

Abstract
Objective
Microglia play a pivotal role in the initiation and progression of Alzheimer's disease (AD). We here tested the therapeutic hypothesis that the Ca2+‐activated potassium channel KCa3.1 constitutes a potential target for treating AD by reducing neuroinflammation.
Methods
To determine if KCa3.1 is relevant to AD, we tested if treating cultured microglia or hippocampal slices with Aβ oligomer (AβO) activated KCa3.1 in microglia, and if microglial KCa3.1 was upregulated in 5xFAD mice and in human AD brains. The expression/activity of KCa3.1 was examined by qPCR, Western blotting, immunohistochemistry, and whole‐cell patch‐clamp. To investigate the role of KCa3.1 in AD pathology, we resynthesized senicapoc, a clinically tested KCa3.1 blocker, and determined its pharmacokinetic properties and its effect on microglial activation, Aβ deposition and hippocampal long‐term potentiation (hLTP) in 5xFAD mice.
Results
We found markedly enhanced microglial KCa3.1 expression/activity in brains of both 5xFAD mice and AD patients. In hippocampal slices, microglial KCa3.1 expression/activity was increased by AβO treatment, and its inhibition diminished the proinflammatory and hLTP‐impairing activities of AβO. Senicapoc exhibited excellent brain penetrance and oral availability, and in 5xFAD mice, reduced neuroinflammation, decreased cerebral amyloid load, and enhanced hippocampal neuronal plasticity.
Interpretation
Our results prompt us to propose repurposing senicapoc for AD clinical trials, as senicapoc has excellent pharmacological properties and was safe and well‐tolerated in a prior phase‐3 clinical trial for sickle cell anemia. Such repurposing has the potential to expedite the urgently needed new drug discovery for AD.
Introduction
Emerging evidence strongly implicates aberrant microglial activation in Alzheimer's disease (AD).1, 2, 3 The functional and pathological diversity of microglia is orchestrated by a complex ensemble of ion channels, receptors, and transporters that regulate intracellular signaling and gene expression. Activities of these surface molecules can be modulated selectively in order to modify microglial activation. One possible target is KCa3.1, an intermediate‐conductance Ca2+‐activated K+ channel expressed in immune cells such as T‐lymphocytes, macrophages and microglia.4, 5 In these cells, KCa3.1 regulates cellular activation, migration and proliferation via K+ efflux, which helps maintain a negative membrane potential to drive Ca2+ influx required for various activities.6, 7 We and others previously demonstrated that rat8 and mouse9 microglia express KCa3.1, but do not display other KCa currents carried by KCa1.1 and KCa2 channels.9 We further demonstrated that microglial activation and neurotoxicity induced by Aβ oligomer (AβO), the most toxic amyloid in AD brains, can be blocked by the specific KCa3.1 blocker TRAM‐34 in vitro, suggesting that KCa3.1 could be a therapeutic target for AD.9, 10
In the current study, we used animal models and human postmortem brain samples to show that KCa3.1 is a biologically relevant and microglial target for AD. We further showed that an orally available KCa3.1 inhibitor called senicapoc was able to mitigate AD‐like deficits in 5xFAD mice. We further found that senicapoc exhibited good brain penetration (Cbrain/Cplasma~5). Senicapoc was previously advanced to a Phase‐3 clinical trial for sickle cell anemia and was found to be safe and well‐tolerated.11, 12, 13 Our results provide a strong rationale to repurpose senicapoc for the treatment of AD and/or mild cognitive impairment.
Methods and Materials
Study approval
All protocols involving mouse models were approved by the Institutional Animal Care and Use Committee. Human brain samples were provided by the University of California Davis Alzheimer's Disease Center, approved by the Institutional Review Board.
Mouse model
Tg6799 5xFAD mice on the C57Bl/6 background were obtained from Dr. Robert Vassar.14 This line co‐expresses human APP695 with the Swedish (K670N, M671L), Florida (I716V), and London (V717I) mutations and human PS1 harboring M146L and L286V mutations. Mice were randomly assigned to treatment groups and phenotyping parameters were evaluated in a blinded manner. Roughly equal numbers of male and females mice were used.
Chemicals and diets
TRAM‐34 and 4‐phenyl‐4H‐pyran (methyl‐5‐acetyl‐4‐(4‐chloro‐3‐(trifluoromethyl)phenyl)‐2,6‐dimethyl‐4H‐pyran‐3‐carboxylate) were synthesized as described.15 Senicapoc was synthesized as shown in Figure 6A and its purity and identity authenticated (Data S1). For intraperitoneal administration of TRAM‐34, animals received either the vehicle Miglyol‐812 (caprylic/capric triglyceride, Spectrum Chemicals) or TRAM‐34 (40 mg/kg) dissolved in Miglyol‐812. For diet administration of senicapoc, animal received either a senicapoc‐medicated diet (2400 ppm, Harlan TD.140594) or a control diet (Harlan TD.140368).
Figure 6.
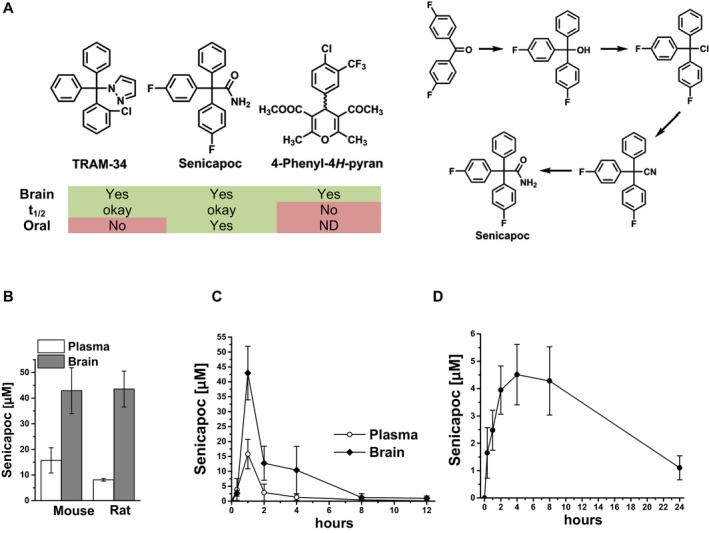
The KCa3.1 inhibitor senicapoc is orally available and has excellent brain penetration. (A) Chemical structures of the three indicated KCa3.1 blockers and the synthetic scheme for senicapoc. (B) Total plasma and brain concentrations in mice (n = 3) and rats (n = 2) at 1 h after intraperitoneal administration of 50 mg/kg senicapoc in 1 mL miglyol per body weight. (C) Time course of total senicapoc brain and plasma concentrations in mice after intraperitoneal administration of 50 mg/kg senicapoc (n = 3 per time point). (D) Total senicapoc plasma concentrations after oral gavage of 50 mg/kg in rats (n = 3). Data are presented as mean ± SD.
Tissue culture and acute isolation of microglia
Primary microglia were prepared from mixed glia cultures as described.9 Hippocampal slice cultures (400 μm thick) were prepared from 7‐day‐old C57BL/6 mice as described9 and cultured for 10 days before use. To study microglia in adult mouse brains without culturing, microglia were acutely isolated as described using anti‐CD11b magnetic beads (Miltenyi Biotec).16
Production of AβO
AβO was prepared and characterized as described.16 To ensure consistency, a random sample from each batch was evaluated for neurotoxic activity and ability to rapidly induce exocytosis of MTT formazan, as described.17, 18
Whole‐cell patch‐clamp
Whole‐cell patch‐clamp studies on cultured primary microglia and isolated adult microglia were conducted as described.9, 16, 19 KCa3.1 conductances were calculated from the slope of the TRAM‐34‐sensitive KCa current between −80 and −75 mV, where, under our recording conditions with an aspartate‐based internal solution, KCa3.1 currents are primarily carried by KCa3.1 and not “contaminated” with Kv1.3 (which activates at voltages more positive than −40 mV), inward rectifier K+ currents (which are appreciable at voltages more negative than −80 mV), or chloride currents.
