

Abstract
High oestradiol (E2) and low dehydroepiandrosterone-sulfate (DHEA-S) levels are risk factors for pulmonary arterial hypertension (PAH) in men, but whether sex hormones are related to PAH in women is unknown.
Post-menopausal women aged ⩾55 years with PAH were matched by age and body mass index to women without cardiovascular disease. Plasma sex hormone levels were measured by immunoassay.
Lower levels of DHEA-S (p<0.001) and higher levels of E2 (p=0.02) were associated with PAH. In PAH cases (n=112), lower DHEA-S levels were associated with worse haemodynamics (all p<0.01) and more right ventricular dilatation and dysfunction (both p=0.001). Lower DHEA-S levels were associated with shorter 6-min walking distance (6MWD) (p=0.01) and worse functional class (p=0.004). Each Ln(1 μg·dL−1) decrease in DHEA-S was associated with a doubling in the risk of death (hazard ratio 2.0, 95% CI 1.5–2.7; p<0.001). Higher levels of E2 were associated with shorter 6MWD (p=0.03) and worse functional class (p=0.01).
High E2 and low DHEA-S levels are associated with the risk and severity of PAH in post-menopausal women. Hormonal modulation should be studied as a treatment strategy in PAH.
Introduction
Pulmonary arterial hypertension (PAH) classically affects young women, but recent registries suggest a female predominance even among older individuals [1]. Women with PAH have better survival than men, which may be explained by differences in right ventricular (RV) adaptation [2]. Temporal changes in sex hormone levels with ageing may further modify the epidemiology of PAH [3]. In post-menopausal women without clinical cardiovascular disease, we have shown that sex hormone levels and genotypic variation in sex hormone metabolism and signalling are associated with RV function, but these relationships have not been extensively studied in PAH [4, 5].
We recently demonstrated that men with PAH have higher circulating oestradiol (E2) levels and lower dehydroepiandrosterone-sulfate (DHEA-S) levels compared to healthy controls and that this profile (high E2/low DHEA-S) had modest associations with worse clinical status [6]. Oestrogen can induce pulmonary hypertension in some animal models [7], but is speculated to have cardioprotective effects [8]. In a pilot trial in which we reduced E2 levels by blocking aromatase with anastrozole, 6-min walking distance (6MWD) was increased [9]. In contrast, DHEA appears to be consistently beneficial in experimental pulmonary hypertension via mechanisms that are independent of oestrogen and directly impact RV function, although these studies include predominantly male animals [10]. A recent study of unbiased metabolomic profiling demonstrated that DHEA-S and its metabolites were reduced in PAH patients compared to healthy controls and that lower circulating levels of DHEA-S were associated with mortality in PAH patients [11]. Given the disparate observations across experimental and clinical studies, the complexities of steroid hormone signalling and the potential interactions between hormonal action, sex, age and other clinical factors, more comprehensive hormone profiling in a discrete PAH patient population is warranted.
We sought to determine whether sex steroid hormone levels (E2, DHEA-S, testosterone, progesterone, sex hormone binding globulin (SHBG)) were associated with PAH in post-menopausal women. We compared circulating plasma levels of these hormones in post-menopausal women with PAH to those of age- and body mass index (BMI)-matched controls without clinical cardiovascular disease. We hypothesised that higher E2 and lower DHEA-S levels would be associated with a higher risk of PAH (as in men), but that this profile would have variable relationships with markers of pulmonary vascular versus RV function.
Methods
Study sample
We performed a case–control study of post-menopausal women with PAH compared with age- and BMI-matched post-menopausal women without clinical cardiovascular disease. Cases were enrolled from the Rhode Island Hospital Pulmonary Hypertension Center at Brown University (Providence, RI, USA), the Pulmonary Vascular Disease Program at Penn (Philadelphia, PA, USA) and the Pulmonary Hypertension Center at Tufts Medical Center (Boston, MA, USA). Controls were selected from the Multi-Ethnic Study of Atherosclerosis (MESA)-RV study, a population-based epidemiological cohort described previously [12]. Exclusion criteria for MESA-RV included weight >300 lb, pregnancy, impediment to long-term participation or to undergo cardiac magnetic resonance imaging (MRI) and clinical cardiovascular disease, defined as a participant having answered “yes” on screening questionnaire to a history of physician-diagnosed heart attack, stroke, transient ischaemic attack, heart failure, angina, current atrial fibrillation or to having undergone any prior cardiovascular procedure.
Post-menopausal status was confirmed for cases and controls using the same approach. In MESA and two of the three pulmonary hypertension centres, participants were considered post-menopausal if they answered “yes” to having undergone menopause and “yes” to a time interval of at least ⩾12 months from their last menstrual period. To ensure post-menopausal status among all subjects, only women aged ⩾55 years old were included in the study sample [13]. Current users of hormone therapy (oestrogen, progesterone, testosterone compounds or DHEA supplements) were excluded.
Case and control definitions
We included post-menopausal women with idiopathic, connective tissue disease (CTD)- or congenital heart disease (CHD)-associated PAH as designated by their treating physicians. In addition, traditional haemodynamic criteria for PAH (mean pulmonary artery pressure (mPAP) ⩾25 mmHg at rest, mean pulmonary capillary wedge pressure ⩽15 mmHg, and a pulmonary vascular resistance (PVR) >3 Wood units) were required. The study included both prevalent (on PAH therapy) and incident (treatment-naïve) patients. Controls were selected from MESA-RV with 1) normal RV and left ventricular measures (ejection fraction, end-diastolic mass and volumes) measured by cardiac MRI; 2) no self-reported history of chronic obstructive lung disease, emphysema or chronic bronchitis; and 3) the absence of obstructive or restrictive ventilatory defects on spirometry [12]. Controls were randomly matched 2:1 to cases by age (within 5 years) and BMI (within 4 kg·m−2).
