

SUMMARY
The mechanisms underlying sterol transport in mammalian cells are poorly understood. In particular, how cholesterol internalized from HDL is made available to the cell for storage or modification is unknown. Here we describe three ER-resident proteins (Aster-A, -B, -C) that bind cholesterol and facilitate its removal from the plasma membrane. The crystal structure of the central domain of Aster-A broadly resembles the sterol-binding fold of mammalian StARD proteins, but sequence differences in the Aster pocket result in a distinct mode of ligand binding. The Aster N-terminal GRAM domain binds phosphatidylserine and mediates Aster recruitment to plasma membrane-ER contact sites in response to cholesterol accumulation in the plasma membrane. Mice lacking Aster-B are deficient in adrenal cholesterol ester storage and steroidogenesis due to an inability to transport cholesterol from SR-BI to the ER. These findings identify a nonvesicular pathway for plasma membrane to ER sterol trafficking in mammals.
Graphical Abstract
ETOC
A nonvesicular pathway for plasma membrane to ER sterol trafficking in mammals is mediated by sterol-binding ER-resident Aster proteins
INTRODUCTION
HDL cholesterol levels in the plasma are inversely associated with coronary heart disease (CHD) risk, but recent genetic studies have strongly implied that HDL cholesterol levels are not causally related to risk for CHD (Rader and Tall, 2012). For example, the P376L mutation in scavenger receptor class B member 1 (SR-BI) is associated with greater cardiovascular disease risk despite high plasma levels of HDL cholesterol (Zanoni et al., 2016). Mounting evidence suggests that the function of HDL, in particular its role in cholesterol movement in cells and tissues, may be key to its role in physiology and disease (Khera et al., 2011; Rader, 2014). HDL is important in the reverse cholesterol transport pathway that brings surplus cholesterol from peripheral tissues to the liver for excretion (Braun et al., 2002). A better understanding of the pathways by which HDL cholesterol moves through cells and tissues may aid in the development of novel diagnostic tools and therapies.
Integral membrane receptors involved in cholesterol uptake at the plasma membrane (PM) have been characterized extensively, but comparatively little is known about mechanisms that traffic cholesterol from the PM to other compartments within the cell (Goldstein and Brown, 1985; Horton et al., 2002; Ikonen, 2008). Scavenger Receptor class B member I (SR-BI) is the principal cell-surface receptor for HDL (Acton et al., 1996). SR-BI is abundant in steroidogenic organs and the liver, where it facilitates the selective uptake of cholesterol from HDL (Glass et al., 1983; Neculai et al., 2013). In the liver, HDL-cholesterol uptake facilitates reverse cholesterol transport by delivering surplus peripheral cholesterol to hepatocytes for secretion into bile or for conversion into bile acids. In steroidogenic organs, HDL-derived cholesterol accumulates in the form of cholesterol ester, and these stores are used for the synthesis of steroid hormones (Connelly, 2009; Hoekstra, 2017). The selective HDL-cholesterol uptake pathway is distinct from the LDLR pathway by which LDL particles are taken up and delivered to lysosomes for degradation (Goldstein and Brown, 2015). HDL-cholesterol uptake does not require clathrin-dependent receptor-mediated uptake or lysosomal targeting, but what happens to HDL-derived cholesterol after SR-BI–mediated uptake is unknown. The pathways downstream of SR-BI that move cholesterol within the cell have not been defined.
Dozens of candidate cholesterol-transfer proteins have been shown to be capable of mediating sterol transfer in vitro (Ikonen, 2008, 2018); however, assigning physiologic functions to those proteins has proven challenging. Three intracellular cholesterol-transfer proteins have been shown to have clear physiologic functions in vivo. The Niemann Pick type C proteins 1 and 2 (NPC1, NPC2) are critical for the export of LDL-derived cholesterol from the lysosome (Infante et al., 2008; Pfisterer et al., 2016). Mutations in either NPC1 or NPC2 lead to accumulation of lysosomal lipids in vivo, explaining the phenotypes associated with Niemann Pick Type C syndrome. Steroid Acute Regulatory Protein (StARD1) is required for the trafficking of cholesterol to the mitochondrial inner membrane (Lin et al., 1995a). Mutations in StARD1 are the major cause of lipoid congenital adrenal hyperplasia.
The ER is known to form membrane contacts with other organelles, and these have been proposed to facilitate the transfer of small molecules between membranes (Chung et al., 2015; Lees et al., 2017; Zhang et al., 2005a). However, no mammalian transporter has yet been shown to be required for the trafficking of cholesterol from the PM to the ER (Ikonen, 2008, 2018). Here we characterize three mammalian proteins (Aster-A, -B, and -C) that bind and transfer cholesterol between membranes. Aster proteins are anchored to the ER by a single transmembrane helix and form ER–PM membrane contacts in response to PM cholesterol loading. We hypothesized that the Asters could be missing links in the trafficking of HDL-derived cholesterol to the ER. We found that Aster-B was selectively enriched in steroidogenic organs and that its expression was required for the storage of HDL-derived cholesterol ester and steroidogenesis in the adrenal cortex. Our findings elucidate a nonvesicular pathway for PM–ER sterol trafficking in mammalian cells and suggest new mechanisms by which HDL-derived cholesterol is mobilized in a variety of physiological contexts.
RESULTS
A Family of Mammalian Lipid-Binding Proteins
We discovered that the Gramd1b gene was regulated by the sterol-responsive Liver X Receptors (LXRs) in mice, suggesting that it could play a role in sterol homeostasis (Janowski et al., 1996). Gramd1b was induced by a synthetic LXR agonist in wild-type (WT) but not LXR-null mouse macrophages (Figure 1A). Moreover, ChIP-seq analyses revealed binding of LXRα and LXRβ to the regulatory regions of Gramd1b, identifying it as a direct transcriptional target (Figure 1B). Gramd1b belongs to a family of highly-conserved genes, designated Gramd1a, -b, and -c in databases. One-to-one orthologs of Gramd1b are present in all vertebrate classes (Supplemental Figure 1A). The predicted amino acid sequence of the human protein is 78% identical to that from Oreochromis niloticus (nile tilapia). These genes have not previously been characterized; their structures are incorrectly annotated in databases; and the function of their protein products is unknown. We determined the correct exon–intron structures of Gramd1a, -b, and -c (Supplemental Figure 1B), which are predicted to encode proteins of 723, 699, and 662 amino acids, respectively. We will refer to the protein products as Aster-A, -B, and -C. The genes are expressed in a tissue-specific manner in mice, with Gramd1a being most abundant in the brain; Gramd1b prominently expressed in steroidogenic tissues and macrophages; and Gramd1c expressed in liver and testes (Supplemental Figure 1C).
Figure 1. Aster Proteins Contain a Lipid-Binding Fold.

(A) Real-time PCR analysis of gene expression from WT (white) and LXR-null (LXRα−/− and LXRβ−/−, red) mouse peritoneal macrophages. Cells were treated with LXR ligand GW3965 (1 µM), with or without RXR ligand LG100754 (100 nM) for 16 h. Values are means ± SEM.
(B) ChIP-Seq bedgraph of LXRα and LXRβ binding patterns in mouse macrophages at the promoter region of the Gramd1b locus (Sallam et al., 2018; Sallam et al., 2016; Zhang et al., 2017). Input (Ctrl) served as a control for LXR enrichment.
(C) Schematic representation of Aster-A, Aster-B, and Aster-C proteins. The N-terminal GRAM (green), central ASTER (blue) and transmembrane (TM, red) domains are indicated.
(D) Purified Aster domains (Aster-A261–576, Aster-B224–560, and Aster-C206–528) bind to 22-NBD-cholesterol but not 6-NBD-cholesterol with nanomolar affinity. Values are means ± SEM.
(E) Aster-B334–562 (1–10 µM) was titrated with 10–3000 nM 22-NBD-cholesterol in PBS and fluorescent ligand binding determined.
(F) Binding of [3H]cholesterol to purified Aster-B224–560 was assessed using a GST-agarose-based assay. Competition assays were performed using increasing concentrations of unlabeled cholesterol as indicated. Results values are means ± SD.
(G) Aster-B334–562 was titrated with 22-NBD-cholesterol in the presence of vehicle, estradiol, or various hydroxycholesterol (HC) sterol competitors as indicated (10 µM). Results values are means ± SD.
(H) Sucrose-loaded heavy PC/Dansyl-PE liposomes and light PC/Dansyl-PE-Cholesterol liposomes were incubated with buffer or with 5 µM albumin, pre-heated Aster-B224–560 (left, 95°C × 10 min), or native Aster-A261–576, Aster-B224–560, and Aster-C206–528 (right) for 20 min at 25°C. Cholesterol transfer to the heavy liposomes was assessed and normalized to dansyl-PE recovery. Values are means ± SEM. All results are representative of at least two independent experiments. See also Supplemental Figures 1E–G.
The Aster proteins contain an N-terminal GRAM domain, which is structurally similar to a pleckstrin homology domain (Begley et al., 2003), and a single transmembrane domain near the C-terminus (Figure 1C). The large central domain of the Aster proteins shows low sequence similarity to structurally characterized proteins. However, modeling programs such as Phyre and I-TASSER predicted that this central domain would resemble the sterol-binding domains from the mammalian StarD proteins and the Starkin domains from the yeast Lam proteins, despite minimal identity at the amino acid level. We named this mammalian START-like domain the ASTER (Greek for “star”) domain.
The ASTER Domain Binds Sterols and Promotes their Transfer Between Membranes
Molecular modeling of the ASTER domain indicated the presence of a hydrophobic pocket capable of accommodating a lipophilic molecule. To test their ability to bind lipids, we expressed and purified the ASTER domains from Aster-A, -B, and -C (Supplemental Figure 1D). Using a fluorescent NBD-cholesterol binding assay, we found that all three Aster domains avidly bound sterols (Petrescu et al., 2001). As shown in Figure 1D, increasing the concentration of NBD-cholesterol while maintaining the concentration of the ASTER-domain constant increased the fluorescence emission of NBD-cholesterol. Fitting the data to a single exponential yielded an average Kd of <100 nM. By contrast, 6-NBD-cholesterol, binding of which would be predicted to place the NBD inside of the binding pocket, did not bind to the ASTER domains (Figure 1D). Using the Aster-B ASTER domain, we further confirmed increased NBD fluorescence in response to increasing protein concentration (Figure 1E). We also confirmed direct binding of [3H]cholesterol to the Aster-B ASTER domain (Figure 1F).
Competition studies further showed that binding of NBD-cholesterol to the ASTER domain was inhibited by 22-R, 25-, and 20α-hydroxycholesterol in addition to cholesterol itself (Figure 1G and Supplemental Figure 1E). However, estradiol, 4β-, 22S-, and 7β-hydroxycholesterols were comparatively poor competitors. The inability of these oxysterols to compete for NBD-cholesterol binding suggests that the different binding affinities are not due to differences in the solubility of the different sterols. Finally, the affinity of the Asters for sterols was comparable to that of the sterol-binding domain from the canonical START domain protein StARD1 (Supplemental Figure 1F).
To test the ability of the ASTER domain to transfer cholesterol between membranes, we optimized an in vitro assay with heavy and light liposomes (Chung et al., 2015). The ASTER domain from Aster-A, -B, and –C, but not BSA, efficiently facilitated cholesterol transfer to the heavy liposomes (Figure 1H). Preheating the ASTER domain to 95°C for 10 min substantially reduced its activity. Interestingly, the ASTER domains were more efficient transporters of cholesterol in this assay than the START domain of the canonical StARD1 protein (Supplemental Figure 1G).