Cell/tissue homogenate preparation, Western blot analysis, and Aβ ELISA
To obtain cell lysates, cells were washed with ice‐cold PBS and incubated and harvested with a lysis buffer (150 mmol/L NaCl, 10 mmol/L NaH2PO4, 1 mmol/L EDTA, 1% TritonX100, 0.5% SDS) with protease inhibitor cocktail and phosphatase inhibitor (Sigma). Mouse and human brain were homogenized in RIPA buffer (ThermoFisher) with protease inhibitor cocktail and phosphatase inhibitor (Sigma) and used for Western blotting as described.9 The following primary antibodies (dilutions) were used: anti‐KCa3.1 P4997 (1:1000, Sigma), anti‐phospho P38MAPK and anti‐P38MAPK (1:1000, Cell Signaling), anti‐β actin (1:3000, Sigma), and anti‐GAPDH (1:2000, Cell Signaling). Secondary antibodies were HRP‐conjugated anti‐rabbit or anti‐mouse antibody (1:1000, GE Healthcare). Brain tissue samples from 5xFAD treated with senicapoc or control diet were fractionated into TBS‐soluble and TBS‐insoluble, SDS‐soluble fractions, which were used for Aβ42 quantification by a Human Aβ42 ELISA kit (Wako) as described.16 Alternatively, Aβ in each fraction was separated in 16.5% Tris/Tricine SDS gel electrophoresis (Bio‐Rad), transferred to PVDF membrane, and detected by anti‐Aβ (6E10, 1:1000, BioLegend).
Quantitative PCR
Total RNA from primary microglia, acutely isolated microglia, and tissue samples were extracted using RNeasy Plus Mini Kit (Qiagen) or RNeasy Plus Universal Mini Kit (Qiagen). RNA samples from acutely isolated microglia were further reverse‐transcribed and pre‐amplified as we previously described.20 The result was normalized to β–actin. The primer sequences used are listed in Table 1. Relative cDNA levels for the target genes were analyzed by the 2‐∆∆Ct method using Actb as the internal control for normalization.
Table 1.
qPCR primer list
| Gene | Forward Primer (5′–>3′} | Reverse Primer (3′–>5′) |
|---|---|---|
| IL‐1β (il1β) | CCCCAAGCAATACCCAAAGA | TACCAGTTGGGGAACTCTG |
| TNF‐α (tnfα) | GACGTGGAACTGGCAGAAGAG | TGCCACAAGCAGGAATGAGA |
| IL‐6 (il6) | GTTCTCTGGGAAATCGTGGA | TTCTGCAAGTGCATCATCGT |
| Arg1 (arg1) | CCAACTCTTGGGAAGACAGC | TATGGTTACCCTCCCGTTGA |
| iNOS (inos2) | CGGATAGGCAGAGATTGGAG | GTGGGGTTGTTGCTGAACTT |
| CD206 (cd206) | TCATCCCTGTCTCTGTTCAGC | ATGGCACTTAGAGCGTCCAC |
| Ym2 (chil3) | AGGAAGCCCTCCTAAGGACA | TGAGTAGCAGCCTTGGAATG |
| IGF‐1 (igf1) | CACAATGCCTGTCTGAGGTG | TGAGTAGCAGCCTTGGAATG |
| CaMK2‐α (camk2α) | CAATATCGTCCGACTCCATG | CATCTGGTGACAGTGTAGC |
| CD11b (cd11b) | AAGGATTCAGCAAGCCAGAA | GGAGGGATGAGAGTCCACAT |
| Nav1.8 (Scn10α) | QuantiTect® Primer Assay (QIAGEN, Mm_Scn10a_1_SG) |
| GFAP (Gfap) | PrimerPCR™ SYBR® Green Assay (Bio‐Rad, qMmuCID0020163) |
| Kca3.1 (Kcnn4) | PrimePCR™ SYBR® Green Assay (Bio‐Rad, qMmuCID0016996) |
| β‐actin (ACTB) | Mouse ACTB (Applied Biosystems, Mm00607939_s1) |
Immunofluorescence staining
Immunofluorescence staining and quantification was performed as described.16 Briefly, microglia cells cultured on cover slip, hippocampal slices and frozen mouse or human brain sections (20 μm) were fixed and stained with anti‐Aβ1‐42 (1:200; Millipore), anti‐CD68 (1:200; Serotec), anti‐CD11b (1:200; AbD Serotec), or anti‐Iba‐1 (1:200, Wako Chemical or Millipore), overnight at 4°C, followed by respective Alexa Fluor‐conjugated secondary antibodies (1:700; Invitrogen). For detection of amyloid plaques, 400 nmol/L FSB (Sigma) was used. To detect mouse KCa3.1, the following antibodies were used: APC064 (1:3000, Alomone Labs), P4997 (1:3000, Sigma), AV35098 (1:3000, Sigma), and SC‐365265 (1:3000, Santa Cruz). Noticeably, SC‐365265 was used in prior studies to support astrocytic expression of KCa3.1.21, 22 However, SC‐365265 highlighted multiple cellular contours throughout the brain in KCa3.1−/− mice, even at a 1:3000 dilution (data not presented), therefore was considered nonspecific and not further used. For human brains, an additional anti‐human KCa3.1 polyclonal antibody (1:300, ThermoFisher PA5:41015) was used. Before FSB (1 μmol/L) staining, sections were incubated with 0.1% Sudan black B (Sigma) to block autofluorescence. Immunostained slides were imaged under a Nikon Eclipse E600 microscope and photographed by a digital camera (SPOT RTke, SPOT Diagnostics). Photomicrographs were randomly taken in a blind fashion and then analyzed by ImageJ as described.16 For co‐localization analysis to determine % of microglia expressing KCa3.1, three photomicrographs from each mouse were analyzed by a Fiji Coloc2 plugin (co‐localization analysis program, https://imagej.net/Coloc_2). The photography and analysis of immunoreactivity were conducted in an investigator‐blinded manner.
Induction of hippocampal long‐term potentiation by high frequency stimulation
See Data S1.
Pharmacokinetic study of senicapoc
Total senicapoc plasma and brain concentrations were determined by LC‐MS analysis using a Waters Acquity UPLC (Waters) interfaced to a TSQ Quantum Access Max mass spectrometer (ThermoFisher Scientific) (Data S1). Basic pharmacokinetic properties (brain/plasma ratios; brain/plasma profiles and oral availability) are shown in Figure 6.
Statistics
All results were expressed as the mean ± S.E, or mean ± S.D. in pharmacokinetics data. Normality of the data distribution was determined using the Shapiro‐Wilk test. For group comparisons in means, paired or unpaired Student's t‐test or one or two‐way ANOVA, as appropriate, were conducted using the SigmaStat 3.1 (Systat Inc.). When the overall ANOVA was significant, Bonferroni's multiple comparison procedure was carried out to maintain the family wise error rate at 0.05. The significance level for the two‐sided analyses was set at P < 0.05. If the assumption of normality was violated, a natural log transform of the outcome was used and reassessed for normality. If normality was still violated, a nonparametric Wilcoxon rank sum test was used instead of the t‐test.
Results
KCa3.1 is upregulation by AβO in microglia
Previously we and others have verified the biophysical and pharmacological signatures of KCa3.1 in primary cultured or acutely isolated adult mouse, rat, and human microglia, and shown that KCa3.1 plays an important role in regulating microglial activation.9, 23, 24 Expression analysis further showed that only microglia expressed significant amounts of KCa3.1 among neural cells (Fig. 1A). In organotypic hippocampal slice cultures, a more physiological model, AβO substantially increased the number of strongly CD11b‐immunoreactive microglia, a large majority of which were also KCa3.1‐positive (Fig. 1B). qPCR (Fig. 1C) and Western blotting (Fig. 1D) further showed parallel increases in the transcript and protein levels of KCa3.1. We did not observe significant co‐localization of the astrocytic marker GFAP with KCa3.1, suggesting that AβO did not upregulate KCa3.1 in astrocytes (Fig. 1B).
Figure 1.
