

Abstract
Practical deployment of cellular therapies requires effective platforms for producing clinically relevant numbers of high-quality cells. This report introduces a materials-based approach to improving activation and expansion of T cells, which are rapidly emerging as an agent for treating cancer and a range of other diseases. Electrospinning is used to create a mesh of poly(ε-caprolactone) fibers, which is used to present activating ligands to CD3 and CD28, which activate T cells for expansion. Incorporation of poly(dimethyl siloxane) elastomer into the fibers reduces substrate rigidity and enhances expansion of mixed populations of human CD4+ and CD8+ T cells. Intriguingly, this platform also rescues expansion of T cells isolated from CLL patients, which often show limited responsiveness and other features resembling exhaustion. By simplifying the process of cell expansion, compared to current bead-based platforms, and improving T cell expansion, the system introduced here may accelerate development of cellular immunotherapy.
Keywords: electrospin, elastomer, cellular therapy, T cell, mechanosensing
Cellular therapies are rapidly emerging as a robust approach to treating of a range of diseases, promising improved persistence, targeting, and efficacy over current biologic agents. In particular, T cell-based adoptive cellular therapy (ACT), combined with chimeric antigen receptor (CAR) technologies, has seen significant successes in treatment of cancer.[1] A central step in T cell ACT is the ex vivo expansion of a smaller starting population, producing therapeutically relevant numbers of cells and allow manipulations that are not practical in vivo. A leading platform for initiating this expansion, the CTS™ Dynabeads® system, consists of microscale plastic beads coated with activating antibodies to CD3 and CD28; engagement of these two receptors on the T cell by mixing them with the beads provides activating and costimulatory signals, initiating functional activation and subsequent proliferation. We described here an alternative platform that provides several advantages over contemporary systems used for T cell activation.
This new platform is based on a fibrous mesh, which, like beads, presents a large surface area for activation but is simpler to remove from the cell growth environment. In addition, this platform takes advantage of recent developments in the area of T cell mechanobiology. The ability of cells to recognize the mechanical rigidity of an underlying substrate has been established predominantly in the context of adherent cells interacting with extracellular matrix proteins. Inspired by this idea, O’Connor et al.[2] demonstrated that primary human T cells respond to the rigidity of planar substrates of poly(dimethyl siloxane) (PDMS, Sylgard 184 by Dow Corning), presenting anti-CD3 and anti-CD28 antibodies. Substrate rigidity was controlled by changing the amount of crosslinker incorporated into the material. Reducing the ratio of crosslinker:elastomer base from the standard formulation of 1:10 to 1:50 decreased the material’s Young’s modulus, E, from several MPa to tens of kPa. T cell proliferation, cytokine secretion, and expansion increased with decreasing rigidity, suggesting that softer materials can provide enhanced manufacturing of cells[3]. The technical goal of this report is thus to fabricate soft materials in a production-compatible, fibrous format to enhance expansion of human T cells.
1. Production of elastomer fibers
Electrospinning, a high-throughput fabrication process,[4] offers many capabilities that are attractive for creating elastomer fibers. However, it is not inherently compatible with PDMS due to the polymerization chemistry of this material.[5] We adapted PDMS for electrospinning by combining the uncured material with poly(ε-caprolactone) (PCL, Mn=80,000), a biocompatible polymer often used in this process.[6] Notably, PCL is stiff, with Young’s modulus ranging from tens to hundreds of MPa,[7] necessitating the inclusion of a soft material to modulate rigidity. Electrospun fibers of PDMS:PCL are smooth and morphologically similar to PCL alone, as illustrated in Figure 1A. The fibers in this example contain S184 prepared at the conventional crosslinker:base ratio of 1:10 (w/w), mixed 1:1 (w/w) with PCL. This was also successful with S184 prepared at ratio as low as 1:50. While it was possible to increase the PDMS content relative to PCL, ratios above 3:1 produced non-uniform fibers with beading; these conditions were not pursued further in this study. FTIR analysis of meshes indicated the presence of both PCL and PDMS[8] chemical groups, confirming incorporation of both polymers (Figure S1). Additionally, PDMS:PCL fibers remained intact following immersion into dichloromethane, which dissolves pure PCL, further demonstrating successful blending of the two polymers. Electrospinning of PDMS:PCL was also achieved using MED-4086 (M4086, NuSil), a soft (E ~ 50 kPa), medical grade elastomer (Figure 1A). Given its lower modulus and suitability for medical device production, the remainder of this report will focus only on M4086. By altering processing parameters, a set of polymer meshes (Table 1) was generated ranging in diameter from sub-micrometer to multi-micrometer (referred to as nanoscale and microscale) and pore radii of up to ~10 µm. Micrometer-scale fibers were further prepared using different PDMS:PCL ratios (series M1 vs. M2) in order to test the effect of PDMS concentration on substrate mechanical properties and cell response. Finally, aligned fibers were produced by electrospinning onto a rotating mandrel rather than a stationary plate. Microscale, unaligned fibers of PCL alone served as controls.