Crystal Structure of the Aster Sterol-binding Domain
To determine the structure and to characterize the mode of sterol binding, we crystalized the ASTER domain from Aster A (aa: 334–562) with 25-hydroxycholesterol. Despite the predicted similarity to the StARD and Lam proteins, solving the structure by molecular replacement proved to be challenging. A solution was found using a truncated model based on the structure of the first start domain from the yeast protein Lam4 (5YQJ) with 23% sequence identity (Tong et al., 2018). The structure of the ASTER domain consists of a highly curved 7-stranded beta-sheet forming a groove to accommodate the hydroxycholesterol ligand. The cavity is closed by a long carboxy-terminal helix and two shorter helices following the amino-terminal beta-strand (Figure 2A). The electron density for the 25-hydroxycholesterol unambiguously defined the position and orientation of the sterol, which was identical in all four molecules within the asymmetric unit (Figure 2B and Supplemental Figure 2A). Interestingly, there was additional volume within the cholesterol-binding cavity adjacent to the C3-OH group of the cholesterol. Within this volume we observed electron density for a glycerol molecule adjacent to the hydroxyl group on the cholesterol (Figure 2C and Supplemental Figure 2A). Glycerol was present during purification of ASTER domain and also used as a cryo-protectant. Interestingly the glycerol is ideally sized to fill the remaining volume of the pocket that is not occupied by the hydroxycholesterol.
Figure 2. Crystal structure of the sterol-binding domain of Aster-A.

(A) The crystal structure of the ASTER domain of the mouse Aster-A. The ribbon representation is colored from blue-red amino-carboxy terminus. The 25-hydroxycholesterol ligand is shown as atomic spheres: grey-carbon, red-oxygen. The right-hand panel is rotated 90°about the indicated axis. The ligand-binding pocket is situated between a concave beta-sheet and a long carboxy-terminal helix.
(B) Details of the 25-hydroxycholesterol ligand-binding pocket. The left-hand panel shows key sidechains within the ligand pocket that mediate interaction with the ligand. Phe405, Tyr524 and Phe525 (cyan) seem to determine the orientation of the ligand and are markedly different in character from the equivalent residues in the yeast Lam proteins (see also Supplemental Figure 3).
(C) Cut-away view of the surface of mouse Aster A showing the ligand-binding pocket. The ligand is completely enclosed with the exception of an opening towards the left of the pocket. The pocket is significantly larger than the ligand beyond the C3-OH group. This additional space is occupied by a glycerol molecule.
(D-E) Potential mechanism for loading cholesterol into the ASTER domain. Structural rearrangements would be essential for cholesterol to gain access to the binding pocket. This is likely to involve the loop comprising amino acids 430–439 that wraps around the ligand. The surface of this region of the ASTER domain is relatively non-polar in character (see labeled amino acids), but with a number of prominent basic residues. It seems likely that this region of the protein will come into contact with the negatively charged / non-polar lipid bilayer into order to facilitate both loading and unloading of the cholesterol ligand. See also Supplemental Figures 2 and 3.
Despite the relatively low sequence identity, the three-dimensional structure of Aster A broadly resembles the START domain fold, and is similar to the START-like domains in the Lam2 and Lam4 proteins (Horenkamp et al., 2018; Jentsch et al., 2018; Tong et al., 2018) (C-alpha RMSD c.2Å; Supplemental Figure 2B). However, sequence differences within the cholesterol-binding pocket result in a different binding mode for the ligand, such that in Aster-A the sterol is rotated by approximately 120° about the long axis of ligand compared with the ligands in the START domains. This appears to be a concerted effect of multiple amino acid differences, but in particular F405, Y524 and F525 in mouse Aster-A seem to influence the ligand orientation (Figure 2B). Interestingly these residues are conserved in all 3 mammalian Aster proteins, but not the yeast Lam proteins (Supplemental Figure 3A,B).
The sterol-binding pocket within the ASTER domain is largely enclosed with the exception of a relatively small opening adjacent to the loop between beta-strands 3 & 4. In order for the sterol to gain access to the pocket it is very likely that this loop will open (Figure 2D and E). In all four complexes within the asymmetric unit, this loop has relatively high B-factors or could not be modeled, consistent with conformational flexibility. Interestingly, there is an abundance of surface-exposed non-polar residues located at the “tip” of the Aster domain around the presumed opening of the sterol-binding cavity (Figure 2C and D). Alongside these non-polar residues, are a number of conserved basic residues. These give the tip of the ASTER domain an overall positive charge but with the opportunity to make non-polar interactions. This conserved surface chemistry of Aster proteins may assist exchange of sterol with negatively-charged / non-polar phospholipid membranes through interaction with and/or partial insertion of the domain into the membrane.
Asters Are Integral ER Proteins Recruited to the PM by Cholesterol
The structure of the Aster proteins suggested that they may promote the transfer of cholesterol between biological membranes. A critical question, therefore, was where these proteins are located within cells. To determine the location of Aster-A, -B, and –C, we expressed N-terminally tagged fusion proteins in A431 and HeLa cells. When cultured in standard lipid-poor conditions (1% lipoprotein-deficient serum, LPDS), Aster proteins displayed a reticular pattern that largely overlapped with the ER marker Sec61β (Figure 3A–C and Supplemental Figure 4A). An Aster-B fusion protein lacking the GRAM domain was also localized to the ER, but one containing the isolated GRAM domain was mainly located within the cytoplasm (Figure 3A).
Figure 3. Asters are ER proteins that localize to the PM in response to cholesterol.

(A) Analysis of the cellular localization of full-length (1–699), B ∆GRAM (225–669), or B GRAM (1–171) Aster-B-GFP constructs with ER marker (Sec61β) in HeLa cells imaged by live-cell confocal microscopy. Scale bar 5 µm.
(B) Analysis of Aster-B-GFP (N-terminal, 1–738) localization and PM (CellMask PM stain) in A431 cells imaged by confocal microscopy. Cells were cultured 5% LPDS or treated with 200 µM cholesterol:cyclodextrin for 1 h. Images were taken from live cells. Scale bar 5 µm. See also Supplemental Movie 1.
(C) Live-cell imaging of GFP-Aster-B localization in A431 cells stably expressing BFP-KDEL (ER marker) and Cherry-CAAX (PM marker). Yellow arrows indicate ER tubules in close proximity to the PM containing foci of Aster-B expression. Results are representative of three independent experiments. Large images, scale bar = 10 µm; insets, scale bar = 2 µm. See also Supplemental Figure 4A–C.
We hypothesized that the ER-localized Aster proteins might facilitate lipid transfer by making transient contacts with another cellular membrane. Remarkably, we found that cholesterol loading by methyl-β-cyclodextrin resulted in the redistribution of all 3 Aster proteins to the periphery of the cell in close proximity to the PM (Figure 3B and Supplemental 4A). We next visualized the location of GFP-Aster-A, -B and -C in A431 cells using live cell Airyscan imaging. When cells were cultured in low-cholesterol media (LPDS), all 3 Aster proteins showed a punctate pattern of distribution in ER structures throughout the cell (Figure 3C and Supplemental Figures 4B and C). However, when cholesterol-cyclodextrin was added to the media, the Aster proteins were found almost exclusively in ER tubules that abutted the PM (yellow arrows).
These findings suggested that Aster proteins were localizing to regions of PM-ER apposition and forming cholesterol-dependent bridges. We assessed the relationship of Aster-PM contacts to membrane contact associated with the proteins ORP5, E-Syt2 and E-Syt3 (Chung et al., 2015; Ghai et al., 2017; Giordano et al., 2013). We analyzed the cellular location of GFP-Aster-B, ORP5, E-Syt2 and E-Syt3 in the presence or absence added cholesterol. In cells cultured in low-cholesterol media, ORP5, E-Syt2 and E-Syt3 were found predominantly in ER tubules located adjacent to the PM (Figure 4A and B and Supplemental Figure 4D). By contrast, Aster-B was located throughout the ER under these conditions and showed minimal colocalization with ORP5, E-Syt2 or E-Syt3. Addition of cholesterol to the cells had little if any effect on the location of ORP5, E-Syt2 or E-Syt3, but caused a dramatic relocalization of Aster-B to ER tubules that were in close proximity to the PM. Moreover, there was substantial, but not complete, overlap of Aster-B signal with signals for ORP5, E-Syt2 and E-Syt3 in cholesterol-loaded cells (quantified in Figure 4C). Interestingly, while ER-PM contacts containing Aster-B were frequently located in the same ER tubules in which ORP5, E-Syt2 or E-Syt3 resided, domains containing only Aster-B could also be readily be identified in proximity to the PM.
Figure 4. Aster-B is recruited to ER–PM contact sites in response to cholesterol.

(A) Live-cell imaging of GFP-Aster-B localization in A431 cells stably expressing BFP-KDEL and Cherry-cultured in 5% LPDS (left) or following cholesterol loading for 20 min (right). Large images, scale bar = 10 µm; insets, scale bar = 2 µm.
(B) Live-cell imaging of GFP-Aster-B localization in A431 cells stably expressing BFP-KDEL and Cherry-E-Syt2 cultured in 5% LPDS (left) or following cholesterol loading for 40 min (right). Large images, scale bar = 10 µm; insets, scale bar = 2 µm.
(C) Quantification of Aster-B colocalization with ER-PM contact proteins. Quantification was done by selecting a square region from the bottom plane (near the PM), thresholding the punctate structures, and calculating their pixel overlap by using a colocalization tool. N = 8–12 cells per construct and treatment from 2 independent experiments.
(D) Time course of Cherry and GFP fluorescence visible in TIRF basal PM-associated fluorescence field of A431 cells expressing with GFP-Aster-B and Cherry-ORP5 after the addition of cyclodextrin-cholesterol to the media.
We further analyzed cholesterol-dependent Aster PM recruitment using total internal reflection (TIRF) microscopy. We analyzed A431 cells expressing GFP-Aster-B and Cherry-ORP5 after loading of the cells with cyclodextrin cholesterol. While ORP5 was detected in the TIRF plane regardless of cellular sterol status, Aster-B was rapidly recruited to the TIRF plane in response to cholesterol (Figure 4D and Supplemental Movie 1). We conclude from these studies that, in contrast to ORP5, E-Syt2 and E-Syt3, which reside in ER-PM contact sites regardless of cellular sterol status, Aster-B is selectively recruited to regions of ER-PM contact in response to excess cholesterol in the PM.
The Gram Domain Mediates Cholesterol-Dependent Aster Localization
The Aster proteins contain an N-terminal GRAM domain, a structural motif that is found in glucosyltransferases, Rab-like GTPase activators, myotubularins, and other membrane-associated proteins (Doerks et al., 2000). Interestingly, the GRAM domains of myotubularins have been shown to interact with phospholipids (Begley et al., 2003). We found that purified Aster-B GRAM domain interacted strongly with both phosphatidylserine (PS) and phosphatidic acid (PA) (Figure 5A). Given that phosphatidylserine is highly enriched in the inner leaflet of the PM, we further determined if the Aster-B GRAM domain was associated with phosphatidylserine-containing liposomes. Co-sedimentation assays revealed that the GRAM domain pelleted selectively with phosphatidylserine-containing liposomes (Figure 5B). The inclusion of cholesterol in PS-containing liposomes did not enhance their association with the GRAM domain.
Figure 5. The GRAM domain mediates cholesterol-dependent Aster recruitment.

(A) Protein-lipid overlay of mouse Aster-B GRAM domain with various phospholipid species.
(B) Purified Aster-B GRAM domain was incubated with liposomes containing Dansyl-PE and 80–85% PC or 80–85% PS +/− 5% Cholesterol. Liposomes were sedimented, washed, and analyzed by immunoblotting for Gram domain association.
(C) Localization of full-length and B ∆GRAM Aster-B-GFP (green) constructs in HeLa cells in the presence or absence of cholesterol loading. Magenta, CellMask PM stain.
(D) Localization of B GRAM-GFP in CHO-K1 cells (left) and A431 cells (right) cultured in LPDS (top) or loaded with cholesterol (bottom). PM marker: PM-cherry; ER marker: mCherry-Sec61β. Scale bar: 5 µM. See also Supplemental Movie 2.
(E) Quantification of Aster-B PH-EGFP intensity in the TIRF plane upon cholesterol loading (left panel; n = 2–3 cells, error bars +/− SD). Representative TIRF images (right panel) from GFP and Aster-B PH-EGFP-expressing cells at the indicated time following cholesterol loading. See also Supplemental Movie S3, S4 and Supplemental Figure 4E. Results are representative at least two independent experiments.