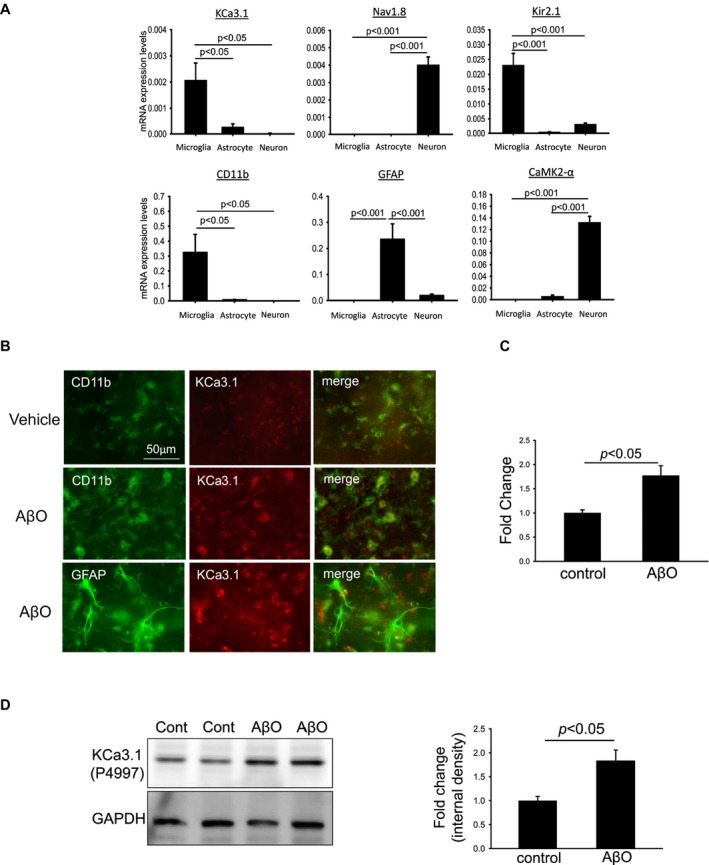
KCa3.1 is expressed in microglia and is upregulated by AβO. (A) qPCR was conducted on RNA extracted from cultured primary mouse microglia, astrocytes and neurons from three independent preparations. KCa3.1, Kir2.1, and CD11b were expressed exclusively in microglia, while Nav1.8 and CAMK2‐α expressions were characteristic of neurons and GFAP expression characteristic of astrocytes. n = 3/group; one‐way ANOVA follow by Bonferroni post hoc test. (B–D) Hippocampal slices were treated with 100 nmol/L AβO for 24 h. (B) Slices were immunostained with CD11b (green) or anti‐GFAP (green) and co‐stained with anti‐KCa3.1 (APC064). AβO treatment caused increased staining of KCa3.1, which was largely colocalized with CD11b, but not with the astrocytic marker GFAP. AβO‐induced KCa3.1 upregulation was further corroborated by the increased transcript (qPCR result in C, n = 4) and protein (representative Western blot and quantification in D, n = 4) levels of KCa3.1. Numerical data are presented by mean ± SE from four independent experiments and were analyzed by two‐sample t‐test.
Microglial KCa3.1 expression/activity is enhanced in 5xFAD mouse and human AD brains
To investigate the in vivo role of microglial KCa3.1, we used 5xFAD mice, which harbor five familial mutations of APP and PSEN1 genes and show robust Aβ production and Aβ‐associated neuroinflammation.14 Soluble AβO made of Aβ42 is a major species of Aβ aggregates in 5xFAD mice25 and could stimulate microglial KCa3.1 expression in vivo. We observed substantial increases in both CD11b and KCa3.1 immunoreactivities in four and ten months‐old 5xFAD brains (Fig. 2A, D, and Fig. S1) compared to age‐matched Wt littermates. Two antibodies, APC064 and P4997, were used and both showed that KCa3.1 was localized to CD11b‐positive microglial cell bodies or processes (Fig. 2A). Higher magnification revealed that KCa3.1 appeared distributed differently from CD11b within microglia. For example, KCa3.1 immunostaining was localized to segments of microglial processes, contiguous (Fig. 2B, arrowheads) or overlapping with CD11b‐positive curvilinear profiles. qPCR (Fig. 2C) and Western blotting (Fig. 2D) confirmed the increased level of KCa3.1 transcript and protein, respectively, in 5xFAD brains. Notably, KCa3.1 expression was not different between Wt and 5xFAD mice at one month of age, but began to increase in 5xFAD mice at 4 months of age. This appears to concomitant with Aβ amyloidosis and microglial activation in 5xFAD mice, which began at 3–4 months of age.
Figure 2.
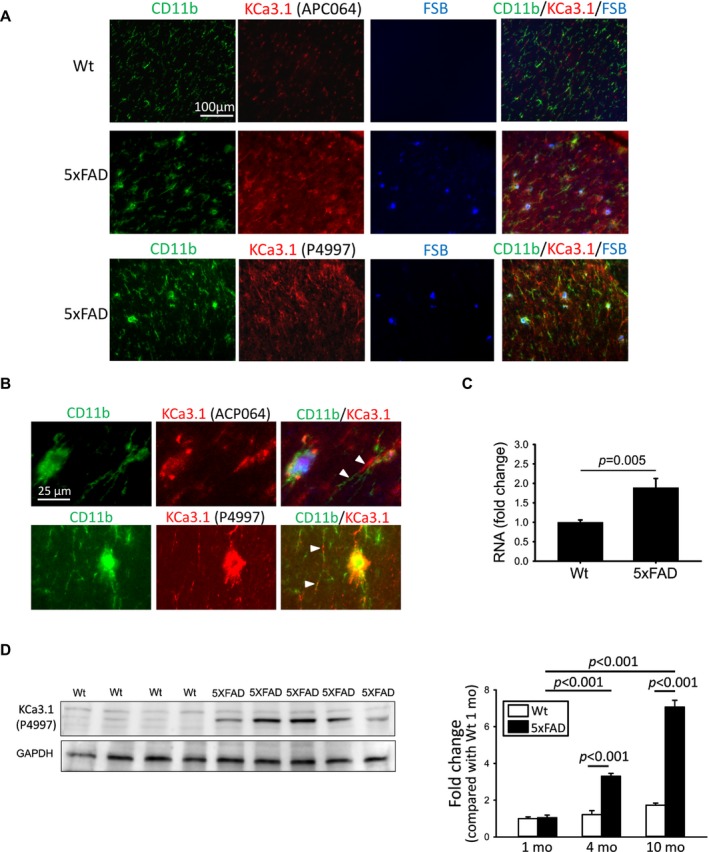
Microglial KCa3.1 expression is elevated in brains of 4‐ and 10‐month‐old 5xFAD mice. (A) Corresponding coronal sections of mouse frontal cortex from 10‐month‐old mice were fluorescently stained with the amyloid dye anti‐CD11b (green), anti‐KCa3.1 (either APC064 or P4997) (red), and FSB (blue). KCa3.1 was localized to activated microglia enriched around FSB‐positive amyloid plaques. (B) Higher magnification images to illustration localization of KCa3.1 to compartments different from but contiguous to or overlapping with CD11b (white arrowheads) in a 5xFAD brain. Quantitative PCR (C) and Western blotting (D) showed increased cerebral transcript and protein levels, respectively, of KCa3.1 in 5xFAD mice. In (D), the left panel shows a representative Western blot using 10‐month‐old mouse samples, and the right panel shows quantified mean intensities of the KCa3.1 band relative to that of 1‐month‐old Wt mice. Numerical data are presented by mean ± SE and were analyzed by two‐sample t‐test or two‐way ANOVA follow by Bonferroni post hoc test. qPCR (10 mo): Wt, n = 9 and 5xFAD, n = 12; Western blotting: (1 mo) Wt, n = 5 and 5xFAD, n = 5; (4 mo) Wt, n = 5 and 5xFAD, n = 5; (10 mo) Wt, n = 8 and 5xFAD, n = 9.
To confirm that KCa3.1 in 5xFAD microglia is functional, we performed whole‐cell patch‐clamp on microglia acutely isolated from brains of 10‐months‐old 5xFAD and Wt littermates. The purity of isolated microglia, determined by flow cytometry, was consistently between 94 to 96%.26 Compared to Wt microglia (Fig. 3A), 5xFAD microglia displayed enhanced expression of a TRAM‐34‐sensitive KCa3.1 current when dialyzed with 1 μmol/L of free Ca2+ (Fig. 3B). The K+ current remaining after application of 1 μmol/L of TRAM‐34 was carried by a combination the voltage‐gated Kv1.3 current (at potentials above −40 mV) and the inward‐rectifier Kir2.1 (at potentials below −90 mV), two other major K+ currents expressed in microglia.16, 27 Quantification of the TRAM‐34‐sensitive current density, which puts the current amplitude in relation to cell size, shows a significant increase in functional KCa3.1 expression in 5xFAD microglia (Fig. 3C).
Figure 3.
