

Abstract
Objective
Dysferlin is a large transmembrane protein that functions in critical processes of membrane repair and vesicle fusion. Dysferlin‐deficiency due to mutations in the dysferlin gene leads to muscular dystrophy (Miyoshi myopathy (MM), limb girdle muscular dystrophy type 2B (LGMD2B), distal myopathy with anterior tibial onset (DMAT)), typically with early adult onset. At least 416 pathogenic dysferlin mutations are known, but for approximately 17% of patients, one or both of their pathogenic variants remain undefined following standard exon sequencing methods that interrogate exons and nearby flanking intronic regions but not the majority of intronic regions.
Methods
We sequenced RNA from myogenic cells to identify a novel dysferlin pathogenic variant in two affected siblings that previously had only one disease‐causing variant identified. We designed antisense oligonucleotides (AONs) to bypass the effects of this mutation on RNA splicing.
Results
We identified a new pathogenic point mutation deep within dysferlin intron 50i. This intronic variant causes aberrant mRNA splicing and inclusion of an additional pseudoexon (PE, we term PE50.1) within the mature dysferlin mRNA. PE50.1 inclusion alters the protein sequence, causing premature translation termination. We identified this mutation in 23 dysferlinopathy patients (seventeen families), revealing it to be one of the more prevalent dysferlin mutations. We used AON‐mediated exon skipping to correct the aberrant PE50.1 splicing events in vitro, which increased normal mRNA production and significantly restored dysferlin protein expression.
Interpretation
Deep intronic mutations can be a common underlying cause of dysferlinopathy, and importantly, could be treatable with AON‐based exon‐skipping strategies.
Introduction
Dysferlinopathy is a degenerative muscle disease caused by insufficient expression of dysferlin protein in skeletal muscles.1, 2 This is a recessively inherited disorder that manifests in late teens to early adulthood as muscle weakness due to muscle degeneration, which progressively worsens, typically leading to significant loss of mobility.3 There is currently no cure for dysferlinopathy, and treatment is limited to palliative care. Clinical trials have recently begun to evaluate a virally mediated gene replacement strategy to restore dysferlin expression, similar to that described,4, 5 but there remains a great need for novel therapeutic approaches to treat this debilitating disease.
Dysferlin, a member of the ferlin family of Ca2+‐dependent phospholipid‐binding proteins, is a large (237 kDa) transmembrane protein important for membrane repair, vesicle trafficking, and T‐tubule structure.6, 7, 8, 9 There are 14 known human isoforms of dysferlin, generated by alternative exon splicing and the use of two different promoters,10 with isoform 8 being the predominant form in muscle. In addition to skeletal muscle, dysferlin is expressed in other tissues, and dysferlin‐deficiency has been associated with immune cell migration changes such as increased motility and phagocytosis by blood monocytes.11, 12, 13 The dysferlin expression level in blood monocytes can be used as a diagnostic tool for dysferlinopathy.14, 15, 16 Clinically, however, the functional requirement for dysferlin is most notable in skeletal muscle.
At least 416 different pathogenic dysferlin variants are listed in the Universal Mutation Database (UMD‐DYSF, http://www.umd.be/DYSF/),17 most of these lying within one of the 55 exons that are spliced together to form isoform 8 that is critical for muscle function. For approximately 17% of patients, a complete understanding of the genetic lesions underlying their disease is lacking; at least one of their pathogenic mutations has not been identified. It is highly likely that these mutations lie somewhere within dysferlin introns or areas other than exons that regulate dysferlin expression. These gene regions are not examined by standard exonic screening methods.
We recently identified one such pathogenic variant deep within dysferlin intron 44i (c.4886 + 1249G>T) in dysferlinopathy patients. This intronic variant induces alternate splicing of the dysferlin transcript, allowing the inclusion of a 177 bp pseudoexon (PE44.1) within the mature transcript, resulting in an in‐frame insertion of 59 amino acids within the C2F domain of the dysferlin protein.18 Here, we report the identification of another deep intronic dysferlin variant, which also causes aberrant mRNA splicing and pathogenic inclusion of a pseudoexon, this one leading to premature translation termination. Antisense oligonucleotides (AONs) can be used to inhibit this pseudoexon splicing event, restoring normal mRNA splicing and dysferlin protein expression.
Methods
Patients
Samples used in this study (blood or skin biopsy samples) were from patients clinically diagnosed as dysferlinopathy patients (MM or LGMD2B). These patients exhibited progressive limb muscle weakness beginning in adolescence or early adulthood, elevated blood creatine kinase levels indicating muscle damage, and demonstrated reduced or absence of dysferlin protein expression in muscle biopsies and/or blood monocyte cells when screened as described.15, 16 Patient materials were collected in accordance with ethical guidelines and protocols approved by the University of Massachusetts Medical School Institutional Review Board, and similar review boards at Emory University School of Medicine, Aix‐Marseille University ,or Institute of Genetic Medicine, Newcastle. For all of the patients described, only one or neither of their pathogenic variants had been defined by exon sequencing. The dysferlinopathy patients initially screened, JF196 and JF23, are siblings participating in an ongoing international “Clinical Outcome Study for Dysferlinopathy”, a collaboration with the Jain Foundation and Newcastle upon Tyne Hospitals. Patient skin fibroblasts were obtained from the Newcastle upon Tyne Hospital Biobank. These patients are heterozygous for one pathogenic dysferlin variant identified by exon sequencing (c.5698_5699delAG; p.Ser1900GlnfsX14, stop codon in exon 51, DYSF C2G domain), but their other pathogenic allele had not been identified. Similarly, dysferlinopathy patient TDM57 had only one pathogenic allele defined by exon sequencing (c.2998T>C; p.Ser1000Pro, inner DysF domain, predicted pathogenic, UMD‐Predictor algorithm, UMD‐DYSF Universal Mutation Database). TDM57 skin biopsy tissue was obtained via the Jain Foundation as part of a diagnostic program at the Centre for Advanced Molecular Diagnostics in Neuromuscular Disorders, Mumbai, Maharashtra, India. TDM57 and other TDM patients reported here (except TDM196 and TDM230) were described previously16; all TDM patient genomic DNA was sequenced at Emory University School of Medicine as described.15 Additional patient genomic DNA sequencing was performed at Aix‐Marseille University or Newcastle upon Tyne Hospitals. Fibroblasts from unrelated individuals served as DNA sequence analysis controls (8597: dysferlinopathy patient with different dysferlin mutations18; RB19895: ALS patient; NHDF‐3: normal).