To determine if the Aster GRAM domain was required for the cholesterol-dependent localization of Asters to the PM, we transfected Hela and A431 cells with mutant form of Aster-B in which the GRAM domain had been deleted (Figure 5C and Supplemental Figure 4E). Loss of the GRAM domain abolished recruitment of Asters to the PM in response to cholesterol. We also expressed the Aster-B GRAM domain alone in CHO-K1 and A431 cells (Figure 5D and Supplemental Movie 2). Under basal culture conditions, the soluble GRAM domain was cytoplasmic, but it was recruited to the PM upon cholesterol loading. TIRF microscopy confirmed that C-terminal GFP-tagged Aster-B GRAM domain was largely cytoplasmic in cells cultured in LPDS but that it was recruited to the PM in a time- and concentration-dependent manner after cholesterol loading (Figure 5E and Supplemental Movies 3 and 4). We conclude that the phosphatidylserine-binding GRAM domain is both necessary and sufficient for cholesterol-dependent Aster redistribution.
Aster-B Is Required for Adrenal Sterol Homeostasis
Movement of cholesterol from the PM to the ER is an important step in the utilization of HDL-derived cholesterol after selective uptake by SR-BI. We hypothesized that Asters may contribute to cholesterol transport from SR-BI at the PM to the ER. To test this idea, we focused on the rodent adrenal gland, which depends on SR-BI–mediated uptake of HDL cholesterol for steroidogenesis (Rigotti et al., 1997). Among the Aster proteins, Aster-B exhibited the highest expression in the adrenal gland, suggesting that it was likely to be the most physiologically relevant Aster family member in this tissue (Supplemental Figure 5A) (Consortium, 2012). We confirmed that Aster-B protein was expressed at high levels in the adrenal, similar to SR-BI (Figure 6A).
Figure 6. Aster-B ablation disrupts adrenal cholesterol homeostasis.

(A) Immunoblot analysis of SR-BI and Aster-B in various tissues from 7-week-old C57BL/6J mice. HMGB1 was used as a loading control.
(B) Representative immunoblot from adrenal lysates of WT and Aster-B KO mice.
(C) Gross appearance of adrenal glands from representative 6-week-old WT and Aster-B knockout mice (1 mm scale). Results are representative of at least ten mice of both genders.
(D) Histological sections of the adrenal cortex stained with oil red O. Representative of eight images per group; 12 µm sections; scale bar: 50 µm.
(E) Representative electron micrographs of adrenal fasciculata cells. Samples were fixed and processed as described in the Methods. Lipid droplets, nuclei and mitochondria are indicated (LD, N, MT; N = 2 mice, 35–52 sections each).
(F) ESI-MS/MS analysis of the abundance of free cholesterol in adrenal glands from WT and Aster-B KO mice (n=7). Statistical analysis was performed using Student’s t-test. Values are mean ± SEM.
(G) ESI-MS/MS analysis of the abundance of cholesterol ester species in adrenal glands (N = 7). Statistical analysis was performed using Student’s t-test. Values are means ± SEM. *p < 0.05; **p < 0.01; ***p<0.001; ****p<0.0001. Results are representative of two independent experiments.
To assess the physiological relevance of cholesterol transport by Aster-B, we generated knockout mice by CRISPR/Cas9 editing (Supplemental Figure 5B and Figure 6B). By visual inspection, the adrenal glands of Aster-B–deficient mice were red, suggesting an absence of cholesterol ester stores (Figure 6C). Indeed, oil red O staining revealed a loss of neutral lipid stores in the adrenal cortex in Aster-B–deficient mice (Figure 6D), and electron microscopy revealed a complete absence of cytosolic lipid droplets (Figure 6E). While levels of free cholesterol in the adrenal gland were not different between genotypes, loss of Aster-B expression led to a dramatic decrease in cholesterol esters (Figure 6F, G) indicating that Aster-B expression is required for tissue cholesterol homeostasis in vivo.
Aster-B Promotes PM to ER Cholesterol Transport
We next investigated the consequence of loss of Aster function for sterol movement in cultured cells. Since Aster-B is poorly expressed in most cultured cell lines, we knocked down Aster-A expression, which is abundant 3T3-L1 cells. We assessed cholesterol movement from the PM to the ER following exogenous delivery by measuring 1. the activity of the SREBP-2 pathway and 2. the formation of cholesterol esters. Treatment of 3T3-L1 cells with an Aster-A-specific ASO (which nearly abolished Gramd1a expression; Figure 7) led to induction of SREBP-2 processing, increased LDLR protein levels, and increased expression of SREBP-2 target genes, including Hmcgr, and Hmgcs (Figure 7A and B). Moreover, the ability of exogenously added cyclodextrin-cholesterol to suppress SREBP-2 processing was clearly delayed in Aster-A-silenced cells. The fact that some suppression of the SREBP-2 pathway at later time points was still observed even with Aster-A ASO was not unexpected, since vesicular sterol transport pathways were presumably still operative. Interestingly, mRNA and protein expression of LXR target gene Abca1 was reciprocally reduced in the absence of Aster-A (Figure 7C), consistent with reduced ER cholesterol availability for oxysterol production. We further found that the rate at which cholesterol delivered to the PM was incorporated into CE was markedly slower in cells in which Aster-A was silenced (Figure 7D). Two hours after cholesterol addition the amount of CE formed in Aster-A-silenced cells was less than 25% of controls cells.
Figure 7. Asters mediate PM to ER cholesterol transport in cells and tissues.

(A) Immunoblot analysis of membrane (top) and nuclear (bottom) protein levels in 3T3-L1 cells treated for 48 h with control or Aster-A-specific ASO and then for the indicated times with cyclodextrin-cholesterol. Calnexin and Lamin A/C served as loading controls.
(B) Real-time PCR analysis of gene expression in 3T3-L1 cells treated for 48 h with control or Aster-A-specific ASO and then for the indicated times with cyclodextrin-cholesterol. ***p<0.001.
(C) Abca1 expression in 3T3-L1 cells treated for 48 h with control or Aster-A-specific ASO and then for the indicated times with cyclodextrin-cholesterol. **p<0.01.; ***p<0.001.
(D) Time course of cholesteryl ester formation in 3T3-L1 cells treated with control or Aster-A-specific ASO for 48 h. *p < 0.05; ***p<0.001.
(E) Expression levels of the indicated genes in adrenal glands from WT and Aster-B KO mice (mean ± SEM; N = 6–8 per group). Statistical analysis was by Student’s t-test. Values are means ± SEM. *p < 0.05; **p < 0.01; ***p<0.001; ****p<0.0001.
(F) Immunoblot analysis of membrane (top) and nuclear (bottom) protein levels in adrenals from WT and Aster-B KO mice (n=5).
(G) Serum cholesterol levels in mice fasted for five h (mean ± SEM; N = 6 per group).
(H) Serum corticosterone levels measured by ELISA in mice that were fed ad-lib or fasted overnight (~16 hours). N = 4 per group. Statistical analysis was by Student’s t-test. Values are mean ± SEM. *p < 0.05; **p < 0.01; ***p<0.001; ****p<0.0001. All results are representative of two independent experiments. See also Supplemental Figure 6.
Rodent adrenal glands rely on the selective uptake of HDL-cholesterol by SR-BI to provide free cholesterol for the generation of cholesterol esters by the ER enzyme ACAT1. Interestingly, the adrenal phenotype of Aster-B knockout mice is virtually identical to that described for mice lacking SR-BI or ACAT1 (Meiner et al., 1996; Rigotti et al., 1997). We therefore assessed whether HDL-mediated cholesterol delivery to cells affected Aster-B localization. Indeed, TIRF microscopy showed that incubation of A431 cells with HDL2 stimulated recruitment of Aster-B to the PM (Supplemental Figure 6A–E).
We next assessed whether Aster-B was important for HDL-cholesterol delivery to adrenal cortical ER in vivo. A failure to transport cholesterol to the ER would render the cells dependent on endogenous cholesterol synthesis for production of cortisol. We analyzed expression of SREBP-2 target genes, whose expression is tightly linked to ER cholesterol content (Brown and Goldstein, 1997). Adrenal SREBP-2 target gene expression was far higher in Aster-B-deficient mice than in WT mice (Figure 7E), indicating that the ER is starved for cholesterol in the absence of Aster-B. In addition, nuclear fractions from Aster-B–deficient adrenal glands showed dramatically increased levels of mature SREBP-2, despite comparable levels of the membrane-bound precursor, indicating that Aster-B deficiency promotes SREBP-2 processing (Figure 7F). Levels of SR-BI were normal in Aster-B–deficient mice, indicating that the phenotype was not a consequence of SR-BI deficiency (Figure 7F).
Cholesterol esters are utilized, particularly during times of stress, for generating corticosteroids. Although serum cholesterol levels did not differ significantly between WT and Aster-B knockout mice (Figure 7G), basal serum corticosterone levels were lower in Aster-B-deficient mice than in controls (Figure 7H). The induction of stress through an overnight fast exacerbated this deficiency (Hoekstra et al., 2008). By contrast, levels of epinephrine and dopamine (non-steroid mediators made by the adrenal medulla) were not different between groups (Supplemental Figure 6F).
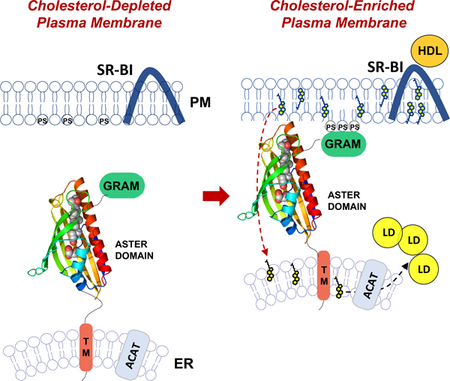
Collectively, our data demonstrate that Aster-B moves cholesterol from the PM to the ER downstream of the HDL receptor SR-BI (Supplemental Figure 7). To our knowledge, Aster-B is the only putative mammalian PM–ER cholesterol transporter to show a loss-of-function phenotype in vivo and to be implicated in the transport of HDL-derived cholesterol.
DISCUSSION
How cholesterol moves between the PM and the ER in mammalian cells has been a longstanding conundrum. Recent studies have emphasized the ability of the ER to sense fluctuations in the “accessible” pool of PM cholesterol and to link these with regulation of the sterol-sensing SREBP-2 pathway (Das et al., 2014; Infante and Radhakrishnan, 2017). Our work reveals that the equilibrium between “accessible” PM cholesterol and the ER pool is maintained, at least in part, through a cholesterol-dependent switch involving the Aster family of ER-anchored transporters. When levels of cholesterol in the PM rise above homeostatic levels, Asters form bridges from the PM to the ER, and the sterol-binding ASTER domain extracts cholesterol from the PM and moves it down the concentration gradient to the ER. One member of this family, Aster-B, is highly expressed in steroidogenic tissues and is required for the ability of HDL-cholesterol to move to the ER in adrenocortical cells. These studies outline a critical function for Aster-B in the nonvesicular transport of cholesterol in vivo, and suggest that members of the Aster family could play important roles in facilitating ER–PM cholesterol movement in other cell types.
Many putative sterol-trafficking proteins have been identified by in vitro experiments. However, defining their physiologic roles has been challenging because of the absence of clear loss-of-function phenotypes. It has been suggested that assigning functions to these proteins may be complicated by redundancy, but an alternative possibility is that the various lipid-binding proteins have not yet been associated with the correct biological function. Defining the physiologic roles of mammalian START proteins has proven particularly difficult. StARD1 is the only family member that has been shown to mediate cholesterol trafficking in vivo in both mice and humans (Caron et al., 1997; Lin et al., 1995b). StARD1 mutations inhibit cholesterol trafficking to the inner mitochondrial membrane and cause a massive accumulation of cholesterol esters in the adrenal—a phenotype opposite to that elicited by Aster-B deficiency.
