

Abstract
Objective
Cerebral amyloid angiopathy (CAA) is caused by cerebrovascular deposition of β‐amyloid fragments leading to cerebrovascular dysfunction and other brain injuries. This phase 2, randomized, double–blind trial in patients with probable CAA assessed the efficacy and safety of ponezumab, a novel monoclonal antibody against Aβ 1–40.
Methods
Thirty‐six participants aged 55–80 years with probable CAA received intravenous placebo (n = 12) or ponezumab (n = 24). The change from baseline to Days 2 and 90 in cerebrovascular reactivity (CVR) was measured in the visual cortex as the natural log of the rising slope of the BOLD fMRI response to a visual stimulus. Safety and tolerability were also assessed.
Results
The mean change from baseline to Day 90 was 0.817 (ponezumab) and 0.958 (placebo): a mean ratio of 0.852 (90% CI 0.735–0.989) representing a trend towards reduced CVR in the ponezumab group. This trend was not present at Day 2. There was one asymptomatic occurrence of amyloid–related imaging abnormality–edema in the ponezumab group. The total number of new cerebral microbleeds from baseline to day 90 did not differ between groups. The ponezumab group had a participant with nonfatal new cerebral hemorrhage with aphasia and a participant with subdural hemorrhage that site investigators deemed to be nondrug related. In the placebo group one participant had a fatal intracerebral hemorrhage and one participant had migraine with aura.
Interpretation
Ponezumab was safe and well‐tolerated. The ponezumab group showed a trend towards treatment effect at Day 90 that was opposite to the hypothesized direction. The prespecified efficacy criteria were thus not met.
Introduction
Cerebral Amyloid Angiopathy (CAA) is a progressive neurovascular disease characterized by deposition of the β‐amyloid peptide, especially Aβ 1–40, in the walls of cortical and leptomeningeal vessels.1 Accumulation of β‐amyloid leads to cerebrovascular dysregulation, lobar cerebral hemorrhages, microbleeds, cortical superficial siderosis, and nonhemorrhagic forms of brain injury such as white matter lesions and microinfarcts. Collectively, these brain injuries result in impaired cognitive and motor function.1 These clinical symptoms can resemble or coexist with those of other neurodegenerative conditions such as Alzheimer's Disease (AD), leading to underestimation of its prevalence.2 Comorbidity of CAA and AD is common, and CAA contributes to the clinical manifestations seen in AD–related dementia3 and to the Amyloid–Related Imaging Abnormalities (ARIA) identified in trials of antiamyloid immunotherapy.4 Estimates suggest that greater than 30% of the individuals over age 65 may have at least some underlying CAA.1, 5 Vascular dysfunction likely mediates ischemic consequences of CAA such as leukoaraiosis, microinfarction, and cortical atrophy6, 7, 8, 9 and thus contribute to CAA–related cognition and gait, impairments.
T2*‐weighted Magnetic Resonance Imaging (MRI) can reveal the presence of characteristic hemorrhages. Another MRI modality, visual stimulus–driven functional MRI (fMRI), has been used to evaluate the markedly impaired vascular regulation that accompanies CAA.10, 11, 12 The fMRI biomarkers constitute radiologic surrogates for vascular dysfunction and open the possibility of detecting neurophysiologic changes in response to treatment in a relatively short period of time.
Passive immunotherapy has been proposed as a means of targeting circulating amyloid and plaques for treatment of degenerative diseases associated with brain amyloid deposition such as AD or CAA.13, 14, 15 It is conceivable that, by decreasing vascular amyloid, vascular dysfunction in CAA could be improved, potentially leading to a decreased ischemic injury and resultant cognitive decline.
Ponezumab is a humanized immunoglobulin G2 (IgG2) monoclonal antibody targeted against an epitope encompassing the C‐terminal amino acids of the Aβ 1–40 peptide derived from the human amyloid precursor protein (APP). Mutations (A330S and P331S) in the IgG2Δa Fc region are intended to minimize the ability of ponezumab to activate complement or support antibody‐dependent cell‐mediated cytotoxicity. As Aβ 1–40 is the predominant species present in blood vessel walls, ponezumab was developed to prevent or reverse β‐amyloid aggregation and deposition and thus prevent or reduce CAA progression with minimal ARIA risk.15
The current study sought to assess the safety, tolerability, and efficacy of intravenous ponezumab versus placebo for improving vascular reactivity in adult participants with CAA (NCT01821118).
Methods
Study design
This was a phase 2, randomized, double–blind, parallel group, placebo–controlled trial examining the effects of intravenous (IV) ponezumab in adults with probable CAA.16, 17 The study was intitiated at eleven sites and conducted at ten sites in five countries: USA (5), Canada (2), UK, Netherlands, and France (1 each). The primary efficacy endpoint was change from baseline to Day 2 or Day 90 in cerebrovascular reactivity measured by blood oxygenation level dependent (BOLD) fMRI in response to visual stimulation. Safety, tolerability, and pharmacokinetics (PK) of ponezumab were also assessed.
Inclusion/exclusion criteria
The study included men and women of nonchildbearing potential ages 55–80 with probable CAA per the modified Boston criteria16, 17 and an acceptable structural MRI scan in the previous 12 months. Study participants were also required to have corrected vision at 20/50 or better on a Snellen chart. Potential participants were excluded if their CAA disease resulted in cognitive or functional deficits as documented by the Principal Investigator (PI) in consultation with the sponsor. Other exclusions included: clinical diagnosis of probable AD dementia or significant cognitive impairment (defined as a score of <26 on the Mini Mental State Examination [MMSE]); history of cancer within the last 5 years (except for excised cutaneous basal or squamous cell cancer resolved, excised colonic polyp, or nonprogressive prostate cancer per investigator's judgment); baseline BOLD fMRI of insufficient quality; uncontrolled hypertension; use of concomitant anticoagulation medications, antiinflammatory treatments given for CAA, or cognition–affecting drugs (anticholinergics and acetylcholinesterase inhibitors/memantine).