Figure 1. Formation of Polydimethylsiloxane (PDMS) fibers.

(A) Electrospinning of blended PDMS : PCL produces fibers with micrometer- to nanometer-scale diameter. These examples illustrate S184 mixed with PCL at a 1:1 (v/v) ratio, and M4086:PCL at a 5:2 ratio. These fibers are morphologically similar to PCL-only fibers. Scale bars = 5 µm. (B) Local rigidity of a M2U mesh, as measured by AFM. Estimated Young’s modulus, E, is encoded over fiber topography as a color skin. (C) Substrate rigidity decreases with increasing PDMS concentration. Data are box plots representing 8 – 11 locations collected in a representative experiment. All conditions are significantly different from the others, as determined by one-way ANOVA (P < 0.01).
Table 1. Series of M4086-based meshes produced by electrospinning.
Fiber diameter and pore size, expressed as mean ± s.d., n = 6 samples per group.
| Mesh | PDMS: PCL | Wt. % | Align | Fiber Diam. (μm) | Pore Radius (μm) |
|---|---|---|---|---|---|
| M1U | 1:1 | 38 | No | 2.12 ± 0.64 | 9.88 ±1.97 |
| M1A | 1:1 | 38 | Yes | 2.28 ± 0.39 | 11.71 ± 2.12 |
| M2U | 5:2 | 38 | No | 2.14 ± 0.50 | 12.21 ± 3.37 |
| M2A | 5:2 | 38 | Yes | 1.02 ± 0.30 | 10.04 ± 4.34 |
| NaU | 3:1 | 19 | No | 0.74 ± 0.27 | 3.07 ± 0.73 |
| NaA | 3:1 | 19 | Yes | 0.68 ± 0.29 | 2.51 ± 0.80 |
| PCL | 0:1 | 19 | No | 1.05 ± 0.22 | 8.19 ± 1.77 |
The effect of incorporating PDMS on fiber rigidity was examined by atomic force microscopy (AFM, in tapping mode with a nanoscale tip). Focusing on microscale fibers, incorporation of M4086 PDMS produced a significant drop in rigidity from the range of 50 MPa (PCL) to hundreds of kPa (M2U), the range identified by O’Connor et al. as important for mechanosensing by human T cells.[2] However, measured rigidity varied as a function of position along individual fibers, as illustrated in Figure 1B; the z-axis of this region shows mesh topography, with local rigidity color mapped onto the substrate surface.
2. Enhance expansion of functional human T cells on PDMS-containing fibers
The ability of electrospun meshes to activate and induce expansion of cells was tested first using human primary T cells from healthy donors. Meshes were coated with an anti-mouse IgG antibody which was then used to capture monoclonal activating antibodies to CD3 and CD28 (clones OKT3 and 9.3, respectively), a two-step approach that provides control over the orientation of the activating antibodies[2]. This method provided similar amounts of captured antibody across the M1U, M2U, and PCL fibers (α = 0.05, Figure S2), as measured by fluorescence microscopy. Furthermore, antibody capture was stable under cell culture conditions, with greater than 75% retention over two days (Figure S2).
Mixed populations of resting CD4+ and CD8+ T cells were seeded onto the prepared substrates in RPMI media supplemented with serum but no additional cytokines. Cells embedded into the meshes (Figure 2A), interacting with individual fibers or junctions between fibers. Small clusters of cells were also observed, attached to the fibers. No attachment was observed in the absence of activating antibodies. By three days after seeding, cells had entered a phase of rapid division and were only loosely attached to the substrate. Cells were then removed from the mesh substrates by gentle aspiration and expanded in plain, unmodified plasticware.[2] Onset of rapid cell division was mirrored by an increase in cell volume from 200 fL to 800–900 fL (Figure S3). Cells returned to a resting state over the next 1 – 2 weeks, accompanied by a decrease in cell volume and slowing of cell division, producing a peak in cell number (expressed as peak doublings) which is used in the following sections as a measure of cell proliferative potential and expansion.