Cell culture
Skin biopsy samples were placed in culture dishes to establish fibroblast cultures as previously described.18 We also obtained normal adult human dermal fibroblasts (we termed NHDF‐2 and NHDF‐3 cells) from the American Type Culture Collection (ATCC, Manassas, VA, USA). Fibroblasts were transduced with lentivirus vectors carrying tamoxifen‐inducible MyoD to generate inducible fibroblast‐derived myogenic cells (iFDMs) as described.18 These cells proliferate as fibroblasts and can be induced to differentiate into myotubes by treatment with 4‐hydroxytamoxifen (TMX). TMX induces MyoD expression and myotube formation when cells are cultured in low serum differentiation medium (DM) (DMEM Glutamax with pyruvate: Medium 199 (Gibco, Grand Island, NY, USA]) (3:1), 2% horse serum (HyClone, Logan, UT, USA), 20 mM HEPES, and 20 μg/mL insulin, 11 μg/mL transferrin, 1.3 μg/mL selenium (2X ITS, Gibco)).18
Nucleic acid purification
Genomic DNA was purified from cells using Gentra Puregene (Qiagen, Valencia, CA, USA) reagents following manufacturers’ protocols. RNA was purified from patient and normal iFDM cells using TRIzol reagent (Life Technologies, Grand Island, NY, USA), digested with DNAse (TURBO DNA‐Free, Ambion, Austin, TX, USA) to remove contaminating DNA, then RNA was reverse transcribed (High Capacity cDNA Reverse Transcription Kit, Applied Biosystems Foster City, CA, USA) using manufacturers protocols to generate cDNA for PCR analyses. For all analyses, we used DYSF mRNA variant 8 (NM_003494.3), the predominant skeletal muscle isoform, as the reference sequence.
RNA analysis
We used an RT‐PCR approach and methods described previously18 for PCR amplification and sequencing of the complete dysferlin cDNA derived from myogenic cell (iFDM cell) RNA. For this, 17 primer sets generating 450–500 bp amplicons that tiled across the DYSF cDNA were used as described18 to sequence and screen the cDNA for mutations. Additionally, 12 primer sets (Table S1) that tiled across DYSF intron 50i were used to sequence and screen the patient genomic DNA for variants as previously done for intron 44i.18 Additional primers (PE50.1‐F and ‐R) that can be used to sequence patient DNA for the mutated sequence are also listed. M13 sequence tails were added to primers to facilitate sequencing (M13‐F, 5′‐GTAAAACGACGGCCAGT ‐3′ on forward primers; M13‐R 5′‐CAGGAAACAGCTATGAC‐3′ on reverse primers).
Quantitative PCR (qPCR) was performed on cDNAs (corresponding to 5 or 10 ng input RNA in reverse transcription reactions) using DyNAmo HS SYBR Green qPCR Kit reagents (Thermo Scientific) and a Bio‐Rad CFX384 C‐1000 Touch Real‐Time PCR Detection System and conditions previously described.18 Primers that distinguish normal and PE50.1‐containing DYSF mRNA were used, along with primers that amplify a region of exons 13–14, common to all DYSF splice forms, to quantitate the total DYSF mRNA expressed (i.e., 100% DYSF expression) (Table S3). Primers for human beta‐2‐microglobulin (B2M, Bio‐Rad) were used to normalize total mRNA levels.
Antisense oligonucleotide treatment of cell cultures
We designed antisense oligonucleotides (AONs) that target potential exonic splicing enhancer (ESE) sequences within PE50.1 using Human Splicing Finder v.3.019 and Rescue‐ESE20 online tools. Blocking these ESEs with AONs could inhibit the splicing of PE50.1 into mature processed mRNAs, thereby leading to the synthesis of normally spliced DYSF transcripts. We used three AONs targeting PE50.1 (Table S2) along with a nonspecific scrambled AON control (SCR) described previously.18 These AONs were synthesized as 2′‐O‐methyl RNA with full‐length phosphorothioate backbones (Integrated DNA Technologies, Coralville, IA, USA). iFDM cells derived from patients JF196, JF23 and normal NHDF‐2 fibroblasts were allowed to differentiate for indicated lengths of time to form myotubes. Cells were transfected with each AON (600 nmol/L or other concentrations as indicated) (or TE buffer as control) using Oligofectamine (Life Technologies) and the manufacturer's protocol. After the indicated times, RNA was extracted and evaluated by RT‐PCR18 using primers 16‐F and 15‐R to amplify either the normal DYSF sequence (88 bp) or the mutant version containing PE50.1 (268 bp). Proteins were extracted from similar cultures and evaluated on western blots using antibodies against the dysferlin C‐terminal region (NCL‐Hamlet, Leica, Buffalo Grove, IL, USA, 1/1000) and GAPDH (G9545, Sigma, St. Louis, MO, USA, 1/1000) as described.18 Protein expression levels were calculated from western blot images using a Li‐COR Odyssey infrared imager and Image Studio Software (Li‐COR).
Statistics
Statistical significance was evaluated using one‐way analysis of variance (ANOVA) with post hoc Tukey tests. For this, we used Prism 5.0 statistical analysis software (GraphPad Software, San Diego, CA, USA).
Results
Identification of a deep intronic mutation in DYSF intron 50i
Skin fibroblasts cultures from sibling dysferlinopathy patients JF196 and JF23 were converted to myogenic iFDM cell lines and induced to differentiate into myotubes. RNA was then isolated and analyzed by RT‐PCR to determine the sequence of any dysferlin expressed in these cells. We analyzed the dysferlin mRNA using PCR primers that generated overlapping amplicons tiling through the entire mRNA. Two of the overlapping amplicon products from these patients were larger than products from normal iFDM cells, indicating an insertion of extra sequence within the dysferlin transcript in these patients (Fig. 1). As shown in Figure 2A, sequence analysis of these RT‐PCR products revealed the insertion of 180 bp of additional sequence between DYSF exons 50 and 51. This sequence is present within intron 50i and is aberrantly spliced into the DYSF mRNA as a novel pseudoexon (PE) which we term pseudoexon 50.1 (PE50.1). Sequence analysis of the remainder of the cDNA products confirmed the other known pathogenic variant in these patients (c.5698_5699delAG, exon 51) and revealed no additional novel variants in the coding sequence. Inclusion of PE50.1 in the mRNA transcript is predicted to lead to insertion of 46 additional amino acids (aa) followed by a stop codon encoded by the pseudoexon sequence (Fig. 2A and C). In this aberrantly spliced DYSF transcript that contains PE50.1, translation is predicted to terminate within the DYSF C2G domain, losing the c‐terminus containing the transmembrane domain, critical for DYSF function.
Figure 1.
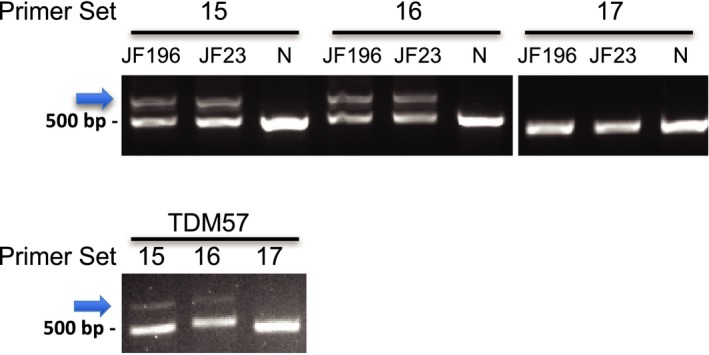
Identification of the DYSF intron 50i mutation. RT‐PCR of differentiated iFDM cDNA from three patients (JF196, JF23, TDM57) shows novel amplicons (arrow) in patient samples that are not in a normal control sample (N). The novel amplicons were produced using two primer sets (15F+R and 16F+R), whose amplicons overlap, but not with any other sets (e.g., 17F+R), demonstrating additional sequence within patient cDNA in this region. Each PCR product was sequenced to identify the inserted sequence.
