

Abstract
PURPOSE
Microsatellite instability-high (MSI-H) colorectal carcinomas (CRCs) show high rates of response to immune checkpoint inhibitors (IOs). B2M mutations and protein loss have been proposed as causes of resistance to IOs, yet they are enriched in MSI-H CRC. We aimed to characterize B2M-mutant, IO-naive CRC.
PATIENTS AND METHODS
All CRCs with results for Memorial Sloan Kettering Integrated Mutation Profiling of Actionable Cancer Targets, a next-generation sequencing assay that interrogates > 400 genes for mutations as well as MSI status, were surveyed for B2M mutations. All B2M-mutant CRCs were assessed for expression of B2M, major histocompatibility complex class I, and programmed death-1 ligand (PD-L1) via immunohistochemistry and average CD3+ and CD8+ tumor-infiltrating lymphocyte counts against a control group of MSI-H B2M wild-type CRCs.
RESULTS
Fifty-nine (3.4%) of 1,751 patients with CRC harbored B2M mutations, with 84% (77 of 92) of the mutations predicted to be truncating. B2M mutations were significantly enriched in MSI-H CRCs, with 44 (24%) of 182 MSI-H CRCs harboring B2M mutations (P < .001). Thirty-two of 44 B2M-mutant CRCs with available material (73%) had complete loss of B2M expression, whereas all 26 CRCs with wild-type B2M retained expression (P < .001). B2M mutation status was not associated with major histocompatibility complex class I expression, KRAS or BRAF mutation, tumor-infiltrating lymphocyte level, or PD-L1 expression after adjustment for MSI status. Of 13 patients with B2M-mutant CRC who received programmed death-1 or PD-L1 IOs, 11 (85%) achieved clinical benefit, defined as stable disease or partial response using Response Evaluation Criteria in Solid Tumors criteria.
CONCLUSION
B2M mutations occur in approximately 24% of MSI-H CRCs and are usually associated with loss of B2M expression. Most patients with B2M-mutant MSI-H CRC with loss of protein expression obtain clinical benefit from IOs.
INTRODUCTION
The B2M gene encodes the protein β2-microglobulin, an extracellular component of major histocompatibility complex (MHC) class I molecules that is present on every nucleated cell in the human body. MHC class I molecules are important for immune system self-recognition. B2M-deficient mice have decreased CD8+ lymphocytes and are susceptible to intracellular pathogens.1,2 With regard to cancer, acquired B2M mutations and loss of B2M expression have been implicated as causes of acquired resistance to immunotherapy in melanoma.3 B2M mutations in immunotherapy-naive colorectal carcinoma (CRC) have recently been implicated as a cause of primary resistance in this disease.4,5
Recently, microsatellite instability-high (MSI-H) CRC has been found to have both high rates of response to immunotherapy6-9 and, interestingly, frequent truncating B2M mutations.10 Here, we sought to define the relationship of B2M mutations in CRC with expression of B2M and MHC class I expression, immunotherapy response, tumor-infiltrating lymphocytes (TILs), and molecular correlates.
PATIENTS AND METHODS
Molecular Analysis
Patients with CRC whose tumors were analyzed using the Memorial Sloan Kettering Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT) assay (a clinically validated, US Food and Drug Administration–cleared, next-generation sequencing assay that interrogates > 400 genes for mutations, copy-number changes, structural variants, and MSI)11-13 between January 1, 2014, and October 31, 2017, were included for B2M, programmed death-1 ligand (PD-L1), MHC, CD3, and CD8 immunohistochemistry (IHC) and molecular analyses. Nonsilent mutations in all coding regions as well as intronic mutations that might disrupt splice sites (up to two base pairs after exon-intron boundary) in B2M were recorded. Presence of loss of heterozygosity (LOH) was assessed via allele-specific copy-number analysis using the Fraction and Allele-Specific Copy Number Estimates from Tumor Sequencing (FACETS) algorithm14 and, in cases of low tumor content, via comparison of B2M mutation variant allele frequency against median variant allele frequency. Clonality of B2M mutations was assessed by calculating the cancer-cell fraction harboring the mutations using FACETS.14 B2M-mutated patient cases were considered clonal if the upper bound of the cancer-cell fraction was > 0.8. Clinical parameters, including primate site (right, cecum to splenic flexure; left, descending colon to rectum), stage at diagnosis, date of distant metastasis, and overall survival (OS), were recorded.