Figure 2. Inclusion of PDMS into fibers enhances T cell expansion.

(A) Mixed CD4+/CD8+ primary human T cells (green, CFSE) bind to individual or small groups of fibers (red, Alexa-568 goat anti-mouse capture antibody). These epifluorescence images illustrate cell attachment 12-hours after seeding, and are presented at two different magnifications (low on top, high on bottom). Scale bars= 25 µm. (B) Timecourse of cell expansion. (C) Comparison of cell division 3-days after seeding. (D) Comparison of peak doublings across stimulation systems. Data in panels C and D represent mean ± s.d. (n = 4 in C, n = 6 in D). Solid overbars group conditions between which no significant difference was detected (ANOVA with Tukey’s HSD, P < 0.05). Dotted lines continue groups joined by solid lines. In all panels, DYN = Dynabeads; NoS = no stimulation. (E) Comparison of IFNγ secretion by CD4+ cells and CD107b mobilization by CD8+ T cells. Data are mean ± s.d., n = 4 independent runs.
Differences in expansion were observed as early as three days following seeding. Most prominently, PDMS:PCL meshes were more effective in inducing T cells to enter proliferation than the rigid, PCL-only control, as measured by CFSE dilution (percent dividing, PDIV, Figure 2C). The average number of divisions exhibited by proliferating cells (proliferative index, PI) was higher for cells activated using the microscale M1U and M1A meshes than those activated on PCL or even Dynabeads (Figure 2C). Longer-term expansion of T cells followed these early readouts, as measured by peak doublings (Figure 2D). In particular, microscale PDMS:PCL meshes induced three- to four- additional doublings than PCL alone, corresponding to almost an order of magnitude more cells. Nanoscale fibers were less effective than their microscale counterparts, but still outperformed the PCL-only meshes. Comparing the M1 and M2 substrates, PDMS:PCL ratio had no significant effect on PDIV, PI, or peak doublings. Fiber alignment also had no detectable effect on these measures. All of the microscale PDMS:PCL fibers induced higher peak doublings than the Dynabeads platform. Together, these results demonstrate that microscale PDMS:PCL fibers, particularly those of the M1 series, provide enhanced production of T cells.
It is increasingly recognized that cell quality, such as the ability of cells to carry out their reactive function, affects the potency and long-term performance of a cellular product. To test such aspects of populations expanded using the mesh substrates, cells were collected when average cell volume decreased below 400 fL then restimulated using CD3/CD28 Dynabeads for analysis of cell response. Secretion of IFNγ was assayed as a measure of CD4+ T cell function. A high percentage of CD4+ T cells in populations activated using the mesh platforms were capable of secreting IFNγ (Figure 2E), with no statistical difference detected across the meshes and Dynabeads (α = 0.05, n = 4 independent runs). Cells that were not restimulated (nostim) showed minimal secretion, demonstrating that cytokine secretion was associated with activation. An additional control (PRI) of resting T cells that had not previously undergone activation exhibited lower IFNγ secretion (P < 0.01). Mobilization of CD107b (LAMP2) to the cell surface was used as a measure of cytotoxic CD8+ T cell function. No difference was observed between cells expanded using any of the meshes or Dynabeads (α = 0.05, n = 4). By these measures, cells expanded on our mesh platforms exhibit similar responses as those produced using Dynabeads. Additional assays comparing phenotypic makeup (based on CCR7 and CD45RO surface expression) also showed no distinct differences as a function of expansion platform (Figure S4).
3. Elastomer fibers rescue expansion of cells from chronic lymphocytic leukemia patients
A current challenge in deployment of ACT is that T cells from individuals undergoing care for cancer often show characteristics of exhaustion,[9] including decreased responsiveness to activation,[10] complicating efficient ex vivo expansion of these cells.[11] Inspired by the enhanced expansion observed on PDMS:PCL meshes in Figure 2, we examined the use of this system with cells from patients undergoing treatment for CLL. Figure 3A reports expansion curves from two donors, showing comparatively successful (D1DYN) and unsuccessful (D2DYN) activation using Dynabeads. For both donors, the M1U and M2U meshes induced a greater number of divisions than either Dynabeads or PCL. Peak doublings from these and two additional CLL donors are compared in Figure 3B, revealing variability between individuals but consistent ability of microscale PDMS:PCL meshes to rescue T cell expansion.
