a significant
percentage of centers at pH 8.5, i.e., 67%. In the remaining 33% of
the centers, recombination between Q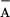 and
P680+ is proposed to occur. In wild-type′ PSII
with preoxidized TyrD, the rate assigned to the recombination between
Q
and
P680+ is proposed to occur. In wild-type′ PSII
with preoxidized TyrD, the rate assigned to the recombination between
Q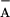 and P680+ occurred in only
12% of the centers. This difference in the amplitude of the
recombination phase between wild-type′ and mutant can in part be
ascribed to the presence of some TyrD⋅ (≈5%) in the TyrZ-less
mutant before the first actinic flash (Fig. 1 A and
B, “dark”). The reason for the remaining 16%
difference is not known.**
and P680+ occurred in only
12% of the centers. This difference in the amplitude of the
recombination phase between wild-type′ and mutant can in part be
ascribed to the presence of some TyrD⋅ (≈5%) in the TyrZ-less
mutant before the first actinic flash (Fig. 1 A and
B, “dark”). The reason for the remaining 16%
difference is not known.**
The amplitude of the sub-μs electron donation from TyrD to
P680+ is pH dependent, with an apparent
pKa close to pH 7.7 (Fig.
3C). At low pH, the TyrD electron donation rate is slower
than the Q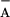 /P680+
recombination (t1/2 > 0.15 ms). This
pKa could represent the
pKa value of a single amino acid
residue. The D2-His-189 residue is a candidate for the protonable
group. Protonation of the D2-His-189 nitrogen proximal to TyrD would be
expected to have drastic effect on TyrD electron donation. In this
case, at low pH, TyrD would have to donate its electron to
P680+either, without releasing its proton, which is
energetically very unfavorable (e.g., ref. 46) or with kinetic
limitations due to deprotonation (and reorientation) of the D2-His-189.
However, this explanation is in contradiction with Fourier transform
infrared (FTIR) measurements, suggesting that D2-His-189 is
deprotonated at pH 6.0 (in the presence of formate and phosphate) (27).
/P680+
recombination (t1/2 > 0.15 ms). This
pKa could represent the
pKa value of a single amino acid
residue. The D2-His-189 residue is a candidate for the protonable
group. Protonation of the D2-His-189 nitrogen proximal to TyrD would be
expected to have drastic effect on TyrD electron donation. In this
case, at low pH, TyrD would have to donate its electron to
P680+either, without releasing its proton, which is
energetically very unfavorable (e.g., ref. 46) or with kinetic
limitations due to deprotonation (and reorientation) of the D2-His-189.
However, this explanation is in contradiction with Fourier transform
infrared (FTIR) measurements, suggesting that D2-His-189 is
deprotonated at pH 6.0 (in the presence of formate and phosphate) (27).
Another possibility, which is in line with the FTIR measurements, is that the protonable group is TyrD itself and that its protonation state is mediated by the protonation state of D2-His-189 at its distal nitrogen. The faster electron-donation rate of TyrD to P680+ at higher pH (pKa of 7.7) is due to the deprotonation of TyrD before oxidation, i.e., the proton is already on the proximal nitrogen of D2-His-189 when the distal nitrogen is deprotonated. At low pH the electron transfer is slow, the TyrD is neutral and H-bonded to the proximal nitrogen of D2-His-189, while the distal nitrogen of the D2-His-189 is protonated. Thus, oxidation of TyrD is limited by H+ transfer to the D2-His-189, which in turn is limited by the release of H+ from its distal nitrogen (see below).