Clinical Response to Immune Checkpoint Inhibitor Therapy
All patients with CRC with MSK-IMPACT data and B2M mutations who underwent therapy with immune checkpoint inhibitors (IOs; durvalumab, nivolumab, or pembrolizumab) before July 2018 were assessed for B2M expression (IHC), response, stable disease (SD), and progressive disease (PD). IOs were administered as standard treatment, in clinical trials, or off label. Formal Response Evaluation Criteria in Solid Tumors (RECIST) scores were assessed via radiologic data as follows: complete response (CR), disappearance of all target lesions, confirmed at 4 weeks; partial response (PR), ≥ 30% decrease, confirmed at 4 weeks; PD, ≥ 20% increase over smallest sum observed; and SD, meeting none of the other criteria. Patients were deemed to have experienced clinical benefit from IOs if RECIST results were SD, PR, or CR.
IHC
IHC staining for B2M using a polyclonal antibody with concentration of 1:6,000 (catalog #A0072; Dako, Santa Clara, CA), MHC class I using a monoclonal antibody with concentration of 1:200 (catalog #14-9958; E-Bioscience, Carlsbad, CA), CD3 using a monoclonal antibody with concentration of 1:200 (catalog #NCL-L-CD3-565; Leica, Lincolnshire, IL), CD8 using a monoclonal antibody with concentration of 1:100 (catalog #M7103; Dako), and PD-L1 using a monoclonal antibody with concentration of 1:100 (catalog #13684; Cell Signaling, Danvers, MA) was performed on all CRCs with B2M mutations with available tissue as well as a set of 26 randomly selected wild-type (WT) CRCs with in-house resection specimens and MSK-IMPACT testing (performed between January 1, 2014, and October 31, 2017) that were matched to the B2M-mutant group for prevalence of MSI status. Levels of CD3+ and CD8+ TILs were assessed via the average of five counted fields per patient case at ×400 original magnification on light microscopy. IHC-positive cells were counted up to a maximum of 150 cells, because counts > 150 per high-powered field (HPF) tended to have clustering, which led to difficulty establishing accurate counts. B2M and MHC class I expression were each recorded as retained or lost for each patient case. Complete loss of B2M on IHC (0% of tumor cells with B2M expression) was interpreted as loss of B2M expression.
Statistical Analyses
Associations were assessed using Pearson’s χ2 test with simulated P value based on 2,000 replicates for low count data. A Cox proportional hazards model was fitted to the data to calculate survival using the covariates of B2M mutation status, age at diagnosis, pathologic stage, MSI status, proximal versus distal status, and KRAS, NRAS, BRAF, and PIK3CA mutation status. These were each assessed through both univariable and multivariable Cox regressions. R survival and survminer software packages were used to perform this analysis (R Foundation, Vienna, Austria).