START-like domains with homology to the ASTER domain have been identified through bioinformatics approaches in plants and other lower organisms (Elbaz-Alon et al., 2015; Gatta et al., 2015; Khafif et al., 2014; Murley et al., 2015). In yeast, a family of six Ltc proteins contain combinations of one or more GRAM domains, START-like domains, and transmembrane segments (Elbaz-Alon et al., 2015; Gatta et al., 2015; Horenkamp et al., 2018; Murley et al., 2015; Tong et al., 2018; Wong and Levine, 2016). Although they share some structural features with yeast START-like domain proteins, one-to-one orthologs for the Asters are restricted to vertebrates, where they appear to have evolved alongside SR-BI and other proteins involved in lipoprotein metabolism. Higher organisms must move lipids between tissues to maintain systemic homeostasis. It is therefore logical that mammals would have evolved specific transporters to facilitate the movement of lipoprotein-derived cholesterol into cells. Indeed, key residues lining the ASTER domain sterol-binding pocket are not conserved in yeast START-like domain proteins, resulting in a distinct mode of ligand binding.
One of the most remarkable features of the Aster proteins is their ability to localize to the PM based on the level of membrane cholesterol. Interestingly, NPC1L1, which facilitates intestinal cholesterol absorption, also relies on a cholesterol-mediated switch (Ge et al., 2008). NPC1L1 is internalized with cholesterol at the PM and then traffics to endosomes–an effect dependent on a sterol-sensing domain in NPC1L1. The mechanism of Aster recruitment by cholesterol is distinct. The Aster GRAM domain, which binds phosphatidylserine, is necessary and sufficient for PM localization in response to cholesterol loading. Phosphatidylserine is enriched on the inner leaflet of PM; hence, phosphatidylserine binding by the GRAM domain could mediate PM localization of Asters. An open question is why the Aster GRAM domain is able to recognize phosphatidylserine only in the presence of excess membrane cholesterol. Additional studies will be needed to test whether cholesterol loading changes the biophysical presentation of phosphatidylserine, stimulates the generation of other lipids, or triggers the production of secondary messengers.
While ER–PM contacts are stable and prominent in yeast, they appear to have more dynamic and tissue-adapted roles in mammalian systems (e.g., STIM1, E-Syts) (Zhang et al., 2005a). While this paper was under review, it was reported that Gramd2 and Gramd1a (Aster A) are localized in ER-PM contact sites (Besprozvannaya et al., 2018). Gramd2 has an N-terminal GRAM domain and is anchored in the ER like the Aster proteins, but lacks the central sterol-binding fold. Asters are unique among known ER-PM contact proteins in their ability to form membrane bridges in a cholesterol-dependent manner. Interestingly, we observed that ER-PM contacts containing Aster-B were frequently located in the same ER tubules in which ORP5, E-Syt2 or E-Syt3 were found, but some contacts appeared to contain only Aster-B, consistent with the idea that they are functionally distinct. The questions of whether other proteins such as E-Syts help to stabilize Aster-dependent contacts and whether lipid transport by ORP5 and Asters is coordinated remain to be addressed.
The phenotype of Aster-B-deficient mice suggests principles that are likely to be relevant to understanding the functions of other Aster family members in other contexts. We expect that trafficking of cholesterol from the PM to the ER and vice versa may be critical for lipid homeostasis in many, if not all, mammalian cell types. Furthermore, the cell type–selective expression of Aster proteins in tissues with active lipid metabolic programs suggests that Asters are likely to play important roles in sterol transport in other tissues. For example, while Aster-B is expressed at low levels in the liver, other Asters are abundant. Given that the liver is an important destination for HDL-cholesterol, it will be of interest to determine whether Aster proteins function downstream of SR-BI in that tissue. The high level of Aster-A expression in the brain suggests that this family member could be particularly important for sterol trafficking in neurons, about which very little is known. Finally, the numerous links between defective cholesterol metabolism and human pathologies underscore the importance of understanding the mechanisms of cellular sterol transport. Uniting the function of individual cholesterol trafficking proteins with their physiologic roles will advance our understanding of physiology and may highlight opportunities to target lipid metabolism in the treatment and diagnosis of human disease.
EXPERIMENTAL PROCEDURES
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Peter Tontonoz (ptontonoz@mednet.ucla.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
All mice were housed in a temperature-controlled room under a 12 hr light–dark cycle and under pathogen-free conditions. Mice were placed on a chow diet. Experiments were performed in male and female mice. Investigators were blinded to group allocation for some but not all studies. Aster-B global knockout mice were generated at Mouse Biology Program facility on a C57BL/6N background using the CRISPR/Cas9 strategy outlined in Supplemental Figure S5B. All animal experiments were approved by the UCLA Institutional Animal Care and Research Advisory Committee. Experimental mice were sacrificed at ages 6–12 weeks unless otherwise specified for histological, serum, lipid, and gene expression analyses.
Cell Culture
A431, CHO-K1, 3t3-L1 and HeLa cells were obtained from the American Type Culture Collection. They have been previously verified by STR testing and were confirmed to be mycoplasma-free by regular testing. CHO-K1 and HeLa cells were transfected with Fugene 6 (Promega) following manufacturer’s protocol. X-tremeGENE HP DNA or Genecellin transfection reagents were used for transfecting A431 cells. A431, 3T3-L1 and HeLa stable cells were grown in monolayer at 37 C in 5% CO2. The cells were maintained in medium A (DMEM containing 100 units/ml penicillin and 100 mg/ml streptomycin sulfate) supplemented with 10% FBS. CHO-K1 cells were grown in medium B (a 1:1 mixture of F-12K medium and Dulbecco’s MEM containing 100 units/ml penicillin and 100 mg/ml streptomycin sulfate) supplemented with 10% FBS. Cholesterol-depleting medium was medium A supplemented with 1–5% lipoprotein-deficient serum (LPDS). Unless otherwise specified, cells were cholesterol loaded using 200 µM cholesterol: methyl-β-cyclodextrin (randomly methylated, Sigma C4555) complexes.
METHOD DETAILS
Construction of Plasmids and Stable Cell Lines
Mouse Aster-A, -B, and –C and truncations were PCR amplified from Mus musculus C57BL/6J cDNA and cloned into pDonr221 by Gateway cloning (BP reaction, ThermoFisher), pEntr4-GFP-C1 or pEntr4-GFP-N2 by Gibson assembly (NEB Gibson Assembly kit) (Gibson et al., 2009). For some studies, human Aster-B (1–738) was used as indicated. For use in transient transfections of GFP-tagged Aster proteins in CHO-K1 cells, pDonr221 plasmids were LR recombined into a pDest53 destination vector containing a CMV promoter and an N-terminal eGFP. For generation of stable C-terminal GFP-tagged Aster protein A431 cells, pEntr-GFP-N2 constructs were LR recombined into a retroviral pBabe DEST vector containing an SV40 promoter. For generation of stable N-terminal GFP-tagged Aster protein A431 cells, pEntr-GFP-C1 constructs were LR recombined into a pLenti Destination vector containing a CMV promoter. After infection, GFP positive cells were selected by flow cytometry based on expression level. PM-mCherry was generated by fusing a plasma membrane targeting sequence of GAP-43 (MLCCMRRTKQVEKNDEDQKI) synthesized as DNA oligo (Biomers, Germany) and inserting into the N-terminus of mCherry (Fig. 5D and Movie S1). SR-BI cDNA (transcript variant 2, NM_001082959.1) was amplified from A431 cDNA with primers SR-BI sense-BglII (atctAGATCTaccATGGGCTGCTCCGCCAAAG) and SR-BI anti-NotI(atttgcggccgcgtgtgtgcaggtgtgcaa), inserted into mammalian expression vector with a EF1a promoter and puromycin selection marker (Fig. 7A–C and Suppl. Fig. 6B). First A431 cells with a stable expression for SR-BI were selected using 1 µg /µl puromycin. These cells were then transfected with GFP-Aster-B (human) and single cell clones with stable expression of SR-BI and GFP-Aster-B were selected with 500 µg / ml G418. Aster-B PH domain-GFP and control GFP constructs were packed into retrovirus to infect A431 cells in the presence of 6 µg/ml polybrene (Millipore). Cells were selected with 1 µg/ml puromycin for 1 week before using in experiment and used as a pool without further subcloning (Fig. 5D, E and Movie S2, S3, S4).
BFP-KDEL (Friedman et al., 2011) was amplified by PCR, inserted into an AAVS1 safe harbor integration vector (Lombardo et al., 2011) with puromycin selection marker. E-syt2 and E-syt3 (Giordano et al., 2013), and OSBPL5 cDNA (from HeLa cDNA, NM_020896.3) amplified by PCR, and the CAAX box (KLNPPDESGPGCMSCKCVLS) synthesized as DNA oligos (Biomers, Germany), were fused to the C-terminus of mCherry, and inserted into the safe harbor vector with blasticidin selection marker.
A431 cell lines double expressing BFP-KDEL plus the mCherry fusion proteins of interest were generated through CRISPR/Cas9 mediated AAVS1 safe harbor co-integration. Briefly, cells were co-transfected with two constructs (BFP-KDEL with puromycin selection and mCherry fusion protein with blasticidin selection) plus a third Cas9/sgRNA construct (backbones kind gifts from Feng Zhang) (Ran et al., 2013) targeting the AAVS1 locus. Transfected cells were selected with 1 µg/ml puromycin and 5 µg/ml blasticidin for 7 days and used as pools for the experiments. Plasmids and plasmid sequences are available upon request.
Protein Expression and Purification
For crystallization studies, the mouse Aster-A protein (334–562) was cloned into pGEX2T (GE Healthcare) with a TEV protease site. Aster-A was expressed in E. Coli Rosetta (DE3) (Novagen) by growing the transformed Rosetta (DE3) at 37° C in 2xTY until A600nm = 0.1, then inducing with 40 micromolar Isopropyl-D-1-thiogalactopyranoside (IPTG) and growth overnight at 20°C. The bacterial cells were lysed by sonication in a buffer containing 1x PBS, 1 mM Dithiothreitol (DTT) and Complete EDTA-free protease inhibitor (Roche). The soluble protein was bound to glutathione sepharose (GE healthcare), and washed with a buffer containing 1x PBS, 0.5% Triton X-100, 0.5mM TCEP. Then the bound protein was washed with TEV cleavage buffer containing 50 mM Tris/Cl pH 7.5, 100 mM NaCl, 5% glycerol and 0.5mM TCEP. The protein was eluted from the resin using TEV protease. After the GST purification excess 25-hydroxy cholesterol was added from a 10 mM stock solution dissolved in ethanol and the complex purified on a Superdex S-200 column in 50 mM Tris/Cl pH 7.5, 100 mM NaCl and 0.5mM TCEP. The peak fractions were concentrated to 11.1 mg/ml and used for the crystallization experiments.
For binding and transfer studies Aster domains (Aster-A261–576, Aster-B224–560, Aster-C206–528) were expressed by baculovirus in Sf-9 insect cells with an N-terminal FLAG tag and a C-terminal 6x His Tag. Proteins were expressed with P3 baculovirus for 48 hours at 27°C. Cells were recovered by centrifugation, lysed by sonication. Insoluble material was pelleted at 16,000xg for 40 minutes. Soluble protein was first purified using a Ni-NTA column (Qiagen, 30210) and eluted with 250mM imidazole PBS buffer after extensive washing. Following dialysis proteins were purified using FLAG M2 affinity gel (Sigma A2220) columns and eluted with 100 µg/mL 1x FLAG peptide after washing for ten column volumes. Proteins were then either dialyzed to remove the FLAG peptide or purified further by size exclusion chromatography. The B Gram domain was expressed in Sf9 cells with a 6xHis tag (Aster-B303–533) and purified as described above. N-terminal GST Aster-B303–533 expression constructs were transformed in Rosetta 2 (DE3) cells (Novagen). LB precultures were diluted into large-scale expression cultures and grown at 37°C to an A600 of 0.6–0.8, then induced with 0.5 mM IPTG at 18°C for 16 hours with shaking. Protein was then purified in PBS + 0.5mM DTT using glutathione agarose resin (Pierce PI16100) and eluted with 10mM GSH peptide in 50mM Tris, 150mM NaCl, pH 8.0. Protein was then dialyzed to remove GSH peptide. HMBP-3C-STAR.66–285 (StAR) was a gift from Nikolai Sluchanko (Addgene plasmid # 100094). Soluble StAR was first purified using a Ni-NTA column (as above) and subsequently purified using amylose resin (NEB E8021S) and eluted with 10 mM maltose in 50mM Tris, 150mM NaCl, pH 8.0. Protein was then dialyzed to remove maltose.
