

Abstract

Engineered biomedical nanoparticles (NPs) administered via intravenous routes are prone to associate to serum proteins. The protein corona can mask the NP surface functionalization and hamper the delivery of the NP to its biological target. The design of corona-free NPs relies on our understanding of the chemical-physical features of the NP surface driving the interaction with serum proteins. Here, we address, by computational means, the interaction between human serum albumin (HSA) and a prototypical monolayer-protected Au nanoparticle. We show that both the chemical composition (charge, hydrophobicity) and the conformational preferences of the ligands decorating the NP surface affect the NP propensity to bind HSA.
Introduction
Nanoparticles (NPs) designed to be administered via intravenous routes are prone to interact with serum proteins, which can stably cluster around the nanoparticle forming a protein corona.1−4 The nonspecific adsorption of proteins on NPs alters their designed function and influences their fate in the body.5−7 The control of protein adsorption8−10 and the minimization of early clearance from the bloodstream are crucial to the clinical integration of synthetic nanoparticles.6 Most often, inorganic NPs designed for diagnostic or therapeutic applications do not expose their bare surface to the biological environment but are functionalized by organic ligands that provide better solubility and specific targeting properties. The density, length, charge, and hydrophobicity of the NP ligands determine the amount and type of proteins that bind to the NP,8,11,12 as well as the reversibility of binding.13−16
One possible route to act on the NP–protein interaction, in the direction of reducing nonspecific adsorption, involves the functionalization of the NPs with proper antifouling functional groups. Poly(ethylene glycol) (PEG) is known to be a good antifouling material,17 and a consistent body of literature has shed light on its action as a stealth agent. Protein-repellent properties of PEG grafted on surfaces are influenced by PEG chain length,18 density, and environment temperature,19,20 although the amount of adsorbed proteins is not always a monotonic function of these parameters.21 The use of PEG as a stealth agent also has some drawbacks, such as its non-biodegradability, immunogenicity,22 and its accumulation in membrane-bound organelles.23 An alternative to PEG is represented by ligands terminated by zwitterionic moieties, which further reduce nonspecific protein adsorption.13,22,24 Zwitterionic groups can thus extend the circulation time of the NPs and increase their ability to effectively penetrate cell membranes.5
The many physical and chemical parameters that characterize the NP–protein interface, on both sides, make it difficult to identify clear correlations between the composition of the NP surface and the composition and stability of the protein corona. The computational approach can contribute to shed light on which factors, on the molecular scale, determine the formation of stable coronas. Molecular simulations face important limitations, although as the corona formation is a process that spans timescales of seconds, the relevant NP sizes for biomedical applications range from a few to hundreds of nanometer, whereas the thickness of the protein corona on metal or metal oxide NPs varies from 20 to 40 nm.8 The simulation, at atomistic or molecularly detailed coarse-grained (CG) resolution and with explicit solvent, of a whole NP + corona complex is still out of reach for current computational resources. The use of implicit solvent schemes has allowed for the simulation of the corona formation on top of model spherical NPs.25,26 Several attempts have been made at the simulation of the interaction of a single NP with a single protein. This has most often required to give away significant details of the NP–protein interface. NPs are often modeled as flat surfaces27 or smooth spherical objects, offering a generic hydrophobic, hydrophilic, or charged surface to the protein.28−30 Proteins, as well, may be treated as rigid bodies29 or polymers with no secondary structure.30
Here, we use molecular dynamics (MD), at coarse-grained (CG) resolution, to investigate the interplay of electrostatics, hydrophobicity, and ligand conformation at shaping NP–protein interactions. Our model combines an atomistic description of the Au core31 to a coarse-grained, explicit solvent model of the rest of the system. The coarse-grained description has submolecular resolution, and it takes into account explicitly the composition of the NP ligand shell, its flexibility, and protein flexibility. We simulate the interaction between human serum albumin (HSA) and monolayer-protected Au NPs. HSA accounts for more than half of the serum proteins in human blood plasma,32 and it is one of the most abundant components of the corona formed around nanoparticles11,12 and specifically ligand-protected Au NPs.14 The Au NPs we consider have the same composition and size as those of NPs synthesized by Moyano et al.13 The Au core has a small diameter of 2 nm (4 nm in the experiments by Moyano et al.13). The Au surface is covalently functionalized by ligands that are terminated by a zwitterionic group and, at the same time, have tunable hydrophobicity. This ligand composition offers the opportunity to monitor the influence of electrostatic and hydrophobic interactions at shaping the NP–protein interaction.