RESULTS
Molecular Findings
We first sought to determine the spectrum of B2M mutations in a cohort of patients with CRC (n = 1,751) with MSK-IMPACT data (Appendix Fig A1). We identified a total of 59 patients with B2M-mutant CRC (3.4%; Fig 1A). Although most B2M mutations were spread throughout the gene, four positions were recurrently altered (Fig 1A): p. L15Ffs*41 (CT dinucleotide repeat ×4), p. X23_splice (c. 68-2A>G), p. V69Wfs*34 (A mononucleotide repeat ×5), and p. T93Hfs*2/Lfs*10 (C mononucleotide repeat ×5). Three of these four hotspots occurred at coding microsatellites (Fig 1A). Next, we classified the samples on the basis of whether they were microsatellite stable (MSS) or unstable (MSI-H) on the basis of genomic data. We identified 182 patients who were MSI-H and 1,569 who were MSS in the overall CRC cohort. Within the MSI-H group, 44 (24.2%) harbored B2M mutations, whereas only 15 (0.9%) of those with MSS CRC harbored B2M mutations, indicating that B2M mutations were significantly enriched in MSI-H CRC (P < .001) even after correcting for differences in total mutation counts in MSI-H versus MSS patient cases. Furthermore, of 8,790 coding microsatellites interrogated within the MSK-IMPACT panel, the B2M p. L15 and p. V69 microsatellites were respectively the ninth and 16th most frequently mutated coding microsatellites in MSI-H CRC. Within the 44 MSI-H B2M-mutant samples, we identified a total of 69 mutations, 61 (88.4%) of which were predicted to be truncating (including frameshift, splicing, and stop-gain events), compared with 16 of the 23 B2M mutations in MSS patient cases (69.6%; P = .03). Of 92 total B2M mutations, 49 were frameshift, 16 were nonsense, 15 were missense, nine were splice-site, and three were translation initiation codon mutations. KRAS and BRAF p. V600E mutations were not significantly associated with B2M mutation status after adjustment for MSI status (Table 1).
FIG 1.
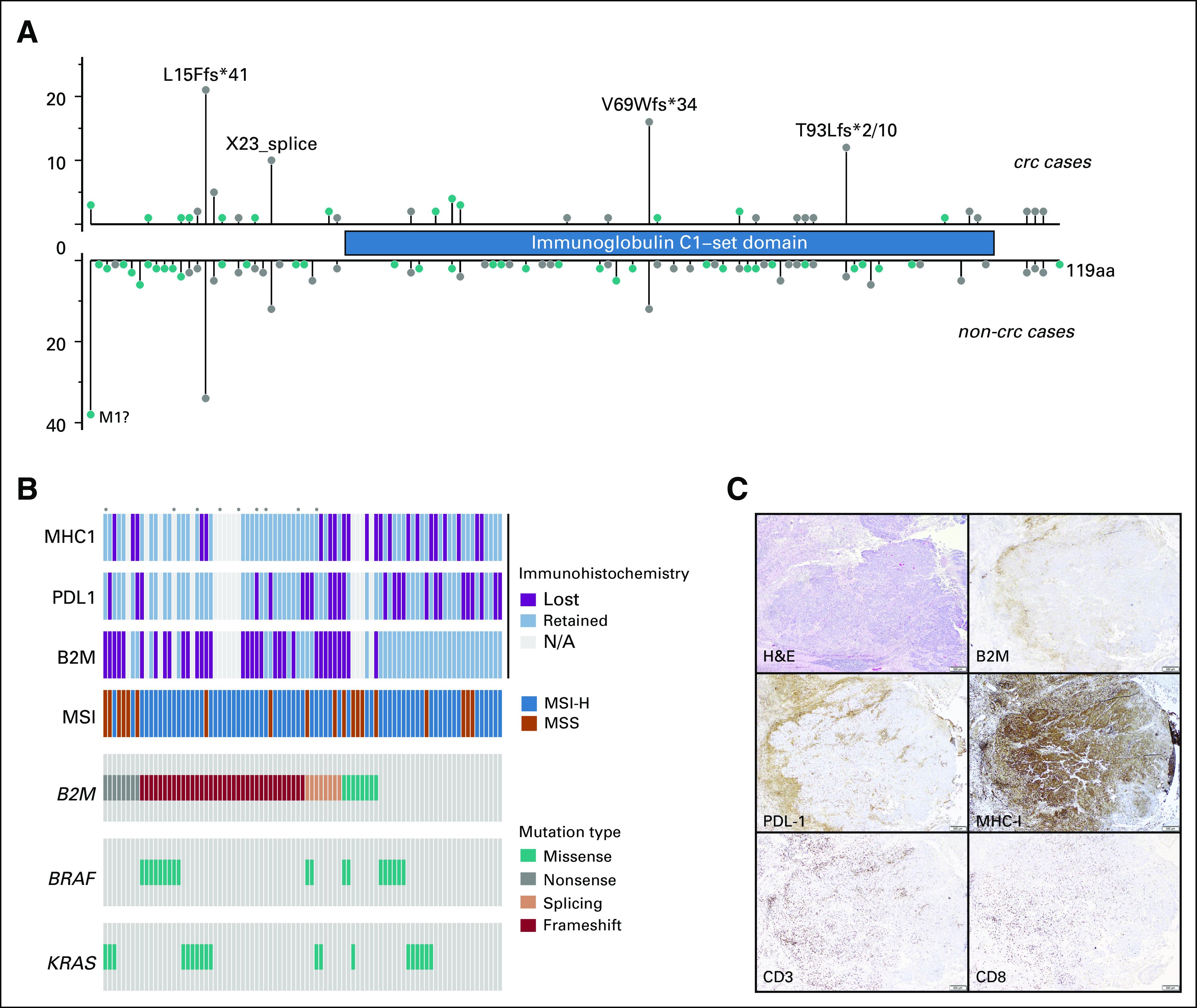
Spectrum of B2M mutations and expression. (A) Fifty-nine patients with colorectal cancer (CRC) harbored B2M mutations. Although several scattered missense (green) mutations were seen, truncating (gray) mutations were more frequent at several hotspots. These included microsatellite loci: 21 mutations at p. L15Ffs*41, 16 at p. V69Wfs*34, 11 at p.T93Hfs*2/Lfs*10, and 10 at the splice site p.X23. (B) Oncoprint summarizing the immunopathologic data with genomic information. Each dot above the sample indicates the patient