

Abstract
Like proteins, some RNAs fold to compact structures. We can model functional RNAs as a series of short, rigid, based-paired elements, connected by non-base-paired nucleotides that serve as junctions. These connecting regions bend and twist, facilitating the formation of tertiary contacts that stabilize compact states. Here, we explore the roles of salt and junction sequence in determining the structures of a ubiquitous connector: an asymmetric internal loop. We focus on the J5/5a junction from the widely studied P4-P6 domain of the Tetrahymena ribozyme. Following the addition of magnesium ions to fold P4-P6, this junction bends dramatically, bringing the two halves of the RNA domain together for tertiary contact engagement. Using single-molecule fluorescence resonance energy transfer (smFRET), we examine the role of sequence and salt on model RNA constructs that contain these junction regions. We explore the wild-type J5/5a junction as well as two sequence variants. These junctions display distinct, salt dependent conformations. Small angle X-ray scattering (SAXS) measurements verify that these effects persist in the full-length P4-P6 domain. These measurements underscore the importance of junction sequence and interactions with ions in facilitating RNA folding.
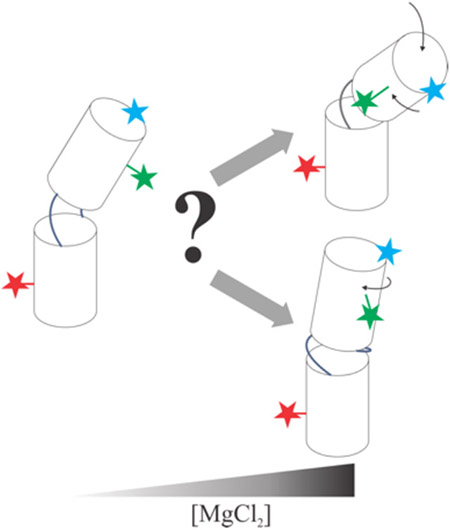
Graphical Abstract
INTRODUCTION
In the past decades, our understanding of the breadth of biological roles played by RNA continues to expand at a remarkable rate1–3. Despite its demonstrated importance, much less is known about RNA than protein conformational dynamics. Because of the many possible interactions between nucleobases, such as stacking and pairing, numerous conformational states can have similar energetics. Subtle changes in environment can tip the balance between different states leading to large conformational changes. This implied rough folding landscape makes it difficult to predict how RNA will respond to environmental clues4–6.
Noting the great strides made by understanding how the simplest systems acquire their structure7, RNA folders have followed the approach introduced by protein folders. Recent efforts to quantify RNA folding use a so-called divide and conquer approach. In some cases, independently folding domains or subdomains of functional RNAs are studied while in others simplified model systems are used to elucidate folding, one interaction or motif at a time8–17. Unlike protein motifs, which often require more structure for stability (for example helical packing), isolated RNA motifs contain base pairs formed by strong hydrogen bonds, thus may retain stability in a wide range of solution conditions.
In vitro RNA folding experiments typically start under conditions where secondary structure is formed without tertiary contacts. These conditions can be realized by dissolving the RNA in solutions containing low concentrations of monovalent ions. In this context, RNA can be described as a series of rigid helical elements connected by non-base-paired junctions18. Folding is typically induced by the addition of divalent Mg2+ ions. These ions reduce the electrostatic repulsion that persists in the lower ionic strength starting state of folding experiments, facilitating the formation of at least some tertiary contacts, and alter the flexibility of non-base paired elements, such as junction regions19. Numerous efforts have targeted many of these distinct processes20–25. However, quantifying the behavior of the most flexible regions, such as unpaired bases that join rigid helical elements, has proven particularly challenging.
It has long been recognized that non-base paired elements, such as internal loops or bulges, facilitate the bending of nucleic acid structures. The bends are dynamic, enabling access to a range of conformations depending on the properties of the junction26. A number of recent studies focus specifically on internal loops, also known as two-way junctions, consisting of non (or non-canonically) base paired connectors between helices. Several groups are exploring the role(s) of connectivity and steric effects, also known as topological constraints, in explaining how a specific linker determines the relative orientations of its flanking helices. Chu et al. demonstrate that connecting DNA helices with two rather than one flexible linker significantly restricts the relative helix conformations24. Notably, a survey of twoway junctions from the PDB outlines a relationship between the length of the linkers and these helical orientations27. While it is appealing to consider only topological factors in determining junction conformation, more subtle interactions are also involved. Mustoe et al. found that junction sequence maximizes non-canonical base pairs, which can lead to structure formation within RNA junctions28. This work also explores the potential importance of interactions with RNA elements outside of the junction in determining junction conformation. For example, tertiary contacts, once formed, might modify the topologically allowed spaces by disrupting base-base interactions to achieve specific junction backbone geometries. Another multidisciplinary study by Daher et al. demonstrates that the design of RNA junctions changes the folding propensity, docking fractions and catalytic activity of a RNA hairpin ribozyme29. Thus, RNA folding over a flexible hinge can be cast as a problem of finding the topologically allowed spaces, the corresponding junction conformations and free energy landscapes. The study by Bisaria et al. adds to our understanding by probing the role of linkers in facilitating the formation of tertiary contacts in model constructs16. A more recent study by Denny et al. demonstrates that non-canonical bases present in the RNA junction determine the junction conformations and energetics of tertiary formations30.
Recently, we investigated how both the sequence and salt dependence of a short RNA junction affects the overall conformations of a small construct. We used a model system, consisting of two short RNA duplexes connected by a single-stranded 5-nucleotide (nt) linker. Notably, the conformation of the junction (the single stranded linker) was found to determine the relative placement of the duplexes, once electrostatic repulsion between the helices was reduced by added salt31. Based on this work, we hypothesize that the interwoven effects of ions and RNA sequence may also play a role in double-stranded RNA junctions. The goal of this work is to expand on such framework to include the sequence and salt-dependence of RNA junction conformation for internal loops.
We couple two distinct experimental methods, single-molecule fluorescence resonance energy transfer32 (smFRET) and size-exclusion chromatography small-angle X-ray scattering33 (SEC-SAXS) to understand how junction conformation impacts the formation of stable RNA structures. The system of interest is the J5/5a junction from the P4-P6 domain of the Tetrahymena ribozyme, a well-studied model system for RNA folding experiments16,19,34–42. The J5/5a junction forms a 180-degree bend near the center of the domain, enabling the formation of tertiary contacts of metal-core and tetraloop. Seminal work on P4-P6 structure suggests that the domain can fold when the native junction sequence is replaced by strands containing uracil nucleobases, but significantly more Mg2+ is required37. More recent studies report slower rates of formation of tertiary contacts in the variant15.