Methods
As a first step, we developed a coarse-grained model of HSA in the framework of the polarizable-water Martini coarse-grained model, which allows realistic large-scale simulations of proteins33−35 and nanoparticles.36,37 The Martini force field does not allow for changes of the protein secondary structure, which is imposed by means of an elastic network connecting the CG beads that are placed on top of the Cα atoms.38 This description of HSA is compatible with the indication that the secondary structure of HSA does not change upon binding to Au surfaces27 and nanoparticles such as fullerenes39 and Au nanoparticles in the 4–40 nm range.14,40 The development of the CG model was based on structural and dynamic parameters obtained from atomistic simulations carried out with the Amberff99SB-ILDN force field.41 Several CG models were tested, with different parameters defining the elastic network. We considered two structural parameters (the root-mean-square deviation (RMSD) of α carbon atoms and the per-residue root-mean-square fluctuation (RMSF)). We also performed principal component analysis (PCA) to quantify the superposition of principal components (PCs) in atomistic and CG simulations. Finally, we selected the CG model most similar to the all-atom model in terms of RMSD and RMSF, with the highest overlap in PCA. All details about the model development are reported in the Supporting Information.
To test the influence of hydrophobicity on the interaction between zwitterionic NPs and HSA, we tested two different NP models. The two NPs have an identical core of 144 Au atoms and differ only for the composition of their 60 ligands (Figure 1). The least hydrophobic NP, referred to as Z, has ligands composed by a short hydrophobic stretch, a sequence of four monomers of PEG, and a zwitterionic sulfobetaine terminal. The most hydrophobic NP, referred to as ZH, has identical ligands except for two additional hydrophobic branches stemming from the zwitterionic terminal group. The details of ligand parameterization are reported in the Supporting Information.
Figure 1.

(a) HSA. On the left, the secondary structure of the protein; on the right, the protein surface colored according to hydrophobicity (hydrophobic residues in blue and charged or polar residues in green); (b) the ligand-protected NP, with gray Au core and S atoms, and Z ligands. (c) Chemical composition of the Z and ZH ligands of the NP. (d) The Z and ZH ligands as represented by the CG model; C1, Q0, and Qda refer to nonbonded types of the Martini force field;42 poly(ethylene oxide) is the Martini type defined in Lee et al.43
Results and Discussion
We characterized the NP–HSA
interaction by means of unbiased
MD runs in which a single NP was allowed to interact with a single
HSA protein. We performed 20 runs at physiological conditions (310
K, atmospheric pressure) with a time step of 20 ns for a total simulated
time of 60 μs for each NP type. All simulations were run with
the GROMACS 5 package. More details on the MD settings can be found
in the Supporting Information. Both NPs
are found to establish transient contacts with the protein. To quantify
the number and temporal stability of NP–protein contacts, we
consider the NP and the protein to be in contact when at least two
of their CG beads are closer than a threshold distance of 0.8 nm.
The ZH NP resides on HSA surface for longer stretches of time (see Figure 2) compared with the
Z NP. For the ZH NP, the total time spent in the bound state is tZHb = 23.8 μs over the simulated trun = 60 μs whereas for the Z NP, tZ = 4.9 μs
over the same trun. The free-energy difference
between the bound and unbound state can be thus estimated as 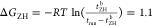 kJ/mol (0.43kBT), and
kJ/mol (0.43kBT), and 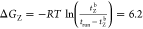 kJ/mol (2.4kBT). We
remark that these energy differences do not
refer to the binding of the NP to a specific site but take effectively
into account all binding mechanisms observed during the simulations.
kJ/mol (2.4kBT). We
remark that these energy differences do not
refer to the binding of the NP to a specific site but take effectively
into account all binding mechanisms observed during the simulations.
Figure 2.