NBD-Cholesterol Binding Experiments
Fluorescent sterol binding assays were carried out as previously described (Petrescu et al., 2001; Wei et al., 2016) in 384-well black flat-bottom plates and equilibrated at room temperature for 1 hour. Measurements were made using a CLARIOstar (BMG LABTECH) microplate reader. The NBD fluorophore was excited with λ(ex) = 470 nm and λ(em) = 525 and plotted using Prism software. Dissociation constants (KD) were determined by nonlinear regression analysis of dose-response curves.
GST Agarose Assay for [3H]Cholesterol Binding
Reactions were carried out in binding buffer (0.003% Triton X-100 in 1 x PBS) containing 150 nM of Aster-B ASTER protein and [3H]cholesterol. After incubation for 30 min at room temperature, the mixture was incubated with pre-equilibrated of glutathione agarose resin (Pierce PI16100) at 4°C for 2 h, then loaded onto a column and washed. The protein-bound [3H]cholesterol was eluted with 10 mM GSH peptide and quantified by scintillation counting. For competition experiments with unlabeled sterols, the assays were carried out in the presence of ethanol containing the indicated unlabeled sterol (0 –10 µM).
Liposome Preparation
Liposomes were generated by drying lipids in glass tube under liquid nitrogen. Lipid films were then resuspended in 50 mM hepes, 120 mM potassium acetate buffer ± 0.75M sucrose where indicated. Lipid suspensions were then vortexed and incubated for 30 minutes at 37°C (2mM total lipid concentration). Suspensions were then snap frozen in liquid nitrogen and thawed rapidly at 37°C five times. Light liposomes were prepared by extruding through 100 nm polycarbonate filters. Heavy liposomes containing sucrose were prepared by extruding through a 400 nm polycarbonate filter, then washing several times in 50 mM hepes, 120 mM potassium acetate buffer with no sucrose. Liposome sizes were confirmed using an N4 Dynamic Light Scattering instrument.
Crystallization and X-ray Structure Determination
Crystals of the Aster-A (334–562):25-hydroxycholesterol complex were obtained using sitting drop vapor diffusion at room temperature. Crystals were grown using 0.2 M NaCl, 0.1 M sodium cacodylate pH 6.0 and 8% PEG 8000 (condition E3 Proplex, Molecular Dimensions). Data were collected to 2.9 Å on the I03 beamline at Diamond Light Source, UK. Data were processed using XDS (within Xia2) and Pointless/Aimless (within CCP4). The structure was solved using molecular replacement using Phaser (within CCP4) and the first StARkin domain of S. cerevisiae Lam4 (pdb code 5YQJ, (Tong et al., 2018) as a model. Model fitting and refinement were performed using Coot, Refmac (within CCP4), PDB-REDO and Phenix.
Live Cell Imaging
For time-lapse fluorescence imaging, cells were plated in poly-d-lysine coated 35 mm glass bottom dishes (Mat-tek) and, when indicated, transfected 48hr prior to imaging. Images were acquired using an Inverted Leica TCS-SP8-SMD Confocal Microscope, equipped with CO2/temperature controlled Tokai Hit system for imaging of live cells at 37°C with 5% CO2. Images were deconvolved using Huygens Professional software. Brightness and contrast were adjusted with ImageJ software. For some experiments cells were sorted for expression of GFP. TIRF imaging was performed in glass bottom µ-slide 4 well plates (Ibidi) with a Nikon Eclipse Ti-E N-STORM microscope, equipped with Andor iXon+ 897 back-illuminated EMCCD camera and × 100 Apo TIRF oil objective NA 1.49, a 65 mW Argon line combined with Quad filter was used for visualization of TIRF and epifluorescence (Fig. 3E). Live cell TIRF imaging was performed similarly at 37 oC, 5% CO2 with EMBL GP168 incubator controller (Fig. 5E and Movie S3, S4).
For automated quantification of TIRF images, cells were fixed with 4% PFA for 15 min, permeabilized with PBS/0.1% Triton for 5 min and stained with DAPI 5 µg/ml and 0.2 µg/ml CellMask Deep Red (Life Technologies) in PBS for 15 min. TIRF images were acquired for GFP-AsterB together with epifluorescent images for DAPI and CellMask Deep Red. These images were automatically quantified using CellProfiler (Carpenter et al., 2006) and the resulting data analyzed with Python/Pandas. For Airyscan superresolution microscopy, cells in 8 well Lab-Tek™ II Chambered coverglass (Thermofisher) were imaged with a Zeiss LSM 880 confocal microscope equipped with an Airyscan detector using a 63 × Plan-apochromat oil objective, NA 1.4. Live cell imaging was performed at 37 oC, 5% CO2 with incubator insert PM S1 and definite focus hardware autofocus system. Images were Airyscan processed automatically using the Zeiss Zen2 software package (Fig. 3D, Movie S1).
For dual color live cell TIRF imaging, A431 cells stably expressing GFP-Aster-B and Cherry-ORP5 were seeded in 4-well LabTek II live cell chamber slides. After two days cells were incubated with DMEM containing 5% LPDS for 8 h. Live cell TIRF imaging was performed in FluoroBrite DMEM containing 5% LPDS: TIRF video microscopy with a frame rate of one image per minute was initiated 50 s after addition of 1 mM cholesterol/cyclodextrin. Images were acquired with a GE Deltavision OMX SR instrument equipped with a 60x Apo-N oil immersion objective. Images were deconvolved using the softWoRx 7.00 software (GE Healthcare).
For imaging Aster-B with PM and other contact site marker proteins, A431 cells stably expressing BFP-KDEL and mCherry fusion proteins of ER/PM contact site markers were seeded in 8-well LabTeK II live cell chamber slides. After 24 h cells were washed with PBS and transiently transfected with GFP-Aster-B expression constructs using X-tremeGENE HP DNA or Genecellin transfection reagents in DMEM containing 5% lipoprotein deficient serum (LPDS). After 24 h cells were switched to FluoroBrite DMEM containing 5% LPDS, with or without 100 – 200 µM cholesterol/cyclodextrin. Cells were imaged 15 to 65 min after cholesterol administration using a Zeiss LSM 880 Airyscan microscope equipped with a 63x Plan-Apochromat oil immersion objective. Images were Airyscan processed and brightness and contrast adjusted with ImageJ. Images from the bottom section of a cell were used for the quantification of the overlap of GFP-Aster-B with contact-site marker proteins. The images were thresholded to select GFP-Aster-B and contact site marker structures and the pixel overlap of the segmented structures was calculated with the JaCOP plugin for ImageJ. For each condition, 9–13 cells from 2 independent experiments were quantified.
Phospholipid Binding Assays
Lipid binding analysis of 6xHis-tagged B GRAM (Aster-B1–337) was conducted using PIP Strips (Echelon Biosciences), with each spot containing 100 pmol of active lipids. Membranes were blocked with PBS Tween (PBST) solution (supplemented with 3% fatty acid free BSA) for 1 hr at room temperature, and incubated with B GRAM fusion protein in blocking buffer for 1 hr. After three washes, the membranes were blotted with anti-His antibody (Biorad, MCA1396GA). The strip contained 15 different types of lipids. LPA, lysophosphatidic acid; LPC, lysophosphatidylcholine; PI, phosphatidylinositol; PI(3)P, phosphatidylinositol 3-phosphate; PI(4)P, phosphatidylinositol 4-phosphate; PI(5)P, phosphatidylinositol 5-phosphate; PE, phosphatidylethanolamine; PC, phosphatidylcholine; S1P, sphingosine-1-phosphate; PI(3,4)P2, phosphatidylinositol 3,4 -phosphate; PI(3,5)P2, phosphatidylinositol 3,5-phosphate; PI(4,5)P2, phosphatidylinositol 4,5-phosphate; PI(3,4,5)P3, phosphatidylinositol 3,4,5-phosphate; PA, phosphatidic acid; PS, phosphatidylserine. Results were confirmed with a FLAG tagged B GRAM Fusion construct.
Gene Expression Analysis
Total RNA was isolated using TRIzol reagent (Invitrogen) and reverse transcribed with the iScript cDNA synthesis kit (Biorad). cDNA was quantified by real-time PCR using SYBR Green Master Mix (Diagenode) on an ABI 7900 instrument. Gene expression levels were determined by using a standard curve. Each gene was normalized to the housekeeping gene 36B4 and was analyzed in duplicate. Primers used for real-time PCR are available upon request.
Protein Analysis
Whole cell lysate or tissue lysate was extracted using RIPA lysis buffer (Boston Bioproducts) supplemented with complete protease inhibitor cocktail (Roche). Proteins were diluted in Nupage loading dye (Invitrogen), heated at 95°C for 5 min, and run on 4–12% NuPAGE Bis-Tris Gel (Invitrogen). Proteins were transferred to hybond ECL membrane (GE Healthcare), blocked with 5% milk (or 5% BSA for anti-SREBP-2) to quench nonspecific protein binding and blotted with the indicated primary antibody. Horseradish peroxidase-conjugated anti-mouse, anti-goat and anti-rabbit IgG (Jackson) were used as secondary antibodies. The immune signal was visualized using the ECL kit (Amersham Biosciences). Nuclei from mouse adrenal glands were prepared by douncing tissue with a motorized overhead stirrer (Caframo Model BDC2002) in 10mM Hepes-KOH, pH 7.4, 10 mM KCl, 1.5 mM MgCl2, 0.5 mM EDTA sodium, 0.5 mM EGTA sodium, 1 mM DTT, and protease inhibitors. Samples were centrifuged at 1,000xg at 4°C for 5 minutes to isolate nuclei. Nuclei were washed and resuspended in 10mM Hepes-KOH, pH 7.4, 0.42 M NaCl, 2.5% glycerol (w/v), 1.5 mM MgCl2, 0.5 mM EDTA sodium, 0.5 mM EGTA sodium, 1 mM DTT, and protease inhibitors. Nuclei were then incubated on ice for 40 minutes with intermittent pipetting, then centrifuged at 10,000xg at 4°C for 10 minutes. The supernatant was then used as nuclear protein extract. The 1,000xg supernatant was used to prepare a membrane lysate by centrifugation at 100,000xg at 4°C for 30 minutes, followed by resuspension in RIPA buffer.
Oil Red O Staining
Oil Red O staining was performed as described (Mehlem et al., 2013). Adrenal glands were dissected carefully and the surrounding fat tissue was removed. After collected the glands were embedded into Tissue-Tek O.C.T. compound (cat No. 4583), placed on dry ice for twenty minutes, then moved to −80°C. Tissue was sectioned (12 µm thick) using a Microm HM 505 E cryostat and sectioned were placed on glass microscope slides (Superfrost plus). Before staining, sections were allowed to equilibrate to room temperature for 10 minutes. Oil Red O solution working solution (Sigma, cat. No. 00625, ~0.4%) was freshly prepared and filtered before covering the sections. Sections were incubated for 10 minutes, then washed under running tap water for 30 minutes. Sections were then mounted on slides with water-soluble mounting medium (Sigma cat. No. GG1) and images were captured on a Zeiss Axioskop 2 plus bright-field light microscope at a x40 magnification. Background was corrected by white balance, selected as a blank area outside the section.
Electron Microscopy
Mice were perfused with 0.1 M Sodium Cacodylate buffer (pH 7.4) and fixed with cold 1.5% Glutaraldehyde, 4% PVP, 0.05% CaCl2 in 0.1 M Sodium Cacodylate buffer (pH 7.4) (Angermuller and Fahimi, 1982). Standard transmission EM ultrastructural analysis was performed on adrenal glands with imidazole staining and visualized with a JEOL JEM-123 40–120kV transmission electron microscope at the Electron Imaging Center for Nanomachines (EICN) at CNSI.