It is known that TyrZ electron donation also shows a pH dependence with a pKa between 7.0 and 8.3 (23, 24, 31), similar to that for TyrD reported here. As for TyrD, titration of the TyrZ itself or the neighboring D1-His-190 were suggested reasons for the pH dependence (23, 24, 31). At high pH, the donation rates of TyrZ and TyrD to P680+ seem to be similar (≈190 ns, see above). In contrast, at low pH, the rates of the dominating kinetic phases are very different, with a t1/2 = 10–30 μs for TyrZ (23, 24, 31) and a t1/2 > 0.2–0.9 ms for TyrD. As suggested above, the deprotonation of the neighboring His at its distal nitrogen could be linked to the protonation of its proximal nitrogen by the proton from the Tyr. This process could be easier with TyrZ than with TyrD, because D1-Glu-189 appears to be (indirectly) associated with a TyrZ deprotonation pathway (47), possibly positioning the proton acceptor for D1-His-190. No equivalent carboxylic acid is conserved in the D2 sequence associated with TyrD, where it is replaced by the acid/base inactive Phe residue (4, 46, 48–51). Another explanation for TyrZ oxidation being faster than TyrD oxidation at low pH could be the possibility that TyrZ⋅ has multiple H-bonding partners (for review, see ref. 3; refs. 25, 26, 52), whereas TyrD⋅ is considered to have D2-His-189 as its unique H-bonding partner (19).
Whereas in the TyrZ-less PSII mutant (D1-Y161F), TyrD is oxidized directly by P680+ (see above), in the wild-type PSII, TyrD could be oxidized in two ways: (i) directly by P680+, and (ii) by rapid formation of a TyrZ/P680+ ⇌ TyrZ⋅(H+)/P680 equilibrium followed by a slower oxidation of TyrD by the equilibrium population of P680+. Because of the lower redox-potential of TyrD (18, 34, 53), it would act as a thermodynamic sink for the electron hole (see Scheme S1). The second of these mechanisms has been the generally accepted view. Our observation of a rapid oxidation of TyrD by P680+ (t1/2 = ≈190 ns) in the D1-Y161F mutant strongly suggests that a direct rapid route also exists in wild type, but it does not rule out the other mechanism.
In this context, an observation of Boussac and Etienne (34) is relevant. They showed in Tris-washed thylakoids with prereduced TyrD that the yield of TyrD⋅ after one flash was almost the same (≈65%) in the absence and in the presence of phenylenediamine, a rapid electron donor to TyrZ⋅. We have performed similar experiments by using Mn-depleted PSII membranes from spinach and confirmed these results (data not shown). Because phenylenediamine donates to TyrZ⋅ with a t1/2 < 1 ms (34, 54), both electron donation rates from TyrD to P680+ and from P680+ to TyrZ⋅ (i.e., kTyrD and kTyrZ back) must be substantially faster than this; otherwise, the yield of TyrD⋅ would decrease dramatically in the presence of phenlyenediamine. Simulation of Scheme S1 using the program GEPASI (see Materials and Methods) in the presence of phenylenediamine (i.e., kDon = 700 s−1 instead of 4 s−1) resulted in a formation of 65% TyrD⋅ only when kTyrD was larger than 1 × 106 s−1 (i.e., t1/2 < 700 ns). Formation of 85% TyrD⋅ needs a kTyrD ≈3.7 × 106 (i.e., t1/2 = ≈190 ns). These simulations provide an argument that TyrD in wild-type PSII is oxidized in the sub-μs range at pH 8.5, and that in a fraction of centers, TyrD is oxidized directly by P680+ (mechanism 1) with a similar rate as in the D1-Y161F mutant.
Before the present work, the reports that electron donation from TyrD to P680+ were slow had been rationalized by assuming a high reorganization energy caused by the trapping of the departing proton from TyrD by D2-His-189 (4). Thus, the more rapid donation from TyrZ to P680+ was taken as indicating that no such charge was accumulated close to TyrZ⋅. This line of thought in turn was used as an argument in favor of the translocation of the proton away from TyrZ⋅, a feature that could be taken as being in favor of certain aspects of the H-atom (electron + proton) abstraction model for oxygen evolution (46). The present work questions the need to invoke such a large reorganization energy associated with the putative charge close to TyrD⋅ and thus weakens this argument for proton translocation away from the TyrZ⋅ site.