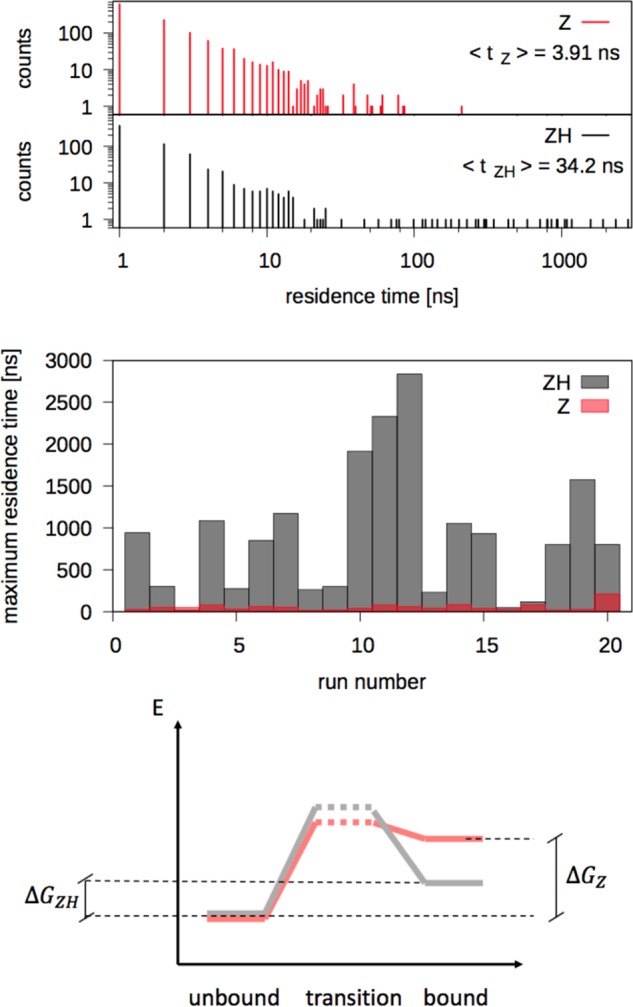
Top: distribution of NP–protein residence times for Z and ZH NPs. Center: maximum residence time during each of the 20 unbiased MD runs, for each NP type. Bottom: sketch of the free-energy barriers for binding and unbinding of ZH and Z NPs (same color code as above).
As both NPs undergo many binding and unbinding events during the simulation time, it is also possible to extract information about the effective free-energy barriers for binding and unbinding. We define the average residence time as the average time duration of a binding event. The average residence time of the ZH NP is ⟨tZH⟩ = 34.2 ± 0.3 ns, whereas for the Z NP, it is ⟨tZ⟩ = 3.91 ± 0.01 ns. On the basis of the mean residence time, we can estimate the difference, Δu‡, between the effective unbinding free-energy barriers for the two NP types
where we have indicated with ΔGiu‡ the height of the unbinding barrier for the NP of type i. Δu‡ results to be equal to 2.25kBT. As for binding, the average time spent by the two NPs in the unbound state, that is in the water phase, ⟨tw⟩, is similar: ⟨tZH⟩ = 51.6 ± 0.2 ns and ⟨tZw⟩ = 42.9 ± 0.1 ns, corresponding to a difference of 0.19kBT between the binding free-energy barriers. Figure 2 shows, in the bottom panel, a sketch of the free energy of the bound, unbound, and transition states for the two NPs.
To further probe the scarce propensity of the zwitterionic NPs to stably bind HSA, we also performed a comparison with a NP functionalized by PEG ligands, with the same density and length as those of the Z and ZH ligands. With PEG ligands, we found that the NP–HSA binding is irreversible on the simulation timescale (3 μs); the result is robust against the use of different PEG parameterizations43,44 (see Figure S1 and the Supporting Information for a detailed description of these simulations). These results are in excellent agreement with the experimental findings by Moyano et al.,13 suggesting that no hard corona is formed on the surface of 4 nm Au NPs with a zwitterionic ligand shell whereas it is formed on NPs functionalized by neutral PEG ligands.13 Moreover, the small difference in the energy barrier for the unbinding of the Z and ZH NPs is consistent with the small, reversible precipitation observed in the experiments for the most hydrophobic NPs.13
The different residence times of the Z and ZH NPs suggest that the increased binding of ZH is due to the contribution of the additional hydrophobic groups on the ZH surface. To verify this hypothesis, we analyzed in more detail the nature of the contacts between the protein and the two NPs. Figure 3 shows that the binding of the Z NP to HSA is quite uniform on the protein surface, whereas two preferential binding sites emerge from the interaction between ZH and HSA. These binding sites have different shapes (one has the form of a protrusion, and the other one, of a pocket) and contain both hydrophobic and charged residues. We classified the contacts between HSA and the NPs as hydrophobic, charged, or polar, depending on the character of the amino acid involved (details on the classification can be found in the Supporting Information). Surprisingly, hydrophobic contacts are roughly the same for Z-HSA and ZH-HSA binding (Figure 3c).