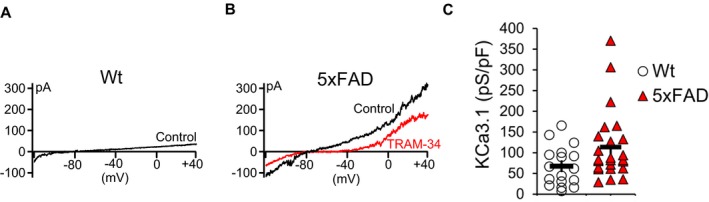
Enhanced KCa3.1 channel membrane expression in 5xFAD microglia. Whole‐cell patch‐clamp was performed on microglia acutely isolated from 10‐month‐old 5xFAD or Wt littermate mice. (A) Representative current elicited by a voltage ramp from ‐120 mV to +40 mV showing a Ca2+‐activated current in Wt microglia in the presence of 1 μmol/L free Ca2+ in the internal recording solution. (B) Increased Ca2+‐activated K+ current recorded from 5xFAD microglia was blocked by the specific KCa3.1 blocker TRAM‐34. (C) Scatter plots showing a statistically greater KCa3.1 channel current density for 5xFAD microglia (n = 20) compared to Wt microglia (n = 20). Shown are mean ± S.E. Statistical difference was determined by Student's t‐test (P = 0.02), following a natural log transform of the outcome which met the assumption of normality.
We next compared KCa3.1 expression in the superior temporal cortex of thirteen pathologically confirmed AD (“high likelihood of AD” according to the National Institute on Aging‐Alzheimer's Association guideline28 and Braak stage 5 and 6 according to the BrainNet Europe criteria29) and nine age‐matched control (cognitively intact with Braak stage 0–2) individuals (Table 2). Immunofluorescence stains highlighted strongly KCa3.1‐positive microglia in AD brains that were much less numerous in control brains (Fig. 4A). KCa3.1 immunoreactivities, assessed by two independent antibodies, were accentuated in or around FSB‐positive amyloid plaques and showed substantial co‐localization with Iba‐1, a microglia marker (Fig. 4B, arrows). Interestingly, KCa3.1 appeared distributed differently from Iba‐1 within same microglia. Substantial co‐localization was also found with CD68, a microglia/macrophage marker, but not with GFAP, an astrocytic marker (Fig. S2). Western blotting showed that human brains express a KCa3.1‐immunoreactive band slightly below the 50 kDa mark, consistent with that seen in mouse brains and a predicted molecular mass of 47.7 kDa (Fig. 4C). Quantitation of KCa3.1 band intensities revealed a substantial increase in AD brains compared to control brains (Fig. 4D).
Table 2.
Summary of human brain samples
| Cases | Alzheimer's disease | Cognitively normal controls |
|---|---|---|
| Number | 13 | 9 |
| Mean Age (± SE) | 81.1 (± 2.2) | 83.4 (± 3.0) |
| % Male | 43.5% | 57.1% |
| Braak Stage | V‐VI | I‐II |
| # with Stroke | 1.0 (7.6%) | 0 |
Figure 4.
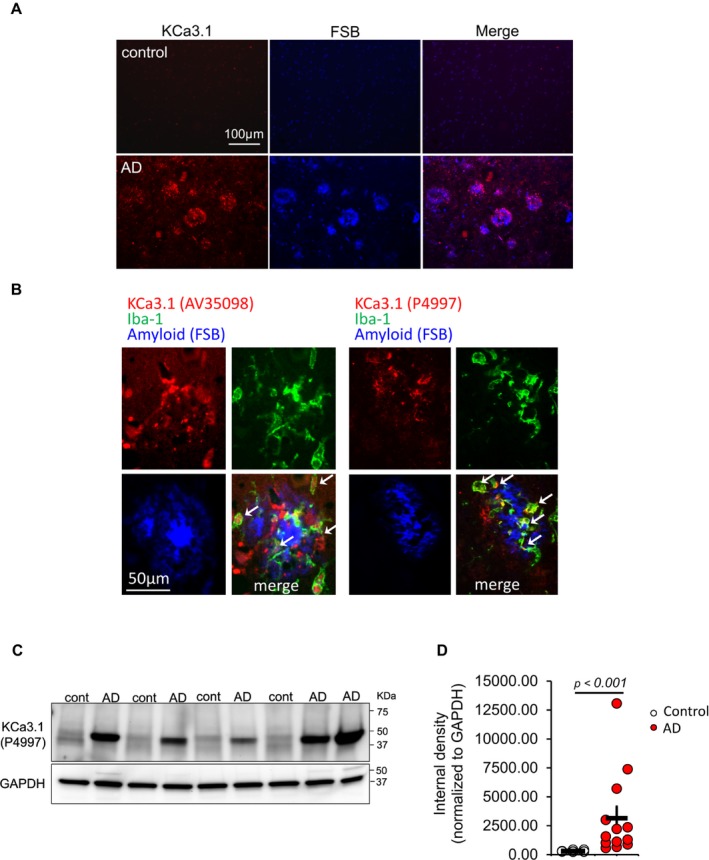
Increased KCa3.1 expression in Alzheimer's disease brains. KCa3.1 expression was assessed in 13 Alzheimer's disease (AD) and nine age‐ and sex‐matched control brain samples from the superior temporal cortex. (A) Sections were fluorescently stained with FSB (blue) and anti‐KCa3.1 (PA5:41015, red). KCa3.1 immunoreactivity is substantially increased in AD brains, and is enriched in or around amyloid plaques. (B) Co‐staining with anti‐KCa3.1 (left panel: AV35098, right panel: P4997), anti‐Iba‐1, and FSB using AD sections again shows enhanced KCa3.1 immunoreactivity in or around amyloid plaques. Many Iba‐1 + cells show KCa3.1 immunoreactivity (arrows). KCa3.1 immunoreactivity does not co‐localized with FSB‐positive fibrillary amyloid. (C–D) Western blot of superior temporal cortex homogenates showed increased cerebral levels of KCa3.1 in AD brains. Statistical analysis was conducted using nonparametric Wilcoxon rank sum test instead of t‐test due to non‐normality of the data.
KCa3.1 inhibition mitigates AβO‐induced impairment in hippocampal long‐term potentiation
hLTP is considered a cellular correlate of learning and memory. Aβ was shown to inhibit induction of NMDA receptor‐dependent hLTP. Notably, minocycline (a tetracycline inhibitor of microglial activation) and iNOS reduction were found to prevent the hLTP disruption by Aβ,30 suggesting that microglial activation and microglial NO production contribute to the detrimental effect of Aβ on synaptic function. Because KCa3.1 inhibition blocks AβO‐induced microglial neurotoxicity as well as NO production in hippocampal slices,9 we hypothesized that KCa3.1 blockade is able to prevent the microglia‐related deleterious effect of AβO on hLTP. Indeed, in the hLTP induced in Schaffer‐collateral‐CA1 synapses following high frequency stimulation, application of AβO to mouse hippocampal slices blocked hLTP induction at 20–100 nmol/L.16 Co‐perfusion of TRAM‐34 recovered hLTP from AβO‐induced reduction (Fig. 5A and B).
Figure 5.