Lipid Analysis
Adrenal glands were weighed and snap-frozen in liquid nitrogen. Blood was centrifuged and serum was snap frozen. 10µl serum was used for analysis. Adrenal glands were pulverized using a hand-held pestle grinder. A modified Bligh-Dyer lipid extraction (Bligh and Dyer, 1959), in the presence of lipid class internal standards including [25,26,26,26-d4]-cholesterol and cholesteryl heptadecanoate, was performed on 5–10 mg of pulverized tissue. Lipid extracts were dried under nitrogen and diluted in chloroform/methanol (2/1, v/v). Molecular species were quantified using ESI/MS on a triple-quadrupole instrument (Thermo Fisher Quantum Ultra) utilizing shotgun lipidomics methodologies (Bowden et al., 2011; Han and Gross, 2005). Free cholesterol was first derivatized with acetyl chloride (Brown et al., 2013) and then quantified in positive ion mode using product ion scanning for 83.03 amu (collision energy = 18 eV). CE molecular species were quantified using neutral loss scanning for 368.5 amu (collision energy = 25 eV). Individual molecular species were calculated by comparing the ion intensities of the molecular species to the ion intensity of the lipid class internal standard as previously described (Bowden et al., 2011; Han and Gross, 2005). Serum corticosterone was measured by ELISA kit following cardiac puncture per manufacturer’s instructions (Cayman Chemical, cat No. 501320).
In vitro antisense oligonucleotide (ASO) studies
Generation 2.5 constrained ethyl ASOs were synthesized as described previously (Seth et al., 2009). For in vitro knockdown studies, control or Aster-A ASO (GTGGAATTTATTCAGG) was used. Undifferentiated murine 3T3-L1 cells were plated in 10% FBS DMEM on Day 0. On Day 1 cells were washed and supplemented with 1% LPDS. Cells were then transfected with ASOs using Dharmafect 1 reagent per the manufacturer’s recommendations for knockdown studies (Dharmacon, 50nM final concentration). On Day 2, the medium was changed to fresh DMEM with 1%LPDS, simvastatin (5 µM) and mevalonate (50 µM). On Day 3 cells were treated as described in the figure legends and samples were collected. The rate of incorporation of [3H]oleate into cholesteryl [3H]oleate was performed on Day 3 as previously described (Goldstein et al., 1983).
Lipoprotein fractionation
Purification of HDL2 particles was performed using potassium bromide density centrifugation from pooled samples of human plasma obtained from the Finnish Red Cross as described previously (Nguyen et al., 2012). Lipid content of HDL2 and LDL fractions was quantified using lipid extraction and thin layer chromatography as described before (Bautista et al., 2014).
QUANTIFICATION AND STATISTICAL ANALYSES
Experimental Replicates and Quantification
Replicates are described in the figure legends. For cellular assays, n corresponds to the number of experimental replicates. For animal experiment, n corresponds to the number of mice used per genotype or condition. Each experiment was repeated using at least two cohorts of mice. All data were represented as mean SEM (standard error) as described in the figure legends.
Statistical Analysis
All data are presented as mean ± SEM and analyzed using Microsoft Excel and Prism (Graphpad). Student’s t test was used for single variable comparison between two groups. Two-way ANOVA followed by Bonferroni posttests was used to examine interactions between multiple variables. Data are presented as ± S.E.M. p <0.05 was considered to be statistically significant and is presented as * p < 0.05, ** p <0.01, *** p <0.001, or **** p <0.0001.
DATA AND SOFTWARE AVAILABILITY
The PDB Code for the crystallography data is 6GQF.
Supplementary Material
(A) Simplified phylogram of Aster-B one-to-one conservation across vertebrates. The scale represents 10% amino acid divergence. Phylogram was made using ClustalW2 and protein sequences were downloaded from Uniprot.
(B) Depiction of annotated intron-exon structure of Gramd1a, Gramd1b, and Gramd1c loci in Mus musculus strain C57BL/6J assembly GRCm38. Coding exons are depicted in black. 5’ and 3’ UTRs are represented with no fill. Exons which correspond to the Gram domain, Aster domain, and Transmembrane (TM) are depicted in green, blue, and red, respectively. Scale bar represents 1kb.
(C) Aster family expression by qPCR from tissues of C57BL/6 mice (n = 5).
(D) Coomassie blue–stained SDS–PAGE gel of purified FLAG-Aster domains (Aster-A261–546, Aster-B224–560, Aster-C206–528) and control (albumin).
(E) Aster-B334–562 was titrated with 22-NBD-cholesterol in the presence of cholesterol competitor as indicated. Results values are means ± SD and representative of at least three independent experiments.
(F) Comparison of NBD-cholesterol binding to Aster-A, Aster-B and StarD1.
(G) Comparison of in vitro cholesterol transport by Aster proteins and StarD1.
Representative video of Aster-B-GFP translocation in response to cholesterol loading. Plasma membrane is indicated by PM-mCherry marker. Time is indicated in minutes after cholesterol loading. Scale bar is 5 µm.
Representative video of B GRAM (Aster-B1–171, C-terminal GFP) translocation in response to cholesterol loading (500 µM). Plasma membrane is indicated by PM-mCherry marker. Time is indicated in minutes after cholesterol loading. Scale bar is 5 µm.
Representative TIRF video of GFP (negative control) translocation in response to cholesterol loading (500 µM). Time is indicated in minutes after cholesterol loading. Scale bar is 5 µm.
Representative TIRF video of B GRAM (Aster-B1–171, C-terminal GFP) translocation in response to cholesterol loading (500 µM). Time is indicated in minutes after cholesterol loading. Scale bar is 5 µm.
(A) Structure of the Aster-A sterol-binding domain showing 25-hydroxycholesterol ligand and a glycerol molecule in the binding cavity.
(B) Comparison of mouse Aster-A, yeast Lam4p-SD2 and StARD5. Despite the low sequence identity (18%), the Aster and Lam domains have a very similar architecture creating a largely non-polar binding cavity for the 25-hydroxycholesterol. However, the orientation of the ligands is quite distinct. The beta-sheets are most similar between Aster-A and yeast Lam4p-SD2. The three helices are somewhat different. In particular, the carboxy-terminal helix in Aster-A is one turn longer at its amino-terminus and shorter at its carboxy-terminus compared with Lam4p-SD2.
(A) Orientation of the 25-hydroxycholesterol ligand. A simulated annealing composite omit map unambiguously identifies the ligand orientation in mouse Aster A (left panel - contoured at 1.0 sigma). The orientation of the 25-hydroxycholesterol ligand in mouse Aster A is markedly different from that in the distantly related yeast Lam4p (right panel - pdbcode 6bym) (Jentsch et al., 2018). The ligand is rotated by approximately 120° about it long axis such that the axial methyl groups on the cholesterol are orientated very differently.
(B) Alignment of the mammalian ASTER domains with the yeast Lam2/4 proteins. Although structurally similar, there is only limited sequence homology between the sterol-binding domains of the Aster and Lam proteins (23% identity between Aster A and Lam4p-1). Residues that are identical, have high, or low similarity are colored red, orange or yellow respectively. Residues colored cyan likely determine the different orientation of the sterol and are very different in character between the Aster and Lam proteins. Cyan asterisks indicate residues lining the pocket in contact with the hydroxycholesterol. Purple and green asterisks indicate surface non-polar and basic residues conserved in the Aster proteins that likely mediate interaction with the phospholipid membrane to facilitate sterol exchange.
(A) Aster-A-GFP, Aster-B-GFP and Aster-C GFP localization in A431 cells imaged by live cell confocal microscopy in 1% LPDS (top) or following cholesterol loading for 1 h (bottom).
(B) Live-cell imaging of GFP-Aster-A localization in A431 cells stably expressing BFP-KDEL (ER marker) and Cherry Lyn (PM marker). Yellow arrows indicate ER tubules in close proximity to the PM containing foci of Aster-A expression.
(C) Live-cell imaging of GFP-Aster-C localization in A431 cells stably expressing BFP-KDEL (ER marker) and Cherry Lyn (PM marker). Yellow arrows indicate ER tubules in close proximity to the PM containing foci of Aster-C expression.
(D) Live-cell imaging of GFP-Aster-B localization in A431 cells stably expressing BFP-KDEL (ER marker) and Cherry-E-Syt3 (ER-PM contact protein) cultured in 5% LPDS (left) or following cholesterol loading for 40 min (right). Large images, scale bar = 10 µm; insets, scale bar = 2 µm.
(E) Localization of full-length Aster-B and B ∆GRAM GFP constructs expressed in A431 cells in the presence or absence of cholesterol loading imaged by live cell confocal microscopy.
(A) RNA-seq expression of Gramd1a, Gramd1b, and Gramd1c in female adult C57BL/6J mouse adrenal gland tissue (10 weeks or age). Data was collected for the ENCODE consortium by the Snyder Lab at Stanford (GEO GSM2453460).
(B) Strategy for generating Crispr-Cas9 mediated global Gramd1b (Aster-B) knockout mice. Coding exons are depicted in black. Exons that correspond to the Gram domain, ASTER domain, and transmembrane (TM) domain are depicted in green, blue, and red respectively. Scale bar represents 1kb.
(A) Immunoblot validation of GFP-Aster-B and SR-BI expression compared to A431 wild-type cells.
(B) Lipid composition of HDL2 purified from human plasma by ultracentrifugation. HDL2 was used for the experiment because of higher lipid content and better SR-B1 interaction compared to HDL3.
(C) A431-SR-BI/Aster-B-GFP cells were lipid depleted for 24 h (LPDS) and treated with control medium (LPDS), 150–200 µg/mL FC-HDL2, or 200 µM cholesterol-cyclodextrin for 1 h. Cells were then fixed and imaged with TIRF microscopy to visualize Aster-B in close proximity to the PM. Dashed lines indicate individual cells. Scale bar 10 µm.
(D) Cells were automatically identified using DAPI (nuclei) and CellMask (cytoplasm) images and used to quantify single cell TIRF intensities.
(E) Quantification of the mean cellular GFP-Aster-B TIRF intensity from cells treated as in (A). N = 254 cells for control, 307 cells for HDL2 and 187 cells for cholesterol from 4 independent experiments.
(F) Epinephrine and dopamine levels in serum.
(A) Model for the recruitment of Aster-B to the plasma membrane. The GRAM domain binds phosphatidylserine and is recruited to the plasma membrane in response to cholesterol loading. Aster-B forms cholesterol-dependent ER–PM contact sites and facilitates the trafficking of cholesterol to the ER.
(B) Model showing PM to ER transport of HDL-derived cholesterol in the presence or absence of Aster-B.
Highlights.