Figure 2.
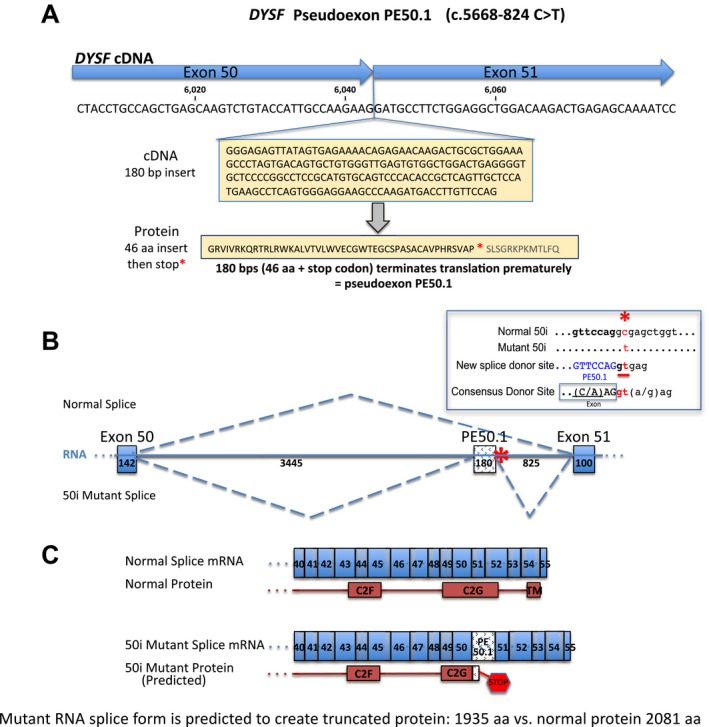
Dysferlin mRNA splicing is altered in patients JF196 and JF23, leading to inclusion of pathogenic pseudoexon PE50.1. (A) cDNA sequencing showed that patients JF196 and JF23 have a 180 nt insertion at the junction of exons 50 and 51, revealing the inclusion of a novel pseudoexon, PE50.1, spliced into the coding sequence. PE50.1 encodes 46 additional amino acids followed by a stop codon. (B) DYSF intron 50i genomic DNA sequencing showed patients are heterozygous for a point mutation (c.5668‐824 C>T) (asterisk *) that creates a novel splice donor consensus site at the 3′ end of the PE50.1 sequence. In these heterozygous cells, both normal and mutant PE50.1‐containing mRNA are expressed, representing the alternative splice forms of the DYSF transcripts. The mRNA structure within DYSF exon 50 ‐ intron 50i ‐ exon 51 and the site of the 50i mutation is shown (insert, upper right), along with the normal and mutant splicing patterns. Numbers along the RNA indicate the size of each element in bp. (C) The normal and mutant DYSF mRNA splice products and proteins in the region of exons 40–55 are shown. PE50.1 inclusion is predicted to result in a 46 amino acids insertion and premature translation termination within the C2G domain and loss of the C‐terminal transmembrane domain (TM), both of which are required for dysferlin function. The predicted truncated protein, if expressed, would be 1935 aa rather than the normal 2081 aa.
Further sequence analysis of genomic DNA from these patient cells revealed that PE50.1 inclusion in the mRNA transcripts is caused by a novel point mutation (c.5668‐824C>T) deep within intron 50i (Fig. S1). As shown in Figure 2B, this point mutation creates a consensus splice donor site that promotes the splicing of the 180 bp PE50.1 sequence between exons 50 and 51. Additionally, a consensus “YAG” splice acceptor sequence exists immediately upstream of the PE50.1 sequence, along with a pyrimidine‐rich region and potential branch points as predicted by Human Splicing Finder, all of which are important for splicing (Fig. S2).
We analyzed genomic DNA and differentiated iFDM cell cDNA derived from an unrelated dysferlinopathy patient, TDM57. This patient also had only one known pathogenic dysferlin variant identified (c.2998T>C). Genomic DNA sequence analysis showed that this patient also carries the c.5668‐824C>T point mutation in intron 50i, and RT‐PCR analysis of iFDM cell mRNA showed the aberrant inclusion of PE50.1 in the mRNA of this patient. Additional sequence variants present within intron 50i of patients JF196, JF23, and TDM57 are summarized in Table 1. Each of these variants are reported in the dbSNP database and have no known pathogenicity.
Table 1.
Genomic DNA sequence variants in DYSF intron 50i of dysferlinopathy patients JF196, JF23 and TDM57
| SNP Name | Ref. | Var. | JF196 | JF23 | TDM57 | NHDF‐2 | nt Position | Sequence | Primers | Global MAF | Chromosomal Location |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Novel | C | T | C|T | C|T | C|T | C|C | c.5668‐824C>T | TTGTTCCAGG[C/T]GAGCTGGTCT | 50i 10 F+R | 2:71673373 | |
| rs3791829 | A | G | G|G | G|G | A|G | G|G | c.5667+199A>G | CTGTCTCAGC[A/G]TTGTGTGATG | 50i 1 F+R |
A = 0.1677/840 A= 0.1425/4148 |
2:71669945 |
| rs3791830 | G | A | A|A | A|A | G|A | A|A | c.5667+1075G>A | TATGCTGGTT[A/G]GAGGCAGAGG | 50i 3 F+R |
G= 0.2298/1151 G= 0.2242/6527 |
2:71670821 |
| rs3791832 | C | A | A|A | A|A | C|A | A|A | c.5668‐2222C>A | GCAGGGGTGG[A/C]GAGGAGGAAG | 50i 6 F+R |
C = 0.2151/1077 C = 0.2033/5918 |
2:71671975 |
| rs72829766 | C | T | C|C | C|C | C|C | C|T | c.5668‐913C>T | CTGAGGGGTG[C/T]TCCCCGGCCT | 50i 9 F+R |
T = 0.0505/253 T = 0.0513/1494 |
2:71673284 |
| rs7604764 | G | A | G|A | G|A | G|A | A|A | c.5668‐642G>A | CCTTACAGCA[A/G]CGTGCTGGGA | 50i 10 F+R |
G = 0.4361/2184 G= 0.4676/13616 |
2:71673555 |
| rs13024390 | T | G | T|G | T|G | T|G | G|G | c.5668‐522T>G | AGAGCAGGAC[G/T]CTGGAACCCA | 50i 10 F+R |
T = 0.4357/2182 T = 0.4674/13610 |
2:71673675 |
| rs882973 | C | T | C|T | C|T | C|T | T|T | c.5668‐366C>T | AGAGGGCCAA[C/T]GCATAGGAAG | 50i 11 F+R |
C = 0.2718/1361 C = 0.2665/7760 |
2:71673831 |
| rs2559081 | C | T | C|T | C|T | C|T | T|T | c.5668‐41C>T | TCTCTCTAAC[C/T]TTGCTTCCTT | 50i 12 F+R |
C = 0.2720/1362 C = 0.2660/7743 |
2:71674156 |
Genomic variations within DYSF intron 50i in patients are listed. The novel mutation c.5668‐824C>T results in PE50.1 inclusion in mature mRNAs. Other variations listed are reported in the dbSNP database and have not been shown to be pathogenic. Global MAF values: top, 1000 Genomes; bottom, TOPMED. The primer sets used to amplify and sequence these regions are shown. The DYSF reference sequence used: GRCh38.p7; mRNA isoform 8, NM_003494.3.