The reassessment of the relative donation rates of TyrZ and TyrD to P680+ has repercussions on the question of the localization of P680+ (for instance, see ref. 45). There are four Chls in the inner reaction center symmetrically positioned around a C2 axis (ref. 6; Protein Databank accession no. 1FE1). It was suggested that the much faster donation rate of TyrZ compared with TyrD could be explained if P680+ were located on the D1 side (i.e., closer to TyrZ than TyrD) of the reaction center (for review, see ref. 45). The similar donation rates found here question this argument. According to Page et al. (55), who describe a correlation between the distance and the rate of an electron transfer in proteins, an electron donation rate of t1/2 = ≈190 ns gives an upper limit of 14 Å for the edge-to-edge distance between the donor and acceptor. In the structural model of PSII, only the Chls on the D2 side (PD2 and ChlD2) are located within this distance from TyrD (the distance PD1 to TyrD is >18 Å edge to edge), and thus a direct electron transfer from TyrD to PD1+ is unlikely to occur with a t1/2 = 190 ns. This argument suggests that P680+ is at least partially localized on the Chl PD2 (located on the D2 side). A recent study, performed in the presence of TyrD⋅, suggested that the cation is predominantly localized on PD1 (D1 side; ref. 12). These results are not contradictory, because the present studies suggest only partial localization of P680+ on PD2 and only in the presence of reduced TyrD (TyrD was oxidized in ref. 12). An effect of the oxidation state of TyrD on the localization of P680+ is conceivable (see below).
In the present work, the measured electron-transfer rates to
P680+ from TyrD and TyrZ were the same in
Mn-depleted PSII at pH 8.5 (t1/2 ≈ 190 ns for
the dominating phase). Thus, when both TyrD and TyrZ are able to donate
electrons to P680+ (e.g., first flash in wild
type), the reduction rate P680+ is expected to be
twice as fast, i.e., t1/2 ≈95 ns. The measured
rate, however, exhibited again a t1/2 ≈190 ns
(Fig. 2, “wt': 1. flash”). The most straightforward explanation
for this finding is that the redox-state of TyrD affects the TyrZ
donation. The following assumptions would allow for such a situation:
(i) the TyrZ donation rate is slower than
t1/2 = 190 ns when both tyrosines are reduced.
This assumption seems reasonable given the environment and
potential of the two tyrosines (see above) (ii) The presence
of TyrD⋅ accelerates the TyrZ to P680+
electron donation. It seems reasonable that the positive charge formed
close to the TyrD site upon its oxidation (for example, the protonated
D2-His-189) has an electrostatic effect on P680+
(55), increasing its potential and perhaps shifting the location
of the positive charge (P680+) over to the D1
side (PD1) (see ref. 56) where it is now supposed
to be (12). A similar role was suggested for D2-Arg-181 (57). Such a
role for TyrD⋅ on the redox-potential of
P680+, and thus on the kinetics of TyrZ
oxidation, could provide a mechanistic raison d'être for TyrD in
addition to its postulated redox role in photoactivation and oxidation
of the lower S states (17). Experimental indications of an interaction
of TyrD and P680 have been reported: (i) TyrD oxidation
induces an electrochromic shift in the absorption spectrum of the P680
Soret band (58); (ii) charge recombination between
Q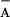 and TyrZ⋅ was slower in wild-type′ PSII (in
the presence of TyrD⋅) than in TyrD-less mutants (59). These
findings can be explained by an electrostatic effect of
TyrD⋅ H+-His on
P680+ that would raise the redox potential of
P680+ and thus depopulate
P680+ (shift the equilibrium of TyrZ⋅ ⇌
P680+ to the left). This depopulation of
P680+, which is an intermediate in the
Q
and TyrZ⋅ was slower in wild-type′ PSII (in
the presence of TyrD⋅) than in TyrD-less mutants (59). These
findings can be explained by an electrostatic effect of
TyrD⋅ H+-His on
P680+ that would raise the redox potential of
P680+ and thus depopulate
P680+ (shift the equilibrium of TyrZ⋅ ⇌
P680+ to the left). This depopulation of
P680+, which is an intermediate in the
Q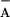 /TyrZ⋅ recombination (45), would slow down
the recombination, as observed. However, Hays et al. (28)
compared the TyrZ donation in wild-type (TyrD⋅ present) and
D2-Y161F mutant at different pHs (7.0–9.0) and found no significant
differences, indicating that the presence of TyrD⋅ does not
affect the TyrZ donation. Further comparative studies on the TyrD-less
and TyrZ-less mutant, as well as on the wild-type′, should clarify
whether the redox chemistry of TyrD has an electrostatic effect on
P680+.