In this study, we used smFRET to measure the distance between two RNA-attached dyes in a model system where the internal loop of interest joins two RNA helices, as in the work by Sutton et al31. Guided by the work of Szewczak et al.37, we chose to explore two particular internal loop sequences: 1) J5/5a, containing the same sequence as the wild-type P4-P6 molecule, and 2) a variant which we call JU4U5, containing the same number of nucleotides as the J5/5a construct, with the nucleobases of the internal loop replaced with uracils. The constructs (Figure 1(a)) contain the junction flanked by a helix on each side and will be referred to as H-J5/5a-H and H-JU4U5-H respectively. In addition to the junction sequence variants, the use of two different fluorescent labeling positions helps to distinguish subtle bending from twisting conformations in these constructs. These measurements, carried out at different salt concentrations, probe the natural response of the J5/5a or JU4U5 junction to changing ionic strength. We also measured the ionic strength dependence of a control molecule with the junction replaced by 5 canonically base paired nucleotides (JBP), which we refer to as H-JBP-H.
Figure 1:

(a) Schematic of three RNA constructs. Constructs consist of two RNA oligonucleotides which are individually labeled with a fluorophore and annealed together. The resulting construct forms either two short duplexes connected by an asymmetric internal loop, or a fully base paired control. Green and red stars represent the dye label positions, which are achieved from either 5’ C12-amino modifier (D1) or an amino-modified dT replacing the uracil nucleotide in the sequence (D2 and A). The constructs are named according to the nature of the junction at the center. H-J5/5a-H: the junction has the native J5/5a sequence found in P4-P6. H-JU4U5-H: both junction strands are replaced by uracil nucleotides. H-JBP-H: the junction contains 5 Watson Crick base pairs. (b) The crystal structure of the full-length wild-type P4-P6 RNA (PDB: 1GID) with the J5/5a junction shown in red. (c) The sequence of the wild-type P4-P6 RNA. The U-mutant P4-P6 replaces the J5/5a junction sequence with 9 uracil nucleotides. The base-paired P4-P6 replaces “ACAU” with “CUGUU” forming 5 canonical base pairs within the junction region.
Due to the nature of these constructs the conformations can be described by bending and twisting motions. While the native J5/5a twists and bends with increasing ionic strength, the JU4U5 junction populates more extended states, consistent with (non-canonical) base pair formation between the strands. To understand how different conformations of these junctions contribute to P4-P6 folding, we perform size-exclusion chromatography small-angle X-ray scattering (SEC-SAXS) experiments on the full length wild-type P4-P6 RNA, as well as variants that contain the same two different junction sequences (which are represented as U-mutant P4-P6 and base-paired P4-P6 respectively), under the same ionic conditions as the smFRET experiments. Whereas wild-type P4-P6 is fully folded following the addition of 5 mM MgCl2, the U-mutant P4-P6 molecule requires higher concentration of MgCl2 to adopt native folded conformations. Our results suggest that the junction sequence is an important factor in properly positioning the participants of the tertiary contacts.
METHODS
RNA Construct Design
smFRET Experiments:
Small RNA constructs consist of two strands annealed together, each with their own fluorophore attachment (Figure 1(a)). The annealed sample contains two RNA helices, each 12 base pairs long, connected on one side by a 4 nt linker, and on the other by a 5 nt linker creating an asymmetric junction. The junction consists of one of three different sequence variations: wild-type J5/5a, the JU4U5 mutant, where all 9 nucleotides in J5/5a are replaced by rUs, and a base-paired control, JBP, where the 4-nt strand is replaced by the complementary strand to the 5-nt one in J5/5a. The dye positions were chosen to ensure that the local environment is the same for all three constructs for direct comparison. The acceptor label site (A in Figure 1(a)) is 8 base pairs (bp) away from the junction region. Two different donor sites were used. The label was placed either at the 5’ end of the helix (site D1) or 8 bp away from the junction (site D2), as shown in Figure 1. RNA molecules were purchased desalted and HPLC purified from IDT (Coralville, IA) with either an amino-modified dT at the desired site for internal labels, or an amine group attached to a C12 linker for the end labels.
SEC-SAXS Experiments:
The wild-type P4-P6 RNA sequence is adopted from Ref. 37. The crystal structure and the diagram of wild-type P4-P6 are shown in Figure 1(b,c). The U-mutant and base-paired P4-P6 required modification of only the corresponding sequences in the construct (described above). The P4-P6 templates and primers were purchased desalted and HPLC purified from IDT (Coralville, IA) and stored at −20 °C.
Sample Preparation
SmFRET:
RNA single strands were first labeled with the desired fluorophore (Alexa Fluor 488 TFP ester for the donor and Cy5 NHS ester for the acceptor)31. Excessive free dye in solution was removed by ethanol precipitation, the two labeled strands were annealed together in a 1:2 donor-acceptor strand ratio at 92 °C for 2 minutes to compensate for the lower yield of Cy5 NHS dye. The samples were slowly cooled to room temperature within an hour. Samples were then buffer exchanged to 100 mM KCl in 50 mM potassium 3-(N-morpholino) propanesulfonic acid (K-MOPS) buffer. This process also removed trace amounts of free dyes which were less efficiently removed by the ethanol precipitation. Samples were aliquoted in small quantities and stored at −20 °C until needed.
SEC-SAXS:
1.0 μL of each P4-P6 DNA template at a concentration of 20 pg/μL was mixed with 2 μL 5 μM forward and reverse primers and 45 μL PCR Supermix (Thermo Fisher Scientific, Waltham, MA) for one-reaction volume. The samples then went through 50 PCR cycles and P4-P6 DNA was purified using Purelink PCR Purification Kit (Thermo Fisher Scientific, Waltham, MA). We used a total of 12 reaction volumes to produce about 20 μg DNA for each P4-P6 sequence. DNA yields and qualities were checked by running an agarose gel. Each 1.0 μg P4-P6 DNA sample was mixed with 10 μL RiboMAX Express T7 2X Buffer, 2 μL T7 Express Enzyme Mix (Promega, Fitchburg, WI) and sufficient RNase-free water to make a one-reaction volume of 20 μL. We used 20 reaction volumes for T7 transcription reaction and the mixtures were kept at 37 °C for 12 hours. The RNA samples were FPLC anion-exchange purified by flowing through HiTrap DEAE FF 5mL column (GE Healthcare, Chicago, IL). The flow-through was collected according to the spectroscopic traces and was concentrated and buffer exchanged to 100 mM KCl with 20 mM K-MOPS and 20 μM Ethylenediaminetetraacetic acid (EDTA). The final P4-P6 RNA yields were 1.2 mg for wild-type and U-mutant sequences and 0.7 mg for base-paired sequence. Directly before SEC-SAXS measurements, three P4-P6 RNA samples were annealed at 95 °C for one minute and cooled to room temperature in 20 minutes. The sample was further diluted to 120 μM with total volume of 100 μL before injecting into the size-exclusion chromatography setup.