Figure 3.

(a) Protein surface colored on the basis of the average number of contacts with the ZH NP. (b) Same colormap, for the Z NP. (c) Protein surface colored on the basis of residue polarity (hydrophobic residues in red, charged in white, and polar in blue). (d) Histogram of protein–NP contacts involving hydrophobic, charged, and polar residues of HSA.
Even more surprising is the picture emerging from the classification of the NP–HSA contacts on the basis of the type of group of the NP ligand bound to the protein, as shown in Figure 4. The main difference between the two NP types is represented by the number of contacts established by the PEG segment of the ligand, significantly higher for the ZH NP. Unexpectedly, the number of hydrophobic contacts is lower for the ZH NP. Why do more hydrophobic ligands make less hydrophobic contacts with the protein? The answer is provided by the radial distribution functions of the different groups composing the NP ligands, as shown in the bottom panel of Figure 4. The terminal groups of the Z NP ligands reach out for the water phase, indicating that the ligands mainly have an extended conformation. On the contrary, the (more hydrophobic) terminal groups of the ZH ligands are found closer to the Au surface, well screened from interactions with water, indicating that the ligands mainly have a folded conformation. Such folding brings the central PEG segment of the ligand chains to the water interface, promoting PEG–HSA contacts. These data highlight that another important physical parameter affects NP–protein interaction: ligand conformation.
Figure 4.

Top: percentage of NP–protein contacts involving different segments of the NP ligands: C1 refers to the hydrophobic groups next to the S atom, PEG refers to the four PEG monomers, Q refers to the charged groups of the zwitterion, and C1T refers to the hydrophobic groups bound to the zwitterionic terminal. The inset shows the difference between the contact percentages of ZH and Z NPs, highlighting the increase of PEG–HSA contacts in the ZH case. Bottom: radial distribution function of the different chemical groups composing the NP ligands. PW stands for polarizable water.
The slight difference between the free-energy barriers for binding observed for the Z and ZH NPs can also be interpreted as a consequence of the different ligand conformations. Indeed, the conformational change induced by the presence of the C1T groups also affects the hydration of the NP. The bottom panel of Figure 4 shows the radial distribution function of water beads (PW) for Z and ZH NPs. ZH NPs, in water, are less hydrated than Z NPs45 (the time-averaged NP–water contacts of the ZH NP amount to 80% of the Z NP–water contacts). Water contacts are further reduced for the charged beads of the zwitterionic groups of ZH nanoparticles, as shown in Table 1, as a consequence of the ligand conformational change. Upon binding, it is the ZH NP that loses the largest number of water contacts, as shown in Table 1, coherently with the presence of a larger free-energy barrier for binding (see also Figure 2).
Table 1. Number of Contacts between NP Beads and Water Beads and between the Charged Beads of the Zwitterionic Groups and Water Beadsa.
| NP | NP–water no NP–protein contact | NP–water during NP–protein contact | zwitterionic group–water no NP–protein contact | zwitterionic group–water during NP–protein contact |
|---|---|---|---|---|
| Z | 4909 ± 1 | 4889 ± 5 (−20) | 1542 ± 1 | 1533 ± 2 (−9) |
| ZH | 3950 ± 1 | 3821 ± 14 (−129) | 1015 ± 1 | 985 ± 4 (−30) |
In parentheses, the difference between the number of contacts in the bound and unbound state.