was treated with a checkpoint inhibitor. (C) Loss of B2M expression and retained major histocompatibility complex (MHC) class I expression in a microsatellite instability-high B2M double-mutant (p. V69Wfs*34, p. S16Afs*27) CRC with high tumor-infiltrating lymphocyte level. A medullary carcinoma of the colon shows immune-cell expression of programmed death-1 ligand at the tumor-stroma interface, loss of B2M expression in tumor cells, retention of strong diffuse MHC class I expression in tumor cells, > 150 CD3+ lymphocytes per high-powered field (HPF), and average of 69 CD8+ cells per HPF.
TABLE 1.
Clinical and Molecular Characteristics of B2M Mutations in CRC

We performed allele-specific copy-number analysis on 45 B2M-mutant specimens where sufficient tumor content was available. Forty-one of 45 B2M-mutant CRCs had at least one clonal B2M mutation on the basis of FACETS analysis (12 samples with one clonal mutation and 29 with > one; Appendix Table A1). Twelve samples showed either LOH or copy-neutral LOH along with a clonal mutation, suggesting biallelic loss.
B2M Expression, MHC Class I Expression, and TIL Level
To evaluate the functional outcome of B2M mutations, we examined protein expression in samples with available tissue (Figs 1B and 1C; Appendix Table A2). Thirty-two (73%) of 44 B2M-mutated CRCs with available tissue had complete loss of B2M expression, whereas the remaining 12 B2M-mutant CRCs had varying proportions of tumor cells expressing B2M (20%, n = 2; 30%, n = 1; 40%, n = 1; 50%, n = 3; and 100%, n = 5). All 26 B2M WT CRCs retained B2M expression. Loss of B2M expression was significantly associated with B2M mutation in immunotherapy-naive CRC (P = .001).
Because B2M protein is an essential part of the MHC complex, we explored the effects of B2M loss on MHC class I expression. MHC class I IHC was performed in 44 B2M-mutated CRCs, of which only 14 (30%) had loss of MHC expression. Of the 26 B2M WT CRCs, 10 (39%) had MHC class I loss. MHC class I expression by IHC did not correlate with either B2M mutation or B2M expression. Because PD-L1 status has been used as a predictive marker of IOs and linked to expression of MHC class 1, we performed PD-L1 IHC in available B2M-mutant and WT patient cases. Thirty-two (73%) of 44 B2M-mutant and 13 (50%) of 26 B2M WT CRCs were positive for PD-L1 expression (immune cells in tumor-stroma interface). The difference in PD-L1 expression between the two groups did not reach statistical significance (P = .07).