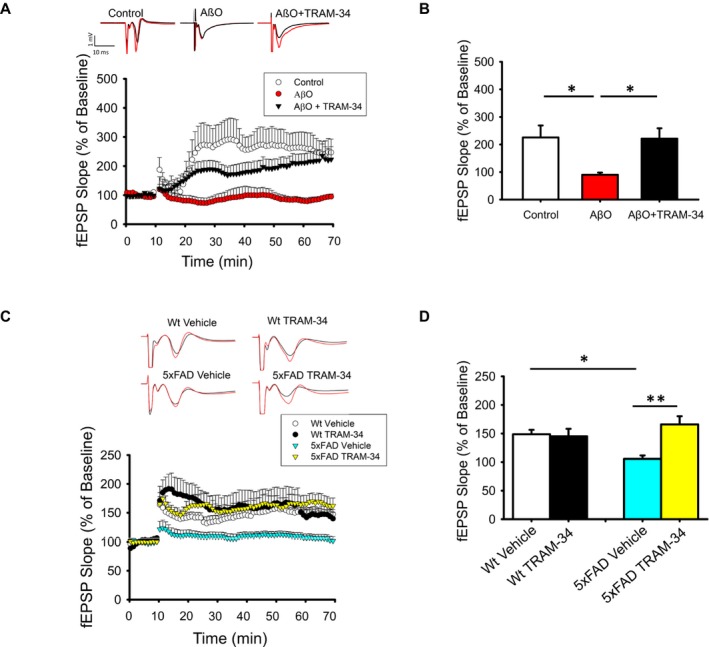
KCa3.1 inhibition mitigates AβO‐induced impairment in hLTP. (A–B) TRAM‐34 co‐perfusion prevented the inhibitory effect of AβO on amplitude of hLTP induced with high‐frequency stimulation (HFS). Hippocampal slices were incubated with TRAM‐34 (1 μmol/L) for 45 min and with AβO (50 nmol/L) for 10 min before recording, and perfused continuously with both throughout the recording. (A) Representative traces show the fEPSP of baseline (black trace) and at 60 min (red trace) after HFS. (B) Summary bar graphs showing the average fEPSP slope between 50 and 60 min after HFS. Data were compiled from recordings using slices obtained from the groups of control (five slices from five mice), AβO (eight slices from six mice), and AβO+TRAM‐34 (eight slices from six mice). AβO alone significantly inhibited the level of hLTP and this effect was prevented by co‐application of TRAM‐34. All data are presented as mean ± SE. One‐way ANOVA follow by Bonferroni post hoc test. *P < 0.005. (C–D) Intraperitoneally injected TRAM‐34 prevented the inhibitory effect of AβO on amplitude of hLTP induced with HFS. Twelve‐month‐old 5xFAD and Wt littermates received intraperitoneal TRAM‐34 (40 mg/kg) or vehicle only and the brain removed 24 h post‐injection for hLTP analysis. (C) Representative traces and time course of hLTP induced with HFS. (D) Summary bar graphs showing the average fEPSP slope between 50 and 60 min, compiled from recordings of Wt/vehicle (10 slices from three mice), Wt/TRAM‐34 (10 slices from three mice), 5xFAD/vehicle (10 slices from three mice), and 5xFAD/RAM‐34 (10 slices from three mice). 5xFAD mice had reduced amplitudes of hLTP but this reduction was mitigated by TRAM‐34 injection. All data are presented as mean ± SE. Two‐way ANOVA follow by Bonferroni post hoc test. *P < 0.05 and **P < 0.001.
To determine if such an effect can be demonstrated in vivo, we used 5xFAD mice, for whom AβO‐related hLTP deficits have been demonstrated.31 We intraperitoneally injected TRAM‐34 (40 mg/kg) or vehicle into 12‐month‐old 5xFAD mice and age‐matched Wt littermates, sacrificed them at 24‐hours post‐injection, and obtained hippocampal slices for recording. The magnitude of hLTP induction was significantly reduced in 5xFAD mice compared to Wt littermates, but was significantly increased to the Wt level in TRAM‐34‐treated 5xFAD mice (Fig. 5C and D). Taken together, our data suggest that KCa3.1 blockade ameliorates neuroinflammation and associated synaptic impairments induced by AβO.
Senicapoc is an orally available, brain penetrant KCa3.1 blocker
Although TRAM‐34 readily passes the blood brain barrier (Cbrain/Cplasma = 1.2)32 and is widely used as a tool compound for proof‐of‐concept rodent studies, it is not optimal for clinical use due to its lack of oral availability and short half‐life.33 To identify a clinically promising KCa3.1 inhibitor, we investigated the pharmacokinetic properties and the brain penetration of two other published KCa3.1 blockers: a phenyl‐pyran, reported by chemists at Bayer,34 and senicapoc35 (Fig. 6A). The Bayer 4‐phenyl‐4H‐pyra exhibited good brain penetration (Cbrain/Cplasma~3) but had a short half‐life of 15 min following intravenous administration at 10 mg/kg in mice (data not shown). We next resynthesized senicapoc (Fig. 6A), an orally available KCa3.1 blocker structurally similar to TRAM‐34.12 The purity of our senicapoc preparations was 96.9% (see Data S1). Despite its long (~7 day) half‐life in humans,12 senicapoc exhibited a relatively short but pharmacologically acceptable half‐life of roughly 1 h in mice and 4 h in rats (data not shown). Its brain concentrations on average were 5–7 times higher than plasma concentrations in both mice and rats (Fig. 6B). At one hour after intraperitoneal injection at 50 mg/kg, brain concentrations averaged 43 ± 8 μmol/L (n = 3) in mice and 43 ± 7 μmol/L (n = 2) in rats. A more detailed time course in mice revealed that senicapoc brain concentrations were significantly higher than plasma concentrations for at least 6 h after intraperitoneal administration (Fig. 6C). Senicapoc further achieved good plasma concentrations following oral gavage administration (Fig. 6D) with an oral availability of roughly 60%.
Senicapoc rectifies Aβ‐related microglial activation and neurotoxicity
Despite senicapoc's structural similarity to TRAM‐34 and its demonstrated selectivity for KCa3.1,35, 36 its effect on microglial activation has not been studied. We next determined if senicapoc rectifies AβO‐induced microglial abnormalities. In primary microglial cultures, senicapoc was able to block AβO‐induced microglial KCa3.1 currents (Fig. 7A), microglial proliferation (Fig. 7B), expression of microglial activation mediators IL‐1β, IL‐6 and iNOS (Fig. 7C), activation of p38MAPK (Fig. 7D), and production of NO (Fig. 7E). These in vitro effects are identical to those reported for TRAM‐34.9
Figure 7.
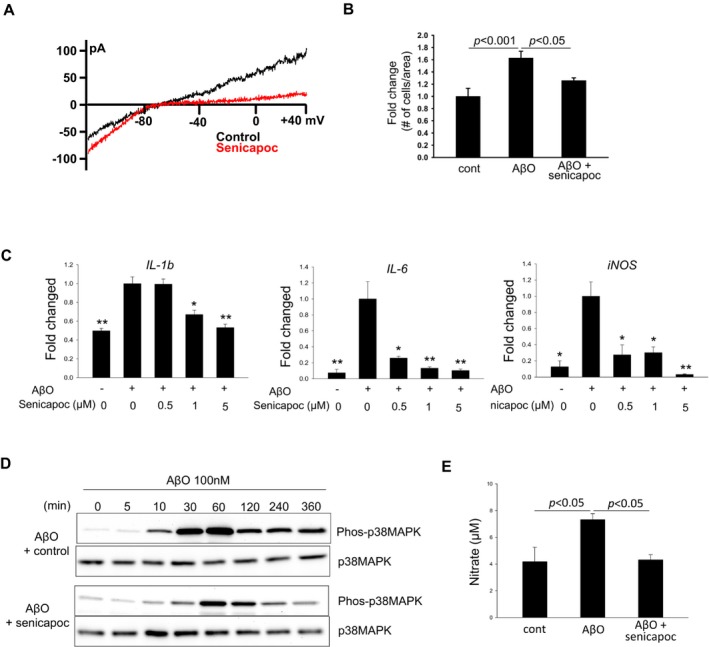
Senicapoc reduces proinflammatory microglial activation in vitro. Cultured primary microglia were activated by AβO for 24 h in the presence of senicapoc or vehicle control. Consistent with our previous observations,9 AβO (100 nmol/L) induced in microglia (A) a large KCa3.1 current as shown by whole‐cell patch clamp; (B) microglial proliferation; (C) increased transcript levels of proinflammatory modulators as shown by qPCR; (D) time‐dependent increases in the level of phosphor‐p38MAPK as shown by Western blotting; and (E) increased NO production. All these proinflammatory changes were significantly reduced by senicapoc (1 μmol/L if not otherwise indicated).
We subsequently tested the in vivo effects of senicapoc by feeding six‐month‐old 5xFAD mice with a senicapoc‐medicated diet. To determine if this medicated diet was able to achieve pharmacologically relevant senicapoc concentrations, we measured the morning (9 am) plasma and brain levels in two weeks after starting the diet. The result showed that brain senicapoc levels ranged between 1 and 2.5 μmol/L (Fig. S3). Considering senicapoc's IC50 of 11 nmol/L for KCa3.1 inhibition and its protein binding of 95%, such brain concentrations are sufficient to exert a pharmacodynamic effect.35, 36 Following ad lib access to the medicated diet for three months, senicapoc curbed microglial activation as evidenced by reduced CD11b immunoreactivity (45% reduction, P < 0.001, Fig. 8A). Similar reductions in Iba‐1 and CD68 immunoreactivities were also observed (Fig. S4). Senicapoc treatment also significantly reduced the number of microglia expressing KCa3.1 (Fig. 8B). qPCR showed significantly reduced expressions of proinflammatory modulators IL‐1β, TNF‐α, IL‐6, and iNOS (Fig. 8C), but no changes in several markers typical of an M2‐like phenotype (Fig. S5). Interestingly, it also resulted in a 39% reduction (P < 0.001) of cerebral amyloid deposits (Fig. 8A). Levels of TBS‐soluble and TBS‐insoluble/SDS‐soluble Aβ142 were reduced 22% and 35%, respectively, when measured by ELISA (Fig. 8D). This observation was corroborated with Western blotting for Aβ following Tricine gel electrophoresis (Fig. 8E).