Study drug administration
Ponezumab or placebo was administered by IV infusion in a total volume of 100 mL over 10–15 min. Ponezumab was administered at a dose of 10 mg/kg at Day 1 followed by 7.5 mg/kg at Days 30 and 60.
Primary and secondary efficacy endpoints
The primary efficacy endpoint was change from baseline to Day 2 or Day 90 in cerebrovascular reactivity as measured by the natural logarithm of the slope of the visual stimulus–driven fMRI BOLD response. The slope was chosen as the primary endpoint based on a linear discriminant analysis of previous study data10 where the modelled amplitude of the hemodynamic response function divided by the time to reach the peak amplitude (on a logarithmic scale) was found to provide better differentiation from controls by incorporating both the reduced amplitude and delayed time to peak associated with CAA. The secondary efficacy endpoints were change from baseline to Day 2 or to Day 90 in cerebrovascular reactivity measured by the individual parameters for time to peak, amplitude, and time to baseline from the time‐course of the visual stimulus–driven fMRI hemodynamic response. The secondary pharmacodynamics (PD) endpoint was change from baseline in total plasma Aβ 1–40 concentrations at Days 1, 2, 30, 90, and 240. Aβ 1–40 concentrations were measured using the V‐Plex™ Aβ Peptide Panel 1 (6E10) Kit (Meso Scale Diagnostics, Rockville, MD, USA) followed by solid phase extraction. Pharmacokinetic (PK) samples were analyzed for ponezumab concentrations using a validated, sensitive, and specific enzyme–linked immunosorbent assay (ICON Laboratory Services Inc., Whitesboro, New York, NY, USA).
Magnetic resonance imaging
BOLD fMRI scans (GE‐EPI acquired at TR = 1.5 sec, TE = 27–30 msec, FA = 75°, res = 3.4 × 3.4 mm2, thickness = 2.9–3.0 mm) were performed during visual stimulation at baseline, Day 2, and Day 90. The details of the acquisition parameters were selected to mimic those previously published10 with modifications in echo time and flip angle values to optimize functional contrast at 3 T instead of 1.5 T. The temporal resolution of the image sampling was not compromised by the modifications and the number of presentations (four per run) and on/off frequency of the stimulus presentation (20 sec on/28 sec off) did not change from the prior reports. The visual stimulus was a standard 8 Hz flashing black‐and‐white radial checkerboard image alternating with a gray screen. Participant attention during the scan was assessed using a color–changing dot in the screen center that changed color at random intervals between 0.25 and 3.75 sec; participants were instructed to press a button when the dot color changed.10, 18 The BOLD fMRI time–course was used to assess vascular reactivity. All efficacy scans were analyzed centrally, according to standardized procedures and quality assurance (QA)/quality control (QC) guidelines (IXICO, Ltd., London, UK).
Structural MRI scans were performed at screening and Days 15, 45, and 90. T2‐weighted, T2*‐weighted, and fluid‐supressed T2‐weighted images were acquired for central detection of cerebral microbleeds (CMBs), infarcts, white matter hyperintensity, or ARIA–hemorrhage type (ARIA‐H) or ARIA–edema type (ARIA‐E) per Alzheimer Association Research Roundtable Working Group standards.4 For T2*‐weighted images, TE was nominally 20–25 msec, with in‐plane resolution approximately 1 mm2 and slice thickness 5 mm. A standard high‐resolution T1‐weighted image, based on ADNI2 parameters,19 was also acquired. All scans were performed on 3T scanners (Siemens, Philips, GE) and all scans analyzed centrally according to rigorous standardization and quality guidelines (IXICO, Ltd., London, UK).
Image analysis
Functional image series were first registered to each participant's high‐resolution T1‐weighted structural image, then warped into Montreal Neurological Institute (MNI) space, and resampled at 2 mm using IRTK software (BioMediaIA, London, UK). Preprocessing continued in the FSL software toolkit (Oxford Centre for Functional MRI of the Brain, Oxford, UK) with motion correction, nonlinear noise reduction (SUSAN), spatial smoothing with a 5‐mm Gaussian kernel, temporal filtering, and prewhitening. The FLOBS tool in FSL was then used to generate a hemodynamic response function basis set with three components that had sufficient flexibility to account for a large array of temporal delays expected in the data, and a general linear model used to fit the time–series data with the FSL tool FEAT. Region of Interest (ROI) analyses were performed using the FSL tool Featquery within two functionally defined regions in the occipital lobe. These ROIs (Fig. 1) were derived from an independent dataset from CAA participants acquired at 1.5 T,10 then mapped into MNI space for use across participants. The primary and secondary ROIs for the analysis were defined by the voxels activated in at least 50% or at least 25%, of the runs across all CAA participants, respectively. The primary ROI for the analysis, ROI1, was used to extract the mean of the modeled time–courses within the ROI. A larger secondary ROI, ROI2, was used to extract a z‐score–weighted mean of the modeled voxel‐wise time‐courses within the ROI. To generate summary metrics of the hemodynamic response, trial–averaged mean and z‐score–weighted mean modeled time courses from ROI1 and ROI2, respectively, were fit with a trapezoid. This strategy was chosen so that hemodynamic response metrics of amplitude, time to peak, and time to baseline could easily be compared with previous work using the same approach.10 To exclude contamination from severely impaired tissue, each ROI was also split into left and right cerebral hemispheres, generating separate unilateral modeled time–courses, which were each fit with a trapezoid. The decision to use the left hemisphere, right hemisphere, or full ROI for longitudinal analysis was determined for each participant based only on activation and trapezoid model fit quality standards applied to baseline functional runs.
Figure 1.