Figure 3. PDMS:PCL meshes rescue expansion of T cells from patients.
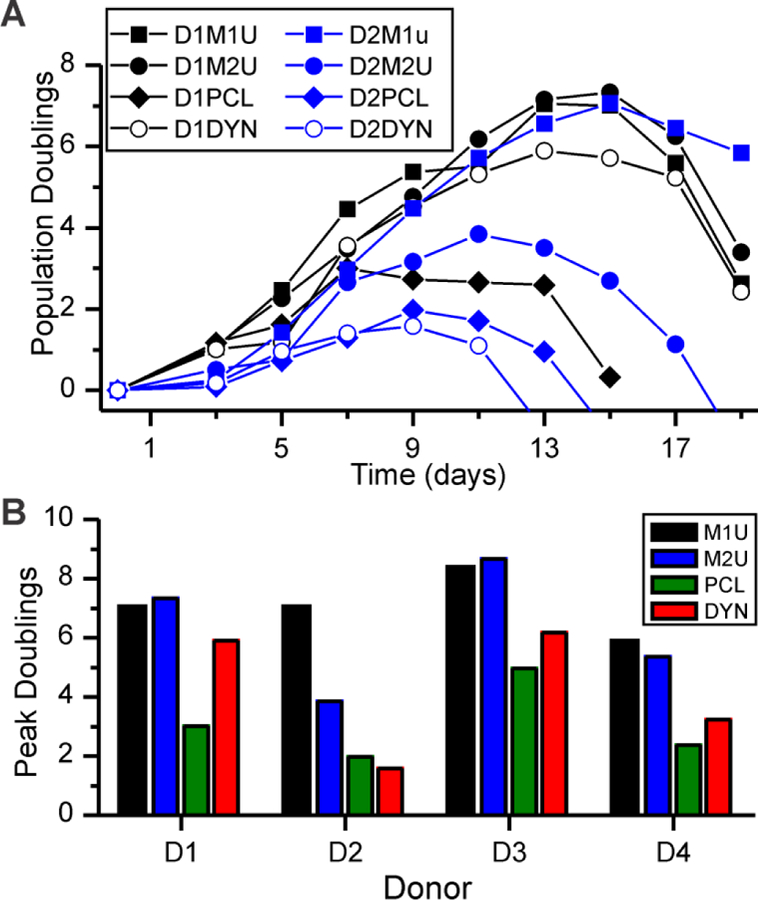
(A) Timecourse of expansion for cells from two donors (D1 and D2), activated on microscale fibers (M1U and M2U). PCL = PCL-only fibers. DYN = Dynabeads. (B) Comparison of peak doublings of cells from four donors as a function of expansion platform. Data are from a single experiment for each combination of donor and substrate.
4. Conclusions
Ex vivo cell expansion currently poses challenges to consistent deployment of T cell-based ACT. Antigen presenting cells provide a physiological approach to initiating this process, but the desire for simpler, manufacturable systems spurred development of alternative platforms.[12] Microscale polystyrene beads emerged as an effective replacement, but it is necessary to remove these particles from the resultant cellular product; beyond physical blockage of fluid flow, introduction of such structures into a patient carries additional risks including uncontrolled activation of T cells. Dynabeads are typically removed using magnets, but this process is incomplete.[13] The mesh platform introduced here, being composed of a single, contiguous construct, elegantly addresses this challenge and provides the added benefit of greater expansion and rescue of exhausted populations by leveraging the ability of T cells to respond to the rigidity of a stimulating surface.
Intriguingly, structuring the PDMS into fibers may play an important role in leveraging T cell mechanosensing. PCL is rigid, so the bulk modulus of even a 3:1 mix of PDMS:PCL would be expected to be tens of MPa, higher than the values identified as important for human T cell expansion.[2] However, T cells are roughly the size of individual fibers, suggesting that cells might respond to local structure of the material. This motivated the use of AFM to examine rigidity, and indeed these measurements report the local modulus of PCL mesh (Figure 1C) as lower than the bulk material. The effect of structure is further seen in Fig. 1C, in that fibers appear stiffest where structures overlap or are bundled and becomes softer by a factor of 2 or 3 along suspended spans. The question of which spatial scales and material structures are important to T cell mechanosensing remains to be fully resolved.