Single-molecule FRET
Prior to smFRET experiments, a fresh sample aliquot was thawed and diluted. Each sample was annealed in a water bath for 2 minutes at 90 °C and then cooled to room temperature over 50 minutes. Samples were diluted by at least 1000 times into the desired measurement buffer. smFRET measurements were performed on freely-diffusing molecules as described in Ref. 31. The experimental energy transfer efficiency is obtained by
| (1) |
where IA and ID are the fluorescent intensities measured in the acceptor and donor channels, corrected for bleedthrough (β) and background. The factor γ accounts for the difference in quantum yields and detection efficiencies between donor and acceptor channels. For our confocal microscope, we found β = 0.027 and γ = 1.2. Histograms of all the detected EFRET values were fit with three normal distributions. The FRET histograms are shown in the Supporting Material Figures S1-S10. The zero-FRET peak represents molecules with an absent or bleached acceptor. The high-FRET peak is caused by the direct excitations of the acceptors. The peak of interest is the concentration-dependent mid-FRET peak. Each measurement was repeated independently at least twice, and the EFRET is converted to dye-dye distance RFRET using the following equation:
| (2) |
where R0 = 52 Å for Alexa Fluor 488 and Cy5 dye pair43. The only correction for R0 is the refractive index, n, for different salts and concentrations. R0 is proportional to n−2/3. Throughout this work, we use the published refractive indices for monovalent and divalent solutions at room temperature44 to correct for the slight variations of R0, which are typically no more than 1.5 Å.
SEC-SAXS
Size-exclusion chromatography small-angle X-ray scattering (SEC-SAXS) was conducted at Cornell High Energy Synchrotron Source (CHESS) beamline G1 using a Pilatus3 X 100K-A detector (Dectris Ltd., Baden-Daettwil, Switzerland) with a sample-to-detector distance of 1.53 meters. The 120 μM 100 μL sample was injected into Superdex® 200 Increase 5/150 GL column (GE Healthcare, Chicago, IL) after equilibration with the desired buffer. The ionic conditions of interest for this work are 200 mM and 500 mM KCl, and 5 mM and 20 mM MgCl2. A background monovalent ion concentration of 100 mM KCl was present in divalent buffers. The in-line SEC column fed a SAXS sample holder, using a total flow rate of 0.2 mL/min. We acquired 1300 frames of data with a 1-second exposure time. The momentum transfer q = (4π/λ) sin(2θ/2), where λ is the X-ray wavelength in Å and 2θ is the scattering angle. A q-range from 0.0107 to 0.277 1/Å was achieved. The SAXS curves were normalized to account for slight variations in beam intensity. Realtime data analysis was carried out by the software package RAW45. Further analysis was done using in-house MATLAB (The MathWorks, Natick, MA) scripts. We averaged the sample scattering profiles with similar values of radius of gyration (Rg), which remained constant with elution volume. Buffer matching was checked by comparing scattering profiles prior to and after the sample passed through the SEC column. If the buffer matching was good, we averaged all the pre and post-buffer frames to obtain the buffer scattering profile. A continuous scaling factor was applied to the subtracted scattering profiles to account for sample dilution out of the SEC column. Final SAXS curves were processed by GNOM46 and DAMMIF47 to determine size parameters and reconstruct the global shape of three different P4-P6 RNA in solutions containing 5 and 20 mM MgCl2.
RESULTS
smFRET Measurements of End- and Side-labeled Junctions
To detect conformational differences induced by the sequence of the internal loop, we first compare RFRET values in titrations of KCl and MgCl2 of the H-J5/5a-H and H-JU4U5-H constructs, labeled with the D1-A dye pair (Figures 2(a) and (b)). (Note: the EFRET values are shown in Supporting Material Figure S11.) Because the donor fluorophore is attached to the end of one RNA helix it is more sensitive to bending than twisting motions, as suggested in Figure 2(e). In KCl solutions (Figure 2(a)) both constructs show similar dye-to-dye distances, with the H-J5/5a-H bending slightly more than H-JU4U5-H over the [K+] studied. In MgCl2 solutions, this effect is magnified (Figure 2(b)) over a roughly comparable range of ionic strength. Notably, H-JU4U5-H displays more resistance to bending than H-J5/5a-H at the lowest [Mg2+] probed. These results suggest that the conformations depend both on the sequence of the internal loop as well as the identity and concentration of excess salt in solution. We note here the challenges of assigning a bend angle based on changes in RFRET response alone. The exact values depend on knowing the placement/orientations of the dyes. In addition, their separation has a non-linear response to the bend angle. In the absence of any other effects, an overall bending of as much as 50 degrees would only yield about a 4 Å decrease in RFRET.
Figure 2:

Measured effects on RFRET as a function of KCl and MgCl2 concentration for the D1-A (end-label) dye pair ((a) and (b)) and the D2-A (side-label) dye pair ((c) and (d)). Data for all three constructs are shown: H-J5/5a-H (blue), H-JU4U5-H (red) and H-JBP-H (orange). The error bars represent the combination of the 95% confidence interval of the fit parameter and the standard deviations of at least two independent measurements. Part (e) shows a cartoon representing the RNA junction constructs and the associated label positions. The D1 label is closer to the helical axis and is more sensitive to overall bend angle, while the D2 label is off-axis and sensitive to both bending and relative twisting.
To tease out additional information about the differences between the constructs described above, we carried out a second series of measurements with a donor dye at position D2 (Figure 2(e)). Because this dye juts out from one side of the helix, it increases our sensitivity to the relative twist angle between the two helices. Importantly, we cannot strictly distinguish bending from twisting using this D2-A dye pair. The RFRET values for the D2-A labeled H-J5/5a-H and H-JU4U5-H constructs in KCl and MgCl2 are shown in Figures 2(c) and 2(d) respectively. With increasing KCl concentration, H-J5/5a-H demonstrates a continuous decrease in RFRET. More subtle change is measured in H-JU4U5-H. In MgCl2, although both constructs display an overall decreasing RFRET with increasing salt, the difference in RFRET values is more pronounced, suggesting that the junction affects the relative rotational positioning of the helices. However, before interpreting any changes in the RFRET values, we must first account for systematic effects: the salt dependence.