Conclusions
In this work, we used coarse-grained molecular dynamics simulations with submolecular resolution to study the interaction of a monolayer-protected Au NP with the most abundant serum protein, HSA. We considered two types of NPs, functionalized by zwitterionic ligands with different degrees of hydrophobicity. Our simulations show that zwitterionic NPs have scarce propensity to form stable complexes with HSA, whereas more hydrophobic ligands interact more strongly with the protein, as measured in experiments by Moyano et al.13 The excellent agreement with the experimental data allows us to interpret the experiments at the molecular level. The ligands terminated by hydrophobic groups interact more stably with the protein not by virtue of hydrophobic interactions but because the hydrophobic moieties are folded toward the center of the NP and the PEG moieties are more exposed to the environment. NP–protein interactions, in this case, are determined by an increase of PEG–protein interactions, compatible with the formation of stable NP–protein complexes such as a hard protein corona.
Our data show that ligand conformation is as relevant as chemical affinity in determining protein–NP interactions. As a result, we propose that the design of protein-repellent NP functionalization should consider carefully the importance of both ligand conformation and ligand chemical composition. Computational models, also at coarse-grained level, are paramount for the prediction of ligand conformations relevant to the NP–protein interface, and we envision that they will contribute more in the future to quantify the relative weight of structural and chemical factors influencing NP–protein interactions.
Acknowledgments
G.R. acknowledges funding from the ERC Starting Grant BioMNP, 677513. L.M. acknowledges funding from INSERM. Calculations were carried out in part at CINES (grant A0040710138 to L.M.) and in part at CINECA (grant HP10BOWTPR to G.R.). The authors thank Elisa Frezza for assistance in PCA analysis and fruitful discussions.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jpcb.8b11204.
NP core and ligand parameterization; human serum albumin parameterization; simulation details; PEG ligands; classification of protein residues; NP–protein contacts; water–NP contacts (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Cedervall T.; Lynch I.; Lindman S.; Berggård T.; Thulin E.; Nilsson H.; Dawson K.; Linse S. Understanding the Nanoparticle-Protein Corona Using Methods to Quantify Exchange Rates and Affinities of Proteins for Nanoparticles. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 2050–2055. 10.1073/pnas.0608582104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch I.; Salvati A.; Dawson Ka. Protein-Nanoparticle Interactions: What Does the Cell See?. Nat. Nanotechnol. 2009, 4, 546–547. 10.1038/nnano.2009.248. [DOI] [PubMed] [Google Scholar]
- Ke P. C.; Lin S.; Parak W. J.; Davis T. P.; Caruso F. A Decade of the Protein Corona. ACS Nano 2017, 11773–11776. 10.1021/acsnano.7b08008. [DOI] [PubMed] [Google Scholar]
- Monopoli M. P.; Åberg C.; Salvati A.; Dawson K. A. Biomolecular Coronas Provide the Biological Identity of Nanosized Materials. Nat. Nanotechnol. 2012, 7, 779–786. 10.1038/nnano.2012.207. [DOI] [PubMed] [Google Scholar]