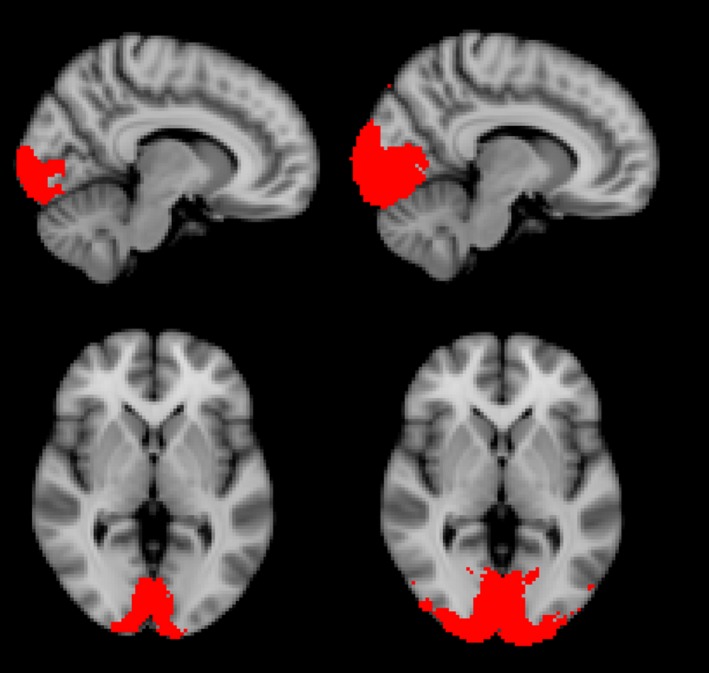
Locations of Regions of Interest (ROI). ROI1 is shown on the left, and ROI2 is shown on the right.
Safety evaluations
The safety endpoints included changes from baseline on physical and neurological assessments, Montreal Cognitive Assessment (MoCA), Columbia Suicide Severity Rating Scale (C‐SSRS), laboratory assessments, 12‐lead electrocardiogram (ECG), vital signs, immunogenicity, and adverse event (AE) monitoring. AEs were judged for likely relationship to treatment by the site investigator and reviewed by the central medical monitor; all determinations were performed without knowledge of treatment assignment. Structural MRI scans were read centrally for abnormalities such as ARIA‐H, ARIA‐E, and changes in CMB counts. CMB assessments were performed visually on standard, multi‐slice axial T2*‐weighted images. To mitigate longitudinal variability in CMB counts and so that emergent CMBs were more readily identifiable, readers blinded to treatment assignment labeled CMBs in their read system and images were viewed side‐by‐side with those from previous visits. All safety scans were interpreted centrally (IXICO, Ltd., London, UK), according to standardized procedures and QA/QC guidelines.
Statistical methods
Sample size calculations were based on an estimate of the variability obtained from previous study data.10 In addition to the variability estimate, these data also provided informative priors for placebo effects at 1 day and 3 months. The Bayesian approach, a statistical technique to incorporate prior information into data analysis, was utilized in the study, and the inclusion of the derived priors was expected to provide an equivalent of nine additional evaluable placebo participants for the study.
Based on the primary endpoint of cerebrovascular reactivity measured by the natural logarithm of the slope in visual stimulus–driven fMRI and utilizing the Bayesian informative prior on placebo, it was estimated that a sample size of 30 evaluable study participants – 20 ponezumab and ten placebo (approximately 1:1 ratio expected in analysis combined with the informative placebo prior) – was required to provide enough precision for a 2‐part predefined criteria for efficacy: C1: point estimate of ponezumab versus placebo effect >20% increase (improvement) in slope; C2: standard error of ponezumab versus placebo effect <60% of the point estimate. Given an expected dropout rate/technical failure of 16.7%, a total of 36 participants were planned for randomization.
For the primary endpoint, the Full Analysis Set (FAS; all participants who received at least one postdose efficacy measurement) was analyzed using an analysis of covariance (ANCOVA) model with treatment as a fixed effect and baseline as a covariate, conducted within a Bayesian framework utilizing the prederived informative prior for the placebo effect. An observed case approach (i.e., missing data excluded) was used, and an outlier–robust model was utilized in Bayesian modelling to down‐weight the influence of potential outliers. Sensitivity analyses using low informative prior only (i.e., essentially without prior information), Last Observation Carried Forward (LOCF), or all equally weighted data (i.e. outliers included), were also performed. The analysis was performed separately for Day 2 and Day 90. Analysis was performed on a natural logarithm (loge) scale.
For the analysis of secondary efficacy endpoints, analysis of change from baseline in time to peak, amplitude, and time to return to baseline was performed on a loge scale. Change from baseline in loge (time to peak) for Days 2 and 90 was analyzed using the same Bayesian method using an observed case approach (i.e., missing data excluded). Analyses were based on the FAS and used an outlier–robust model. The other secondary endpoints from the visual stimulus–driven fMRI (change from baseline in loge[amplitude] and loge[time to return to baseline]) were analyzed using ANCOVA with loge(baseline value) as a covariate. These analyses were also based on the FAS and analyses were performed separately for Days 2 and 90.
Ponezumab plasma concentrations were summarized and plotted for participants in the PK analysis set: all participants in the FAS for whom there was at least one ponezumab plasma concentration. The PD analysis set consisted of all participants in the FAS with at least one plasma Aβ concentration. Plasma Aβ was plotted against plasma ponezumab concentrations and the relationship between ponezumab plasma concentration and percent change from baseline to Days 2 and 90 for slope and time to peak were assessed by linear regression. For immunogenicity analysis, the proportion of participants with a measurable antibody response to ponezumab was summarized for each visit. Safety data were evaluated using descriptive statistics. The statistical analysis software package (SAS version 9.2, SAS Institute, Cary, NC, USA; OpenBUGS 3.2.3, rev 1012, Members of the OpenBUGS Project Management Group, Medical Research Council Biostatistics Unit, Cambridge UK, and Imperial College School of medicine, London UK) was used for all analyses.