Additional dysferlinopathy patients with the intron 50i c.5668‐824C>T mutation
We examined genomic DNA from additional dysferlinopathy patients that had only one or neither of their pathogenic variants identified to determine if they carried the 50i c.5668‐824C>T intronic variant, including two additional siblings of JF196 and JF23. As summarized in Table 2, a total of 22 patients from 17 families were found to carry this mutation, nine of them being homozygous. As an indication of prevalence, this mutation was found in 7/25 patients from India screened in Atlanta (Dastur et al.), 3/32 patients screened in Marseille; 8/56 patients screened in Newcastle.
Table 2.
Summary of patients with the new c.5668‐824C>T mutation in intron 50i
| Patient ID | Patient Origin | Intron 50i Mutation | Second DYSF Mutation | Second DYSF Mutation Effects | Laboratory | Sibling Groups |
|---|---|---|---|---|---|---|
| JF23 | UK | c.5668‐824C>T | c.5698_5699delAG |
p.Ser1900GlnfsX14 Stop at p.1913, C2 domain G |
1, 5 | A |
| JF196 | UK | c.5668‐824C>T | c.5698_5699delAG |
p.Ser1900GlnfsX14 Stop at p.1913, C2 domain G |
1, 5 | A |
| IGM7 | UK | c.5668‐824C>T | c.5698_5699delAG |
p.Ser1900GlnfsX14 Stop at p.1913, C2 domain G |
5 | A |
| IGM8 | UK | c.5668‐824C>T | c.5698_5699delAG |
p.Ser1900GlnfsX14 Stop at p.1913, C2 domain G |
5 | A |
| JF404 | USA | c.5668‐824C>T | c.2997G>T |
p.Trp999Cys Missense, DysF domain |
2, 4 | B |
| JF1729 | USA | c.5668‐824C>T | c.2997G>T |
p.Trp999Cys Missense, DysF domain |
2 | B |
| JF4228 | USA | c.5668‐824C>T | c.5668‐824C>T | 2 | ||
| TDM48 | India | c.5668‐824C>T | c.5668‐824C>T | 3 | ||
| TDM57 | India | c.5668‐824C>T | c.2998T>C |
p.Ser1000Pro Missense, inner DysF domain |
1, 3 | C |
| TDM58 | India | c.5668‐824C>T | c.2998T>C |
p.Ser1000Pro Missense, DysF domain |
3 | C |
| TDM63 | India | c.5668‐824C>T | c.5668‐824C>T | 3 | ||
| TDM93 | India | c.5668‐824C>T | c.5668‐824C>T | 3 | ||
| TDM180 | India | c.5668‐824C>T | c.5668‐824C>T | 3 | ||
| TDM182 | India | c.5668‐824C>T | c.5668‐824C>T | 3 | ||
| TDM196 | India | c.5668‐824C>T | c.5668‐824C>T | 2 | ||
| TDM230 | India | c.5668‐824C>T | c.1911C>G |
p.Tyr637X Stop at p.637 |
2 | |
| F1‐170‐1‐2 | France | c.5668‐824C>T | c.855+1delG |
p.285 Exon 8 +1 delG, splicing C2 domain B |
4 | D |
| F1‐170‐2‐2 | France | c.5668‐824C>T | c.855+1delG |
p.285 Exon 8 +1 delG, splicing C2 domain B |
4 | D |
| F1‐436‐1‐0 | India | c.5668‐824C>T | c.5668‐824C>T | 4 | ||
| IGM1 | UK | c.5668‐824C>T | c.1911c>G |
p.Tyr637X Stop at p.637 |
5 | |
| IGM2 | UK | c.5668‐824C>T | c.5668‐824C>T | 5 | ||
| IGM3 | UK | c.5668‐824C>T | c.2434dup |
p.811 Ferlin domain B |
5 | |
| IGM4 | UK | c.5668‐824C>T | c.937+1G>A |
p.312 Exon 10 +1G>A, splicing Ferlin domain |
5 |
- Univ. Massachusetts Medical School, Worcester.
- Emory Univ. School of Medicine, Atlanta.
- Emory Univ. School of Medicine, Atlanta, described in Dastur et al. 2017.
- Marseille Medical Genetics, Aix‐Marseille University, Marseille.
- Institute of Genetic Medicine, Newcastle.
Antisense oligonucleotides targeting PE50.1 restore production of normal DYSF mRNA
Given that aberrant splicing, caused by the c.5668‐824C>T intron 50i mutation, leads to inclusion of the disruptive PE50.1 sequence, blocking this aberrant splicing should restore normal DYSF RNA and protein. We designed three antisense oligonucleotides targeting different possible exonic splice enhancer (ESE) regions within PE50.1 that could promote PE50.1 splicing into the mature mRNA; targeting these ESEs with AONs would prevent PE50.1 inclusion. RT‐PCR analysis of patient's iFDM myotubes treated for 2 days with AONs reveals that AONs indeed inhibit the expression of the mutant PE50.1 DYSF mRNA spliced form and restore higher levels of the normal mRNA splice form that lacks PE50.1 (Fig. 3). The normal splice form could represent both normal splicing (i.e., PE50.1 skipping) of the intron 50i mutant allele as well as the normal splicing of the other DYSF mutant allele expressed in these cells. The presence and absence of PE50.1 in the mRNA transcripts were confirmed by sequencing the larger and smaller RT‐PCR products, respectively. These effects of AONs 1, 2, and 3 on PE50.1 skipping have been confirmed in independent cell culture experiments (Two experiments for AON1, five for AON2, four for AON3), and the effects observed in cells from all three patients (JF196, TDM57, JF23; Figs. 3, 4, 5).
Figure 3.