Comparison with Base-Paired Control Molecule
Both Alexa Fluor 488 and Cy5 dyes are negatively charged and connected to the RNA by flexible linkers, thus may display a salt dependence that is unrelated to structural changes. To address concerns about possible variations in dye response due to changing salt concentrations, in addition to the correction resulting from the salt dependent refractive index, we measured RFRET for the D2-A dye pair using a fully base-paired RNA helix (H-JBP-H), for which we expect minimal structural changes across concentrations and ions. Figure 2(c) and 2(d) show these RFRET values as a function of KCl and MgCl2 concentration, respectively. (We could not carry out comparable measurements for the D1-A dye pair, as the distances are too large to provide significant energy transfer.) Surprisingly, an overall decrease in RFRET with salt is observed for the H-JBP-H and RFRET only approaches the Accessible Volume (AV) simulated value of 52.3 Å (Figure S12) at extremely high salt concentrations.
Since the change of RFRET in both KCl and MgCl2 for H-JBP-H is comparable to the changes observed for the other constructs, we compute the differences in RFRET between H-JBP-H and each of the others, shown in Figure 3(a) and 3(b). These difference plots allow us to eliminate helical and fluorophore dynamics, leaving only the junction effects. In KCl solutions, while the difference in RFRET between H-JBP-H and H-J5/5a-H levels out at 5.5 Å, comparison between H-JBP-H and H-JU4U5-H shows a further reduction, asymptotically approaching 3 Å. Above 5 mM MgCl2, these differences stabilize around 6.6 Å and 3 Å respectively. Interestingly, the 3 Å distance difference between RFRET in H-JBP-H and H-JU4U5-H is nearly identical to the change in length induced by the presence/absence of one extra base pair in an RNA A-form helix. Thus, H-JU4U5-H appears to be duplex like, while the H-J5/5a-H construct assumes different structures, presumably reflecting the bending suggested by the D1-A dye pair.
Figure 3:

The difference between RFRET for the base paired (H-JBP-H) control and H-J-H in (a) KCl and (b) MgCl2, where J represents either J5/5a or JU4U5. Dashed red lines mark a salt concentration above which one of the difference values remains constant. Note the RH –JBP – H – RH – JU4U5 – H does not approach a constant value at high concentration of KCl. Above 5 mM MgCl2 the H-JU4U5-H construct is 3 Å shorter than the base paired construct. This value is close to the height contributed by one base pair (2.8 Å) to an A-form RNA duplex.
The Folding of P4-P6 Domain
To interpret the behavior of the junction in the context of a folding molecule, we used SAXS to monitor the global conformations of full length wild-type P4-P6 in addition to two variants, created by modifying the junction sequence to match those shown in Figure 1. SAXS profiles acquired under different ionic conditions are shown in Figure 4 and 5. Kratky plots of I(q)q2 vs. q, where I(q) is the measured scattering intensity, emphasize scattering intensity near the middle of the measured q range, and are extremely useful in distinguishing folded from extended macromolecular states: a more pronounced peak reflects a more compact conformation48. We first compare the three P4-P6 sequences in different [K+] and [Mg2+] to assess the impact of modifying junction sequences (Figure 4). The wild-type P4-P6 remains unfolded (extended) in solutions containing up to 500 mM KCl, consistent with past studies42. With the addition of MgCl2, the appearance of a pronounced peak in the Kratky plot confirms that the domain folds to compact structures (Figure 4(a)). The formation of native tertiary contacts is facilitated by the large bend angle of the J5/5a junction, and the subsequent formation of the (Mg2+-dependent) tertiary contacts. Like the wild-type, the U-mutant P4-P6 remains extended in all measured KCl concentrations, shown in Figure 4(b). It folds partially in 5 mM MgCl2 and nearly completely in 20 mM MgCl2, consistent with the gel analysis results of Ref. 37. The base-paired P4-P6 remains extended/unfolded in all salt solutions tested, as shown in Figure 4(c). By design, replacement of a short flexible junction with fully base-paired stem precludes formation of the native tertiary contacts. The small peaks in the Kratky plot of base-paired P4-P6 (Figure 4(c)) likely result from the folding of a subdomain (P5abc) that can form without the bend (see Figure 1). They vary subtly with increasing MgCl2 concentration.
Figure 4:

Kratky plots of SAXS data reporting the salt dependence of (a) wild-type P4-P6 (b) U-mutant P4-P6 and (c) base-paired P4-P6. These plots show a more pronounced peak when the molecule is in a more compact state. The wild-type P4-P6 shows two states, unfolded (black, green, showing no peak) in KCl and folded (blue, magenta, peak appears) in MgCl2 respectively. The U-mutant P4-P6 appears unfolded in KCl, partially folded in 5 mM MgCl2 and fully folded in 20 mM MgCl2. By design the base-paired P4-P6 remains unfolded across all the conditions.
Figure 5:

Comparison between the three different P4-P6 sequences in (a) 5mM MgCl2 and (b) 20mM MgCl2. (c) The reconstructions of the SAXS profiles in 5mM and 20mM MgCl2 (described in Material and Method section), color coded by the same scheme, assuming a homogeneous population. Global shape parameters are also reported.
Figure 5 provides more in-depth information about the Mg2+ induced structural changes in three P4-P6 molecules with different junction sequences. In addition to the Kratky plots, Figures 5(a) and 5(b), SAXS also provides information about global structural parameters, such as radius of gyration and maximum molecular dimension, Rg and Dmax respectively. These global shape parameters for each molecule at the two different MgCl2 concentrations, as well as the ab initio reconstructions for each concentration are shown in Figure 5(c) with the mesh surfaces in the same coloring scheme. Because our mutations do not alter the tertiary contacts or the P5abc independently folding subdomain, differences in the SAXS profiles reflect the different folding propensities imposed by the junction sequences. As [Mg2+] increases to 20 mM, there is no change in the conformation of either the wild-type or base-paired P4-P6: the wild-type P4-P6 remains in the native compact state while the base-paired P4-P6 remains unfolded. Interestingly, the U-mutant P4-P6 folds more completely, with similar global size and shape to the folded wild-type.
DISCUSSION
We first address the unexpected salt-dependence of RFRET measurements in the H-JBP-H construct shown in Figures 2(c) and 2(d). Next, we provide a simple model to explain the bending and twisting of H-J5/5a-H and H-JU4U5-H. Finally, we connect the behavior of the isolated junction constructs to P4-P6 RNA folding.