Figure 8.
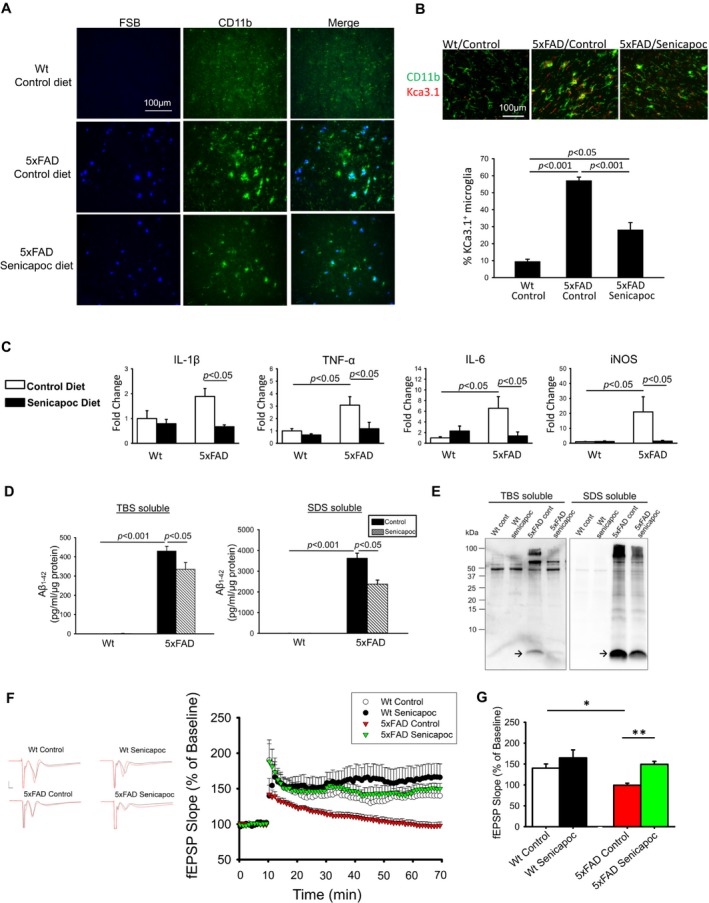
The effect of chronic oral senicapoc treatment in 5xFAD mice. Six‐month‐old 5xFAD and Wt littermates were fed ad lib senicapoc‐medicated or control diet (n = 9 per group) for three months and the brains removed for analysis. (A) Frontal cortex sections co‐stained with anti‐CD11b and FSB showed that senicapoc diet reduced the CD11b and FSB reactivities in 5xFAD brains. (B) Frontal cortex sections co‐stained with anti‐CD11b and anti‐KCa3.1 (APC064) were analyzed by co‐localization analysis. 5xFAD mice showed increased % of microglia expressing KCa3.1, which was reduced by senicapoc treatment. (C) The transcript levels of indicated proinflammatory modulators in acutely isolated microglia were increased in 5xFAD mice consuming control diet, compared to the Wt littermates. Senicapoc diet reduced the expression of all four modulators. Two‐way ANOVA follow by Bonferroni post hoc test. (D) The fresh brains were fractionated into TBS‐soluble and TBS‐insoluble, SDS‐soluble fractions, which were used for ELISA quantification of Aβ42. Senicapoc diet significantly reduced the levels of Aβ42 in both fractions. Two‐way ANOVA follow by Bonferroni post hoc test. (E) The ELISA result was corroborated with Tricine gel electrophoresis/Western blotting, which showed reduced Aβ band (arrow) intensities in both TBS‐solule and SDS‐soluble fractions of 5xFAD brains. (F) Traces and time course of hLTP induced with HFS. (G) Summary bar graphs showing the average fEPSP slope between 50 and 60 min, compiled from recordings of Wt/control (eight slices from five mice), Wt/Senicapoc (eight slices from five mice), 5xFAD/control (12 slices from six mice), and 5xFAD/senicapoc (10 slices from six mice). Data are expressed as mean percent change in fEPSP slope ± SE. from baseline. Two‐way ANOVA follow by Bonferroni post hoc test. *P < 0.005, **P < 0.001. 5xFAD mice on control diet had reduced amplitudes of hLTP but this reduction was mitigated by senicapoc diet.
As reductions of soluble Aβ species and microglial activation following senicapoc treatment may help mitigate synaptic impairment, we further tested if KCa3.1 inhibition by senicapoc in vivo would improve hLTP.31 Indeed, we found that senicapoc treatment rectified the reduced induction of hLTP in 5xFAD mice to the level of Wt mice (Fig. 8F and G).
Discussion
Therapies aimed at microglia face several challenges as activated microglia can exert both beneficial and harmful effects in AD. Such therapies should ideally: (1) achieve reduced neurotoxicity while maintaining or enhancing neuroprotective functions of microglia, such as phagocytosis of Aβ aggregates; (2) be relatively specific to microglia to avoid adversely affecting important neuronal functions; (3) not be broadly immunosuppressive. We consider inhibition of KCa3.1 would selectively modulate microglial activation by “fine‐tuning” Ca2+ signaling and selectively modulating activation signaling pathways. Evidence from our laboratory and others’ indicates that KCa3.1 blockers inhibit microglia‐mediated neurotoxicity without affecting microglial migration and phagocytosis.4, 5, 6, 7, 8, 9, 10 Moreover, KCa3.1 blockers are very mild immunosuppressants that do not affect the ability of rodents to clear infections.10 Therefore in this study, we intended to use animal models and human postmortem brain samples to show that KCa3.1 is a biologically relevant and microglial target for AD. Several conclusions can be drawn: (1) soluble AβO, the highly toxic Aβ species implicated as one of the principal initiators of AD phenotypes, enhances microglial KCa3.1 activity; (2) KCa3.1 is required for AβO‐induced microglial proinflammatory activation and neurotoxicity; (3) KCa3.1 expression/activity is upregulated in transgenic AD animals and human AD brains; and (4) pharmacological inhibition of KCa3.1 with TRAM‐34 or senicapoc in mice mitigates some key AD‐like phenotypes including neuroinflammation, hLTP deficit and amyloid pathology. Our study supports that KCa3.1 is a candidate therapeutic target for AD.
KCa3.1 appears particularly upregulated in amyloid plaque‐associated microglia, notably in human AD brains (Fig. 4). Recent evidence suggests that plaque‐associated microglia and plaque‐distant microglia are in different activation states,37 with plaque‐associated microglia exhibiting a “hyperactive” state.38 Plescher et al. found that, in acute cerebral slices of the TgCRND8 mouse model of AD, plaque‐associated microglia expressed significant Kv and Kir currents with the latter most likely attributed to Kir2.1..37 This result is consistent with our previous finding of enhanced microglial Kv and Kir2.1 activity/expression in 5xFAD mice,16 but the role of KCa3.1 was not investigated. It would be interesting in future experiments using single‐cell approaches to determine the KCa3.1 expression and activity in plaque‐associated microglia or in “disease‐associated microglia (DAM)”, a unique microglia‐type recently identified in 5xFAD mice that has the potential to restrict neurodegeneration,39 and to test if inhibition of KCa3.1 in such subgroups of microglia affects their disease‐related phenotype.
The significance of microglia in modulating AD‐like amyloid pathology remains controversial. Our in vivo data (Fig. 8A–E) of substantial reductions of soluble and insoluble Aβ species in mice chronically treated with senicapoc suggest that KCa3.1 inhibition may enhance the microglial Aβ clearance capacity that is suppressed in AD transgenic mice.40, 41 Our result is consistent with several reports showing that modulation of specific microglial signaling pathways alters Aβ deposition and clearance,40, 42, 43, 44, 45, 46, 47 but appears contradictory to reports showing that inducible microglia depletion by genetic or pharmacological means fails to affect amyloid pathology in transgenic AD models.48, 49 Although further studies are needed to resolve this controversy, our data using K+ channel expression as a readout show that the microglial activation profiles in transgenic AD mice are complex.16 Therefore, differences in the timing and approaches of neuro‐immunomodulation to alter microglia quantity or signaling may differentially influence the outcomes.