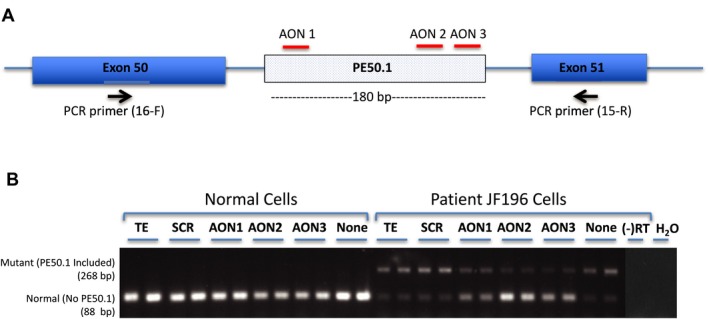
Antisense oligonucleotide‐mediated skipping of PE50.1 in patient iFDM cells. (A) Pre‐mRNA DYSF transcript and AON1, AON2 and AON3 (Table S2), which target potential exonic splicing enhancers in PE50.1 as shown. Primers in exon 50 (16F) and 51(15R) amplify cDNAs to distinguish normal mRNA transcripts (88 bp product containing exon 50 + 51) from mutant PE50.1 transcripts (268 bp product containing exon 50 + PE50.1 + 51). (B) RT‐PCR analysis of mRNA splicing. Patient JF196 iFDM cells treated with AON1, AON2 and AON3 (600 nmol/L, duplicate cultures for each) expressed reduced amounts of PE50.1 mutant mRNA and higher normal DYSF mRNA compared with a non‐specific scrambled (SCR, 600 nmol/L) AON‐treated, TE‐treated, or no treatment cells. Normal (NHDF‐2) iFDMs only expressed normal DYSF transcripts. iFDMs were allowed to differentiate in DM for 6 days then treated with AONs in DM for an additional 2 days. (‐RT), no reverse transcriptase; H2O, no RNA used in RT reactions.
Figure 4.
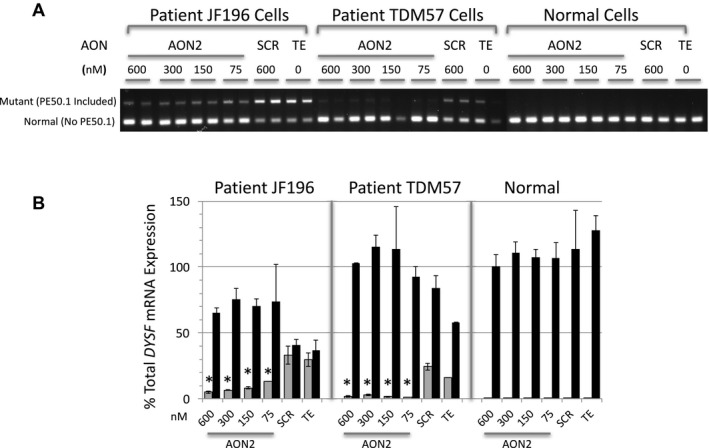
Antisense oligonucleotides at a variety of concentrations induce skipping of PE50.1 in patient iFDM cells. (A) Using experimental conditions in Figure 3, iFDM cells (duplicate cultures for each treatment) from patients JF196, TDM57 and a normal (NHDF‐2) control were treated with AON2 or scrambled (SCR) control AONs at the concentrations shown or TE alone. In both patient cell lines, AON2 treatment at each concentration tested reduced PE50.1 mutant mRNA expression compared with SCR or TE controls. (B) Quantitative RT‐PCR analysis of the RNAs in panel A shows that treatment of patient iFDMs with AON2 significantly reduces the expression of the mutant transcripts compared to SCR and TE controls (*P < 0.05) (gray bars, mutant transcripts; black bars, normal transcripts; mean ± SD). The relative expression of each form is calculated using the amplification of a PCR product spanning the exon 13‐14 junction as representative of 100% DYSF expression.
Figure 5.
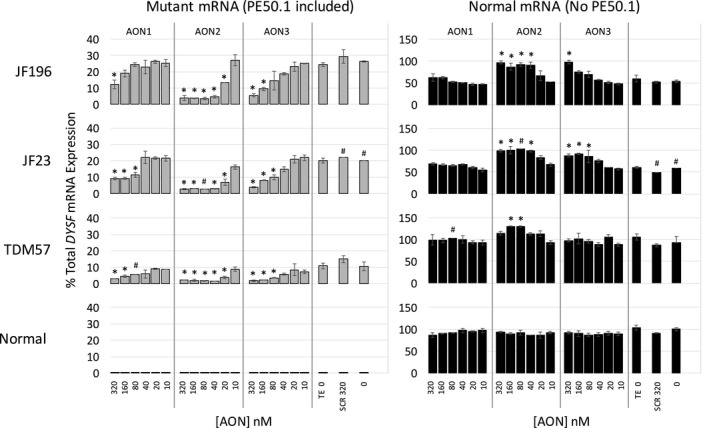
AON2 is more effective than AON1 and AON3 for inducing PE50.1 skipping in all three patient cell lines. Using experimental conditions and analyses as in Figures 3 and 4, iFDM cells from patients JF196, JF23, and TDM57 and a normal (NHDF‐2) control were treated with AON1, AON2 or AON3 at the concentrations show, scrambled control AONs (SCR 320 nmol/L), TE (TE 0) or not treated (0). RT‐Q‐PCR shows that in all three cell lines, each of the AONs at the highest concentration can significantly reduce the expression of the mutant transcripts compared to TE controls (*P < 0.05). AON2 shows significant effects on PE50.1 expression at lower concentrations than other AONs in all three cell lines. Data indicate the relative effectiveness for inducing PE50.1 skipping is AON2 > AON3 > AON1 (gray bars, mutant transcripts; black bars, normal transcripts; duplicate cultures for each treatment (mean ± SD) except for four samples (marked #) in which a single culture was used due to sample loss or poor RNA quality.
As shown in Figure 4A, a decrease in the mutant PE50.1 mRNA splice form is observed across a range of AON2 concentrations tested in both JF196 and TDM57 patient cells, representing two different families with this mutation. Quantitation of this effect is shown in Figure 4B. In a separate experiment, a broader AON concentration range was used to evaluate the relative effectiveness of AONS 1, 2, and 3 on PE50.1 skipping in iFDM cells from three patients (JF196, JF23, and TDM57) and a normal control (Fig. 5). In all three patient cell lines, AON2 was most effective in reducing the expression of the mutant PE50.1 splice form (i.e., inducing PE50.1 skipping) and increasing levels of the normal splice form. AON3 was less effective and AON1 least effective in altering mutant PE50.1 mRNA levels. In all three cell lines, as little as 20 nmol/L AON2 significantly reduced the mutant PE50.1 mRNA levels compared with TE‐treated controls (Fig. 5, left panels). AON3 significantly reduced mutant PE50.1 expression at 80 nmol/L for two of the three cell lines and at 160 nmol/L, for the third, while AON1 significantly reduced expression in only one line at 80 nM, and at 160–320 nmol/L for the other lines. A similar trend in relative potency of the three AONs was observed for the increase in normal transcripts (Fig. 5 right panels). Consistent with previous experiments, a scrambled AON control (SCR) did not affect mutant PE50.1 mRNA in any cells, and no PE50.1 mRNA was detected in normal cells.