/TyrZ⋅ recombination (45), would slow down
the recombination, as observed. However, Hays et al. (28)
compared the TyrZ donation in wild-type (TyrD⋅ present) and
D2-Y161F mutant at different pHs (7.0–9.0) and found no significant
differences, indicating that the presence of TyrD⋅ does not
affect the TyrZ donation. Further comparative studies on the TyrD-less
and TyrZ-less mutant, as well as on the wild-type′, should clarify
whether the redox chemistry of TyrD has an electrostatic effect on
P680+.
Acknowledgments
We thank Dr. P. Sétif for providing the software for the kinetic analyses; A. P. Nguyen for isolating the thylakoid membranes from and maintaining the wild-type′ and mutant cultures of Synechocystis 6803; Drs. F. Rappaport, S. Un, T. Mattioli, A. Ivancich, C. Berthomieu, and R. Hienerwadel for helpful discussions; Drs. B. Diner, E. Schlodder, P. Nixon, W. Coleman, F. Rappaport, J. Lavergne, W. Vermaas, and D. Chisholm for providing us with a preprint of ref. 12. The research was supported by a grant from the Swiss National Science Foundation (to P.F.), a European Union Training and Mobility of Researchers (TMR) Research Network grant (FMRX-CT98–2014), a cooperation grant “metalloproteins Japan” from the Ministre des Affaires Étrangers (PO N°2000/1577), and National Institutes of Health Grant GM43496 (to R.J.D.).
Abbreviations
- Mes
2-(N-morpholino)ethanesulfonic acid
- Chl
chlorophyll
- P680
photooxidizable chlorophyll
- PD1
PD2, two central chlorophylls bound to polypeptide D1 and D2, respectively
- PSII
photosystem II
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
In Fig. 1F, TyrZ⋅ is detected in almost all of the PSII centers. The apparent smaller amplitude of the reversible signal in Fig. 1D is only due to a lower time resolution.
We obtained similar values for the yield of TyrD oxidation after one flash at pH 8.5 for PSII from Synechococcus elongatus (i.e., 85%) and spinach (i.e., 55%) (data not shown). For chloroplasts from spinach, values from 65–85% were reported (34, 35).
Two possible explanations were tested: (i) Working in total darkness and blocking the measuring light beam until 2.5 ms before the measurement did not change the results. Thus it is unlikely that TyrD was oxidized to a significant extent by the measuring and/or stay light before the measurement occurred (ii) The incubation time (20 h instead of 2 h) at pH 8.5 before the measurement did not affect the result, indicating the TyrD site was pH equilibrated.