- Jiang Y.; Huo S.; Mizuhara T.; Das R.; Lee Y.; Hou S.; Moyano D. F.; Duncan B.; Liang X.; Rotello V. M. The Interplay of Size and Surface Functionality on the Cellular Uptake. ACS Nano 2015, 9, 9986–9993. 10.1021/acsnano.5b03521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decuzzi P. Facilitating the Clinical Integration of Nanomedicines: The Roles of Theoretical and Computational Scientists. ACS Nano 2016, 10, 8133–8138. 10.1021/acsnano.6b05536. [DOI] [PubMed] [Google Scholar]
- Melby E. S.; Lohse S. E.; Park J. E.; Vartanian A. M.; Putans R. A.; Abbott H. B.; Hamers R. J.; Murphy C. J.; Pedersen J. A. Cascading Effects of Nanoparticle Coatings: Surface Functionalization Dictates the Assemblage of Complexed Proteins and Subsequent Interaction with Model Cell Membranes. ACS Nano 2017, 11, 5489–5499. 10.1021/acsnano.7b00231. [DOI] [PubMed] [Google Scholar]
- Walkey C. D.; Chan W. C. W. Understanding and Controlling the Interaction of Nanomaterials with Proteins in a Physiological Environment. Chem. Soc. Rev. 2012, 41, 2780–2799. 10.1039/C1CS15233E. [DOI] [PubMed] [Google Scholar]
- Herda L. M.; Hristov D. R.; Giudice M. C. L.; Polo E.; Dawson K. A. Mapping of Molecular Structure of the Nanoscale Surface in Bionanoparticles. J. Am. Chem. Soc. 2017, 139, 111–114. 10.1021/jacs.6b12297. [DOI] [PubMed] [Google Scholar]
- Kelly P. M.; Åberg C.; Polo E.; O’Connell A.; Cookman J.; Fallon J.; Krpetić Ž.; Dawson K. A. Mapping Protein Binding Sites on the Biomolecular Corona of Nanoparticles. Nat. Nanotechnol. 2015, 10, 472–479. 10.1038/nnano.2015.47. [DOI] [PubMed] [Google Scholar]
- Caracciolo G.; Pozzi D.; Capriotti A. L.; Cavaliere C.; Piovesana S.; Amenitsch H.; Laganà A. Lipid Composition: A “Key Factor” for the Rational Manipulation of the Liposome – Protein Corona by Liposome Design. RSC Adv. 2015, 5967–5975. 10.1039/C4RA13335H. [DOI] [Google Scholar]
- Tenzer S.; Docter D.; Kuharev J.; Musyanovych A.; Fetz V.; Hecht R.; Schlenk F.; Fischer D.; Kiouptsi K.; Reinhardt C.; et al. Rapid Formation of Plasma Protein Corona Critically Affects Nanoparticle Pathophysiology. Nat. Nanotechnol. 2013, 8, 772–781. 10.1038/nnano.2013.181. [DOI] [PubMed] [Google Scholar]
- Moyano D. F.; Saha K.; Prakash G.; Yan B.; Kong H.; Yazdani M.; Rotello V. M. Fabrication of Corona-Free Nanoparticles with Tunable Hydrophobicity. ACS Nano 2014, 8, 6748–6755. 10.1021/nn5006478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casals E.; Pfaller T.; Duschl A.; Oostingh G. J.; Puntes V. Time Evolution of Nanoparticle Protein Corona. ACS Nano 2010, 4, 3623–3632. 10.1021/nn901372t. [DOI] [PubMed] [Google Scholar]
- Lundqvist M.; Stigler J.; Cedervall T.; Berggård T.; Flanagan M. B.; Lynch I.; Elia G.; Dawson K. The Evolution of the Protein Corona around Nanoparticles: A Test Study. ACS Nano 2011, 5, 7503–7509. 10.1021/nn202458g. [DOI] [PubMed] [Google Scholar]
- del Pino P.; Pelaz B.; Zhang Q.; Maffre P.; Nienhaus G. U.; Parak W. J. Protein Corona Formation around Nanoparticles – from the Past to the Future. Mater. Horiz. 2014, 1, 301–313. 10.1039/C3MH00106G. [DOI] [Google Scholar]
- Knop K.; Hoogenboom R.; Fischer D.; Schubert U. S. Poly(Ethylene Glycol) in Drug Delivery: Pros and Cons as Well as Potential Alternatives. Angew. Chem., Int. Ed. 2010, 49, 6288–6308. 10.1002/anie.200902672. [DOI] [PubMed] [Google Scholar]
- Norde W.; Gags D. Interaction of Bovine Serum Albumin and Human Blood Plasma with PEO-Tethered Surfaces: Influence of PEO Chain Length, Grafting Density, and Temperature. Langmuir 2004, 20, 4162–4167. 10.1021/la030417t. [DOI] [PubMed] [Google Scholar]