Asters are ER-resident proteins that mediate nonvesicular sterol transport
The sterol-binding ASTER domain is structurally related to the START domain
Asters are dynamically recruited to PM-ER contacts in response to cholesterol
Aster-B is required for delivery of HDL-cholesterol from SR-BI to the ER in adrenals
ACKNOWLEDGMENTS
We thank J. Kim, S. Munday, P. Rajbhandari, and T. Sallam for technical assistance and experimental guidance. J.S. was supported by NIH GM08042, NIH T32HL69766 and the P. Whitcome Fellowship. J.O. was supported by NIH 5T34GM008563. This research was also supported by NIH grants HL066088, DK100627, S10RR019232 and GM115553, Academy of Finland grants 307415, 312491 and 275964, and grants from Sigrid Juselius Foundation, and the Helsinki Institute of Life Science Imaging Unit and Biomedicum Functional Genomics Unit. J.W.R.S. is a Wellcome Trust Senior Investigator (WT100237) and Royal Society Wolfson Research Merit Award Holder. Confocal microscopy was performed at the California NanoSystems Institute Advanced Light Microscopy/Spectroscopy Facility. We thank Diamond Light Source for beamtime (proposal MX14692), and the staff of beamlines I04–1, I03 and I24 for assistance with crystal testing and data collection. We dedicate this manuscript to the memory of Jaspal Singh Sandhu. His courageous battle with coronary artery disease has been a key inspiration to this work.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
REFERENCES
- Acton S, Rigotti A, Landschulz KT, Xu S, Hobbs HH, and Krieger M (1996). Identification of scavenger receptor SR-BI as a high density lipoprotein receptor. Science 271, 518–520. [DOI] [PubMed] [Google Scholar]
- Angermuller S, and Fahimi HD (1982). Imidazole-buffered osmium tetroxide: an excellent stain for visualization of lipids in transmission electron microscopy. Histochem J 14, 823–835. [DOI] [PubMed] [Google Scholar]
- Bautista G, Pfisterer SG, Huttunen MJ, Ranjan S, Kanerva K, Ikonen E, and Kauranen M (2014). Polarized THG microscopy identifies compositionally different lipid droplets in mammalian cells. Biophys J 107, 2230–2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begley MJ, Taylor GS, Kim SA, Veine DM, Dixon JE, and Stuckey JA (2003). Crystal structure of a phosphoinositide phosphatase, MTMR2: insights into myotubular myopathy and Charcot-Marie-Tooth syndrome. Mol Cell 12, 1391–1402. [DOI] [PubMed] [Google Scholar]
- Besprozvannaya M, Dickson E, Li H, Ginburg KS, Bers DM, Auwerx J, and Nunnari J (2018). GRAM domain proteins specialize functionally distinct ER-PM contact sites in human cells. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bligh EG, and Dyer WJ (1959). A rapid method of total lipid extraction and purification. Can J Biochem Physiol 37, 911–917. [DOI] [PubMed] [Google Scholar]
- Bowden JA, Shao F, Albert CJ, Lally JW, Brown RJ, Procknow JD, Stephenson AH, and Ford DA (2011). Electrospray ionization tandem mass spectrometry of sodiated adducts of cholesteryl esters. Lipids 46, 1169–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun A, Trigatti BL, Post MJ, Sato K, Simons M, Edelberg JM, Rosenberg RD, Schrenzel M, and Krieger M (2002). Loss of SR-BI expression leads to the early onset of occlusive atherosclerotic coronary artery disease, spontaneous myocardial infarctions, severe cardiac dysfunction, and premature death in apolipoprotein E-deficient mice. Circ Res 90, 270–276. [DOI] [PubMed] [Google Scholar]
- Brown MS, and Goldstein JL (1997). The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell 89, 331–340. [DOI] [PubMed] [Google Scholar]
- Brown RJ, Shao F, Baldan A, Albert CJ, and Ford DA (2013). Cholesterol efflux analyses using stable isotopes and mass spectrometry. Anal Biochem 433, 56–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caron KM, Soo SC, Wetsel WC, Stocco DM, Clark BJ, and Parker KL (1997). Targeted disruption of the mouse gene encoding steroidogenic acute regulatory protein provides insights into congenital lipoid adrenal hyperplasia. Proc Natl Acad Sci U S A 94, 11540–11545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter AE, Jones TR, Lamprecht MR, Clarke C, Kang IH, Friman O, Guertin DA, Chang JH, Lindquist RA, Moffat J, et al. (2006). CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol 7, R100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung J, Torta F, Masai K, Lucast L, Czapla H, Tanner LB, Narayanaswamy P, Wenk MR, Nakatsu F, and De Camilli P (2015). INTRACELLULAR TRANSPORT. PI4P/phosphatidylserine countertransport at ORP5- and ORP8-mediated ER-plasma membrane contacts. Science (New York, NY) 349, 428–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium EP (2012). An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das A, Brown MS, Anderson DD, Goldstein JL, and Radhakrishnan A (2014). Three pools of plasma membrane cholesterol and their relation to cholesterol homeostasis. eLife 3, 19316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doerks T, Strauss M, Brendel M, and Bork P (2000). GRAM, a novel domain in glucosyltransferases, myotubularins and other putative membrane-associated proteins. Trends Biochem Sci 25, 483–485. [DOI] [PubMed] [Google Scholar]
- Elbaz-Alon Y, Eisenberg-Bord M, Shinder V, Stiller SB, Shimoni E, Wiedemann N, Geiger T, and Schuldiner M (2015). Lam6 Regulates the Extent of Contacts between Organelles. Cell Rep 12, 7–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman JR, Lackner LL, West M, Dibenedetto JR, Nunnari J, and Voeltz GK (2011). ER tubules mark sites of mitochondrial division. Science (New York, NY) 334, 358–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatta AT, Wong LH, Sere YY, Calderon-Norena DM, Cockcroft S, Menon AK, and Levine TP (2015). A new family of StART domain proteins at membrane contact sites has a role in ER-PM sterol transport. Elife 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge L, Wang J, Qi W, Miao HH, Cao J, Qu YX, Li BL, and Song BL (2008). The cholesterol absorption inhibitor ezetimibe acts by blocking the sterol-induced internalization of NPC1L1. Cell Metab 7, 508–519. [DOI] [PubMed] [Google Scholar]
- Ghai R, Du X, Wang H, Dong J, Ferguson C, Brown AJ, Parton RG, Wu JW, and Yang H (2017). ORP5 and ORP8 bind phosphatidylinositol-4, 5-biphosphate (PtdIns(4,5)P 2) and regulate its level at the plasma membrane. Nat Commun 8, 757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA 3rd, and Smith HO (2009). Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 6, 343–345. [DOI] [PubMed] [Google Scholar]
- Giordano F, Saheki Y, Idevall-Hagren O, Colombo SF, Pirruccello M, Milosevic I, Gracheva EO, Bagriantsev SN, Borgese N, and De Camilli P (2013). PI(4,5)P(2)-dependent and Ca(2+)-regulated ER-PM interactions mediated by the extended synaptotagmins. Cell 153, 1494–1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass C, Pittman RC, Weinstein DB, and Steinberg D (1983). Dissociation of tissue uptake of cholesterol ester from that of apoprotein A-I of rat plasma high density lipoprotein: selective delivery of cholesterol ester to liver, adrenal, and gonad. Proc Natl Acad Sci U S A 80, 5435–5439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein JL, Basu SK, and Brown MS (1983). Receptor-mediated endocytosis of low-density lipoprotein in cultured cells. Methods Enzymol 98, 241–260. [DOI] [PubMed] [Google Scholar]
- Goldstein JL, and Brown MS (1985). The LDL receptor and the regulation of cellular cholesterol metabolism. J Cell Sci Suppl 3, 131–137. [DOI] [PubMed] [Google Scholar]
- Goldstein JL, and Brown MS (2015). A century of cholesterol and coronaries: from plaques to genes to statins. Cell 161, 161–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han X, and Gross RW (2005). Shotgun lipidomics: electrospray ionization mass spectrometric analysis and quantitation of cellular lipidomes directly from crude extracts of biological samples. Mass Spectrom Rev 24, 367–412. [DOI] [PubMed] [Google Scholar]
- Hoekstra M (2017). SR-BI as target in atherosclerosis and cardiovascular disease - A comprehensive appraisal of the cellular functions of SR-BI in physiology and disease. Atherosclerosis 258, 153–161. [DOI] [PubMed] [Google Scholar]
- Hoekstra M, Meurs I, Koenders M, Out R, Hildebrand RB, Kruijt JK, Van Eck M, and Van Berkel TJ (2008). Absence of HDL cholesteryl ester uptake in mice via SR-BI impairs an adequate adrenal glucocorticoid-mediated stress response to fasting. J Lipid Res 49, 738–745. [DOI] [PubMed] [Google Scholar]
- Horenkamp FA, Valverde DP, Nunnari J, and Reinisch KM (2018). Molecular basis for sterol transport by StART-like lipid transfer domains. EMBO J 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton JD, Goldstein JL, and Brown MS (2002). SREBPs: transcriptional mediators of lipid homeostasis. Cold Spring Harb Symp Quant Biol 67, 491–498. [DOI] [PubMed] [Google Scholar]
- Ikonen E (2008). Cellular cholesterol trafficking and compartmentalization. Nat Rev Mol Cell Biol 9, 125–138. [DOI] [PubMed] [Google Scholar]
- Ikonen E (2018). Mechanisms of cellular cholesterol compartmentalization: recent insights. Curr Opin Cell Biol 53, 77–83. [DOI] [PubMed] [Google Scholar]
- Infante RE, Abi-Mosleh L, Radhakrishnan A, Dale JD, Brown MS, and Goldstein JL (2008a). Purified NPC1 protein. I. Binding of cholesterol and oxysterols to a 1278-amino acid membrane protein. J Biol Chem 283, 1052–1063. [DOI] [PubMed] [Google Scholar]
- Infante RE, and Radhakrishnan A (2017). Continuous transport of a small fraction of plasma membrane cholesterol to endoplasmic reticulum regulates total cellular cholesterol. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Infante RE, Radhakrishnan A, Abi-Mosleh L, Kinch LN, Wang ML, Grishin NV, Goldstein JL, and Brown MS (2008b). Purified NPC1 protein: II. Localization of sterol binding to a 240-amino acid soluble luminal loop. J Biol Chem 283, 1064–1075. [DOI] [PubMed] [Google Scholar]
- Janowski BA, Willy PJ, Devi TR, Falck JR, and Mangelsdorf DJ (1996). An oxysterol signalling pathway mediated by the nuclear receptor LXR alpha. Nature 383, 728–731. [DOI] [PubMed] [Google Scholar]
- Jentsch JA, Kiburu I, Pandey K, Timme M, Ramlall T, Levkau B, Wu J, Eliezer D, Boudker O, and Menon AK (2018). Structural basis of sterol binding and transport by a yeast StARkin domain. J Biol Chem 293, 5522–5531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khafif M, Cottret L, Balague C, and Raffaele S (2014). Identification and phylogenetic analyses of VASt, an uncharacterized protein domain associated with lipid-binding domains in Eukaryotes. BMC Bioinformatics 15, 222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khera AV, Cuchel M, de la Llera-Moya M, Rodrigues A, Burke MF, Jafri K, French BC, Phillips JA, Mucksavage ML, Wilensky RL, et al. (2011). Cholesterol efflux capacity, high-density lipoprotein function, and atherosclerosis. N Engl J Med 364, 127–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lees JA, Messa M, Sun EW, Wheeler H, Torta F, Wenk MR, De Camilli P, and Reinisch KM (2017). Lipid transport by TMEM24 at ER-plasma membrane contacts regulates pulsatile insulin secretion. Science (New York, NY) 355, eaah6171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin D, Sugawara T, Strauss JF 3rd, Clark BJ, Stocco DM, Saenger P, Rogol A, and Miller WL (1995a). Role of steroidogenic acute regulatory protein in adrenal and gonadal steroidogenesis. Science 267, 1828–1831. [DOI] [PubMed] [Google Scholar]
- Lin D, Sugawara T, Strauss JF, Clark BJ, Stocco DM, Saenger P, Rogol A, and Miller WL (1995b). Role of steroidogenic acute regulatory protein in adrenal and gonadal steroidogenesis. Science (New York, NY) 267, 1828–1831. [DOI] [PubMed] [Google Scholar]
- Lombardo A, Cesana D, Genovese P, Di Stefano B, Provasi E, Colombo DF, Neri M, Magnani Z, Cantore A, Lo Riso P, et al. (2011). Site-specific integration and tailoring of cassette design for sustainable gene transfer. Nat Methods 8, 861–869. [DOI] [PubMed] [Google Scholar]