Clinical variables
Medical records or the local research registry were reviewed for clinical data for subjects with PAH. Functional class, 6MWD, haemodynamics, echocardiography, brain natriuretic peptide (BNP) levels and PAH therapies were collected at the time of (or as close as possible to) blood collection for sex hormone analysis. Survival status and dates of death (when applicable) were tracked and reported by the centre. The median time from blood sampling to last known visit was 34.5 months (95% CI 27.5–43.0 months).
Steroid hormone levels
Blood samples were drawn and stored using standardised procedures. Plasma sex hormones from PAH cases and controls were measured using the Roche e411 system (F. Hoffman-La Roche, Basel, Switzerland) at the Laboratory for Clinical Biochemistry Research at the University of Vermont (Burlington, VT, USA). Inter-assay coefficients of variation for E2 were 2.2–10.5%, DHEA-S 4.6–6.5%, testosterone 2.3–5.6%, progesterone 9.5–11.5% and SHBG 3.0–4.7%.
Informed consent was obtained from all PAH and MESA participants. Each centre had institutional review board (IRB) approval for this study (Rhode Island Hospital: IRB registration #021911; Penn: IRB registration #706091; Tufts: IRB registration #7437). The protocols for MESA were approved by the IRBs of all MESA sites and the National Heart, Lung, and Blood Institute.
Statistical analysis
All analyses were conducted using SAS software 9.4 (SAS, Cary, NC, USA). Continuous data were presented as median (interquartile range) and categorical data presented as n (%). Sex hormone levels were natural log-transformed. Modelling was accomplished using the GLIMMIX procedure. Case–control status was modelled by hormone levels using generalised estimating equations (GEE) with classic sandwich estimation assuming a binary distribution, where observations were nested within match group. Likewise, hormone levels were modelled by case–control status using a GEE and assuming a log-normal distribution. Case–control random matching for age and BMI was accomplished using a macro designed for SAS software. The relationships between each sex hormone level and clinical markers in PAH cases were examined with multivariable linear or binomial regression as appropriate. Cox hazard regression was used to model the relationship between hormone levels with survival using the PHREG procedure with the likelihood ratio test. The supremum test was used to test the proportional hazards assumption and Harrell’s c-statistic for Cox proportional-hazards models was calculated using a SAS macro [14]. Kaplan–Meier estimation was used to estimate time until death by increments of DHEA-S levels with the LIFETEST procedure. Last known follow-up was used for right censoring. Final models were further adjusted for the exact values of age and BMI. All hypotheses were tested using two-tailed tests and interval estimates were calculated for 95% confidence. α was established at the 0.05 level.
Results
A total of 112 post-menopausal women with PAH (cases) were included in the study (table 1). The majority had CTD-associated PAH (58%), most of whom (n=48, 74%) had systemic sclerosis, followed by idiopathic PAH (35%). Subjects tended to have moderate haemodynamic impairment and 50% were functional class III or IV. After matching for age and BMI, 78 cases were matched with 149 controls for the comparative analyses. Because of the multi-ethnic nature of the MESA-RV cohort and exclusion of individuals with non-normative cardiopulmonary measures, there were imbalances in race/ethnicity, particularly among white (86% PAH cases, 53% controls) and Asian (1% PAH cases, 28% controls) subjects. Clinical characteristics of the total PAH cohort (n=112) and the cases for the comparative analyses (n=78) were nearly identical.
TABLE 1.
Baseline characteristics of pulmonary arterial hypertension (PAH) cases and controls
| Matched controls | Matched PAH cases | All PAH cases | |
|---|---|---|---|
| Subjects | 149 | 78 | 112 |
| Age years | 68 (62–74) | 67 (60–71) | 68 (61–74) |
| Race | |||
| White | 79 (53) | 67 (86) | 97 (86) |
| Black | 29 (19) | 9 (12) | 13 (12) |
| Asian | 41 (28) | 1 (1) | 1 (1) |
| Other | 1 (1) | 1 (1) | |
| BMI kg·m−2 | 26 (23–28) | 27 (23–30) | 28 (24–33) |
| Diagnosis | |||
| Idiopathic PAH | 26 (33) | 39 (35) | |
| CTD-associated PAH | 46 (59) | 65 (58) | |
| CHD-associated PAH | 6 (8) | 8 (7) | |
| WHO functional class | |||
| Subjects with available data | 53 | 81 | |
| I | 1 (2) | 1 (1) | |
| II | 15 (28) | 24 (30) | |
| III | 19 (36) | 33 (41) | |
| IV | 18 (34) | 23 (28) | |
| 6-min walking distance m | 368 (244–419) | 324 (198–396) | |
| Haemodynamics | |||
| Right atrial pressure mmHg | 7 (4–10) | 7 (4–13) | |
| Mean pulmonary artery pressure mmHg | 44 (35–52) | 44 (35–53) | |
| Cardiac output L·min−1 | 4.7 (3.8–5.6) | 4.7 (3.7–5.8) | |
| Pulmonary capillary wedge pressure mmHg | 11 (8–13) | 11 (8–14) | |
| Pulmonary vascular resistance Wood units | 6.6 (4.4–9.1) | 6.8 (4.4–9.4) | |
| Echocardiographic parameters | |||
| RVSP mmHg | 60 (40–75) | 60 (43–75) | |
| Subjects with available data | 45 | 70 | |
| RV enlargement | |||
| Subjects with available data | 71 | 103 | |
| Mild | 15 (21) | 36 (35) | |
| Moderate | 14 (20) | 19 (18) | |
| Severe | 17 (24) | 24 (23) | |
| RV dysfunction | |||
| Subjects with available data | 71 | 104 | |
| Mild | 10 (14) | 13 (13) | |
| Moderate | 15 (21) | 21 (20) | |
| Severe | 11 (15) | 19 (18) | |
| BNP pg·mL−1 | 139 (53–295) | 144 (55–366) | |
| Subjects with available data | 48 | 70 | |
| PAH therapies | |||
| Calcium channel blockers | 13 (17) | 20 (18) | |
| Phosphodiesterase type 5 inhibitors | 50 (64) | 67 (60) | |
| Endothelin receptor antagonists | 40 (51) | 53 (47) | |
| Prostacyclin analogues | 27 (35) | 35 (31) | |
| Combination therapy | 67 (86) | 90 (80) |
Data are presented as n, median (interquartile range) or n (%). BMI: body mass index; CTD: connective tissue disease; CHD: congenital heart disease; WHO: World Health Organization; RVSP: right ventricular systolic pressure; RV: right ventricle; BNP: brain natriuretic peptide.