The intracellular molecular mechanisms allowing T cell mechanosensing during activation by TCR/CD28 are also not completely understood. Early studies demonstrated that the TCR complex could be activated by applying cellular-level forces to a T cell.[14] Complementary studies showed that T cells can apply significant traction forces to an underlying substrate through the TCR complex.[15] This suggests a feedback loop through which resistance to cell-generated forces sustains activation, allowing for rigidity sensing; in this direction, actomyosin contractility is required for a range of T cell functions including mechanosensing and overall stability of cellular interfaces.[16] Identification of the specific molecular linkages between these stages is an area of active research.
Finally, the enhanced expansion seen on the mesh platform was attained by changing the bulk material. In comparison to other substrate manipulations that have been proposed to modulate T cell production, including micro-/nano-patterning of an activating surface,[17] electrospinning of an elastomer mesh as described here is highly scalable. In conjunction with simplified removal from the cell growth system compared to beads, the mesh platform provides a strategic combination of properties that are needed for reliable production of clinically relevant numbers of T cells.
Experimental Section
Primary T cells:
Mixed CD4+ and CD8+ T cells were prepared from healthy adult donors (Leukopacks, New York Blood Center) or patients receiving care for CLL (whole blood, Dana-Farber Cancer Institute) using the RosetteSep system (Stemcell Technologies). Cells were stored in liquid nitrogen in complete media + 10% v/v DMSO.
Mesh fabrication and functionalization:
Fibrous meshes were synthesized via electrospinning. For PDMS:PCL blends, NuSil MED-4086 PDMS (M4086) or Sylgard 184 PDMS (S184) and PCL (Mn = 80,000; Sigma) were solubilized in 3:1 (v/v) dichloromethane (DCM) : N,N-dimethylformamide (DMF) to concentrations listed in Table 1. Solutions dispensed through a 21-gauge stainless steel blunt tipped needle. Unaligned meshes were captured onto a grounded static metal collector plate at 10–12 kV at a distance of 12 cm and flow rate of 1 ml/hr. Aligned meshes were spun onto a rotating mandrel (2100 rpm). Meshes were coated with goat-anti-mouse IgG (MP Biomedicals, 2 µg/mL) at room temperature for 2 h followed by a 1:4 mol/mol mix of anti-human CD3 (clone OKT3, Bio X Cell) to anti-human CD28 (9.3, Bio X Cell) for 2 h (2 µg/mL total solution). Meshes were then blocked with 5% bovine serum albumin (BSA, Sigma).
Meshes were characterized by Fourier transform infrared (FTIR, DigiLab Excalibur) spectroscopy and scanning electron microscopy (SEM, Hitachi S-4700, 2 kV, 10 µA). Meshes were pre-coated with gold-palladium for SEM. Mesh rigidity was estimated using atomic force microscopy (AFM, Bruker MultiMode 8-HR, 5 – 20 nm ScanAsyst tips).
T cell expansion and phenotype:
Meshes presenting goat-anti-mouse IgG were coated with 1:5 mol/mol ratio of FITC-tagged mouse IgG-anti-human CD3 (Biolegend, clone OKT3) to non-fluorescent mouse IgG-anti-human CD3 (Bio X Cell, clone OKT3), then incubated in RPMI media at 37 °C. Protein concentration was quantified by widefield fluorescence microscopy.
T cell culture was carried out using RPMI 1640 media (Gibco) supplemented with L-glutamine (20 µM, Gibco), fetal bovine serum (FBS) (5%, GE Healthcare) and HEPES (100 µM, Sigma). For expansions, cells were thawed then rested in complete media for 12–14 hours (37° C, 5% CO2 / 95 % air) prior to use. Positive and negative controls consisted of stimulation with Dynabeads (CTS Dynabeads CD3/CD28, Thermo Fisher) and uncoated, non-activating culture wells. T cells were seeded at 1 × 106 cells in a 1 mL volume into 15.6-mm diameter wells and incubated under standard cell culture conditions. Every second day, starting on day 3, cells were reseeded at 1 × 106 cells / mL. Cells were frozen when mean volume dropped below 400 fL.