Results
Between 25 June 2013 and 20 January 2015, 67 participants were screened for study entry and 36 were randomized to treatment: 24 to ponezumab and 12 to placebo (Fig. 2). Thirty‐five completed the study. All 36 randomized participants were included in the FAS and the safety analyses.
Figure 2.
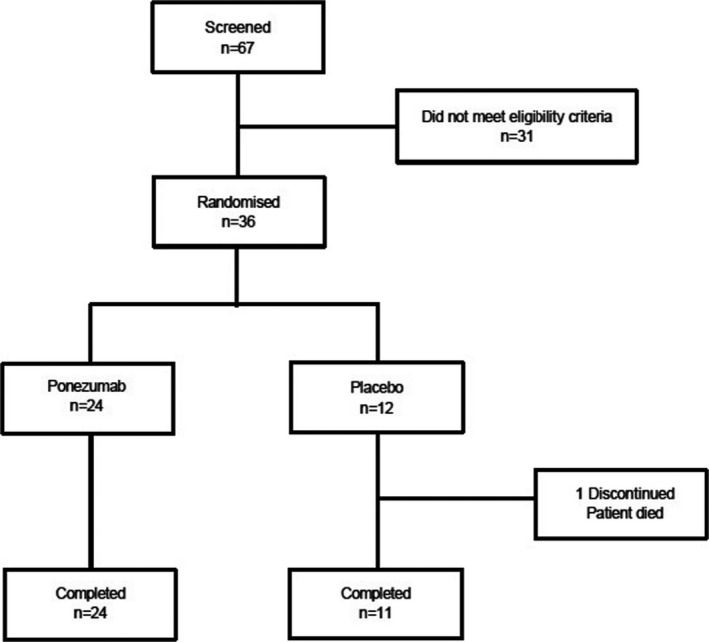
CONSORT flow diagram.
Demographic and baseline characteristics are listed in Table 1. A majority of participants were male (23/36), ≥65 years (24/36 participants), and white (35/36 participants). Compared with the placebo group, nonsignificantly greater numbers of participants in the ponezumab group had moderate baseline white matter hyperintensities (13/24 ponezumab vs. 5/12 placebo) and baseline CMB counts > 300 (6/24 ponezumab vs. 1/12 placebo). There was also an imbalance in apolipoprotein E genotype, with 50% of the ponezumab participants with the E3/E4 or E4/E4 genotype versus 25% in the placebo group.
Table 1.
Demographics and baseline characteristics
| Ponezumab (n = 24) | Placebo (n = 12) | |
|---|---|---|
| Gender, Male (%) | 16 (66.7) | 7 (58.3) |
| Age, Mean (SD) | 68.8 (6.8) | 65.0 (5.7) |
| Weight, Mean (SD) kg | 75.3 (14.5) | 73.5 (12.1) |
| BMI, Mean (SD) kg/m2 | 25.9 (3.0) | 25.2 (3.7) |
| Height, Mean (SD), cm | 169.6 (10.5) | 170.6 (11.9) |
| Lobar CMB (%) | ||
| 0–10 | 8 (33.3) | 5 (41.7) |
| 11–40 | 4 (16.7) | 3 (25.0) |
| 41–100 | 2 (8.3) | 1 (8.3) |
| 101–300 | 4 (16.7) | 2 (16.7) |
| >300 | 6 (25.0) | 1 (8.3) |
| Overall (>0) | 23 (95.8) | 12 (100.0) |
| Intracranial hemorrhage | 13 (54.2) | 6 (50.0) |
| Superficial siderosis | 14 (58.3) | 7 (58.3) |
| White matter hyperintensities | ||
| Absent | 1 (4.2) | 2 (16.7) |
| Mild | 8 (33.3) | 4 (33.3) |
| Moderate | 13 (54.2) | 5 (41.7) |
| Severe | 2 (8.3) | 1 (8.3) |
| Apolipoprotein E genotype: | ||
| E2/E3 | 2 (8.3) | 0 |
| E2/E4 | 4 (16.7) | 2 (16.7) |
| E3/E3 | 6 (25.0) | 7 (58.3) |
| E3/E4 | 5 (20.8) | 1 (8.3) |
| E4/E4 | 7 (29.2) | 2 (16.7) |
| MMSE, mean (SD) | 28.8 (1.24) | 28.8 (1.06) |
| MoCA, mean (SD) | 25.5 (3.41) | 25.9 (3.34) |
| Stable use of antiepileptic drug | 3 (12.5) | 0 |
| Stable use of antiinflammatory drug: | ||
| For treatment of CAAa | 0 | 0 |
| For treatment of other conditionb | 3 (12.5) | 1 (8.3) |
| Baseline BOLD fMRI parametersc, mean (SD) | ||
| Slope (percent/second) | 0.12 (0.08) | 0.15 (0.06) |
| Time to peak (seconds) | 11.92 (1.85) | 11.39 (2.12) |
| Amplitude (percent) | 1.36 (0.71) | 1.69 (0.59) |
| Time to return to baseline (seconds) | 12.17 (1.63) | 11.91 (1.95) |
Abbreviations: BOLD, blood oxygenation level dependent; CAA, cerebral amyloid angiopathy; CMB, cerebral microbleeds; fMRI, functional magnetic resonance imaging; MMSE, Mini Mental State Examination; MoCA, Montreal Cognitive Assessment; n, number of subjects; ROI, region of interest; SD, standard deviation.
Include aspirin and oral/intravenous steroids.
Include any nonsteroidal antiinflammatory drugs and topical/oral/intravenous steroids.
BOLD fMRI parameters were from ROI1.