The relative proportion of the mutant PE50.1 mRNA splice form expressed in patient cells was calculated using pooled control (TE and no treatment cells) RT‐qPCR data from Figures 4 and 5 (patient JF196 n = 6, patient JF23 n = 3, patient TDM57 n = 5 cultures). PCR amplicons that span DYSF exons 13–14 were used to quantify total DYSF expression in each sample, which overall is reduced to similar extents in each of the mutant lines compared to normal cells: patients JF196, JF23, and TDM 57 DYSF mRNA levels are ~38%, ~31%, and ~43% that of normal NHDF2 cells, respectively. Mutant PE50.1 transcripts represent ~26.9 ± 3.4%, 20.2 ± 1%, and ~11.8 ± 3.0% (mean ± SD) of total DYSF transcripts in JF196, JF23, and TDM57 cells, respectively. It is not clear if this variation in the proportion of mutant transcripts reflects true differences in PE50.1 expression between patients or simply cell line variation. Quantitation of PE50.1 expression in muscle tissue would more accurately address mutant PE50.1 mRNA expression variation among patients with the intron 50i mutation, but unfortunately, muscle tissues were not available from any patients in this study. The relative abundance of mutant PE50.1 mRNA could reflect a combination of i) the efficiencies of mutant and normal splicing reactions that generate mutant and normal mRNA from pre‐mRNA carrying the intron 50i mutation, ii) the rate of mRNA decay, likely accelerated by stop codons such as that encoded in PE50.1 (nonsense‐mediated decay), and iii) the production and stability of transcripts derived from the patients’ other mutant alleles. For patients JF196 and JF23, the second DYSF mutation creates a stop codon at p.1913, while patient TDM57 has a missense mutation (p.Ser1000Pro) (Table 2). These second mutations, combined with the intron 50i mutation in these patients, lead to the overall reduction in DYSF mRNA expression.
Antisense oligonucleotide treatment for 3 days (followed by 4 days without AONs) also restored DYSF protein expression to levels similar to that in normal myotubes (Fig. 6), likely as a result of the higher amounts of normally spliced transcripts induced by PE50.1 skipping. The premature translational stop codon within PE50.1 likely leads to nonsense‐mediated mRNA decay, whereby mRNA carrying a premature stop codon is transcribed and spliced but degraded during the translation process,21 leading to reduced mRNA levels (typically detectable using sensitive RT‐PCR methods) along with aborted protein synthesis. The overall reduced levels of RT‐PCR products we observed in untreated or control‐treated patient cells compared with normal cells (described above) are consistent with this. It is unlikely that the mutation leads to significant synthesis of a truncated protein (as predicted from the sequence, shown in Fig. 2C) because we did not detect the expression of a shorter DYSF protein on western blots using an antibody specific to the DYSF N‐terminal region (Abcam JAI‐1‐49‐3, Romeo, data not shown). However, our current data cannot rule out the possibility of low level expression of a truncated, possible unstable protein, or possibly low levels of a toxic protein species. Studies overexpressing the truncated protein form could determine if it affects muscle cells, which could have a bearing on AON therapeutic strategies because it would be important to block the production of a toxic protein production as much as possible.
Figure 6.
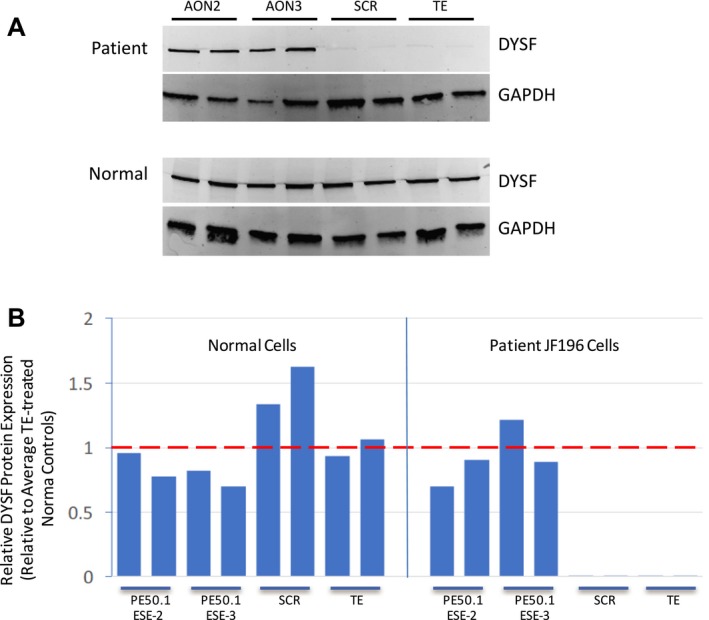
Treatment of patient iFDM cells with AON2 and AON3 directed to PE50.1 induces dysferlin protein expression. JF196 iFDM cells (duplicate cultures) were allowed to differentiate in DM for 6 days then treated with AONs in DM (or TE buffer as controls) for 3 days, and cells collected for protein analysis 7 days after AON addition. (A) Western blots show that after 7 days of AON3 treatment (4 days after AON removal) there was a dramatic increase in DYSF protein expression compared with control cells. GAPDH expression served as a control for protein loading (5 μg protein/lane). Protein levels in normal control iFDM cultures are not affected by AON treatments. (B) Quantitation of DYSF protein expressed in panel A, normalized to GAPDH levels and shown relative to normal cells treated with TE. The mean relative DYSF expression in patient cells was significantly higher in both AON2‐ and AON3‐treated cells compared with SCR‐ or TE‐treated controls (P < 0.05, one‐way ANOVA, Tukey's Multiple Comparison Test).
Discussion
Using myogenic cells derived from patient skin fibroblasts, we identified a novel point mutation deep within DYSF intron 50i (c.5668‐824C>T) that causes aberrant dysferlin mRNA splicing and the inclusion of a pathogenic pseudoexon (PE50.1) in dysferlin mRNA. This is the second such deep intronic pseudoexon‐generating point mutation we have identified using this approach; the previous one was identified within DYSF intron 44i (PE44.1).18 Initially identified in two siblings with dysferlinopathy (JF196, JF23), we found a total of 23 patients from 17 families that carry this intron 50i mutation, making it one of the more prevalent pathogenic DYSF mutations when compared with those in the Universal Mutation Database (UMD‐DYSF, http://www.umd.be/DYSF/).17
Known pathogenic variants in genes are typically identified by screening patients’ genomic DNA using exon sequencing methods that interrogate the coding regions and intronic sequences near the exon junctions. Whole genome sequencing can identify sequence variants in other noncoding regions (e.g., introns, promoters, and untranslated regions), but the pathological significance of these might not be directly evident. We find that screening RNA from patient cells (i.e., iFDM cells derived from dysferlinopathy patients) for sequence variations is a useful approach to identifying mutations that alter the protein coding sequence. Our current studies, as with our previous work,18 demonstrate that point mutations deep within dysferlin introns can alter splicing and drive the inclusion of pathogenic pseudoexons.