Salt-dependence of Base-paired RNA Duplex in smFRET
Despite our expectation that the H-JBP-H molecule would exhibit no conformational changes with change in salt concentration, we measure a change in RFRET comparable to that of the other RNA junction constructs (Figure 2). We first consider the most obvious explanation: the dependence of the fluorophore environment on salt concentration. Since RFRET for the H-JBP-H approaches the AV-simulated RFRET for an A-form helix at high MgCl2 concentrations, repulsion between the RNA phosphate backbone and the fluorophores could restrict the dye environments at low salt to a subset of states obtained from the simulations. This electrostatic repulsion could restrict the dyes to the environment around the ends of the helix at low salt. AV simulations would not capture this behavior because they only account for steric constraints. Increasing the salt concentration may allow the dyes to more closely approach the RNA helix, moving inwards and decreasing their average separation. To test this hypothesis, we carried out experiments using identically labeled DNA of comparable sequence (Figure S13). No changes in RFRET were detected as the solution salt content varied. This result suggests that the FRET labels are measuring salt dependent changes in the structure of the RNA duplex.
The salt dependent structure of a 25 bp RNA duplex was confirmed in recent work, using solution small and wide angle X-ray scattering (S/WAXS) to show that multivalent ions, such as cobalt(III) hexamine, stabilize the RNA helix, rendering a more A-form-like conformation49. Simulations suggest that the length of the helix decreases with increasing salt, as the structure more closely resembles the ideal A-form. We also observe similar phenomena in S/WAXS experiments using different concentrations of KCl and MgCl2 Thus, the salt induced deviation of the A-form RNA helix appears to affect the topologically allowed space28.
Despite the unexpected changes discussed above, results on the H-JBP-H construct can provide insight into the structure of the H-JU4U5-H variant. Figure 3(b) shows that the RFRET difference between H-JU4U5-H and H-JBP-H approaches 3 Å above 5 mM MgCl2. This is consistent with a picture where four base pairs in the JU4U5 junction are nearly fully formed and stacked, therefore, roughly coaxial (The H-JBP-H contains five pairs in the modified junction.). In contrast, results shown in Figure 3(a) imply that any comparable base-pairing and stacking would require much higher [K+] since the difference in RFRET does not stabilize over the measured range. It has been reported that symmetric loops tend to pair the nucleobases to form A-helix-like conformations26. Because the JU4U5 internal loop is asymmetric, a fully paired junction would only have four base pairs, leaving one rU base flipped away from the central axis, consistent with reports from other studies50–52.
Sequence- and Concentration- Dependence of Bending and Twisting
By combining the end and side-labeled RFRET data we construct a model suggesting how junction sequence affects the sampling of conformational space. The use of the two separate donor label positions allows us to partially distinguish bending from twisting: the D1-A dye pair is only sensitive to bending while the D2-A dye pair is affected by the combination of bending and twisting. The data interpretation must also account for the intrinsic salt dependent conformations of the RNA duplex, identified through the use of the H-JBP-H molecule.
In general, the isolated junction constructs display similar trends in KCl solutions; the RFRET values and their variations with salt are comparable. Thus, junction sequence contributes modestly to junction conformation when KCl is present. More significant differences are found when MgCl2 is introduced: the H-J5/5a-H construct has a lower overall RFRET value at any given [Mg2+] for both dye pairs. Because the D1-A pair (Figure 2b) reports bending motions, this reduction suggests that H-J5/5a-H is more compact than H-JU4U5-H. Above 5 mM MgCl2, where the latter construct favors formation of an A-form duplex, RFRET decreases more rapidly with [Mg2+] for the H-J5/5a-H. Changes in the RFRET values reported by the D2-A dye pair are harder to interpret, as they may signal twisting in addition to bending. Interpretation of D2-A distances are aided by measurements on a fully base paired construct. The decrease in RFRET for the H-JBP-H construct (Figure 2d) suggests that the RNA helical structure twists with increasing salt. This trend is mirrored in H-JU4U5-H (Figure 3b) at MgCl2 concentrations above 5 mM. The fixed offset relative to H-JBP-H can be accounted for by the one-base-pair difference in the junction. In summary, with added MgCl2, the J5/5a junction bends, while the JU4U5 junction twists as it pairs and stacks. Figure 6 presents a model of our results.
Figure 6:

Schematic representing salt concentration and sequence effects of RNA junction conformations. At low salt, the H-J5/5a-H and H-JU4U5-H have different helical twists (shown by the side label RFRET data), but similar bend angles (shown by the end label RFRET data). As [MgCl2] increases, the difference between sequences also increases, demonstrating an interplay between sequence and salt dependence.
The proposed conformation of JU4U5 at high concentrations of MgCl2 is consistent with the suggestion from Mustoe et al. that non-canonical base pairs tend to form within internal loop regions28: the bases on each strand stack and are therefore primed for base pair formation. While some studies show single stranded poly(rU) RNA has a low base stacking propensity53–56, other work suggests ion dependent structuring57. It is also known that unpaired uracil nucleobases are more responsive to changes in [Mg2+] than other nucleotides (for example A’s)58. This work suggests that the [Mg2+] dependent behavior of the two poly(rU) strands determines the conformations. The U-U mismatch (along with the G-A mismatch) is more stable than many other non-canonical base pairs59. The most likely junction structures as predicted by MC-fold60 suggest that all but one rU base in JU4U5 is paired, while there are only two base pairs in the wild-type J5/5a sequence. Moreover, these four U-U non-canonical base pairs form more easily in two poly(rU) strands because the junction can flip any one of the five uracil nucleobases according to subtly different backbone geometries. On the other hand, the J5/5a junction has to adopt specific conformations to ensure the formation of two lone base pairs. Therefore, we propose that the JU4U5 junction is partially stabilized by stacking interactions and non-canonical base-pairing. With increasing [Mg2+], this junction is A-form-like with one non-paired base. The exact behavior of this extra base can be critical in defining the overall conformation of the construct regardless of its exact location in the poly(rU)5 strand. If it “loops out”, the remaining bases pair and stack, continuing the A-form structures of the adjacent helices. If it “loops in”, it can disrupt the helical conformation and promote a bend. This process is likely dynamic and may result in changes of the junction structure.