For the primary efficacy endpoint, the geometric mean change from baseline in the BOLD fMRI slope on Day 2 was 0.954 in the ponezumab group compared with 0.969 in the placebo group giving a geometric mean ratio (ponezumab vs. placebo) of 0.984 with a 90% credible interval of 0.820–1.184. For Day 90, the geometric mean change from baseline in the BOLD fMRI slope was 0.817 in the ponezumab group compared with 0.958 in the placebo group, giving a geometric mean ratio for ponezumab versus placebo of 0.852 with a 90% credible interval of 0.735–0.989 (Fig. 3 and Table 2) – that is, the slope of the fMRI response was shallower in the ponezumab group than the placebo group, contrary to the prespecified hypothesis. Thus, the predefined efficacy criteria for increased vascular reactivity were not met either at Day 2 or Day 90. Similarly, for the secondary fMRI endpoints (change in time to peak, amplitude, and time to return to baseline), ponezumab treatment showed negligible mean changes at Day 2 while at Day 90, the mean observed differences were opposite to the hypothesized direction; none of these values were statistically significant at the 5% level. Sensitivity analyses on primary or secondary ROIs, with or without informative Bayesian priors, were also conducted and provided consistent results. Post hoc analysis in which participants with CMB counts >300 were excluded did not change the findings for the primary endpoint. Additionally, post hoc analyses with apolipoprotein E status, CMB count (log‐transformed), or white matter hyperintense lesion percentage as covariates did not change the primary results. In analyses that used a Bayesian approach, the study placebo group yielded responses consistent with the prespecified placebo priors derived from previous fMRI study data.10
Figure 3.
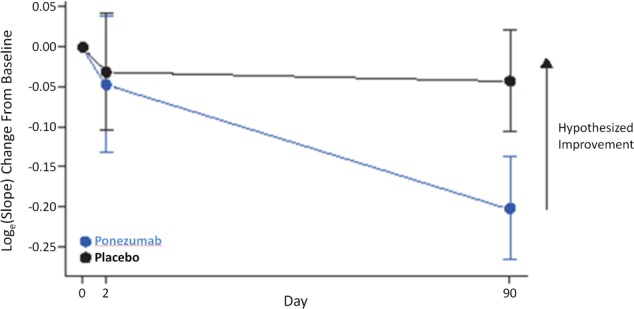
Primary outcome for loge(slope) change from baseline, mean ± SE.
Table 2.
Change from baseline in the primary efficacy endpoint of BOLD fMRI slope at Days 2 and 90, ROI1
| Treatment group | n | Geo‐metric mean | SEa | 90% CIb | Geo‐metricmean ratio | SEa | 90% CIb | Mean/SEa | Probability (Treatment Effect>x), where x= | C1c achieved | C2d achieved | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0% | 20% | |||||||||||
| Day 2 | ||||||||||||
| Ponezumab | 20 | 0.954 | 0.085 | (0.831, 1.096) | 0.984 | 0.112 | (0.820, 1.184) | −0.146 | 0.4367 | 0.0390 | No | No |
| Placebo | 11 | 0.969 | 0.073 | (0.861, 1.092) | ||||||||
| Day 90 | ||||||||||||
| Ponezumab | 20 | 0.817 | 0.064 | (0.736, 0.908) | 0.852 | 0.091 | (0.735, 0.989) | –1.761 | 0.0390 | 0.0002 | No | No |
| Placebo | 10 | 0.958 | 0.063 | (0.864, 1.064) | ||||||||
The units for slope are percent/second.
Abbreviations: BOLD, blood oxygenation level dependent; CI, credible interval; fMRI, functional magnetic resonance imaging; n, number of subjects; ROI, region of interest; SE, standard error.
Log scale value presented.
A credible interval was defined as a posterior probability interval.
Point estimate of ponezumab versus placebo effect >20% increase (improvement) in slope.
Standard error of ponezumab versus placebo effect <60% of the point estimate.
Ponezumab plasma concentrations peaked 1 h postdose with mean concentration 239.1 μg/mL on Day 1. Mean trough concentrations on Days 30, 60, and 90 were 46.1 μg/mL, 60.37 μg/mL, and 83.02 μg/mL, respectively. These findings were consistent with those from previous ponezumab AD trials.20 Robust and cumulative increases from baseline in plasma Aβ 1–40 were observed after ponezumab dosing in all participants, whereas plasma Aβ 1–40 levels in the placebo group were low and stable as expected (Fig. 4; mean (range): 4747.6 (3054–7154) pg/mL and 87710.8 (59418–131756) pg/mL for Day1 8 h post dose and Day 90 respectively in the ponezumab group, and −8.4 (−55–42) pg/mL and 0.7 (−61–88) pg/mL for Day1 8 h post dose and Day 90 respectively in the placebo group). The mean plasma Aβ 1–40 concentration, presented as placebo–adjusted change from baseline at Day 1 time zero, was 68.0 ng/mL and 87.7 ng/mL at Day 30 and Day 90, respectively. A hysteresis relationship was observed between ponezumab plasma concentrations and plasma Aβ 1‐40 levels, likely due to a longer terminal half‐life of plasma Aβ 1–40. Qualitative assessment of exposure–response analysis revealed no relationship between trough ponezumab plasma concentration and BOLD fMRI response slope or time to peak change.
Figure 4.
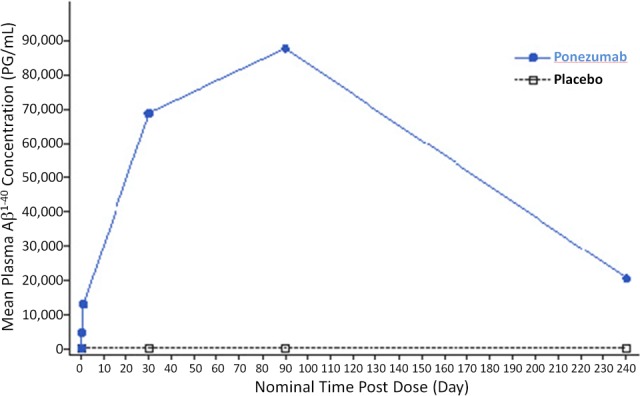
Mean plasma Aβ 1–40 concentration versus time.