TIL level has been directly correlated with prognosis and implicated as a marker of neoantigen level and potential immunotherapy response.15 We examined the number of TILs using the pan–T-cell marker CD3 and the cytotoxic T-cell marker CD8. Average median CD3+ count per HPF was 22.3 in B2M-mutant and 37.3 in B2M WT CRCs, whereas average median CD8+ count per HPF was 15.1 and 49 for B2M-mutant and B2M WT CRCs, respectively. In comparison with B2M WT CRCs matched for MSI status, B2M-mutant CRCs tended to have lower average levels of CD3 and CD8 per patient case (Fig 2A), but these differences did not reach statistical significance. These findings are summarized in Appendix Table A2.
FIG 2.

Comparison of tumor-infiltrating lymphocyte (TIL) levels and predictors of overall survival (OS). (A) Box plot graphs of average CD3 (left) and CD8 (right) counts in B2M-mutant vs wild-type (WT) colorectal carcinoma (CRC) show that median average CD3 and CD8 trend toward higher values in B2M WT carcinoma. (B) Multivariable model showing the clinical variables associated with OS of patients with CRC in our cohort. Error bars indicate 95% CI for hazard ratios. MSI-H, microsatellite instability-high. Asterisks indicate level of significance.
Clinical Findings
Median age, male-to-female ratio, and percentage of patients with early-stage (stage I to III) disease were 58.3 years, 1.36, and 77.19% in B2M-mutant and 55.3 years, 1.18, and 39.52% in B2M WT CRCs, respectively. On multivariable analysis, B2M mutation status was not associated with OS from date of metastasis (P = .90). Age, stage, and KRAS, NRAS, and BRAF status were associated with OS, whereas PIK3CA mutation status, MSI status, and proximal versus distal location were not associated with OS (Fig 2B; Appendix Table A2).
Response to Immunotherapy
Because B2M mutations were identified as a possible resistance mechanism for checkpoint inhibition, we identified 13 (MSI-H, n = 11; MSS, n = 2) patients with CRC with B2M mutations who subsequently received IO therapy, and we evaluated their response to treatment. This group of patients predominantly consisted of those with MSI-H tumors but included two patients with MSS disease. One patient’s tumor was hypermutated because of an exonucelase domain mutation in the POLE gene. IOs administered to patients included PD-L1 inhibitors (n = 3 patients) and programmed death-1 inhibitors (n = 10 patients). Because some patients received treatment in ongoing clinical trials, specific IOs are not listed. Analysis of their tumor response by RECIST criteria demonstrated PR in five patients, SD in six patients, and PD in one patient, likely pseudoprogression (Fig 3A; Appendix Table A3). B2M mutation was biallelic in half the patients and did not lead to a significant difference in response to treatment. One patient experienced progression without radiologic studies, and RECIST evaluation could not be completed. This response rate is in line with recent publications of IOs in MSI-H CRC.8,9 Of the two patients with MSS tumors, one had an ultramutated POLE hotspot–mutated tumor and experienced tumor growth with IO treatment but was able to continue treatment for 1 year, because growth was thought likely to be pseudoprogression, and the other patient had SD during treatment with a combination of IO treatment and targeted therapy (after progression with targeted therapy alone). All patients except this MSS patient were treated with immunotherapy alone. Median treatment time was 5 months (Fig 3B); three of the 12 patients stopped treatment because of toxicity.
FIG 3.

Immune checkpoint inhibitor (IO) response in B2M-mutant colorectal cancer (CRC). Waterfall plot of IO response in B2M-mutant CRC (colored by microsatellite instability [MSI] status, type of B2M mutation, and type of IO received). (B) Duration of treatment, with different colors indicating if the patient is still undergoing treatment or has stopped receiving IOs. Patient P-0025883 was not included in this graph because of nonmeasurable disease. MSI-H, MSI-high; MSS, microsatellite stable; PD-1, programmed death-1; PD-L1, PD-1 ligand.