References
- 1.Stubbe J, van der Donk W A. Chem Rev (Washington, DC) 1998;98:705–762. doi: 10.1021/cr9400875. [DOI] [PubMed] [Google Scholar]
- 2.Diner B A. Biochim Biophys Acta. 2001;1503:147–163. doi: 10.1016/s0005-2728(00)00220-6. [DOI] [PubMed] [Google Scholar]
- 3.Debus R J. Biochim Biophys Acta. 2001;1503:164–186. doi: 10.1016/s0005-2728(00)00221-8. [DOI] [PubMed] [Google Scholar]
- 4.Babcock G T, Espe M, Hoganson C, Lydakis-Simantriris N, McCracken J, Shi W, Styring S, Tommos C, Warncke K. Acta Chem Scand. 1997;51:533–540. doi: 10.3891/acta.chem.scand.51-0533. [DOI] [PubMed] [Google Scholar]
- 5.Rappaport F, Lavergne J. Biochim Biophys Acta. 2001;1503:246–259. doi: 10.1016/s0005-2728(00)00228-0. [DOI] [PubMed] [Google Scholar]
- 6.Zouni A, Witt H-T, Kern J, Fromme P, Krauss N, Saenger W, Orth P. Nature (London) 2001;409:739–743. doi: 10.1038/35055589. [DOI] [PubMed] [Google Scholar]
- 7.Debus R J, Barry B A, Babcock J T, McIntosh L. Proc Natl Acad Sci USA. 1988;85:427–430. doi: 10.1073/pnas.85.2.427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vermass W F J, Rutherford A W, Hansson O. Proc Natl Acad Sci USA. 1988;85:8477–8481. doi: 10.1073/pnas.85.22.8477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hirsh D J, Brudvig G W. J Phys Chem. 1993;97:13216–13222. [Google Scholar]
- 10.Koulougliotis D, Tang X-S, Diner B A, Brudvig G W. Biochemistry. 1995;34:2850–2856. doi: 10.1021/bi00009a015. [DOI] [PubMed] [Google Scholar]
- 11.Astashkin A V, Kodera Y, Kawamori A. Biochim Biophys Acta. 1994;1187:89–93. [Google Scholar]
- 12.Diner B A, Schlodder E, Nixon P J, Coleman W J, Rappaport F, Lavergne J, Vermaas W F J, Chisholm D A. Biochemistry. 2001;40:9265–9281. doi: 10.1021/bi010121r. [DOI] [PubMed] [Google Scholar]
- 13.Rutherford A W, Nitschke W. In: Origin and Evolution of Biological Energy Conversion. Baltscheffsky H, editor. New York: VCH; 1996. pp. 143–75. [Google Scholar]
- 14.Babcock G T, Sauer K. Biochim Biophys Acta. 1975;376:315–328. doi: 10.1016/0005-2728(75)90024-9. [DOI] [PubMed] [Google Scholar]
- 15.Brettel K, Schlodder E, Witt H T. Biochim Biophys Acta. 1984;766:403–415. [Google Scholar]
- 16.Babcock G T, Sauer K. Biochim Biophys Acta. 1973;325:483–503. doi: 10.1016/0005-2728(73)90209-0. [DOI] [PubMed] [Google Scholar]
- 17.Styring S, Rutherford A W. Biochemistry. 1987;26:2401–2405. [Google Scholar]
- 18.Vass I, Styring S. Biochemistry. 1991;30:830–839. doi: 10.1021/bi00217a037. [DOI] [PubMed] [Google Scholar]
- 19.Campell K A, Peloquin J M, Diner B A, Tang X S, Chisholm D A, Britt R D. J Am Chem Soc. 1997;119:4787–4788. [Google Scholar]
- 20.Rodriguez I D, Chandrashekar T K, Babcock G T. In: Progress in Photosynthesis Research. Biggins J, editor. I. Dordrecht, The Netherlands: Martinus Nijhoff; 1987. pp. 471–474. [Google Scholar]