Insights from isolated junctions into P4-P6 Folding
We can extrapolate the results of smFRET studies on isolated junction constructs to explain differences in the observed folding of three P4-P6 sequence: wild-type, U-mutant and base-paired. As seen in previous work on P4-P615,37,61, it is not surprising that variations in the J5/5a junction alter the folding properties. In the wild-type P4-P6, the J5/5a junction acts as a hinge, allowing two halves of the P4-P6 domain to form the two native tertiary contacts indicated in Figure 1(b). Mg2+ is required for tertiary contact formation; however, its effect on the hinge has not been directly explored. Our smFRET experiments on the isolated J5/5a junction show a definitive response to Mg2+: the hinge bends and (possibly) twists independent of the distal tertiary contacts present in the P4-P6 molecule. We postulate that these movements are carefully calibrated to precisely position the two sides of the tertiary interactions for contact, when they come into proximity. At the other extreme, the base-paired P4-P6 is designed to remain unfolded. The five canonical base pairs present in the junction preclude hinge bending.
In between these two extremes, the U-mutant P4-P6 resists folding. At 5mM MgCl2, the scattering profile is intermediate between extended and folded states, likely reflecting that a fraction of the population is in each state, though we cannot rule out occupation of a partially folded intermediate. At 20 mM MgCl2 its conformation closely resembles the native folded state of the wildtype P4-P6. At and above 5 mM Mg2+, smFRET suggests that the junction is non-canonically base-paired, an extension of the A-form duplexes that flank it. From the SAXS data, the small degree of compaction at 5 mM MgCl2, and the more significant compaction seen at 20 mM imply that the U-mutant P4-P6 overcomes the energy barrier imposed by the non-canonical base-pairing and base-stacking interactions suggested by the FRET dataset. The subsequent formation of tertiary contacts dramatically reduces the overall folding free energy. Two conditions must be fulfilled for this molecule to fold: first, there must be enough conformational variation in the hinge (bending) to initiate tertiary contact formation. Subsequently, the two sides of the contacts must be accurately positioned to engage. The first requires a transient disruption of the junction, presumably by base flipping either into or out of the stack. The stability of the smFRET results suggests little conformational difference between 5 and 20 mM MgCl2. We therefore speculate that the difference in folding between 5 and 20 mM MgCl2 results from a [Mg2+] dependent twisting of the junction that affects the initial transient alignment of the two sides of the tertiary contact. The hinge bends, but the contacts are imperfectly aligned (in contrast to the wild-type J5/5a junction, where the sequence is tuned to preposition them accurately). Although the results of Figure 3 show a constant difference between the H-JBP-H and the H-JU4U5-H containing construct, the control itself varies with [Mg2+] suggesting a subtle helical twist with respect to the central axis.
CONCLUSION
We apply the divide-and-conquer strategy to study how the varying sequence of a small junction influences the folding of the P4-P6 domain of the Tetrahymena ribozyme. smFRET measurements of the junctions, placed in a small HJH construct, show that they sample different regions of conformational space, depending on their sequences, identities and concentration of the salt ion used. Different labeling sites allow us to distinguish bending and twisting degrees of freedom. The wildtype junction tends to bend with increasing salt. A JU4U5 mutant with all junction residues replaced by U’s appears to pair and stack, creating a continuous A-form helix through the junction region. Both effects are magnified in Mg2+ relative to K+ salts. We use SEC-SAXS to show how different conformational space sampling of the junction alters the folding of the entire P4-P6 domain. While the wildtype junction sequence is calibrated to exhibit optimal bend-twist combination, the U-mutant appears to resist folding due to mis-alignment of tertiary contacts until higher Mg2+ modifies the twist. Overall these studies suggest that junction sequence is an important factor in RNA macromolecular folding and can be subtly tuned by ionic strength.
Supplementary Material
ACKNOWLEDGEMENT
We acknowledge funding support from NIH Grant R01 GM085062 and R35 GM122514. The authors thank Richard Gillilan and Suzette A. Pabit for experimental assistance. The authors also thank Joshua Tokuda and Pollack Lab Members for helpful discussions. SEC-SAXS data were acquired at the Cornell High Energy Synchrotron Source (CHESS), G-1 beamline. CHESS is supported by the NSF and NIH/NIGMS via NSF Award No. DMR-1332208, and the MacCHESS resource is funded by NIGMS Award No. GM-103485.
Footnotes
SUPPORTING INFORMATION
See supporting information for raw smFRET data, AV simulation results and DNA/RNA duplex comparisons as a function of changing salt conditions.
REFERENCES
- 1.Sharp PA. Essay The Centrality of RNA. 2009:577–580. doi: 10.1016/j.cell.2009.02.007. [DOI] [PubMed] [Google Scholar]
- 2.Morris KV, Mattick JS. The rise of regulatory RNA. Nat Publ Gr. 2014;15(6):423–437. doi: 10.1038/nrg3722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cech TR, Steitz JA. Review The Noncoding RNA Revolution — Trashing Old Rules to Forge New Ones. Cell. 2014;157(1):77–94. doi: 10.1016/j.cell.2014.03.008. [DOI] [PubMed] [Google Scholar]
- 4.Schroeder R, Barta A, Semrad K. Strategies for RNA folding and assembly. Nat Rev Mol Cell Biol. 2004;5(11):908–919. doi: 10.1038/nrm1497. [DOI] [PubMed] [Google Scholar]
- 5.Al-Hashimi HM, Walter NG. RNA dynamics: it is about time. Curr Opin Struct Biol. 2008;18(3):321–329. doi: 10.1016/j.sbi.2008.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Solomatin SV, Greenfeld M, Chu S, Herschlag D. Multiple native states reveal persistent ruggedness of an RNA folding landscape. Nature. 2010;463(7281):681–684. doi: 10.1038/nature08717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eaton WA, Munoz V, Hagen SJ, et al. Fast kinetics and mechanisms in protein folding. Annu Rev Biophys Biomol Struct. 2000;29:327–359. doi: 10.1146/annurev.biophys.29.1.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tinoco I, Bustamante C. How RNA folds. JMol Biol. 1999;293(2):271–281. doi: 10.1006/jmbi.1999.3001. [DOI] [PubMed] [Google Scholar]