The safety profile of ponezumab was consistent with previous studies in AD and consistent with the profile of the general CAA population.20 In the ponezumab group one participant had nonfatal cerebral hemorrhage associated with aphasia and one participant had a subdural hematoma, both of which were determined by the site investigators not to be treatment related. In the placebo group there was one fatal occurrence of intracerebral hemorrhage and one occurance of migraine with aura. At screening, the median (range) lobar CMB count was 40.5 (0.0–881.0) and 19.5 (2.0–1113.0) in the ponezumab and placebo groups, respectively. No notable changes in CMB incidence from screening to Day 90 were observed (Table 3). There was one instance of asymptomatic ARIA‐E at Day 90 in the ponezumab group (Fig. 5) in which cognition was unchanged, as measured by MoCA. No participants developed antidrug antibodies. There were no notable changes in laboratory analysis, vital signs, ECGs, physical examinations, structural brain MRI findings, cognition, or suicidality.
Table 3.
Summary of cerebral microhemorrhage frequency
| Placebo | Ponezumab | |||||||
|---|---|---|---|---|---|---|---|---|
| (N = 12) | (N = 24) | |||||||
| Day 0 | Day 15 | Day 45 | Day 90 | Day 0 | Day 15 | Day 45 | Day 90 | |
| Median | 19.5 | 19.5 | 19.5 | 20.5 | 40.5 | 41.5 | 41.5 | 44.0 |
| Range | 2–1113 | 2–1114 | 2–1114 | 2–1114 | 0–881 | 0–881 | 0–881 | 0–883 |
Figure 5.
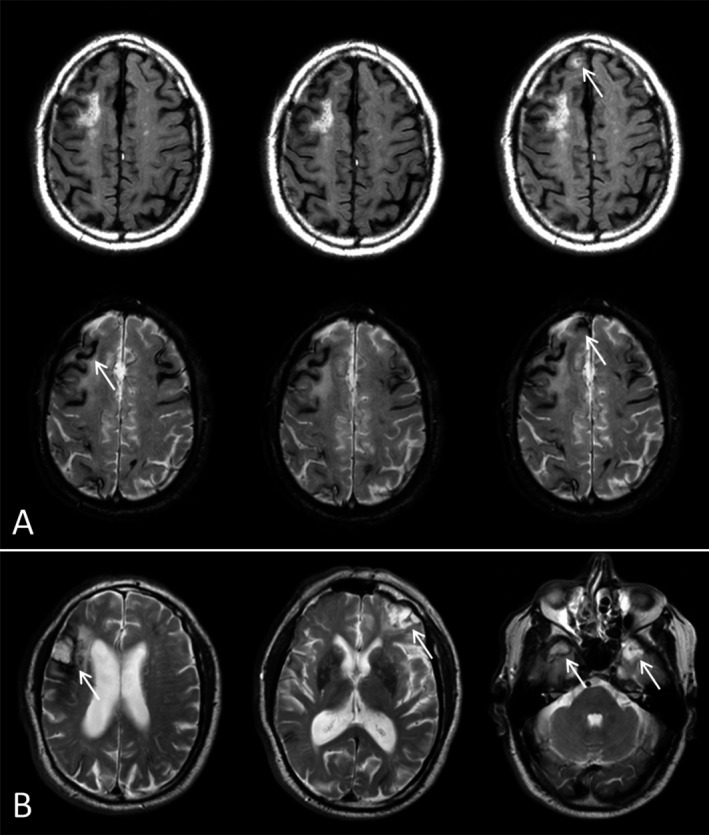
FLAIR and T2*‐weighted images of ARIA‐E event. (A) FLAIR (top row) and T2*‐weighted images (bottom row) taken at baseline (left), Day 45 (middle) and Day 90 (right). The arrow in the Day 90 FLAIR image indicates the presence of new ARIA‐E, and the associated T2*‐weighted image at Day 90 shows a new colocalized subarachnoid hemorrhage. Baseline findings in this subject included multiple areas of superficial siderosis of both cerebral hemispheres. (B) Baseline findings also included four old parenchymal macro‐hemorrhages, as show in three different levels in the baseline T2‐weighted images.
Discussion
In this study in adults with probable CAA per the modified Boston criteria, treatment with the humanized anti‐Aβ 1–40 antibody, ponezumab, was safe and well‐tolerated, with minimal safety concerns. There were no deaths or treatment–related serious adverse events, as judged by the site investigator and central medical monitor, in the ponezumab treatment group. Advanced CAA is associated with a spontaneous inflammatory syndrome resembling ARIA‐E – apparently driven by anti‐Aβ autoantibodies in cerebrospinal fluid21, 22 – that has been postulated to occur by similar mechanisms as treatment–related ARIA‐E.4 Exogenous anti‐Aβ antibody treatment of CAA patients might therefore have been expected to trigger a significant increase in ARIA‐E. There was little evidence of ARIA‐E in these participants with probable CAA, however, with a single occurrence of asymptomatic ARIA‐E at Day 90 in the ponezumab group in an apolipoprotein Eε4 noncarrier deemed by the site investigator not to be treatment‐related, and no apparent increase in ARIA‐H. A possible contributing factors to the low incidence of treatment–related ARIA‐E in this trial may be the particular characteristics of the monoclonal antibody ponezumab.
Although there was no apparent evidence for treatment–associated increase in ARIA‐H, it should be noted that consistent enumeration of microbleeds can be challenging, particularly when the burden is high, the microbleeds are small, or are in close proximity to each other or other susceptibility–related signal voids. The partial volume of small microbleeds within the relatively thick T2*‐weighted slices also means that small differences in participant positioning and slice angulation can reduce the consistency of the microbleed count. While every effort was made to ensure consistent head placement and image slice planning, small differences are largely unavoidable. Automating microbleed detection may improve consistency in future studies.