The association of hormone levels with PAH status are presented in table 2, online supplementary table S1, and figure 1 and online supplementary figure S1. There were significant differences in all hormone levels between PAH cases and controls, except for progesterone. DHEA-S levels were 60% lower in PAH cases as compared to matched controls (p<0.001) (figure 1 and online supplementary table S1). Conversely, higher levels of E2 were observed in PAH compared to matched controls (p=0.02) (figure 1 and online supplementary table S1). Lower levels of DHEA-S, total testosterone, bioavailable testosterone and SHBG were associated with increased odds of PAH. For every unit decrease in DHEA-S, total testosterone, bioavailable testosterone and SHBG, the odds of PAH increased three- to four-fold (table 2). These results were unchanged when age and BMI were added as continuous parameters to the models.
TABLE 2.
Generalised estimating equation models for the prediction of pulmonary arterial hypertension (PAH) case status with decreasing sex hormone levels
| OR# (95% CI) | p-value | Effect size (each 1-unit decrease, change in odds of PAH) | |
|---|---|---|---|
| Oestradiol | 0.65 (0.38–1.11) | 0.11 | |
| DHEA-S | 3.21 (1.63–6.33) | 0.001 | 3.20-fold increase |
| Total testosterone | 2.97 (1.86–4.75) | <0.001 | 2.97-fold increase |
| Bioavailable testosterone | 2.80 (1.74–4.50) | <0.001 | 2.80-fold increase |
| Progesterone | 0.79 (0.47–1.33) | 0.37 | |
| SHBG | 3.69 (1.35–10.04) | 0.01 | 3.70-fold increase |
Subjects were matched on age within 5 years and body mass index (BMI) within 4 kg·m−2 and adjusted for age and BMI. DHEA-S: dehydroepiandrosterone-sulfate, SHBG: sex hormone-binding globulin.
per 1-Ln decrease.
FIGURE 1.

Adjusted parameter estimates for hormone levels by case status after matching for age and body mass index. a) Oestradiol; b) dehydroepiandrosterone-sulfate (DHEA-S). Data are presented as median (95% CI). PAH: pulmonary arterial hypertension.
The relationship between circulating hormone levels and markers of disease severity were investigated for all available cases (n=112) and adjusted for age and BMI. Lower levels of DHEA-S were associated with more severe disease assessed by functional class, 6MWD, haemodynamics (including higher right atrial pressure (RAP), mPAP and PVR), and greater RV dilatation and dysfunction by echocardiography (table 3, figures 2 and 3). For example, each Ln(1 μg·dL−1) decrease in DHEA-S was associated with a shorter 6MWD (−30 m, 95% CI −54 to −6.5 m; p=0.01). These relationships were consistent across indicators of pulmonary vascular and right heart function. Lower DHEA-S levels may have been associated with lower cardiac output (p=0.07) and higher BNP levels (p=0.06). Finally, lower increments of DHEA-S levels were associated with increased mortality (figure 4). Each Ln(1 μg·dL−1) decrease in DHEA-S was associated with a doubling in the risk of death (hazard ratio 2.00, 95% CI 1.46–2.72; p<0.001) after adjustment for age and BMI (table 4). Cox models were further adjusted for type of PAH, 6MWD, RAP and functional class and the estimate for DHEA-S was unchanged, although precision was poorer due to smaller sample size. One subject was lost to follow-up. Median follow-up time was 24.8 months (95% CI 19.6–30.0 months), and because there were so few deaths in this window, the median survival time could not be calculated. The first 25% expired at 25 months (95% CI 16.0–35.6 months), with 36 deaths and 74 patients still alive (censored) at this time point.
TABLE 3.