Cells frozen at the end of expansion were thawed, allowed to rest, and restimulated using Dynabeads. Cells were assayed for IFNγ (Secretion assay, Miltenyi Biotech) and CD107b mobilization (anti-CD107b clone H4B4, Biolegend) at four and twelve hours after restimulation. Cells were counterstained for CD4 and CD8, as described for phenotype analysis.
Statistical Analysis:
Quantitative data collected over multiple, independent experiments are presented as mean ± standard deviation (s.d.), with the number of replicates indicated in the figure caption. No data was excluded on basis of appearance as an outlier. Normally distributed, homoscedastic data was analyzed by ANOVA followed by Tukey post-hoc methods. In all cases, significance was defined at a two-tailed level of α = 0.05. Data were analyzed using the MATLAB software suite (Mathworks).
Supplementary Material
Acknowledgements
This work was supported in part by the National Institutes of Health (AI110593 to L.C.K.), the National Science Foundation (through a GRFP to S.D. and A.P.D.), and the Columbia-Coulter Translational Research Partnership. J.R.B. has been supported by the Leukemia and Lymphoma Society (TRP# 6289-13) and the American Cancer Society (RSG-13-002-01-CCE), with the DFCI CLL Biorepository particularly supported by the Melton Family Fund for CLL Research, the Susan and Gary Rosenbach Fund for Lymphoma Research, and the Okonow Lipton Family Lymphoma Research Fund.
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Contributor Information
Alex Dang, Department of Biomedical Engineering, Columbia University, New York, NY 10027, USA.
Dr. Sarah De Leo, Department of Biomedical Engineering, Columbia University, New York, NY 10027, USA
Dr. Danielle R. Bogdanowicz, Department of Biomedical Engineering, Columbia University, New York, NY 10027, USA
Dennis J. Yuan, Department of Biomedical Engineering, Columbia University, New York, NY 10027, USA
Stacey M. Fernandes, Department of Medical Oncology, Dana-Farber Cancer Institute, Harvard Medical School, Boston, MA 02215, USA
Prof. Jennifer R. Brown, Department of Medical Oncology, Dana-Farber Cancer Institute, Harvard Medical School, Boston, MA 02215, USA
Prof. Helen H. Lu, Department of Biomedical Engineering, Columbia University, New York, NY 10027, USA
Prof. Lance C. Kam, Department of Biomedical Engineering, Columbia University, New York, NY 10027, USA, lk2141@columbia.edu
References
- [1].Restifo NP, Dudley ME, Rosenberg SA, Nat. Rev. Immunol 2012, 12, 269; [DOI] [PMC free article] [PubMed] [Google Scholar]; Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, June CH, Sci. Transl. Med 2011, 3, 95ra73; [DOI] [PMC free article] [PubMed] [Google Scholar]; Porter DL, Levine BL, Kalos M, Bagg A, June CH, N. Engl. J. Med 2011, 365, 725; [DOI] [PMC free article] [PubMed] [Google Scholar]; Carpenito C, Milone MC, Hassan R, Simonet JC, Lakhal M, Suhoski MM, Varela-Rohena A, Haines KM, Heitjan DF, Albelda SM, Carroll RG, Riley JL, Pastan I, June CH, Proc. Natl. Acad. Sci. U. S. A 2009, 106, 3360; [DOI] [PMC free article] [PubMed] [Google Scholar]; Brentjens RJ, Rivière I, Park JH, Davila ML, Wang X, Stefanski J, Taylor C, Yeh R, Bartido S, Borquez-Ojeda O, Olszewska M, Bernal Y, Pegram H, Przybylowski M, Hollyman D, Usachenko Y, Pirraglia D, Hosey J, Santos E, Halton E, Maslak P, Scheinberg D, Jurcic J, Heaney M, Heller G, Frattini M, Sadelain M, Blood 2011, 