One limitation of our study is that we could not demonstrate significant memory deficits in 5xFAD/control diet mice compared to Wt mice at 9 months of age despite their large cerebral amyloid load, therefore the cognitive benefit of senicapoc could not be demonstrated in our current trial. Two tasks were tested: novel object recognition and step‐through passive avoidance. Using these two tests, we did successfully demonstrated deficits in 10‐month‐old APP/PS1 [B6.Cg‐Tg(APPswe,PSEN1dE9)85Dbo/Mmjax] mice in our prior study,16 arguing against technical issues. Future therapeutic trials using alternative models and larger numbers of animals are warranted.
Based on our data, here we propose repurposing senicapoc for AD treatment. Drug repurposing offers a rapid and economic route to the clinic than new drug discovery, which on average takes at least 15 years and $1.5 billion to bring a new successful drug to the market. It is therefore advantageous to reposition known targets, such as KCa3.1, for which a wealth of pharmacological knowledge has been accumulated, and safety has been demonstrated. Our finding that senicapoc is brain penetrant (Cbrain/Cplasma~5) and orally available makes it suitable for the potential treatment of AD. Senicapoc was initially developed for sickle cell anemia. Since K+ efflux through KCa3.1 is a major pathway for erythrocyte dehydration during sickling,50 it appeared very promising to test KCa3.1 inhibitors, which had been shown to prevent erythrocyte dehydration in both patients51 and mouse models.52 The Phase‐2 clinical trial showed that senicapoc reduced hemolysis and increased hemoglobin levels.12 However, in Phase‐3, senicapoc failed to achieve its primary clinical end‐point, which was reduction in the rate of vaso‐occlusive pain crisis, despite clearly improving all hematological parameters.53 Since senicapoc were demonstrated safe and well‐tolerated in humans, it was deposited in the NIH National Center for Advancing Translational Research (NCAT) library as PF‐05416266, making it available for investigator‐initiated clinical trials. In conclusion, our preclinical data support that repurposing of senicapoc has the potential to expedite the urgently needed new drug discovery for AD.
Conflict of Interest
The authors declare no conflicts of interest.
Author Contributions
L‐WJ, HW, and IM conceived and designed the experiments. Performed the experiments: LWJ, JDL, HMN, VS, LS, MC, TB, HW, and IM performed the experiments. L‐WJ, JDL, VS, LS, HMN, HW, and IM analyzed the data. L‐WJ, HW, and IM wrote the paper.
Supporting information
Figure S1. Microglial KCa3.1 expression is elevated in brains of 4‐month‐old 5xFAD mice.
Figure S2. Microglial KCa3.1 expression in human AD brains is located to microglia but not astrocytes.
Figure S3. Treatment with senicapoc‐medicated diet resulted in therapeutically active brain levels.
Figure S4. The effect of chronic oral senicapoc treatment in 5xFAD mice on Iba‐1 and CD68 immunoreactivities.
Figure S5. Senicapoc treatment did not significantly affect markers typically associated with an M2‐like phenotype.
Data S1. Supplementary Methods.
Acknowledgments
This work was supported by a grant from the BrightFocus Foundation (A2013414S) to L‐W.J, I.M., and H.W, a grant from the UC Davis Department of Pathology and Laboratory Medicine to I.M., and U.S. National Institutes of Health (NIH) grants R01 AG043788 (NIA) to I.M. and R01 NS100294 (NINDS) to H.W. This work was also supported in part by P30 AG10129 (NIA) to L‐W.J.. We thank Dr. Danielle Harvey for assistance in statistical analysis.
Funding Information
This work was supported by a grant from the BrightFocus Foundation (A2013414S) to L‐W.J, I.M., and H.W, a grant from the UC Davis Department of Pathology and Laboratory Medicine to I.M., and U.S. National Institutes of Health (NIH) grants R01 AG043788 (NIA) to I.M. and R01 NS100294 (NINDS) to H.W. This work was also supported in part by P30 AG10129 (NIA) to L‐W.J. and I.M. We thank Dr. Danielle Harvey for assistance in statistical analysis.
Funding Statement
This work was funded by BrightFocus Foundation grant A2013414S; UC Davis Department of Pathology and Laboratory Medicine grant ; U.S. National Institutes of Health (NIH) grant ; NIA grants R01 AG043788 and P30 AG10129; NINDS grant R01 NS100294.
References
- 1. Zhang B, Gaiteri C, Bodea LG, et al. Integrated systems approach identifies genetic nodes and networks in late‐onset Alzheimer's disease. Cell 2013;153:707–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Seyfried NT, Dammer EB, Swarup V, et al. A multi‐network approach identifies protein‐specific co‐expression in asymptomatic and symptomatic Alzheimer's disease. Cell Systems 2017;4:60–72.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Karch CM, Cruchaga C, Goate AM. Alzheimer's disease genetics: from the bench to the clinic. Neuron 2014;83:11–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wulff H, Castle NA. Therapeutic potential of KCa3.1 blockers: recent advances and promising trends. Expert Rev Clin Pharmacol 2010;3:385–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Feske S, Wulff H, Skolnik EY. Ion channels in innate and adaptive immunity. Annu Rev Immunol 2015;33:291–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Di L, Srivastava S, Zhdanova O, et al. Inhibition of the K+ channel KCa3.1 ameliorates T cell‐mediated colitis. Proc Natl Acad Sci U S A 2010;107:1541–1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ghanshani S, Wulff H, Miller MJ, et al. Up‐regulation of the IKCa1 potassium channel during T‐cell activation. Molecular mechanism and functional consequences. J Biol Chem 2000;275:37137–37149. [DOI] [PubMed] [Google Scholar]
- 8. Kaushal V, Koeberle PD, Wang Y, Schlichter LC. The Ca2+‐activated K+ channel KCNN4/KCa3.1 contributes to microglia activation and nitric oxide‐dependent neurodegeneration. J Neurosci 2007;27:234–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Maezawa I, Zimin PI, Wulff H, Jin LW. Amyloid‐β protein oligomer at low nanomolar concentrations activates microglia and induces microglial neurotoxicity. J Biol Chem 2011;286:3693–3706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Maezawa I, Jenkins DP, Jin BE, Wulff H. Microglial KCa3.1 channels as a potential therapeutic target for Alzheimer's disease. Int J Alz Dis 2012;2012:868972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ataga KI, Orringer EP, Styles L, et al. Dose‐escalation study of ICA‐17043 in patients with sickle cell disease. Pharmacotherapy 2006;26:1557–1564. [DOI] [PubMed] [Google Scholar]
- 12. Ataga KI, Smith WR, De Castro LM, et al. Efficacy and safety of the Gardos channel blocker, senicapoc (ICA‐17043), in patients with sickle cell anemia. Blood 2008;111:3991–3997. [DOI] [PubMed] [Google Scholar]
- 13. Ataga KI, Stocker J. Senicapoc (ICA‐17043): a potential therapy for the prevention and treatment of hemolysis‐associated complications in sickle cell anemia. Expert Opin Investig Drugs 2009;18:231–239. [DOI] [PubMed] [Google Scholar]
- 14. Oakley H, Cole SL, Logan S, et al. Intraneuronal beta‐amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer's disease mutations: potential factors in amyloid plaque formation. J Neurosci 2006;26:10129–10140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wulff H, Miller MJ, Haensel W, et al. Design of a potent and selective inhibitor of the intermediate‐conductance Ca2+‐activated K+ channel, IKCa1: A potential immunosuppressant. Proc Natl Acad Sci USA 2000;97:8151–8156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Maezawa I, Nguyen HM, Di Lucente J, et al. Kv1.3 inhibition as a potential microglia‐targeted therapy for Alzheimer's disease: preclinical proof of concept. Brain 2018;141:596–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Maezawa I, Hong HS, Liu R, et al. Congo red and thioflavin‐T analogs detect Abeta oligomers. J Neurochem 2008;104:457–468. [DOI] [PubMed] [Google Scholar]