DISCUSSION
In this study, we show that B2M mutations occurred in approximately 24% of immunotherapy-naive MSI-H CRCs and were associated with B2M loss in 93% of patient cases (usually without loss of MHC class I), and 85% of B2M-mutant CRCs demonstrated some clinical benefit from IOs. As IO therapy becomes standard treatment in advanced MSI-H CRC, the importance of identifying additional predictors of response and resistance to checkpoint inhibitors has also grown.
Our molecular findings, including enrichment of B2M mutations in MSI-H CRC and B2M mutations occurring at certain hotspots, are consistent with previous reports. Both findings likely result from the fact that there are several coding mono- and dinucleotide microsatellites within B2M. In addition, B2M mutations may confer a growth advantage for MSI-H CRC tumors, as suggested by the facts that B2M mutations were statically significantly enriched in MSI-H CRC after adjustment for total mutation count and that two coding microsatellites in B2M are within the top 20 most frequently mutated coding microsatellites of 8,790 microsatellites assessed. That B2M mutations in CRC are usually associated with loss of B2M expression indicates that a second hit or LOH occurs. The association of B2M mutations with complete loss of expression on whole sections of tumor also argues against the idea that mutation subclonality may be responsible for response to immunotherapy. Indeed, in the 42 B2M-mutant CRCs analyzed by FACETS, 60% had evidence of either two clonal B2M mutations or one clonal B2M mutation and LOH (Appendix Table A1). It is possible that the remaining B2M-mutant CRCs have epigenetic modifications as a mechanism of B2M silencing.
Unlike previous studies,3 we found that B2M protein loss is not correlated with loss of MHC class I expression. Although previous studies have focused on patients with resistance to immunotherapy, the patients in this study were immunotherapy naive. We show that B2M mutation and loss do not correlate with MHC class I loss of expression, although the effect of B2M loss on the functional competence of MHC class I is known to be deleterious.16
Limitations to our study include the retrospective analysis and relatively small number of patients treated with IOs, as well as limitations in molecular testing for epigenetic issues and allele specificity of B2M mutations.
Most importantly, our study shows that B2M mutation and loss in immunotherapy-naive CRC do not predict primary resistance to IOs. We focused on whether patients receiving IOs can benefit from treatment if their tumors have B2M mutations and protein loss. We saw that most patients had some degree of regression with treatment; larger, prospective studies are needed to clarify if the response rate and duration of response vary by B2M mutation status. However, our data indicate that patients with CRC whose tumors harbor B2M mutations should not be excluded from IO treatment. Giannakis et al15 have shown that TIL level predicts neoantigen load. The clinical benefit demonstrated in patients with B2M-deficient CRC who received IOs may have resulted from the fact that despite B2M loss, there was still evidence of functional neoantigen recognition, as indicated by the high number of TILs (median, 22.3 per HPF) still present in B2M-deficient MSI-H CRC. Thus, B2M loss may not be sufficient to lead to primary resistance to immunotherapy in MSI-H CRC.
Appendix
FIG A1.

CONSORT diagram. Mutation and immunohistochemistry analyses included data from 1,751 patients with colorectal cancer (CRC) with Memorial Sloan Kettering Integrated Mutation Profiling of Actionable Cancer Targets testing. Fifteen of 1,569 microsatellite stable (MSS) CRCs harbored B2M mutations, whereas 138 of 182 microsatellite instability-high (MSI-H) CRCs harbored B2M mutations. WT, wild type.
TABLE A1.
Predicted B2M Mutation Clonality by Cellularity-Corrected Allele Fraction via FACETS and Corresponding Protein Expression (IHC)

TABLE A2.
IHC Expression of Immune and MHC Markers in B2M-Mutant Versus WT CRC

TABLE A3.
Response Data of Patients With B2M-Mutant CRC Who Received IOs

Footnotes
Supported by the National Cancer Institute under Memorial Sloan Kettering Cancer Center Support Grant/Core Grant No. P30 CA008748.