Relationship between decreasing sex hormone levels and clinical measures in pulmonary arterial hypertension (PAH) cases
| Estimate (95% CI) | p-value | |
|---|---|---|
| 6MWD m | ||
| Ln(oestradiol) | 32.4 (3.5 to 61.4) | 0.03 |
| Ln(DHEA-S) | −30.2 (−54.0 to −6.5) | 0.01 |
| Ln(total testosterone) | −0.8 (−26.6 to 24.9) | 0.95 |
| Ln(SHBG) | 24.5 (−30.9 to 79.9) | 0.38 |
| Right atrial pressure mmHg | ||
| Ln(oestradiol) | −1.6 (−2.8 to −0.3) | 0.02 |
| Ln(DHEA-S) | 1.6 (0.8 to 2.4) | <0.001 |
| Ln(total testosterone) | 0.5 (−0.7 to 1.6) | 0.43 |
| Ln(SHBG) | −0.4 (−2.5 to 1.8) | 0.73 |
| mPAP mmHg | ||
| Ln(oestradiol) | 0.6 (−2.3 to 3.6) | 0.68 |
| Ln(DHEA-S) | 3.6 (1.8 to 5.4) | <0.001 |
| Ln(total testosterone) | 2.8 (0.0 to −5.5) | 0.05 |
| Ln(SHBG) | 3.0 (−2.5 to 8.4) | 0.28 |
| Cardiac output L·min−1 | ||
| Ln(oestradiol) | 0.0 (−0.3 to 0.3) | 0.85 |
| Ln(DHEA-S) | −0.2 (−0.4 to 0.0) | 0.07 |
| Ln(total testosterone) | 0.1 (−0.1 to 0.4) | 0.30 |
| Ln(SHBG) | −0.3 (−0.8 to 0.3) | 0.37 |
| PVR Wood units | ||
| Ln(oestradiol) | 0.4 (−0.3 to 1.0) | 0.27 |
| Ln(DHEA-S) | 0.9 (0.3 to 1.5) | 0.002 |
| Ln(total testosterone) | 0.4 (−0.4 to 1.1) | 0.36 |
| Ln(SHBG) | 0.8 (−0.7 to 2.3) | 0.31 |
| RVSP mmHg | ||
| Ln(oestradiol) | −0.9 (−6.6 to 4.7) | 0.74 |
| Ln(DHEA-S) | 5.6 (1.3 to 9.9) | 0.01 |
| Ln(total testosterone) | 3.0 (−2.9 to 9.0) | 0.31 |
| Ln(SHBG) | −2.9 (−14.4 to 8.5) | 0.61 |
| BNP pg·mL−1 | ||
| Ln(oestradiol) | −101.4 (−241.0 to 38.4) | 0.15 |
| Ln(DHEA-S) | 70.1 (1.5 to 141.8) | 0.06 |
| Ln(total testosterone) | 71.8 (−9.4 to 153.0) | 0.08 |
| Ln(SHBG) | −97.6 (−314.9 to 119.6) | 0.37 |
Adjusted for age and body mass index. 6MWD: 6-min walking distance; DHEA-S: dehydroepiandrosterone-sulfate; SHBG: sex hormone-binding globulin; mPAP: mean pulmonary artery pressure; PVR: pulmonary vascular resistance; RVSP: right ventricular systolic pressure; BNP: brain natriuretic peptide.
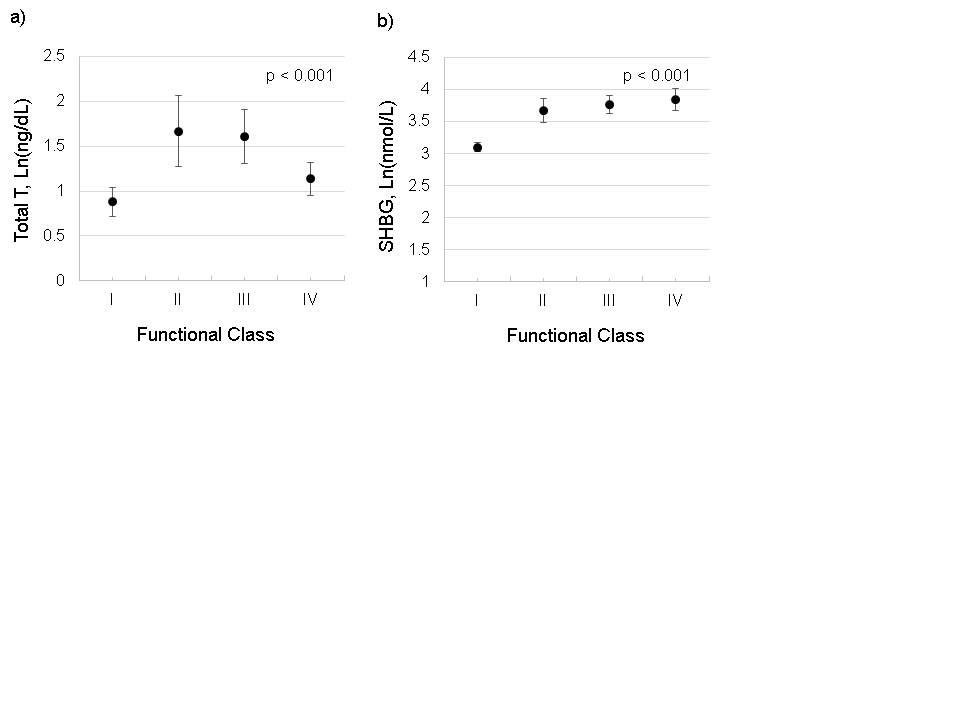
FIGURE 2.

Adjusted parameter estimates for hormone levels by functional class in pulmonary arterial hypertension (PAH) cases. a) Oestradiol; b) dehydroepiandrosterone-sulfate (DHEA-S). Data are presented as median (95% CI). p-value: test for trend across category.
FIGURE 3.