- 21.Tang X-S, Chisholm D A, Dismukes G C, Brudvig G W, Diner B A. Biochemistry. 1993;32:13742–13748. doi: 10.1021/bi00212a045. [DOI] [PubMed] [Google Scholar]
- 22.Tommos C, McCracken J, Styring S, Babcock G T. J Am Chem Soc. 1998;120:10441–10452. [Google Scholar]
- 23.Diner B A, Force D A, Randall D W, Britt R D. Biochemistry. 1998;37:17931–17943. doi: 10.1021/bi981894r. [DOI] [PubMed] [Google Scholar]
- 24.Ahlbrink R, Haumann M, Cherapanov D, Bogenshausen O, Mulkidjanian A, Junge W. Biochemistry. 1998;37:1131–1142. doi: 10.1021/bi9719152. [DOI] [PubMed] [Google Scholar]
- 25.Force D A, Randall D W, Britt R D, Tang X S, Diner B A. J Am Chem Soc. 1995;117:10547–10554. [Google Scholar]
- 26.Tommos C, Tang X S, Warncke K, Hoganson C W, Styring S, McCracken J, Diner B A. J Am Chem Soc. 1995;117:10325–10335. [Google Scholar]
- 27.Hienerwadel R, Boussac A, Breton J, Diner B, Berthomieu C. Biochemistry. 1997;36:14712–14723. doi: 10.1021/bi971521a. [DOI] [PubMed] [Google Scholar]
- 28.Berthomieu C, Hienerwadel R, Boussac A, Breton J, Diner B. Biochemistry. 1998;37:10547–10554. doi: 10.1021/bi980788m. [DOI] [PubMed] [Google Scholar]
- 29.Conjeaud H, Mathis P. Biochim Biophys Acta. 1980;590:353–359. doi: 10.1016/0005-2728(80)90206-6. [DOI] [PubMed] [Google Scholar]
- 30.Schlodder E, Meyer B. Biochim Biophys Acta. 1987;890:23–31. [Google Scholar]
- 31.Hays A A, Vassiliev I R, Golbeck J H, Debus R J. Biochemistry. 1999;38:11851–11865. doi: 10.1021/bi990716a. [DOI] [PubMed] [Google Scholar]
- 32.Hays A A, Vassiliev I R, Golbeck J H, Debus R J. Biochemistry. 1998;37:11352–11365. doi: 10.1021/bi980510u. [DOI] [PubMed] [Google Scholar]
- 33.Buser C A, Thompson L K, Diner B A, Brudvig G W. Biochemistry. 1990;29:8977–8985. doi: 10.1021/bi00490a014. [DOI] [PubMed] [Google Scholar]
- 34.Boussac A, Etienne A L. Biochem Biophys Res Commun. 1982;109:1200–1205. doi: 10.1016/0006-291x(82)91904-0. [DOI] [PubMed] [Google Scholar]
- 35.Boussac A, Etienne A L. FEBS Lett. 1982;148:113–116. [Google Scholar]
- 36.Debus R J, Barry B A, Sithole I, Babcock G T, McIntosh L. Biochemistry. 1988;27:9071–9074. doi: 10.1021/bi00426a001. [DOI] [PubMed] [Google Scholar]
- 37.Debus R J, Nguyen A P, Conway A B. In: Current Research in Photosynthesis. Baltscheffsky M, editor. I. Dordrecht, The Netherlands: Kluwer; 1990. pp. 829–832. [Google Scholar]
- 38.Debus R J, Campbell K A, Gregor W, Li Z-L, Burnap R L, Britt R D. Biochemistry. 2001;40:3690–3699. doi: 10.1021/bi002394c. [DOI] [PubMed] [Google Scholar]
- 39.Mendes P. Trends Biochem Sci. 1997;22:361–363. doi: 10.1016/s0968-0004(97)01103-1. [DOI] [PubMed] [Google Scholar]
- 40.Yerkes C T, Babcock G T, Crofts A R. Biochim Biophys Acta. 1983;158:359–363. [Google Scholar]