- 9.Klein DJ, Schmeing TM, Moore PB, Steitz TA. The kink-turn: A new RNA secondary structure motif. EMBO J. 2001;20(15):4214–4221. doi: 10.1093/emboj/20.15.4214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lukavsky PJ, Kim I, Otto GA, Puglisi JD. Structure of HCV IRES domain II determined by NMR. Nat Struct Biol. 2003;10(12):1033–1038. doi: 10.1038/nsb1004. [DOI] [PubMed] [Google Scholar]
- 11.Wu L, Chai D, Fraser ME, Zimmerly S. Structural Variation and Uniformity among Tetraloop- Receptor Interactions and Other Loop-Helix Interactions in RNA Crystal Structures. PLoS One. 2012;7(11). doi: 10.1371/journal.pone.0049225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Costa M, Michel F. Frequent use of the same tertiary motif by self-folding RNAs. EMBO J. 1995; 14(6): 1276–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Geary C, Baudrey S, Jaeger L. Comprehensive features of natural and in vitro selected GNRA tetraloop-binding receptors. Nucleic Acids Res. 2008;36(4): 1138–1152. doi: 10.1093/nar/gkm1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nissen P, Ippolito JA, Ban N, Moore PB, Steitz TA. RNA tertiary interactions in the large ribosomal subunit: The A-minor motif. Proc Natl Acad Sci. 2001;98(9):4899–4903. doi: 10.1073/pnas.081082398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bisaria N, Greenfeld M, Limouse C, Pavlichin DS, Mabuchi H, Herschlag D. Kinetic and thermodynamic framework for P4-P6 RNA reveals tertiary motif modularity and modulation of the folding preferred pathway. Proc Natl Acad Sci. 2016;113(34):E4956–E4965. doi: 10.1073/pnas.1525082113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bisaria N, Greenfeld M, Limouse C, Mabuchi H, Herschlag D. Quantitative Tests of a Reconstitution Model for RNA Folding Thermodynamics and Kinetics. Proc Natl Acad Sci. 2017;(in press). doi: 10.1073/pnas.1703507114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pollack L Time resolved SAXS and RNA folding. Biopolymers. 2011;95(8):543–549. doi: 10.1002/bip.21604. [DOI] [PubMed] [Google Scholar]
- 18.Butcher SE, Pyle AM. The molecular interactions that stabilize RNA tertiary structure: RNA motifs, patterns, and networks. Acc Chem Res. 2011;44(12): 1302–1311. doi: 10.1021/ar200098t. [DOI] [PubMed] [Google Scholar]
- 19.Schlatterer JC, Kwok LW, Lamb JS, et al. Hinge Stiffness Is a Barrier to RNA Folding. J Mol Biol. 2008;379(4):859–870. doi: 10.1016/j.jmb.2008.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Das R, Kwok LW, Millett IS, et al. The fastest global events in RNA folding: Electrostatic relaxation and tertiary collapse of the tetrahymena ribozyme. J Mol Biol. 2003;332(2):311–319. doi: 10.1016/S0022-2836(03)00854-4. [DOI] [PubMed] [Google Scholar]
- 21.Greenfeld M, Herschlag D, Chu VB, et al. Quantitative and Comprehensive Decomposition of the Ion Atmosphere around Nucleic Acids. 2007;129:14981–14988. doi: 10.1021/ja075020g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bai Y, Chu VB, Lipfert J, Pande VS, Herschlag D, Doniach S. Critical Assessment of Nucleic Acid Electrostatics via Experimental and Computational Investigation of an Unfolded State Ensemble. 2008;(4):12334–12341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chu VB, Bai Y, Lipfert J, Herschlag D, Doniach S. A repulsive field: advances in the electrostatics of the ion atmosphere. Curr Opin Chem Biol. 2008;12(6):619–625. doi: 10.1016/j.cbpa.2008.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chu VB, Lipfert J, Bai Y, Pande VS, Doniach S, Herschlag D. Do conformational biases of simple helical junctions influence RNA folding stability and specificity? RNA. 2009;15(12):2195–2205. doi: 10.1261/rna.1747509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Solomatin SV, Greenfeld M, Chu S, Herschlag D. Multiple native states reveal persistent ruggedness of an RNA folding landscape. Nature. 2010;463(7281):681–684. doi: 10.1038/nature08717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bailor MH, Mustoe AM, Iii CLB, Al-Hashimi HM, Brooks CL, Al-Hashimi HM. Topological constraints: Using RNA secondary structure to model 3D conformation, folding pathways, and dynamic adaptation. Curr Opin Struct Biol. 2011;21(3):296–305. doi: 10.1016/j.sbi.2011.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bailor MH, Sun X, Al-Hashimi HM. Topology Links RNA Secondary Structure with Global Conformation, Dynamics, and Adaptation. Science (80-). 2010;327(5962):202–206. doi: 10.1126/science.1181085. [DOI] [PubMed] [Google Scholar]
- 28.Mustoe AM, Bailor MH, Teixeira RM, et al. New insights into the fundamental role of topological constraints as a determinant of two-way junction conformation. Nucleic Acids Res. 2012;40(2):892–904. doi: 10.1093/nar/gkr751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Daher M, Mustoe AM, Morriss-Andrews A, Brooks CL, Walter NG. Tuning RNA folding and function through rational design of junction topology. Nucleic Acids Res. 2017;45(16):9706–9715. doi: 10.1093/nar/gkx614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sarah Knight Denny A, Bisaria N, David Yesselman J, et al. High-Throughput Investigation of Diverse Junction Elements in RNA Tertiary Folding. Cell. 2018;174:1–14. doi: 10.1016/j.cell.2018.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sutton JL, Pollack L. Tuning RNA Flexibility with Helix Length and Junction Sequence. Biophys J. 2015;109(12):2644–2653. doi: 10.1016/j.bpj.2015.10.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rahul R, Hohng S, Ha T. A Practical Guide to Single Molecule FRET. Nat Methods. 2008;5(6):507–516. doi: 10.1038/nmeth.1208.A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Watanabe Y, Inoko Y. Size-exclusion chromatography combined with small-angle X-ray scattering optics. J Chromatogr A. 2009;1216(44):7461–7465. doi: 10.1016/j.chroma.2009.02.053. [DOI] [PubMed] [Google Scholar]