Adjusted parameter estimates for hormone levels by degree of right ventricular (RV) dilatation (a, c) and dysfunction (b, d) on echocardiogram in pulmonary arterial hypertension (PAH) cases. a, b) Oestradiol; c, d) dehydroepiandrosterone-sulfate (DHEA-S). Data are presented as median (95% CI). p-value: test for trend across category.
FIGURE 4.

Kaplan–Meier survival estimates and 95% confidence limits in pulmonary arterial hypertension cases. Numbers of subjects at risk as indicated over the follow-up period. DHEA-S: dehydroepiandrosterone-sulfate.
TABLE 4.
Adjusted# Cox proportional hazards estimate for death by unit decrease in sex hormone level in pulmonary arterial hypertension (PAH) cases
| Sex hormone | Hazard ratio (95% CI) | p-value | Harrell’s c-statistic |
|---|---|---|---|
| Oestradiol pg·mL−1 | 0.91 (0.54–1.53) | 0.71 | 0.52 |
| DHEA-S μg·dL−1 | 2.00 (1.46–2.72) | <0.001 | 0.71 |
| Total testosterone ng·dL−1 | 1.16 (0.72–1.86) | 0.54 | 0.52 |
| Bioavailable testosterone ng·dL−1 | 1.20 (0.74–1.97) | 0.45 | 0.57 |
| Progesterone ng·mL−1 | 0.67 (0.37–1.20) | 0.17 | 0.54 |
| SHBG nmol·L−1 | 0.62 (0.28–1.38) | 0.23 | 0.56 |
Interpretation is by unit decrease. The null for all proportional hazard assumptions tests were not rejected for any model. DHEA-S: dehydroepiandrosterone-sulfate; SHBG: sex hormone-binding globulin.
adjusted for age and body mass index.
Higher levels of E2 were associated with shorter 6MWD and higher RAP (table 3). Each Ln(1 pg·mL−1) increase in E2 was associated with a 32 m shorter 6MWD (95% CI −62 to −4 m; p=0.03) that was also reflected in worse functional class (figure 2). While there were some significant relationships between other circulating hormones and disease markers (e.g. lower testosterone and higher mPAP and BNP levels), these findings were inconsistent or less robust (online supplementary figures S2 and S3). DHEA-S was the only hormone associated with survival.
As DHEA is a prohormone of E2 and testosterone (and testosterone can be aromatised to E2), we investigated whether there were correlations between hormones. First, there were no significant correlations between E2 and DHEA-S levels (r=0.17) or testosterone (r=0.22). There was a modest correlation between DHEA-S levels and testosterone levels (r=0.55). Second, the significant associations between DHEA-S levels and the risk of PAH and all clinical severity measures were unaffected when E2 or testosterone was added to the models (data not shown), indicating the independent nature of the DHEA-S findings. Third, there was no evidence of significant moderation when E2 and testosterone were added to the models for DHEA-S and E2 was added to the models for total testosterone (data not shown).
Finally, in sensitivity analyses in which case–control comparisons were limited to only white and black subjects, results were identical and there was no evidence of effect modification (or interaction) by race in the models between hormone levels and clinical outcomes (data not shown). When we limited our analyses to the subgroup of cases with idiopathic PAH, results were unchanged for DHEA-S, including the relationship between lower DHEA-S levels and mortality (p=0.02). However, the difference in E2 levels between cases and controls was no longer apparent, with this smaller sample size (n=26) (data not shown). Higher levels of E2 remained significantly associated with lower 6MWD (−31 m, 95% CI −55 to −8 m; p=0.01) in those with idiopathic PAH. The number of incident (untreated) PAH patients was small (n=11, 14% for matched cases; n=22, 20% for unmatched cases). Results were unchanged when we excluded these subjects from analyses and included only prevalent (those on PAH-specific treatment) cases (data not shown).
Discussion
We have found that post-menopausal women with idiopathic, CTD- and CHD-associated PAH are characterised by lower levels of DHEA-S, testosterone and SHBG and higher levels of E2 compared to age- and BMI-matched controls without clinical cardiopulmonary disease. Lower levels of DHEA-S had strong associations with multiple measures showing greater clinical severity in PAH including exercise tolerance (6MWD, functional class), RV afterload (mPAP, PVR) and RV function (RAP, RV dilatation and dysfunction, possibly cardiac output and BNP). Lower levels of DHEA-S were associated with an increased risk of death. Higher levels of E2 were associated with shorter 6MWD, higher RAP and worse functional class, but were not associated with survival. The low DHEA-S/high E2 profile seen in post-menopausal women is similar to our study in men with PAH [6], although the associations in post-menopausal women were more robust, probably in part due to differences in sample size. Surprisingly, the associations with testosterone levels in post-menopausal women with PAH were not detected in men with PAH. The findings with DHEA-S, E2 and testosterone were not interrelated, implying that each may have direct effects on the pulmonary circulation and, for DHEA-S, the RV.
Preclinical and observational studies suggest that DHEA-S may play a major role in cardiopulmonary function in PAH. A DHEA receptor is coupled to endothelial nitric oxide synthase, and DHEA regulates endothelin-1 synthesis and inhibits endothelin-1 promoter activity [15, 16], two major pathologic and therapeutic targets in PAH. Human pulmonary artery endothelial cells (PAECs) actively metabolise DHEA and treatment of PAECs from PAH patients with DHEA decreases signal transducer and activator of transcription 3 (STAT3) [17], an important mediator of pulmonary vascular remodelling. Lower DHEA-S levels have been linked to systemic endothelial dysfunction and vascular remodelling, and DHEA supplementation may restore normal vascular function in men and post-menopausal women [18, 19]. Restoring DHEA levels could be effective in mitigating pulmonary afterload since PAH is characterised by low circulating levels of DHEA-S in both sexes and these low levels correlate with worse haemodynamics (including higher PVR).