Pseudoexons such as these, caused by mutations that introduce novel splice donor or acceptor sites, or affect exonic splicing enhancer interactions, have been identified in many other genes (reviewed in22), among them DMD, the gene for dystrophin, causing Duchenne muscular dystrophy (reviewed in 23), and recently COL6A1, causing collagen VI‐related dystrophy.24 Given that many dysferlinopathy patients do not have both of their pathogenic variants identified through standard exome sequencing, it is likely that identification of other novel DYSF mutations in noncoding regions could be identified by analysis of RNA sequence variations. This could be done either using our approach of direct sequencing of RT‐PCR products, or through RNAseq analysis, recently used to characterize splicing patterns of normal human dystrophin transcripts25 and to identify splicing abnormalities in this and a number of other muscle disease‐related genes.24, 26
While our studies used RNA from myogenic cells (iFDMs) derived from skin biopsy fibroblasts, future diagnostic pipelines could potentially use blood monocytes, which also express dysferlin.14 Analysis of dysferlin protein expression in monocytes is a useful diagnostic tool for primary dysferlinopathies.15, 16 Different isoforms of dysferlin are expressed in blood cells compared with muscle,10 but these differences involve variable splicing of only 3 of the 55 dysferlin exons (5a, 17, 40a), using two different promoters. Therefore, the majority of exon splicing events are likely to be common between blood and muscle cells; blood cell RNA screening could detect aberrant spicing events in a large proportion of the DYSF transcripts. In preliminary studies (not shown), we have generated amplicons that span the DYSF transcript using RNA from blood cells and our overlapping primer sets. RNA sequence analysis of blood monocytes could provide a less invasive and more cost‐effective primary screen for elusive pathogenic variants not identified by standard exon sequencing methods.
The aberrant, pathogenic inclusion of a pseudoexon in the mature transcript of a gene, such as DYSF PE50.1, provides an ideal target for AON‐mediated exon‐skipping therapeutics. As with our previous studies targeting the pathogenic pseudoexon DYSF PE44.1,18 treatment of cells with AONs targeting PE50.1 induces skipping of this pseudoexon, synthesis of the normal DYSF mRNA transcript, and the restoration of near normal DYSF protein levels. The minimum level of DYSF protein required for normal muscle function has not been determined, but in one documented case a 70‐year‐old patient with only mild dystrophic symptoms was found to express dysferlin at approximately 10% of normal levels, indicating that this is sufficient for significant muscle function. This patient carried one severe DYSF mutation that produced no protein and a second intronic DYSF mutation that altered splicing, causing omission of exon 32 but maintaining the reading frame, possibly generating a partially functional protein.27 Data suggested that the DYSF protein expressed was largely from the aberrantly spliced mRNA, but authors noted that expression of the normally spliced form could not be excluded. It will be important to evaluate the efficacy of AON treatments in altering the expression of DYSF protein in vivo, for example in homozygous mutant animal models expressing DYSF PE50.1, to determine the level of protein achieved in diseased muscle tissue, the kinetics and efficiency of this process, and the therapeutic impact on disease progression and muscle restoration at later stages of disease.
Author Contributions
J.A.D, L.R., and R.H.B. designed the study. J.A.D., O.U., acquired and interpreted data to identify and characterize the pathogenic variant and data related to altered RNA splicing. D.M‐Y., B.R.R.N., V.K., M.B., N.L., J.H., T.E., H.L. M.K. L.R., and M.H. provided critical input on patients and acquired patient data including genomic DNA variant screening. J.A.D. drafted the manuscript. All authors critically edited the content of the article and approved the final version.
Conflict of Interest
J.A.D. and R.H.B. are employed by the University of Massachusetts Medical School and co‐inventors on a patent application for antisense sequences and exon‐skipping technology targeting dysferlinopathies. M.K., N.L. and M.B. are co‐inventors on a patent for dysferlin exon skipping.
Supporting information
Figure S1. Analysis of genomic DNA from patients JF196, JF23 and TDM57 revealed that they are heterozygous for the c.5668‐824C>T mutation deep within DYSF intron 50i. Genomic DNAs from patients' fibroblasts were amplified and sequenced using primer sets that tiled through 50i (Supplementary Table 1), revealing this mutation in these dysferlinopathy patient cells but not in fibroblasts from unrelated patients (8597: patient with other dysferlin mutations18; RB19895: ALS patient) or normal human dermal fibroblasts (NHDF‐3). Samples shown here were amplified using primers PE50.1‐F and PE50.1‐R then sequenced in the forward (F) and reverse (R) directions as indicated.
Figure S2. The intronic sequence upstream of pseudoexon PE50.1 contains additional consensus sites required for mRNA splicing. These include a splice acceptor sequence (ag) at the 5′ end of PE50.1, an adjacent pyrimidine‐rich region and two potential lariat branch point consensus sequences (underlined) that could be used to promote splicing. These sequences, in the presence of c.5668‐824 C>T mutation, likely allow PE50.1 to be spliced between exons 50 and 51.
Table S1. Primers (forward (F) and reverse (R)) used to generate overlapping amplicons that span dysferlin intron 50i, and an additional primer set (PE50.1‐F, PE50.1‐R) useful for genotyping.
Table S2. AONs targeting human exonic splicing enhancer sequences (ESE) in DYSF PE50.1. AONs for in vitro studies are 2′‐O‐methyl RNA with full‐length phosphorothioate backbones.
Table S3. Primers (forward (F) and reverse (R)) used in quantitative PCR assays to analyze DYSF mRNA expression.
Acknowledgments
We thank the Cecil B. Day Foundation for supporting this work. R. H. B.'s laboratory also receives support from the NINDS: R01 NS073873, R01 NS104022. M. H acknowledges support from the Jain Foundation.
Funding Information
We thank the Cecil B. Day Foundation for supporting this work. R. H. B.'s laboratory also receives support from the NINDS: R01 NS073873, R01 NS104022. M. H acknowledges support from the Jain Foundation.
Funding Statement
This work was funded by NINDS grants R01 NS073873 and R01 NS104022; Jain Foundation grant .