- 18. Hong HS, Maezawa I, Yao N, et al. Combining the rapid MTT formazan exocytosis assay and the MC65 protection assay led to the discovery of carbazole analogs as small molecule inhibitors of Abeta oligomer‐induced cytotoxicity. Brain Res 2007;1130:223–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chen YJ, Nguyen HM, Maezawa I, et al. The potassium channel KCa3.1 constitutes a pharmacological target for neuroinflammation associated with ischemia/reperfusion stroke. J Cereb Blood Flow Metab 2016;36:2146–2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Horiuchi M, Smith L, Maezawa I, Jin LW. CX3CR1 ablation ameliorates motor and respiratory dysfunctions and improves survival of a Rett syndrome mouse model. Brain Behav Immun 2017;60:106–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yi M, Yu P, Lu Q, et al. KCa3.1 constitutes a pharmacological target for astrogliosis associated with Alzheimer's disease. Mol Cell Neurosci 2016;76:21–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wei T, Yi M, Gu W, et al. The potassium channel KCa3.1 represents a valid pharmacological target for astrogliosis‐induced neuronal impairment in a mouse model of Alzheimer's disease. Front Pharmacol 2016;7:528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Blomster LV, Strobaek D, Hougaard C, et al. Quantification of the functional expression of the Ca2+ ‐activated K+ channel KCa 3.1 on microglia from adult human neocortical tissue. Glia 2016;64:2065–2078. [DOI] [PubMed] [Google Scholar]
- 24. Nguyen HM, Blomster LV, Christophersen P, Wulff H. Potassium channel expression and function in microglia: plasticity and possible species variations. Channels 2017;11:305–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hong HS, Maezawa I, Budamagunta M, et al. Candidate anti‐Aβ fluorene compounds selected from analogs of amyloid imaging agents. Neurobiol Aging 2010;31:1690–1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jin LW, Horiuchi M, Wulff H, et al. Dysregulation of glutamine transporter SNAT1 in Rett syndrome microglia: a mechanism for mitochondrial dysfunction and neurotoxicity. J Neurosci 2015;35:2516–2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nguyen HM, Grossinger EM, Horiuchi M, et al. Differential Kv1.3, KCa3.1, and Kir2.1 expression in “classically” and “alternatively” activated microglia. Glia 2017;65:106–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hyman BT, Phelps CH, Beach TG, et al. National Institute on Aging‐Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease. Alzheimers Dement 2012;8:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Alafuzoff I, Arzberger T, Al‐Sarraj S, et al. Staging of neurofibrillary pathology in Alzheimer's disease: a study of the BrainNet Europe Consortium. Brain Pathol 2008;18:484–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wang Q, Rowan MJ, Anwyl R. Beta‐amyloid‐mediated inhibition of NMDA receptor‐dependent long‐term potentiation induction involves activation of microglia and stimulation of inducible nitric oxide synthase and superoxide. J Neurosci 2004;24:6049–6056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Maezawa I, Zou B, Di Lucente J, et al. The anti‐amyloid‐β and neuroprotective properties of a novel tricyclic pyrone molecule. J Alzheimers Dis 2017;58:559–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chen YJ, Raman G, Bodendiek S, et al. The KCa3.1 blocker TRAM‐34 reduces infarction and neurological deficit in a rat model of ischemia/reperfusion stroke. J Cereb Blood Flow Metab 2011;31:2363–2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Al‐Ghananeem AM, Abbassi M, Shrestha S, et al. Formulation‐based approach to support early drug discovery and development efforts: a case study with enteric microencapsulation dosage form development for a triarylmethane derivative TRAM‐34; a novel potential immunosuppressant. Drug Dev Ind Pharm 2010;36:563–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Urbahns K, Horvath E, Stasch JP, Mauler F. 4‐Phenyl‐4H‐pyrans as IK(Ca) channel blockers. Bioorg Med Chem Lett 2003;13:2637–2639. [DOI] [PubMed] [Google Scholar]
- 35. McNaughton‐Smith GA, Burns JF, Stocker JW, et al. Novel inhibitors of the Gardos channel for the treatment of sickle cell disease. J Med Chem 2008;51:976–982. [DOI] [PubMed] [Google Scholar]
- 36. Stocker JW, De Franceschi L, McNaughton‐Smith GA, et al. ICA‐17043, a novel Gardos channel blocker, prevents sickled red blood cell dehydration in vitro and in vivo in SAD mice. Blood 2003;101:2412–2418. [DOI] [PubMed] [Google Scholar]
- 37. Plescher M, Seifert G, Hansen JN, et al. Plaque‐dependent morphological and electrophysiological heterogeneity of microglia in an Alzheimer's disease mouse model. Glia 2018;66:1464–1480. [DOI] [PubMed] [Google Scholar]
- 38. Yin Z, Raj D, Saiepour N, et al. Immune hyperreactivity of Aβ plaque‐associated microglia in Alzheimer's disease. Neurobiol Aging 2017;55:115–122. [DOI] [PubMed] [Google Scholar]
- 39. Keren‐Shaul H, Spinrad A, Weiner A, et al. A unique microglia type associated with restricting development of Alzheimer's disease. Cell 2017;169:1276–12790.e17. [DOI] [PubMed] [Google Scholar]
- 40. Hellwig S, Masuch A, Nestel S, et al. Forebrain microglia from wild‐type but not adult 5xFAD mice prevent amyloid‐beta plaque formation in organotypic hippocampal slice cultures. Sci Rep 2015;5:14624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hickman SE, Allison EK, El Khoury J. Microglial dysfunction and defective beta‐amyloid clearance pathways in aging Alzheimer's disease mice. J Neurosci 2008;28:8354–8360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Guillot‐Sestier MV, Doty KR, Gate D, et al. Il10 deficiency rebalances innate immunity to mitigate Alzheimer‐like pathology. Neuron 2015;85:534–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Krauthausen M, Kummer MP, Zimmermann J, et al. CXCR3 promotes plaque formation and behavioral deficits in an Alzheimer's disease model. J Clin Invest 2015;125:365–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. El Khoury J, Toft M, Hickman SE, et al. Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer‐like disease. Nat Med 2007;13:432–438. [DOI] [PubMed] [Google Scholar]
- 45. Johansson JU, Woodling NS, Wang Q, et al. Prostaglandin signaling suppresses beneficial microglial function in Alzheimer's disease models. J Clin Invest 2015;125:350–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Cameron B, Tse W, Lamb R, et al. Loss of interleukin receptor‐associated kinase 4 signaling suppresses amyloid pathology and alters microglial phenotype in a mouse model of Alzheimer's disease. J Neurosci 2012;32:15112–15123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Heneka MT, Kummer MP, Stutz A, et al. NLRP3 is activated in Alzheimer's disease and contributes to pathology in APP/PS1 mice. Nature 2013;493:674–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Grathwohl SA, Kalin RE, Bolmont T, et al. Formation and maintenance of Alzheimer's disease beta‐amyloid plaques in the absence of microglia. Nature Neurosci 2009;12:1361–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Spangenberg EE, Lee RJ, Najafi AR, et al. Eliminating microglia in Alzheimer's mice prevents neuronal loss without modulating amyloid‐beta pathology. Brain 2016;139:1265–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Brugnara C. Sickle cell disease: from membrane pathophysiology to novel therapies for prevention of erythrocyte dehydration. J Pediatr Hematol Oncol 2003;25:927–933. [DOI] [PubMed] [Google Scholar]
- 51. Brugnara C, Gee B, Armsby CC, et al. Therapy with oral clotrimazole induces inhibition of the Gardos channel and reduction of erythrocyte dehydration in patients with sickle cell disease. J Clin Invest 1996;97:1227–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. De Franceschi L, Saadane N, Trudel M, et al. Treatment with oral clotrimazole blocks Ca(2 + )‐activated K+ transport and reverses erythrocyte dehydration in transgenic SAD mice. A model for therapy of sickle cell disease. J Clin Invest 1994;93:1670–1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ataga KI, Reid M, Ballas SK, et al. Improvements in haemolysis and indicators of erythrocyte survival do not correlate with acute vaso‐occlusive crises in patients with sickle cell disease: a phase III randomized, placebo‐controlled, double‐blind study of the Gardos channel blocker senicapoc (ICA‐17043). Br J Haematol 2011;153:92–104. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Microglial KCa3.1 expression is elevated in brains of 4‐month‐old 5xFAD mice.
Figure S2. Microglial KCa3.1 expression in human AD brains is located to microglia but not astrocytes.
Figure S3. Treatment with senicapoc‐medicated diet resulted in therapeutically active brain levels.
Figure S4. The effect of chronic oral senicapoc treatment in 5xFAD mice on Iba‐1 and CD68 immunoreactivities.
Figure S5. Senicapoc treatment did not significantly affect markers typically associated with an M2‐like phenotype.
Data S1. Supplementary Methods.