In addition, lower levels of DHEA-S were associated with worse RV function, which may explain the relationships with 6MWD and functional class. Low DHEA-S levels have been linked to clinical severity and an increased risk of death in left heart failure [20]. DHEA reverses cardiomyocyte hypertrophy and improves tissue levels of collagen and fibronectin [21] decreasing myocardial fibrosis, a key phenotypic change in RV remodelling and decompensation in PAH [22]. In the Sugen/hypoxia model of pulmonary hypertension, DHEA treatment improves cardiac index and inhibits RV capillary rarefaction, fibrosis and oxidative stress [10], suggesting that DHEA may have direct beneficial effects on the RV as well as the pulmonary vasculature.
Lower DHEA-S levels are tied to PAH risk and severity of disease in men and post-menopausal women, but how sex hormone levels interact with sex, treatment response and outcomes in PAH is unknown. DHEA’s key links with nitric oxide, endothelin-1 and RV remodelling could explain sex-based differences in prevalence and survival, as well as differential therapeutic responses by sex in patients treated with phosphodiesterase type 5 inhibitors and endothelin receptor antagonists [23, 24]. The high rate of combination therapy (>80%) in this study prohibited the analysis of treatment-specific interactions. A recent study using unbiased metabolomic profiling demonstrated DHEA-S and its metabolites were reduced in PAH patients compared to unmatched controls and that lower circulating levels of DHEA-S were associated with an increased risk of death [11], but relationships between DHEA-S levels and clinical measures were not reported. In eight patients with pulmonary hypertension related to chronic obstructive pulmonary disease, open-label treatment with 3 months of DHEA was associated with a significant increase in 6MWD and improvements in haemodynamics [25]. Together, these observations suggest that DHEA should be studied as a potential therapy for PAH.
There has been much focus on the role of oestrogen in pulmonary vascular disease, given the strong female predominance and important intersections between oestrogen and the bone morphogenetic receptor type II pathway [26]. In men and in this study, higher E2 levels were observed in PAH as compared to controls, and higher levels were associated with some clinical severity markers, but there was no relationship between E2 levels and mortality, and an attenuated signal when we limited our analyses to idiopathic PAH. This may be because circulating levels of E2 do not reflect production, metabolism or effects in the pulmonary circulation, all shown to be important in pulmonary vascular disease. Alternatively, E2 may have variable effects across the pulmonary vascular–RV circuit, leading to a dilution of the relationship with disease metrics. In a small placebo-controlled, double-blind, randomised clinical trial, we have shown that anastrozole, an aromatase inhibitor, reduced E2 levels and increased 6MWD, but had no effect on RV function [9]. Anastrozole reduced circulating E2 levels, but to a lesser extent than has been reported in breast cancer [27]. Peripheral levels may not reflect local cardiopulmonary activity, and therefore may explain the less robust associations seen here. In post-menopausal women with waning oestrogen levels, more sensitive assays of E2 and its metabolites may be more informative.
Since steroid signalling is complex and individual hormones are highly biologically interrelated, we sought to determine whether some of our findings were collinear. Progesterone, the parent hormone for E2 and testosterone, did not vary across cases and controls and no association was observed with any clinical measures. Similarly, there was no evidence of correlation or effect modification in our cases among biologically related hormones (E2 and testosterone; DHEA-S and E2; and DHEA-S and testosterone). Anastrozole reduced levels of E2 in our trial, but had no effect on levels of DHEA-S, testosterone, bioavailable testosterone or SHBG [9]. This suggests that, as has been seen in experimental models [19, 28], DHEA-S, E2 and possibly testosterone have unique and direct effects in PAH that should be delineated. It is also true that, given the complexity of both the exposure (sex steroid synthesis, metabolism and signalling) and the disease, discrete hormone analysis may miss important signals that would be revealed with a systems biology approach for the entire “hormonosome” [29].
Limitations of this study are that hormone levels were measured only once in predominantly treated PAH patients and that cases and MESA controls were derived from two different sampling populations. In PAH cases, clinical data were collected at the time of or as close as possible to the blood draw for sex hormone levels, and were comprehensive in scope. Differences in the timing of hormone measurement and clinical assessments in PAH cases would have biased to the null. While longitudinal assessment of sex hormones is likely to be even more revealing, the significant associations seen particularly for DHEA-S and to a lesser extent E2 across the spectrum of exercise tolerance, haemodynamics and echocardiography as well as the consistency of the high E2/low DHEA-S signal observed for both sexes strongly argues these relationships are not just epiphenomena, but are important for disease pathobiology. Hormone measurements have been shown to be stable over several years in healthy post-menopausal women [30, 31], but it is not known whether sex hormone levels change more rapidly in PAH. There were more Asian subjects in the control sample than PAH cases; sensitivity analyses without Asian subjects yielded identical results. While sex hormones (and their effects) can vary by race [5, 30], the associations with disease metrics and lack of effect modification by race suggest that the differences seen between hormone levels in cases and controls are not due to race, but are related to PAH. Given the post-menopausal nature of the cohort, most cases had CTD-associated PAH. Like PAH, CTDs affect mostly women. Two small studies in women with systemic sclerosis have demonstrated high levels of E2 and low levels of DHEA-S [32, 33]; adjustment for PAH subtype and sensitivity analyses including only idiopathic PAH subjects did not change our results. Additional and more informative metrics of RV function (i.e. MRI, N-terminal pro-hormone BNP) were not available clinically.