References
- 1. Liu J, Aoki M, Illa I, et al. Dysferlin, a novel skeletal muscle gene, is mutated in Miyoshi myopathy and limb girdle muscular dystrophy. Nat Genet 1998;20:31–36. [DOI] [PubMed] [Google Scholar]
- 2. Bashir R, Britton S, Strachan T, et al. A gene related to Caenorhabditis elegans spermatogenesis factor fer‐1 is mutated in limb‐girdle muscular dystrophy type 2B. Nat Genet 1998;20:37–42. [DOI] [PubMed] [Google Scholar]
- 3. Harris E, Bladen CL, Mayhew A, et al. The Clinical Outcome Study for dysferlinopathy: an international multicenter study. Neurol Genet 2016;2:e89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sondergaard PC, Griffin DA, Pozsgai ER, et al. AAV Dysferlin overlap vectors restore function in dysferlinopathy animal models. Ann Clin Transl Neurol 2015;2:256–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Potter RA, Griffin DA, Sondergaard PC, et al. Systemic delivery of dysferlin overlap vectors provides long‐term gene expression and functional improvement for dysferlinopathy. Hum Gene Ther 2018;29:749–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Glover L, Brown RH Jr. Dysferlin in membrane trafficking and patch repair. Traffic 2007;8:785–794. [DOI] [PubMed] [Google Scholar]
- 7. Han R, Campbell KP. Dysferlin and muscle membrane repair. Curr Opin Cell Biol 2007;19:409–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Klinge L, Laval S, Keers S, et al. From T‐tubule to sarcolemma: damage‐induced dysferlin translocation in early myogenesis. FASEB J 2007;21:1768–1776. [DOI] [PubMed] [Google Scholar]
- 9. Klinge L, Harris J, Sewry C, et al. Dysferlin associates with the developing T‐tubule system in rodent and human skeletal muscle. Muscle Nerve 2010;41:166–173. [DOI] [PubMed] [Google Scholar]
- 10. Pramono ZA, Tan CL, Seah IA, et al. Identification and characterisation of human dysferlin transcript variants: implications for dysferlin mutational screening and isoforms. Hum Genet 2009;125:413–420. [DOI] [PubMed] [Google Scholar]
- 11. Nagaraju K, Rawat R, Veszelovszky E, et al. Dysferlin deficiency enhances monocyte phagocytosis: a model for the inflammatory onset of limb‐girdle muscular dystrophy 2B. Am J Pathol 2008;172:774–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wein N, Krahn M, Courrier S, et al. Immunolabelling and flow cytometry as new tools to explore dysferlinopathies. Neuromuscul Disord 2010;20:57–60. [DOI] [PubMed] [Google Scholar]
- 13. de Morree A, Flix B, Bagaric I, et al. Dysferlin regulates cell adhesion in human monocytes. J Biol Chem 2013;288:14147–14157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ho M, Gallardo E, McKenna‐Yasek D, et al. A novel, blood‐based diagnostic assay for limb girdle muscular dystrophy 2B and Miyoshi myopathy. Ann Neurol 2002;51:129–133. [DOI] [PubMed] [Google Scholar]
- 15. Ankala A, Nallamilli BR, Rufibach LE, et al. Diagnostic overview of blood‐based dysferlin protein assay for dysferlinopathies. Muscle Nerve 2014;50:333–339. [DOI] [PubMed] [Google Scholar]
- 16. Dastur RS, Gaitonde PS, Kachwala M, et al. Detection of dysferlin gene pathogenic variants in the indian population in patients predicted to have a dysferlinopathy using a blood‐based monocyte assay and clinical algorithm: a model for accurate and cost‐effective diagnosis. Ann Indian Acad Neurol 2017;20:302–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Blandin G, Beroud C, Labelle V, et al. UMD‐DYSF, a novel locus specific database for the compilation and interactive analysis of mutations in the dysferlin gene. Hum Mutat 2012;33:E2317–E2331. [DOI] [PubMed] [Google Scholar]
- 18. Dominov JA, Uyan Ö, Sapp PC, et al. A novel dysferlin mutant pseudoexon bypassed with antisense oligonucleotides. Ann Clin Transl Neurol 2014;1:703–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Desmet FO, Hamroun D, Lalande M, et al. Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res 2009;37:e67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fairbrother WG, Yeh RF, Sharp PA, Burge CB. Predictive identification of exonic splicing enhancers in human genes. Science 2002;297:1007–1013. [DOI] [PubMed] [Google Scholar]
- 21. Brogna S, McLeod T, Petric M. The Meaning of NMD: translate or Perish. Trends Genet 2016;32:395–407. [DOI] [PubMed] [Google Scholar]
- 22. Vaz‐Drago R, Custodio N, Carmo‐Fonseca M. Deep intronic mutations and human disease. Hum Genet 2017;136:1093–1111. [DOI] [PubMed] [Google Scholar]
- 23. Tuffery‐Giraud S, Miro J, Koenig M, Claustres M. Normal and altered pre‐mRNA processing in the DMD gene. Hum Genet 2017;136:1155–1172. [DOI] [PubMed] [Google Scholar]
- 24. Cummings BB, Marshall JL, Tukiainen T, et al. Improving genetic diagnosis in Mendelian disease with transcriptome sequencing. Sci Transl Med 2017;9:5209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bouge AL, Murauer E, Beyne E, et al. Targeted RNA‐Seq profiling of splicing pattern in the DMD gene: exons are mostly constitutively spliced in human skeletal muscle. Sci Rep 2017;3:39094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gonorazky H, Liang M, Cummings B, et al. RNAseq analysis for the diagnosis of muscular dystrophy. Ann Clin Transl Neurol. 2016;3:55–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sinnreich M, Therrien C, Karpati G. Lariat branch point mutation in the dysferlin gene with mild limb‐girdle muscular dystrophy. Neurology 2006;66:1114–1116. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Analysis of genomic DNA from patients JF196, JF23 and TDM57 revealed that they are heterozygous for the c.5668‐824C>T mutation deep within DYSF intron 50i. Genomic DNAs from patients' fibroblasts were amplified and sequenced using primer sets that tiled through 50i (Supplementary Table 1), revealing this mutation in these dysferlinopathy patient cells but not in fibroblasts from unrelated patients (8597: patient with other dysferlin mutations18; RB19895: ALS patient) or normal human dermal fibroblasts (NHDF‐3). Samples shown here were amplified using primers PE50.1‐F and PE50.1‐R then sequenced in the forward (F) and reverse (R) directions as indicated.
Figure S2. The intronic sequence upstream of pseudoexon PE50.1 contains additional consensus sites required for mRNA splicing. These include a splice acceptor sequence (ag) at the 5′ end of PE50.1, an adjacent pyrimidine‐rich region and two potential lariat branch point consensus sequences (underlined) that could be used to promote splicing. These sequences, in the presence of c.5668‐824 C>T mutation, likely allow PE50.1 to be spliced between exons 50 and 51.
Table S1. Primers (forward (F) and reverse (R)) used to generate overlapping amplicons that span dysferlin intron 50i, and an additional primer set (PE50.1‐F, PE50.1‐R) useful for genotyping.
Table S2. AONs targeting human exonic splicing enhancer sequences (ESE) in DYSF PE50.1. AONs for in vitro studies are 2′‐O‐methyl RNA with full‐length phosphorothioate backbones.
Table S3. Primers (forward (F) and reverse (R)) used in quantitative PCR assays to analyze DYSF mRNA expression.