- 41.Rappaport F, Lavergne J. Biochemistry. 1997;36:15294–15303. doi: 10.1021/bi971287o. [DOI] [PubMed] [Google Scholar]
- 42.Visser J W M, Rijgersberg C P, Gast P. Biochim Biophys Acta. 1977;460:36–46. doi: 10.1016/0005-2728(77)90149-9. [DOI] [PubMed] [Google Scholar]
- 43.Hanley J, Deligiannakis Y, Pascal A, Faller P, Rutherford A W. Biochemistry. 1999;38:8189–8185. doi: 10.1021/bi990633u. [DOI] [PubMed] [Google Scholar]
- 44.Blankenship R E, Babcock G T, Warden J T, Sauer K. FEBS Lett. 1975;51:287–293. doi: 10.1016/0014-5793(75)80909-4. [DOI] [PubMed] [Google Scholar]
- 45.Diner B A, Babcock G T. In: Oxygenic Photosynthesis: The Light Reactions. Ort D R, Yocum C F, editors. Dordrecht: Kluwer; 1996. pp. 213–247. [Google Scholar]
- 46.Tommos C, Babcock G T. Biochim Biophys Acta. 2000;1458:199–219. doi: 10.1016/s0005-2728(00)00069-4. [DOI] [PubMed] [Google Scholar]
- 47.Debus R J, Campbell K A, Pham D P, Hays A-M A, Britt R D. Biochemistry. 2000;39:627–6287. doi: 10.1021/bi992749w. [DOI] [PubMed] [Google Scholar]
- 48.Svensson B, Vass I, Cedergren E, Styring S. EMBO J. 1990;9:2051–2059. doi: 10.1002/j.1460-2075.1990.tb07372.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ruffle S, Donnelly D, Blundell T L, Nugent J H A. Photosynth Res. 1992;34:287–300. doi: 10.1007/BF00033446. [DOI] [PubMed] [Google Scholar]
- 50.Chu H A, Nguyen A P, Debus R J. Biochemistry. 1995;34:5839–5858. doi: 10.1021/bi00017a016. [DOI] [PubMed] [Google Scholar]
- 51.Svensson B, Etchebest C, Tuffery P, van Kan P, Smith J, Styring S. Biochemistry. 1996;35:14486–14502. doi: 10.1021/bi960764k. [DOI] [PubMed] [Google Scholar]
- 52.Un S, Tang X S, Diner B A. Biochemistry. 1996;35:679–684. doi: 10.1021/bi9523769. [DOI] [PubMed] [Google Scholar]
- 53.Boussac A, Etienne A L. Biochim Biophys Acta. 1984;766:576–584. [Google Scholar]
- 54.Diner B A. In: Proceedings of the 4thInternational Congress on Photosynthesis. Hall D O, Coombs J, Goodwin T W, editors. London: Biochemical Society; 1977. pp. 359–372. [Google Scholar]
- 55.Page C C, Moser C C, Chen X, Dutton P L. Nature (London) 1999;402:47–52. doi: 10.1038/46972. [DOI] [PubMed] [Google Scholar]
- 56.Nugent J H A, Bratt P J, Evans M C W, MacLachlan D J, Rigby S E J, Ruffle S V, Turconi S. Biochem Soc Trans. 1994;22:327–331. doi: 10.1042/bst0220327. [DOI] [PubMed] [Google Scholar]
- 57.Mulkidjanian A. Biochim Biophys Acta. 1999;1410:1–6. doi: 10.1016/s0005-2728(98)00174-1. [DOI] [PubMed] [Google Scholar]
- 58.Diner B, Tang X-S, Zheng M, Dismukes G C, AnnForce D, Randall D W, Britt R D. In: Photosynthesis: From Light to Biosphere. Mathis P, editor. Dordrecht, The Netherlands: Kluwer; 1995. pp. 229–234. [Google Scholar]
- 59.Boerner R J, Bixby K A, Nguyen A P, Noren G H, Debus R J, Barry B A. J Biol Chem. 1993;268:1817–1823. [PubMed] [Google Scholar]