AUTHOR CONTRIBUTIONS
Conception and design: Sumit Middha, Marc Ladanyi, Ahmet Zehir, Jaclyn F. Hechtman
Collection and assembly of data: Sumit Middha, Rona Yaeger, Jinru Shia, Sarah King, Shanna Guercio, Victoriya Paroder, David D.B. Bates, Neil Segal, Ahmet Zehir, Jaclyn F. Hechtman
Data analysis and interpretation: Sumit Middha, Rona Yaeger, Jinru Shia, Zsofia K. Stadler, Victoriya Paroder, Satshil Rana, Luis A. Diaz Jr, Leonard Saltz, Marc Ladanyi, Ahmet Zehir, Jaclyn F. Hechtman
Manuscript writing: All authors
Final approval of manuscript: All authors
AUTHORS’ DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST AND DATA AVAILABILITY STATEMENT
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Rona Yaeger
Research Funding: Array BioPharma, GlaxoSmithKline, Novartis
Travel, Accommodations, Expenses: Array BioPharma
Zsofia K. Stadler
Consulting or Advisory Role: Allergan (I), Genentech/Roche (I), Regeneron (I), Optos (I), Adverum (I), Biomarin (I), Alimera Sciences (I), Novartis (I), Spark Therapeutics (I), Fortress Biotech (I), Regenxbio (I)
Luis A. Diaz Jr
Leadership: Personal Genome Diagnostics, Jounce Therapeutics
Stock and Other Ownership Interests: PapGene, Personal Genome Diagnostics, Jounce Therapeutics, Zydecom
Consulting or Advisory Role: Merck, Personal Genome Diagnostics, Cell Design Labs, Phoremost, Lyndra, Caris Life Sciences, Genocea Biosciences, Zydecom
Research Funding: Merck (Inst)
Patents, Royalties, Other Intellectual Property: US-2010041048-A1 Circulating mutant DNA to assess tumor dynamics; US-2015344970-A1 personalized tumor biomarkers; WO-2010118016-A2 digital quantification of DNA methylation; US-2005202465-A1 thymidylate synthase gene and metastasis; US-2014227271-A1 somatic mutations in ATRX in brain cancer; WO-2012094401-A2 genes frequently altered in pancreatic neuroendocrine tumors; US-2013323167-A1 detecting and treating solid tumors through selective disruption of tumor vasculature; EP-2912468-B1 Papanicolaou test for ovarian and endometrial cancers; US-9976184-B2 mutations in pancreatic neoplasms; US-2017267760-A1 checkpoint blockade and microsatellite instability; US-2018171413-A1 head and neck squamous cell carcinoma assays; US-2018086832-A1 HLA-restricted epitopes encoded by somatically mutated genes; US-2018258490-A1 assaying ovarian cyst fluid; US-2016208340-A1 TERT promoter mutations in urothelial neoplasia; US-2015252415-A1 Arid1b and neuroblastoma; WO-2018071796-A2 compositions and methods for identifying functional antitumor T-cell responses; EP-3322824-A1 detection of tumor-derived DNA in cerebrospinal fluid; US-2016273049-A1 systems and methods for analyzing nucleic acid (Inst); US-2018135044-A1 nonunique barcodes in a genotyping assay (Inst); US-2017016075-A1 neoantigen analysis (Inst)
Travel, Accommodations, Expenses: Merck
Leonard Saltz
Consulting or Advisory Role: McNeil (I)
Research Funding: Taiho Pharmaceutical
Neil Segal
Consulting or Advisory Role: Bristol-Myers Squibb, Pfizer, AstraZeneca/MedImmune, Imugene, Roche/Genentech, Pieris Pharmaceuticals, Synlogic, Aduro Biotech, Kyn Therapeutics, Boehringer Ingelheim, Merck, Puretech, Horizon Pharma, EMD Serono, Gritstone Oncology, Chugai Pharma, TRM Oncology, IFM Therapeutics, PsiOxus Therapeutics
Research Funding: MedImmune, Bristol-Myers Squibb, Pfizer, Roche/Genentech, Merck, Incyte
Marc Ladanyi
Honoraria: Merck (I)
Consulting or Advisory Role: National Comprehensive Cancer Network/AstraZeneca Tagrisso RFP Advisory Committee, Takeda Pharmaceuticals, Bristol-Myers Squibb, Bayer HealthCare Pharmaceuticals, Merck (I)
Research Funding: Loxo (Inst), Helsinn Therapeutics
Ahmet Zehir
No relationship to disclose
Jaclyn F. Hechtman
Honoraria: Medscape
Consulting or Advisory Role: Navigant Consulting, Axiom Biotechnologies
Research Funding: Bayer
No other potential conflicts of interest were reported.