Plasma ponezumab concentrations were consistent with those observed in previous trials of ponezumab in AD at similar dose regimen.20 Compared with placebo, plasma Aβ 1‐40 concentrations increased cumulatively after multiple ponezumab administrations, with peak concentrations observed on Day 90 (30 days post last dose). These findings were consistent with previous observations showing transient mobilization and stabilization of Aβ 1–40 deposits by ponezumab peripherally.20, 23, 24
For the primary endpoint of change from baseline in natural logarithm of the slope of visual stimulus‐driven fMRI responses, ponezumab showed negligible mean treatment effect at Day 2 and marginally negative mean treatment effect at Day 90. At both timepoints, the placebo group remained stable, but the direction of any mean treatment effect was opposite to the hypothesized direction. As such, the treatment did not produce the expected improvement in the radiologic surrogate of vascular dysfunction, and the prespecified criteria for efficacy were not met at either Day 2 or 90. The individual fMRI secondary endpoints were in line with primary endpoint observations.
The failure to meet the primary endpoint may indicate that vascular amyloid was not cleared, or perhaps not sufficiently cleared, to improve vascular reactivity. The unexpected trend towards a decrease in vascular reactivity estimated via fMRI could be due to mobilization of amyloid plaque,25 mobilization of vascular amyloid with resultant vascular damage, the possibility of a random chance finding associated with a small sample size, or a currently unidentified effect of antiamyloid immunotherapy.
The large gamut of clinical, radiologic, and laboratory data obtained longitudinally did not show any concerning changes in the ponezumab group, so there is no indication that the reduced slope observed on fMRI represents a clinically meaningful worsening. As the BOLD fMRI signal is influenced by a combination of neurometabolic, vascular, and hemodynamic responses to the visual stimulus, a definitive biological interpretation of the observed fMRI time–course is not currently possible. Further, the timing and direction of the vascular reactivity response to amyloid–removing therapy is unknown, as this is the first study of this kind. It is possible that the removal of amyloid from the vessel wall triggers a transient increase in cerebrovascular dysfunction. Future studies employing this fMRI technique could consider additional time points to more fully characterize the response.
The consistency of baseline and placebo data in this study, together with the fMRI results from previously published studies,12 suggest that it is feasible to use the technique in a consistent manner across multiple study timepoints and sites, so it may become a viable biomarker for future studies.10, 12 Our results were unique in suggesting that a pharmacologic intervention might cause a consistent change in these fMRI markers. The exact physiologic determinants of these markers and the clinical implications of altering them remain to be determined.
Limitations of this study include the relatively small sample size, relatively short duration of treatment, and limited number of timepoints. Another potential limitation is misdiagnosis of CAA, though we note that the modified Boston criteria appear to have high specificity (87.5% to 100%) among symptomatic patients. The novel finding of a tendency towards apparent reduced vascular reactivity with little evidence of ARIA or other overt inflammatory effects raises the possibility of a previously unidentified effect of antiamyloid immunotherapy that may be worthy of further investigation.
Future CAA studies should employ advanced brain imaging to disentangle the potential cause–effect relationships between amyloid load and vascular function and may help us understand how pharmacologic interventions can modify these important parameters to obtain a beneficial clinical effect.
Author Contributions
CL, JG, YZ, PH, JP, EG, MB, ES, and SG contributed to conception and design of the study; CL, JG, YZ, PH, JP, EG, ML, LF, AV, SS, MB, ES, and SG contributed to acquisition and analysis of data, and CL, JG, YZ, PH, JP, EG, ML, LF, SS, AV, MB, ES, and SG contributed to drafting of the manuscript and/or figures.
Ponezumab Trial Study Group:
Anand Viswanathan (Massachusetts General Hospital),
Charlotte Cordonnier (Lille University),
David John Werring (University College London),
Sandra Elizabeth Black (University of Toronto),
Lawrence Sterling Honig (Columbia Univeristy),
Catharina J.M. Klijn (University Medical Center Utrecht),
Jin‐Moo Lee (Washington University in St. Louis),
Lucas Restrepo (University of California at Los Angeles),
Eric Edward Smith (University of Calgary),
Carlos Kase (Boston Univeristy),
Sean I. Savitz (University of Texas).
[Correction added on 17 April 2019 after first online publication on 18 March 2019: The Author Contributions has been corrected.]
Conflict of Interest
Claire Leurent, James A. Goodman, Yao Zhang, Ping He, Monica Lindsay, Linda Frattura, U. S. Sohur, and Martin M. Bednar were employees of Pfizer. Steven M. Greenberg served as a consultant to Pfizer and GlaxoSmithKline and on safety monitoring committees for amyloid immunotherapy studies conducted by Roche, Biogen, and DIAN‐TU. Massachusetts General Hospital participated in this study under a Clinical Research Support Agreement with Pfizer. Anand Viswanathan served on safety monitoring committees for amyloid immunotherapy studies conducted by Roche.
Funding Information
This study was funded by Pfizer, Inc (Clinicaltrials.gov identifier NCT01821118). Medical writing and editorial assistance in the preparation of this manuscript was provided by Paul Hassan, of Engage Scientific (Horsham, UK) and was funded by Pfizer.
[Correction added on 17 April 2019 after first online publication on 18 March 2019: Ponezumab Trial Study Group has been added in the author list.]
Funding Statement
This work was funded by Pfizer, Inc grant .