- Gref R.; Lück M.; Quellec P.; Marchand M.; Dellacherie E.; Harnisch S.; Blunk T.; Müller R. H. “Stealth” Corona-Core Nanoparticles Surface Modified by Polyethylene Glycol (PEG): Influences of the Corona (PEG Chain Length and Surface Density) and of the Core Composition on Phagocytic Uptake and Plasma Protein Adsorption. Colloids Surf., B 2000, 18, 301–313. 10.1016/S0927-7765(99)00156-3. [DOI] [PubMed] [Google Scholar]
- Pelaz B.; Del Pino P.; Maffre P.; Hartmann R.; Gallego M.; Rivera-Fernández S.; De La Fuente J. M.; Nienhaus G. U.; Parak W. J. Surface Functionalization of Nanoparticles with Polyethylene Glycol: Effects on Protein Adsorption and Cellular Uptake. ACS Nano 2015, 9, 6996–7008. 10.1021/acsnano.5b01326. [DOI] [PubMed] [Google Scholar]
- Bernhard C.; Roeters S. J.; Franz J.; Weidner T.; Bonn M.; Gonella G. Repelling and Ordering: The Influence of Poly(Ethylene Glycol) on Protein Adsorption. Phys. Chem. Chem. Phys. 2017, 19, 28182–28188. 10.1039/C7CP05445A. [DOI] [PubMed] [Google Scholar]
- Wang M.; Siddiqui G.; Gustafsson O. J. R.; Käkinen A.; Javed I.; Voelcker N. H.; Creek D. J.; Ke P. C.; Davis T. P. Plasma Proteome Association and Catalytic Activity of Stealth Polymer-Grafted Iron Oxide Nanoparticles. Small 2017, 13, 1–11. 10.1002/smll.201701528. [DOI] [PubMed] [Google Scholar]
- Butcher N. J.; Mortimer G. M.; Minchin R. F. Drug Delivery: Unravelling the Stealth Effect. Nat. Nanotechnol. 2016, 11, 310–311. 10.1038/nnano.2016.6. [DOI] [PubMed] [Google Scholar]
- Safavi-Sohi R.; Maghari S.; Raoufi M.; Jalali S. A.; Hajipour M. J.; Ghassempour A.; Mahmoudi M. Bypassing Protein Corona Issue on Active Targeting: Zwitterionic Coatings Dictate Specific Interactions of Targeting Moieties and Cell Receptors. ACS Appl. Mater. Interfaces 2016, 8, 22808–22818. 10.1021/acsami.6b05099. [DOI] [PubMed] [Google Scholar]
- Ding F.; Radic S.; Chen R.; Chen P.; Geitner N. K.; Brown J. M.; Ke P. C. Direct Observation of a Single Nanoparticle–Ubiquitin Corona Formation. Nanoscale 2013, 5, 9162. 10.1039/c3nr02147e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao Q.; Hall C. K. Protein Adsorption on Nanoparticles: Model Development Using Computer Simulation. J. Phys.: Condens. Matter 2016, 28, 414019 10.1088/0953-8984/28/41/414019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan S.; Gupta A.; Nandi C. K. Controlling the Fate of Protein Corona by Tuning Surface Properties of Nanoparticles. J. Phys. Chem. Lett. 2013, 4, 3747–3752. 10.1021/jz401874u. [DOI] [Google Scholar]
- Yu S.; Perálvarez-Marín A.; Minelli C.; Faraudo J.; Roig A.; Laromaine A. Albumin-Coated SPIONs: An Experimental and Theoretical Evaluation of Protein Conformation, Binding Affinity and Competition with Serum Proteins. Nanoscale 2016, 8, 14393–14405. 10.1039/C6NR01732K. [DOI] [PubMed] [Google Scholar]
- Lopez H.; Lobaskin V. Coarse-Grained Model of Adsorption of Blood Plasma Proteins onto Nanoparticles. J. Chem. Phys. 2015, 143, 243138 10.1063/1.4936908. [DOI] [PubMed] [Google Scholar]
- Ding H.-M.; Ma Y.-Q. Computer Simulation of the Role of Protein Corona in Cellular Delivery of Nanoparticles. Biomaterials 2014, 35, 8703–8710. 10.1016/j.biomaterials.2014.06.033. [DOI] [PubMed] [Google Scholar]
- Torchi A.; Simonelli F.; Ferrando R.; Rossi G. Local Enhancement of Lipid Membrane Permeability Induced by Irradiated Gold Nanoparticles. ACS Nano 2017, 11, 12553–12561. 10.1021/acsnano.7b06690. [DOI] [PubMed] [Google Scholar]