- 34.Lamb J, Kwok L, Qiu X, Andresen K, Park HY, Pollack L. Reconstructing three-dimensional shape envelopes from time-resolved small-angle X-ray scattering data. J Appl Crystallogr. 2008;41(6):1046–1052. doi: 10.1107/S0021889808028264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cate JH, Gooding AR, Podell E, et al. Crystal Structure of a Group I Ribozyme Domain: Principles of RNA Packing. Science (80-). 1996;273(5282):1678–1685. [DOI] [PubMed] [Google Scholar]
- 36.Doherty EA, Doudna JA. The P4-P6 domain directs higher order folding of the Tetrahymena ribozyme core. Biochemistry. 1997;36(11):3159–3169. doi: 10.1021/bi962428+. [DOI] [PubMed] [Google Scholar]
- 37.Szewczak AA, Cech TR. An RNA internal loop acts as a hinge to facilitate ribozyme folding and catalysis. RNA. 1997;3(8):838–849. http://www.ncbi.nlm.nih.gov/pubmed/9257643%5Cnhttp://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=PMC1369529. [PMC free article] [PubMed] [Google Scholar]
- 38.Szewczak AA, Podell ER, Bevilacqua PC, Cech TR. Thermodynamic Stability of the P4-P6 Domain RNA Tertiary Structure Measured by Temperature Gradient Gel Electrophoresis † Biochemistry. 1998;37(32): 11162–11170. doi: 10.1021/bi980633e. [DOI] [PubMed] [Google Scholar]
- 39.Silverman SK, Deras ML, Woodson SA, Scaringe SA, Cech TR. Multiple folding pathways for the P4-P6 RNA domain. Biochemistry. 2000;39(40):12465–12475. doi: 10.1021/bi000828y. [DOI] [PubMed] [Google Scholar]
- 40.Deras ML, Brenowitz M, Ralston CY, Chance MR, Woodson SA. Folding mechanism of the Tetrahymena ribozyme P4-P6 domain. Biochemistry. 2000;39(36):10975–10985. doi: 10.1021/bi0010118. [DOI] [PubMed] [Google Scholar]
- 41.Russell R, Zhuang X, Babcock HP, et al. Exploring the folding landscape of a structured RNA. Proc Natl Acad Sci USA. 2002;99(1):155–160. doi: 10.1073/pnas.221593598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Takamoto K, Das R, He Q, et al. Principles of RNA compaction: Insights from the equilibrium folding pathway of the P4-P6 RNA domain in monovalent cations. J Mol Biol. 2004;343(5): 1195–1206. doi: 10.1016/j.jmb.2004.08.080. [DOI] [PubMed] [Google Scholar]
- 43.Sindbert S, Kalinin S, Nguyen H, et al. Accurate distance determination of nucleic acids via förster resonance energy transfer: Implications of dye Linker length and rigidity. J Am Chem Soc. 2011;133(8):2463–2480. doi: 10.1021/ja105725e. [DOI] [PubMed] [Google Scholar]
- 44.Tan CY, Huang YX. Dependence of Refractive Index on Concentration and Temperature in Electrolyte Solution, Polar Solution, Nonpolar Solution, and Protein Solution. J Chem Eng Data. 2015;60(10):2827–2833. doi: 10.1021/acs.jced.5b00018. [DOI] [Google Scholar]
- 45.Hopkins JB, Gillilan RE, Skou S. BioXTAS RAW: Improvements to a free open-source program for small-angle X-ray scattering data reduction and analysis. J Appl Crystallogr. 2017;50(5): 1545–1553. doi: 10.1107/S1600576717011438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Svergun DI. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J Appl Crystallogr. 1992;25(pt 4):495–503. doi: 10.1107/S0021889892001663. [DOI] [Google Scholar]
- 47.Franke D, Svergun DI. DAMMIF, a program for rapid ab-initio shape determination in small- angle scattering. J Appl Crystallogr. 2009;42(2):342–346. doi: 10.1107/S0021889809000338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Svergun DI, Koch MHJ. Small-angle scattering studies of biological macromolecules in solution. Reports Prog Phys. 2003;66(10):1735–1782. doi: 10.1088/0034-4885/66/10/R05. [DOI] [Google Scholar]
- 49.Pabit SA, Katz AM, Tolokh IS, et al. Understanding nucleic acid structural changes by comparing wide-angle x-ray scattering (WAXS) experiments to molecular dynamics simulations. J Chem Phys. 2016;144(20). doi: 10.1063/1.4950814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yang X, Gerczei T, Glover L, Correll CC. Crystal structures of restrictocin-inhibitor complexes with implications for RNA recognition and base flipping. Nat Struct Biol. 2001;8(11):968–973. doi: 10.1038/nsb1101-968. [DOI] [PubMed] [Google Scholar]
- 51.Reiter NJ, Blad H, Abildgaard F, Butcher SE. Dynamics in the U6 RNA intramolecular stem-loop: A base flipping conformational change. Biochemistry. 2004;43(43):13739–13747. doi: 10.1021/bi048815y. [DOI] [PubMed] [Google Scholar]
- 52.Matthews MM, Thomas JM, Zheng Y, et al. Structures of human ADAR2 bound to dsRNA reveal base-flipping mechanism and basis for site selectivity. Nat Struct Mol Biol. 2016;23(5):426–433. doi: 10.1038/nsmb.3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jacques R Molecular Properties and Conformation of Polyribouridylic Acid. Biopolymers. 1963;1:431–446. [Google Scholar]
- 54.Inners LD, Felsenfeld G. Conformation of polyribouridylic acid in solution. J Mol Biol. 1970;50(2):373–389. doi: 10.1016/0022-2836(70)90199-3. [DOI] [PubMed] [Google Scholar]
- 55.Condon DE, Kennedy SD, Mort BC, Kierzek R, Yildirim I, Turner DH. Stacking in RNA: NMR of four tetramers benchmark molecular dynamics. J Chem Theory Comput. 2015;11(6):2729–2742. doi: 10.1021/ct501025q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Brown RF, Andrews CT, Elcock AH. Stacking free energies of all DNA and RNA nucleoside pairs and dinucleoside-monophosphates computed using recently revised AMBER parameters and compared with experiment. J Chem Theory Comput. 2015; 11(5):2315–2328. doi: 10.1021/ct501170h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tan D, Piana S, Dirks RM, Shaw DE. RNA force field with accuracy comparable to state-of-the-art protein force fields. Proc Natl Acad Sci. 2018:201713027. doi: 10.1073/pnas.1713027115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zacharias M, Hagerman PJ. Bulge-Induced Bends in RNA: Quantification by Transient Electric Birefringence. J Mol Biol. 1995;247(3):486–500. doi: 10.1006/jmbi.1995.0155. [DOI] [PubMed] [Google Scholar]
- 59.Schroeder S, Kim J, Turner DH. G.A and U.U Mismatches Can Stabilize RNA Internal Loops of Three Nucleotides. Biochemistry. 1996;35(50): 16105–16109. doi: 10.1021/bi961789m. [DOI] [PubMed] [Google Scholar]
- 60.Parisien M, Major F. The MC-Fold and MC-Sym pipeline infers RNA structure from sequence data. Nature. 2008;452(7183):51–55. doi: 10.1038/nature06684. [DOI] [PubMed] [Google Scholar]
- 61.Matsumura S, Ikawa Y, Inoue T. Biochemical characterization of the kink-turn RNA motif. Nucleic Acids Res. 2003;31(19):5544–5551. doi: 10.1093/nar/gkg760. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.