Contributor Information
James A. Goodman, Email: james.goodman@pfizer.com
Steven M. Greenberg, Email: sgreenberg@mgh.harvard.edu
the Ponezumab Trial Study Group:
Charlotte Cordonnier, David John Werring, Sandra Elizabeth Black, Lawrence Sterling Honig, Catharina J.M. Klijn, Jin‐Moo Lee, Lucas Restrepo, Carlos Kase, and Sean I. Savitz
References
- 1. Viswanathan A, Greenberg SM. Cerebral amyloid angiopathy in the elderly. Ann Neurol 2011;70:871–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Yamada M. Cerebral amyloid angiopathy: emerging concepts. J Stroke 2015;17:17–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Launer LJ, Hughes TM, White LR. Microinfarcts, brain atrophy, and cognitive function: the Honolulu Asia Aging Study Autopsy Study. Ann Neurol 2011;70:774–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sperling RA, Jack CR Jr, Black SE, et al. Amyloid‐related imaging abnormalities in amyloid‐modifying therapeutic trials: recommendations from the Alzheimer's Association Research Roundtable Workgroup. Alzheimer's Dement 2011;7:367–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Arvanitakis Z, Leurgans SE, Wang Z, et al. Cerebral amyloid angiopathy pathology and cognitive domains in older persons. Ann Neurol 2011;69:320–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fotiadis P, van Rooden S, van der Grond J, et al. Cortical atrophy in patients with cerebral amyloid angiopathy: a case‐control study. Lancet Neurol 2016;15:811–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gregoire SM, Charidimou A, Gadapa N, et al. Acute ischaemic brain lesions in intracerebral haemorrhage: multicentre cross‐sectional magnetic resonance imaging study. Brain 2011;134(Pt 8):2376–2386. [DOI] [PubMed] [Google Scholar]
- 8. Gurol ME, Viswanathan A, Gidicsin C, et al. Cerebral amyloid angiopathy burden associated with leukoaraiosis: a positron emission tomography/magnetic resonance imaging study. Ann Neurol 2013;73:529–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Soontornniyomkij V, Choi C, Pomakian J, Vinters HV. High‐definition characterization of cerebral β‐amyloid angiopathy in Alzheimer's disease. Hum Pathol 2010;41:1601–1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dumas A, Dierksen GA, Gurol ME, et al. Functional magnetic resonance imaging detection of vascular reactivity in cerebral amyloid angiopathy. Ann Neurol 2012;72:76–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Peca S, McCreary CR, Donaldson E, et al. Neurovascular decoupling is associated with severity of cerebral amyloid angiopathy. Neurology 2013;81:1659–1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Switzer AR, McCreary C, Batool S, et al. Longitudinal decrease in blood oxygenation level dependent response in cerebral amyloid angiopathy. Neuroimage Clin 2016;11:461–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wilcock DM, Rojiani A, Rosenthal A, et al. Passive immunotherapy against Abeta in aged APP‐transgenic mice reverses cognitive deficits and depletes parenchymal amyloid deposits in spite of increased vascular amyloid and microhemorrhage. J Neuroinflammation 2004;1:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wilcock DM, Alamed J, Gottschall PE, et al. Deglycosylated anti‐amyloid‐beta antibodies eliminate cognitive deficits and reduce parenchymal amyloid with minimal vascular consequences in aged amyloid precursor protein transgenic mice. J Neurosci 2006;26:5340–5346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bales KR, O'Neill SM, Pozdnyakov N, et al. Passive immunotherapy targeting amyloid‐beta reduces cerebral amyloid angiopathy and improves vascular reactivity. Brain 2016;139(Pt 2):563–577. [DOI] [PubMed] [Google Scholar]
- 16. Knudsen KA, Rosand J, Karluk D, Greenberg SM. Clinical diagnosis of cerebral amyloid angiopathy: validation of the Boston criteria. Neurology 2001;56:537–539. [DOI] [PubMed] [Google Scholar]
- 17. Linn J, Halpin A, Demaerel P, et al. Prevalence of superficial siderosis in patients with cerebral amyloid angiopathy. Neurology 2010;74:1346–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhang L, Hennig J, Zhong K, Speck O. Reduced Spurious Activation by Continued Controlled Attention in fMRI Measurements of Primary Sensory Areas 2006;14:1129. [Google Scholar]
- 19. Jack CR Jr, Bernstein MA, Borowski BJ, et al. Update on the magnetic resonance imaging core of the Alzheimer's disease neuroimaging initiative. Alzheimer's & Dement 2010;6:212–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Burstein AH, Zhao Q, Ross J, et al. Safety and pharmacology of ponezumab (PF‐04360365) after a single 10‐minute intravenous infusion in subjects with mild to moderate Alzheimer disease. Clin Neuropharmacol 2013;36:8–13. [DOI] [PubMed] [Google Scholar]
- 21. Eng JA, Frosch MP, Choi K, et al. Clinical manifestations of cerebral amyloid angiopathy‐related inflammation. Ann Neurol 2004;55:250–256. [DOI] [PubMed] [Google Scholar]
- 22. Piazza F, Greenberg SM, Savoiardo M, et al. Anti‐amyloid β autoantibodies in cerebral amyloid angiopathy‐related inflammation: implications for amyloid‐modifying therapies. Ann Neurol 2013;73:449–458. [DOI] [PubMed] [Google Scholar]
- 23. Landen JW, Zhao Q, Cohen S, et al. Safety and pharmacology of a single intravenous dose of ponezumab in subjects with mild‐to‐moderate Alzheimer disease: a phase I, randomized, placebo‐controlled, double‐blind, dose‐escalation study. Clin Neuropharmacol 2013;36:14–23. [DOI] [PubMed] [Google Scholar]
- 24. Miyoshi I, Fujimoto Y, Yamada M, et al. Safety and pharmacokinetics of PF‐04360365 following a single‐dose intravenous infusion in Japanese subjects with mild‐to‐moderate Alzheimer's disease: a multicenter, randomized, double‐blind, placebo‐controlled, dose‐escalation study. Int J Clin Pharmacol Ther 2013;51:911–923. [DOI] [PubMed] [Google Scholar]
- 25. Boche D, Zotova E, Weller RO, et al. Consequence of Abeta immunization on the vasculature of human Alzheimer's disease brain. Brain 2008;131(Pt 12):3299–3310. [DOI] [PubMed] [Google Scholar]