REFERENCES
- 1.Roberts AD, Ordway DJ, Orme IM. Listeria monocytogenes infection in beta 2 microglobulin-deficient mice. Infect Immun. 1993;61:1113–1116. doi: 10.1128/iai.61.3.1113-1116.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schaible UE, Collins HL, Priem F, et al. Correction of the iron overload defect in beta-2-microglobulin knockout mice by lactoferrin abolishes their increased susceptibility to tuberculosis. J Exp Med. 2002;196:1507–1513. doi: 10.1084/jem.20020897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sade-Feldman M, Jiao YJ, Chen JH, et al. Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat Commun. 2017;8:1136. doi: 10.1038/s41467-017-01062-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Janikovits J, Müller M, Krzykalla J, et al. High numbers of PDCD1 (PD-1)-positive T cells and B2M mutations in microsatellite-unstable colorectal cancer. OncoImmunology. 2017;7:e1390640. doi: 10.1080/2162402X.2017.1390640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Grasso CS, Giannakis M, Wells DK, et al. Genetic mechanisms of immune evasion in colorectal cancer. Cancer Discov. 2018;8:730–749. doi: 10.1158/2159-8290.CD-17-1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Le DT, Uram JN, Wang H, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;372:2509–2520. doi: 10.1056/NEJMoa1500596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Riaz N, Havel JJ, Makarov V, et al. Tumor and microenvironment evolution during immunotherapy with nivolumab. Cell. 2017;171:934–949.e16. doi: 10.1016/j.cell.2017.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Le DT, Durham JN, Smith KN, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science. 2017;357:409–413. doi: 10.1126/science.aan6733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Overman MJ, McDermott R, Leach JL, et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): An open-label, multicentre, phase 2 study. Lancet Oncol. 2017;18:1182–1191. doi: 10.1016/S1470-2045(17)30422-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Clendenning M, Huang A, Jayasekara H, et al. Somatic mutations of the coding microsatellites within the beta-2-microglobulin gene in mismatch repair-deficient colorectal cancers and adenomas. Fam Cancer. 2018;17:91–100. doi: 10.1007/s10689-017-0013-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zehir A, Benayed R, Shah RH, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med. 2017;23:703–713. doi: 10.1038/nm.4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cheng DT, Mitchell TN, Zehir A, et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A hybridization capture-based next-generation sequencing clinical assay for solid tumor molecular oncology. J Mol Diagn. 2015;17:251–264. doi: 10.1016/j.jmoldx.2014.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Middha S, Zhang L, Naka K et al: Reliable pan-cancer microsatellite instability assessment by using targeted next-generation sequencing data. JCO Precis Oncol 10.1200/PO.17.00084, 2017. [DOI] [PMC free article] [PubMed]
- 14.Shen R, Seshan VE. FACETS: allele-specific copy number and clonal heterogeneity analysis tool for high-throughput DNA sequencing. Nucleic Acids Res. 2016;44:e131. doi: 10.1093/nar/gkw520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. doi: 10.1016/j.celrep.2016.03.075. Giannakis M, Mu XJ, Shukla SA, et al: Genomic correlates of immune-cell infiltrates in colorectal carcinoma. Cell Reports 15:857-865, 2016 [Erratum: Cell Rep 17:1206, 2016] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hulpke S, Tampé R. The MHC I loading complex: A multitasking machinery in adaptive immunity. Trends Biochem Sci. 2013;38:412–420. doi: 10.1016/j.tibs.2013.06.003. [DOI] [PubMed] [Google Scholar]