Conclusions
Lower levels of DHEA-S predict risk of disease, clinical severity and death in post-menopausal women with PAH. In addition, lower levels of testosterone and higher levels of E2 were observed among cases and tracked with some clinical metrics, but these relationships were less robust and did not predict outcome. The relationships demonstrated differed for each hormone and did not appear to be interrelated. Together, these observations support the importance of sex steroids in the development of PAH and RV failure and suggest that hormonal modulation may be a viable treatment strategy in pulmonary vascular disease.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Acknowledgements:
This manuscript has been reviewed by all investigators for scientific content and consistency of data interpretation with previous MESA publications. Significant comments have been incorporated prior to submission for publication. The authors thank the other investigators, the staff, PAH patients and the participants of the MESA study for their valuable contributions. A full list of participating MESA investigators and institutions can be found at www.mesa-nhlbi.org
Support statement: This work was completed with support from an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health (P20 GM103652), 11FTF7400032 from the American Heart Association, and K24-HL103844 from the National Heart, Lung, and Blood Institute. Support for MESA is provided by contracts R01-HL086719, R01-HL077612, HHSN268201500003I, N01-HC-95159, N01-HC-95160, N01-HC-95161, N01-HC-95162, N01-HC-95163, N01-HC-95164, N01-HC-95165, N01-HC-95166, N01-HC-95167, N01-HC-95168 and N01-HC-95169 from the National Heart, Lung and Blood Institute, and by grants UL1-TR-000040, UL1-TR-001079 and UL1-TR-001420 from the National Center for Advancing Translational Sciences. Funding information for this article has been deposited with the Crossref Funder Registry.
Conflict of interest: S.M. Kawut reports grants (for research and CME) from Actelion, United Therapeutics, Gilead, Lung Biotech, Bayer and the Cardiovascular Medical Research and Education Fund, and personal fees (for travel) from the American Thoracic Society, outside the submitted work. J.R. Klinger reports grants from Actelion, United Therapeutics, Gilead, Eiger and Bayer, outside the submitted work. H.I. Palevsky reports personal fees from Actelion, Janssen, Bayer, GSK and United Therapeutics, outside the submitted work. I.R. Preston reports grants and personal fees from Actelion, grants from Bayer and Gilead, and personal fees from Arena, United Therapeutics and Pfizer, outside the submitted work. A. Smith reports grants from Actelion, Gilead and United Therapeutics, outside the submitted work. M. Whittenhall reports personal fees from Actelion, Bayer, Gilead and United Therapeutics, outside the submitted work. C.E. Ventetuolo reports personal fees from Bayer, Acceleron and United Therapeutics and grants from the CHEST Foundation (Actelion sponsored) and Eiger paid to her institution, outside the submitted work. C. Archer-Chicko reports personal fees from Actelion, outside the submitted work.
Footnotes
This article has supplementary material available from erj.ersjournals.com
References
- 1.Ling Y, Johnson MK, Kiely DG, et al. Changing demographics, epidemiology, and survival of incident pulmonary arterial hypertension: results from the pulmonary hypertension registry of the United Kingdom and Ireland. Am J Respir Crit Care Med 2012; 186: 790–796. [DOI] [PubMed] [Google Scholar]
- 2.Jacobs W, van de Veerdonk MC, Trip P, et al. The right ventricle explains sex differences in survival in idiopathic pulmonary arterial hypertension. Chest 2014; 145: 1230–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ventetuolo CE, Praestgaard A, Palevsky HI, et al. Sex and haemodynamics in pulmonary arterial hypertension. Eur Respir J 2014; 43: 523–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ventetuolo CE, Ouyang P, Bluemke DA, et al. Sex hormones are associated with right ventricular structure and function: the MESA-Right Ventricle study. Am J Respir Crit Care Med 2011; 183: 659–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ventetuolo CE, Mitra N, Wan F, et al. Oestradiol metabolism and androgen receptor genotypes are associated with right ventricular function. Eur Respir J 2016; 47: 553–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ventetuolo CE, Baird GL, Barr RG, et al. Higher estradiol and lower dehydroepiandrosterone-sulfate levels are associated with pulmonary arterial hypertension in men. Am J Respir Crit Care Med 2016; 193: 1168–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.White K, Johansen AK, Nilsen M, et al. Activity of the estrogen-metabolizing enzyme cytochrome P450 1B1 influences the development of pulmonary arterial hypertension. Circulation 2012; 126: 1087–1098. [DOI] [PubMed] [Google Scholar]
- 8.Lahm T, Frump AL, Albrecht ME, et al. 17β-Estradiol mediates superior adaptation of right ventricular function to acute strenuous exercise in female rats with severe pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 2016; 311: L375–L388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kawut SM, Archer-Chicko CL, DeMichele A, et al. Anastrozole in pulmonary arterial hypertension. A randomized, double-blind, placebo-controlled trial. Am J Respir Crit Care Med 2017; 195: 360–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Alzoubi A, Toba M, Abe K, et al. Dehydroepiandrosterone restores right ventricular structure and function in rats with severe pulmonary arterial hypertension. Am J Physiol Heart Circ Physiol 2013; 304: H1708–H1718. [DOI] [PubMed] [Google Scholar]
- 11.Rhodes CJ, Ghataorhe P, Wharton J, et al. Plasma metabolomics implicates modified transfer RNAs and altered bioenergetics in the outcomes of pulmonary arterial hypertension. Circulation 2017; 135: 460–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kawut SM, Lima JAC, Barr RG, et al. Sex and race differences in right ventricular structure and function: the Multi-Ethnic Study of Atherosclerosis-Right Ventricle study. Circulation 2011; 123: 2542–2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Randolph JF Jr, Zheng H, Sowers MR, et al. Change in follicle-stimulating hormone and estradiol across the menopausal transition: effect of age at the final menstrual period. J Clin Endocrinol Metab 2011; 96: 746–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kremers W Biomedical Statistics and Informatics. Locally Written SAS Macros. www.mayo.edu/research/departments-divisions/department-health-sciences-research/division-biomedical-statistics-informatics/software/locally-written-sas-macros Date last updated: August 2009. Date last accessed: March 2018.