- Mangelsdorf DJ, Thummel C, Beato M, Herrlich P, Schutz G, Umesono K, Blumberg B, Kastner P, Mark M, Chambon P, et al. (1995). The nuclear receptor superfamily: the second decade. Cell 83, 835–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehlem A, Hagberg CE, Muhl L, Eriksson U, and Falkevall A (2013). Imaging of neutral lipids by oil red O for analyzing the metabolic status in health and disease. Nat Protoc 8, 1149–1154. [DOI] [PubMed] [Google Scholar]
- Meiner VL, Cases S, Myers HM, Sande ER, Bellosta S, Schambelan M, Pitas RE, McGuire J, Herz J, and Farese RV Jr. (1996). Disruption of the acyl-CoA:cholesterol acyltransferase gene in mice: evidence suggesting multiple cholesterol esterification enzymes in mammals. Proc Natl Acad Sci U S A 93, 14041–14046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murley A, Sarsam RD, Toulmay A, Yamada J, Prinz WA, and Nunnari J (2015). Ltc1 is an ER-localized sterol transporter and a component of ER-mitochondria and ER-vacuole contacts. J Cell Biol 209, 539–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neculai D, Schwake M, Ravichandran M, Zunke F, Collins RF, Peters J, Neculai M, Plumb J, Loppnau P, Pizarro JC, et al. (2013). Structure of LIMP-2 provides functional insights with implications for SR-BI and CD36. Nature 504, 172–176. [DOI] [PubMed] [Google Scholar]
- Nguyen SD, Oorni K, Lee-Rueckert M, Pihlajamaa T, Metso J, Jauhiainen M, and Kovanen PT (2012). Spontaneous remodeling of HDL particles at acidic pH enhances their capacity to induce cholesterol efflux from human macrophage foam cells. J Lipid Res 53, 2115–2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrescu AD, Gallegos AM, Okamura Y, Strauss JF 3rd, and Schroeder F (2001). Steroidogenic acute regulatory protein binds cholesterol and modulates mitochondrial membrane sterol domain dynamics. J Biol Chem 276, 36970–36982. [DOI] [PubMed] [Google Scholar]
- Pfisterer SG, Peranen J, and Ikonen E (2016). LDL-cholesterol transport to the endoplasmic reticulum: current concepts. Curr Opin Lipidol 27, 282–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rader DJ (2014). Spotlight on HDL biology: new insights in metabolism, function, and translation. Cardiovascular Research 103, 337–340. [DOI] [PubMed] [Google Scholar]
- Rader DJ, and Tall AR (2012). The not-so-simple HDL story: Is it time to revise the HDL cholesterol hypothesis? Nat Med 18, 1344–1346. [DOI] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, and Zhang F (2013). Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigotti A, Trigatti BL, Penman M, Rayburn H, Herz J, and Krieger M (1997). A targeted mutation in the murine gene encoding the high density lipoprotein (HDL) receptor scavenger receptor class B type I reveals its key role in HDL metabolism. Proc Natl Acad Sci U S A 94, 12610–12615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sallam T, Jones M, Thomas BJ, Wu X, Gilliland T, Qian K, Eskin A, Casero D, Zhang Z, Sandhu J, et al. (2018). Transcriptional regulation of macrophage cholesterol efflux and atherogenesis by a long noncoding RNA. Nat Med 24, 304–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sallam T, Jones MC, Gilliland T, Zhang L, Wu X, Eskin A, Sandhu J, Casero D, Vallim TQ, Hong C, et al. (2016). Feedback modulation of cholesterol metabolism by the lipid-responsive non-coding RNA LeXis. Nature 534, 124–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seth PP, Siwkowski A, Allerson CR, Vasquez G, Lee S, Prakash TP, Wancewicz EV, Witchell D, and Swayze EE (2009). Short antisense oligonucleotides with novel 2’−4’ conformationaly restricted nucleoside analogues show improved potency without increased toxicity in animals. J Med Chem 52, 10–13. [DOI] [PubMed] [Google Scholar]
- Tong J, Manik MK, and Im YJ (2018). Structural basis of sterol recognition and nonvesicular transport by lipid transfer proteins anchored at membrane contact sites. Proc Natl Acad Sci U S A 115, E856–E865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei W, Schwaid AG, Wang X, Wang X, Chen S, Chu Q, Saghatelian A, and Wan Y (2016). Ligand Activation of ERRalpha by Cholesterol Mediates Statin and Bisphosphonate Effects. Cell Metab 23, 479–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong LH, and Levine TP (2016). Lipid transfer proteins do their thing anchored at membrane contact sites... but what is their thing? Biochem Soc Trans 44, 517–527. [DOI] [PubMed] [Google Scholar]
- Zanoni P, Khetarpal SA, Larach DB, Hancock-Cerutti WF, Millar JS, Cuchel M, DerOhannessian S, Kontush A, Surendran P, Saleheen D, et al. (2016). Rare variant in scavenger receptor BI raises HDL cholesterol and increases risk of coronary heart disease. Science 351, 1166–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Rajbhandari P, Priest C, Sandhu J, Wu X, Temel R, Castrillo A, de Aguiar Vallim TQ, Sallam T, and Tontonoz P (2017). Inhibition of cholesterol biosynthesis through RNF145-dependent ubiquitination of SCAP. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang SL, Yu Y, Roos J, Kozak JA, Deerinck TJ, Ellisman MH, Stauderman KA, and Cahalan MD (2005). STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature 437, 902–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Simplified phylogram of Aster-B one-to-one conservation across vertebrates. The scale represents 10% amino acid divergence. Phylogram was made using ClustalW2 and protein sequences were downloaded from Uniprot.
(B) Depiction of annotated intron-exon structure of Gramd1a, Gramd1b, and Gramd1c loci in Mus musculus strain C57BL/6J assembly GRCm38. Coding exons are depicted in black. 5’ and 3’ UTRs are represented with no fill. Exons which correspond to the Gram domain, Aster domain, and Transmembrane (TM) are depicted in green, blue, and red, respectively. Scale bar represents 1kb.
(C) Aster family expression by qPCR from tissues of C57BL/6 mice (n = 5).
(D) Coomassie blue–stained SDS–PAGE gel of purified FLAG-Aster domains (Aster-A261–546, Aster-B224–560, Aster-C206–528) and control (albumin).
(E) Aster-B334–562 was titrated with 22-NBD-cholesterol in the presence of cholesterol competitor as indicated. Results values are means ± SD and representative of at least three independent experiments.
(F) Comparison of NBD-cholesterol binding to Aster-A, Aster-B and StarD1.
(G) Comparison of in vitro cholesterol transport by Aster proteins and StarD1.
Representative video of Aster-B-GFP translocation in response to cholesterol loading. Plasma membrane is indicated by PM-mCherry marker. Time is indicated in minutes after cholesterol loading. Scale bar is 5 µm.
Representative video of B GRAM (Aster-B1–171, C-terminal GFP) translocation in response to cholesterol loading (500 µM). Plasma membrane is indicated by PM-mCherry marker. Time is indicated in minutes after cholesterol loading. Scale bar is 5 µm.
Representative TIRF video of GFP (negative control) translocation in response to cholesterol loading (500 µM). Time is indicated in minutes after cholesterol loading. Scale bar is 5 µm.
Representative TIRF video of B GRAM (Aster-B1–171, C-terminal GFP) translocation in response to cholesterol loading (500 µM). Time is indicated in minutes after cholesterol loading. Scale bar is 5 µm.
(A) Structure of the Aster-A sterol-binding domain showing 25-hydroxycholesterol ligand and a glycerol molecule in the binding cavity.
(B) Comparison of mouse Aster-A, yeast Lam4p-SD2 and StARD5. Despite the low sequence identity (18%), the Aster and Lam domains have a very similar architecture creating a largely non-polar binding cavity for the 25-hydroxycholesterol. However, the orientation of the ligands is quite distinct. The beta-sheets are most similar between Aster-A and yeast Lam4p-SD2. The three helices are somewhat different. In particular, the carboxy-terminal helix in Aster-A is one turn longer at its amino-terminus and shorter at its carboxy-terminus compared with Lam4p-SD2.
(A) Orientation of the 25-hydroxycholesterol ligand. A simulated annealing composite omit map unambiguously identifies the ligand orientation in mouse Aster A (left panel - contoured at 1.0 sigma). The orientation of the 25-hydroxycholesterol ligand in mouse Aster A is markedly different from that in the distantly related yeast Lam4p (right panel - pdbcode 6bym) (Jentsch et al., 2018). The ligand is rotated by approximately 120° about it long axis such that the axial methyl groups on the cholesterol are orientated very differently.
(B) Alignment of the mammalian ASTER domains with the yeast Lam2/4 proteins. Although structurally similar, there is only limited sequence homology between the sterol-binding domains of the Aster and Lam proteins (23% identity between Aster A and Lam4p-1). Residues that are identical, have high, or low similarity are colored red, orange or yellow respectively. Residues colored cyan likely determine the different orientation of the sterol and are very different in character between the Aster and Lam proteins. Cyan asterisks indicate residues lining the pocket in contact with the hydroxycholesterol. Purple and green asterisks indicate surface non-polar and basic residues conserved in the Aster proteins that likely mediate interaction with the phospholipid membrane to facilitate sterol exchange.
(A) Aster-A-GFP, Aster-B-GFP and Aster-C GFP localization in A431 cells imaged by live cell confocal microscopy in 1% LPDS (top) or following cholesterol loading for 1 h (bottom).
(B) Live-cell imaging of GFP-Aster-A localization in A431 cells stably expressing BFP-KDEL (ER marker) and Cherry Lyn (PM marker). Yellow arrows indicate ER tubules in close proximity to the PM containing foci of Aster-A expression.
(C) Live-cell imaging of GFP-Aster-C localization in A431 cells stably expressing BFP-KDEL (ER marker) and Cherry Lyn (PM marker). Yellow arrows indicate ER tubules in close proximity to the PM containing foci of Aster-C expression.
(D) Live-cell imaging of GFP-Aster-B localization in A431 cells stably expressing BFP-KDEL (ER marker) and Cherry-E-Syt3 (ER-PM contact protein) cultured in 5% LPDS (left) or following cholesterol loading for 40 min (right). Large images, scale bar = 10 µm; insets, scale bar = 2 µm.
(E) Localization of full-length Aster-B and B ∆GRAM GFP constructs expressed in A431 cells in the presence or absence of cholesterol loading imaged by live cell confocal microscopy.
(A) RNA-seq expression of Gramd1a, Gramd1b, and Gramd1c in female adult C57BL/6J mouse adrenal gland tissue (10 weeks or age). Data was collected for the ENCODE consortium by the Snyder Lab at Stanford (GEO GSM2453460).
(B) Strategy for generating Crispr-Cas9 mediated global Gramd1b (Aster-B) knockout mice. Coding exons are depicted in black. Exons that correspond to the Gram domain, ASTER domain, and transmembrane (TM) domain are depicted in green, blue, and red respectively. Scale bar represents 1kb.
(A) Immunoblot validation of GFP-Aster-B and SR-BI expression compared to A431 wild-type cells.
(B) Lipid composition of HDL2 purified from human plasma by ultracentrifugation. HDL2 was used for the experiment because of higher lipid content and better SR-B1 interaction compared to HDL3.
(C) A431-SR-BI/Aster-B-GFP cells were lipid depleted for 24 h (LPDS) and treated with control medium (LPDS), 150–200 µg/mL FC-HDL2, or 200 µM cholesterol-cyclodextrin for 1 h. Cells were then fixed and imaged with TIRF microscopy to visualize Aster-B in close proximity to the PM. Dashed lines indicate individual cells. Scale bar 10 µm.
(D) Cells were automatically identified using DAPI (nuclei) and CellMask (cytoplasm) images and used to quantify single cell TIRF intensities.
(E) Quantification of the mean cellular GFP-Aster-B TIRF intensity from cells treated as in (A). N = 254 cells for control, 307 cells for HDL2 and 187 cells for cholesterol from 4 independent experiments.
(F) Epinephrine and dopamine levels in serum.
(A) Model for the recruitment of Aster-B to the plasma membrane. The GRAM domain binds phosphatidylserine and is recruited to the plasma membrane in response to cholesterol loading. Aster-B forms cholesterol-dependent ER–PM contact sites and facilitates the trafficking of cholesterol to the ER.
(B) Model showing PM to ER transport of HDL-derived cholesterol in the presence or absence of Aster-B.
Data Availability Statement
The PDB Code for the crystallography data is 6GQF.