- He X. M.; Carter D. C. Atomic Structure and Chemistry of Human Serum Albumin. Nature 1992, 358, 209–215. 10.1038/358209a0. [DOI] [PubMed] [Google Scholar]
- Monticelli L.; Kandasamy S. K.; Periole X.; Larson R. G.; Tieleman D. P.; Marrink S.-J. The MARTINI Coarse-Grained Force Field: Extension to Proteins. J. Chem. Theory Comput. 2008, 4, 819–834. 10.1021/ct700324x. [DOI] [PubMed] [Google Scholar]
- De Jong D. H.; Singh G.; Bennett W. F. D.; Arnarez C.; Wassenaar T. A.; Schäfer L. V.; Periole X.; Tieleman D. P.; Marrink S. J. Improved Parameters for the Martini Coarse-Grained Protein Force Field. J. Chem. Theory Comput. 2013, 9, 687–697. 10.1021/ct300646g. [DOI] [PubMed] [Google Scholar]
- Yesylevskyy S. O.; Schäfer L. V.; Sengupta D.; Marrink S. J. Polarizable Water Model for the Coarse-Grained MARTINI Force Field. PLoS Comput. Biol. 2010, 6, e1000810 10.1371/journal.pcbi.1000810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salassi S.; Simonelli F.; Bochicchio D.; Ferrando R.; Rossi G. Au Nanoparticles in Lipid Bilayers: A Comparison between Atomistic and Coarse-Grained Models. J. Phys. Chem. C 2017, 121, 10927–10935. 10.1021/acs.jpcc.6b12148. [DOI] [Google Scholar]
- Rossi G.; Monticelli L. Gold Nanoparticles in Model Biological Membranes: A Computational Perspective. Biochim. Biophys. Acta, Biomembr. 2016, 1858, 2380–2389. 10.1016/j.bbamem.2016.04.001. [DOI] [PubMed] [Google Scholar]
- Periole X.; Cavalli M.; Marrink S.-J.; Ceruso M. Combining an Elastic Network With a Coarse-Grained Molecular Force Field: Structure, Dynamics, and Intermolecular Recognition. J. Chem. Theory Comput. 2009, 5, 2531–2543. 10.1021/ct9002114. [DOI] [PubMed] [Google Scholar]
- Leonis G.; Avramopoulos A.; Papavasileiou K. D.; Reis H.; Steinbrecher T.; Papadopoulos M. G. A Comprehensive Computational Study of the Interaction between Human Serum Albumin and Fullerenes. J. Phys. Chem. B 2015, 119, 14971–14985. 10.1021/acs.jpcb.5b05998. [DOI] [PubMed] [Google Scholar]
- Huang R.; Carney R. P.; Ikuma K.; Stellacci F.; Lau B. L. T. Effects of Surface Compositional and Structural Heterogeneity on Nanoparticle-Protein Interactions: Different Protein Configurations. ACS Nano 2014, 8, 5402–5412. 10.1021/nn501203k. [DOI] [PubMed] [Google Scholar]
- Lindorff-Larsen K.; Piana S.; Palmo K.; Maragakis P.; Klepeis J. L.; Dror R. O.; Shaw D. E. Improved Side-Chain Torsion Potentials for the Amber Ff99SB Protein Force Field. Proteins: Struct., Funct., Bioinf. 2010, 78, 1950–1958. 10.1002/prot.22711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrink S. J.; Risselada H. J.; Yefimov S.; Tieleman D. P.; de Vries A. H. The MARTINI Force Field: Coarse Grained Model for Biomolecular Simulations. J. Phys. Chem. B 2007, 111, 7812–7824. 10.1021/jp071097f. [DOI] [PubMed] [Google Scholar]
- Lee H.; Pastor R. W. Coarse-Grained Model for PEGylated Lipids: Effect of PEGylation on the Size and Shape of Self-Assembled Structures. J. Phys. Chem. B 2011, 115, 7830–7837. 10.1021/jp2020148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grunewald F.; Rossi G.; De Vries A. H.; Marrink S. J.; Monticelli L. A Transferable MARTINI Model of Polyethylene Oxide. J. Phys. Chem. B 2018, 122, 7436–7449. 10.1021/acs.jpcb.8b04760. [DOI] [PubMed] [Google Scholar]
- Shao Q.; He Y.; White A. D.; Jiang S. Difference in Hydration between Carboxybetaine and Sulfobetaine. J. Phys. Chem. B 2010, 114, 16625–16631. 10.1021/jp107272n. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.