- 15.Chen H, Lin AS, Li Y, et al. Dehydroepiandrosterone stimulates phosphorylation of FoxO1 in vascular endothelial cells via phosphatidylinositol 3-kinase- and protein kinase A-dependent signaling pathways to regulate ET-1 synthesis and secretion. J Biol Chem 2008; 283: 29228–29238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu D, Dillon JS. Dehydroepiandrosterone activates endothelial cell nitric-oxide synthase by a specific plasma membrane receptor coupled to Gαi2,3. J Biol Chem 2002; 277: 21379–21388. [DOI] [PubMed] [Google Scholar]
- 17.Paulin R, Meloche J, Jacob MH, et al. Dehydroepiandrosterone inhibits the Src/STAT3 constitutive activation in pulmonary arterial hypertension. Am J Physiol Heart Circ Physiol 2011; 301: H1798–H1809. [DOI] [PubMed] [Google Scholar]
- 18.Kawano H, Yasue H, Kitagawa A, et al. Dehydroepiandrosterone supplementation improves endothelial function and insulin sensitivity in men. J Clin Endocrinol Metab 2003; 88: 3190–3195. [DOI] [PubMed] [Google Scholar]
- 19.Bonnet S, Paulin R, Sutendra G, et al. Dehydroepiandrosterone reverses systemic vascular remodeling through the inhibition of the Akt/GSK3-β/NFAT axis. Circulation 2009; 120: 1231–1240. [DOI] [PubMed] [Google Scholar]
- 20.Moriyama Y, Yasue H, Yoshimura M, et al. The plasma levels of dehydroepiandrosterone sulfate are decreased in patients with chronic heart failure in proportion to the severity. J Clin Endocrinol Metab 2000; 85: 1834–1840. [DOI] [PubMed] [Google Scholar]
- 21.Aragno M, Mastrocola R, Alloatti G, et al. Oxidative stress triggers cardiac fibrosis in the heart of diabetic rats. Endocrinology 2008; 149: 380–388. [DOI] [PubMed] [Google Scholar]
- 22.Rain S, Andersen S, Najafi A, et al. Right ventricular myocardial stiffness in experimental pulmonary arterial hypertension: relative contribution of fibrosis and myofibril stiffness. Circ Heart Fail 2016; 9: e002636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mathai SC, Hassoun PM, Puhan MA, et al. Sex differences in response to tadalafil in pulmonary arterial hypertension. Chest 2015; 147: 188–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gabler NB, French B, Strom BL, et al. Race and sex differences in response to endothelin receptor antagonists for pulmonary arterial hypertension. Chest 2012; 141: 20–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dumas de La Roque E, Savineau JP, Metivier AC, et al. Dehydroepiandrosterone (DHEA) improves pulmonary hypertension in chronic obstructive pulmonary disease (COPD): a pilot study. Ann Endocrinol 2012; 73: 20–25. [DOI] [PubMed] [Google Scholar]
- 26.Chen X, Austin ED, Talati M, et al. Oestrogen inhibition reverses pulmonary arterial hypertension and associated metabolic defects. Eur Respir J 2017; 50: 1602337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kyvernitakis I, Albert US, Kalder M, et al. Effect of anastrozole on hormone levels in postmenopausal women with early breast cancer. Climacteric 2015; 18: 63–68. [DOI] [PubMed] [Google Scholar]
- 28.Liu D, Dillon JS. Dehydroepiandrosterone stimulates nitric oxide release in vascular endothelial cells: evidence for a cell surface receptor. Steroids 2004; 69: 279–289. [DOI] [PubMed] [Google Scholar]
- 29.Maron BA, Leopold JA. Systems biology: an emerging strategy for discovering novel pathogenetic mechanisms that promote cardiovascular disease. Glob Cardiol Sci Pract 2016; 2016: e201627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lasley BL, Santoro N, Randolf JF, et al. The relationship of circulating dehydroepiandrosterone, testosterone, and estradiol to stages of the menopausal transition and ethnicity. J Clin Endocrinol Metab 2002; 87: 3760–3767. [DOI] [PubMed] [Google Scholar]
- 31.Hankinson SE, Manson JE, Spiegelman D, et al. Reproducibility of plasma hormone levels in postmenopausal women over a 2–3-year period. Cancer Epidemiol Biomarkers Prev 1995; 4: 649–654. [PubMed] [Google Scholar]
- 32.La Montagna G, Baruffo A, Buono G, et al. Dehydroepiandrosterone sulphate serum levels in systemic sclerosis. Clin Exp Rheumatol 2001; 19: 21–26. [PubMed] [Google Scholar]
- 33.Aida-Yasuoka K, Peoples C, Yasuoka H, et al. Estradiol promotes the development of a fibrotic phenotype and is increased in the serum of patients with systemic sclerosis. Arthritis Res Ther 2013; 15: R10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.