118, 4817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].O’Connor RS, Hao X, Shen K, Bashour K, Akimova T, Hancock WW, Kam LC, Milone MC, J. Immunol 2012, 189, 1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Lambert LH, Goebrecht GK, De Leo SE, O’Connor RS, Nunez-Cruz S, Li TD, Yuan J, Milone MC, Kam LC, Nano Lett 2017, 17, 821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Doshi J, Reneker DH, Electrost J 1995, 35, 151; [Google Scholar]; Subramony SD, Su A, Yeager K, Lu HH, J. Biomech 2014, 47, 2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Kim YB, Cho D, Park WH, J. Appl. Polym. Sci 2009, 114, 3870; [Google Scholar]; Yang D, Liu X, Jin Y, Zhu Y, Zeng D, Jiang X, Ma H, Biomacromolecules 2009, 10, 3335. [DOI] [PubMed] [Google Scholar]
- [6].Kurniawan D, Nor FM, Lee HY, Lim JY, Proc. Inst. Mech. Eng., Part H 2011, 225, 1015; [DOI] [PubMed] [Google Scholar]; Lee KH, Kim HY, Khil MS, Ra YM, Lee DR, Polymer 2003, 44, 1287; [Google Scholar]; Li WJ, Tuli R, Okafor C, Derfoul A, Danielson KG, Hall DJ, Tuan RS, Biomaterials 2005, 26, 599; [DOI] [PubMed] [Google Scholar]; Yoshimoto H, Shin YM, Terai H, Vacanti JP, Biomaterials 2003, 24, 2077. [DOI] [PubMed] [Google Scholar]
- [7].Broz ME, VanderHart DL, Washburn NR, Biomaterials 2003, 24, 4181; [DOI] [PubMed] [Google Scholar]; Kim GH, Biomed. Mater 2008, 3, 025010. [DOI] [PubMed] [Google Scholar]
- [8].Efimenko K, Wallace WE, Genzer J, J. Colloid Interface Sci 2002, 254, 306. [DOI] [PubMed] [Google Scholar]
- [9].Gallimore A, Glithero A, Godkin A, Tissot AC, Pluckthun A, Elliott T, Hengartner H, Zinkernagel R, J. Exp. Med 1998, 187, 1383; [DOI] [PMC free article] [PubMed] [Google Scholar]; Zajac AJ, Blattman JN, Murali-Krishna K, Sourdive DJ, Suresh M, Altman JD, Ahmed R, J. Exp. Med 1998, 188, 2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Riches JC, Davies JK, McClanahan F, Fatah R, Iqbal S, Agrawal S, Ramsay AG, Gribben JG, Blood 2013, 121, 1612; [DOI] [PMC free article] [PubMed] [Google Scholar]; Ramsay AG, Johnson AJ, Lee AM, Gorgun G, Le Dieu R, Blum W, Byrd JC, Gribben JG, J. Clin. Invest 2008, 118, 2427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Wherry EJ, Kurachi M, Nat. Rev. Immunol 2015, 15, 486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Andres PG, Howland KC, Dresnek D, Edmondson S, Abbas AK, Krummel MF, J. Immunol 2004, 172, 5880; [DOI] [PubMed] [Google Scholar]; Ratta M, Fagnoni F, Curti A, Vescovini R, Sansoni P, Oliviero B, Fogli M, Ferri E, Della Cuna GR, Tura S, Baccarani M, Lemoli RM, Blood 2002, 100, 230; [DOI] [PubMed] [Google Scholar]; Zheng CY, Ostad M, Andersson M, Celsing F, Holm G, Sundblad A, Br. J. Haematol 2002, 118, 778; [DOI] [PubMed] [Google Scholar]; Kim K, An HJ, Jun SH, Kim TJ, Lim SA, Park G, Na HB, Park YI, Hyeon T, Yee C, Bluestone JA, Kim J, Lee KM, Nano Lett 2012, 12, 4018. [DOI] [PubMed] [Google Scholar]
- [13].Levine BL, Cotte J, Small CC, Carroll RG, Riley JL, Bernstein WB, Van Epps DE, Hardwick RA, June CH, J. Hematother 1998, 7, 437. [DOI] [PubMed] [Google Scholar]
- [14].Kim ST, Takeuchi K, Sun ZY, Touma M, Castro CE, Fahmy A, Lang MJ, Wagner G, Reinherz EL, J. Biol. Chem 2009, 284, 31028; [DOI] [PMC free article] [PubMed] [Google Scholar]; Li YC, Chen BM, Wu PC, Cheng TL, Kao LS, Tao MH, Lieber A, Roffler SR, J. Immunol 2010, 184, 5959. [DOI] [PubMed] [Google Scholar]
- [15].Bashour KT, Gondarenko A, Chen H, Shen K, Liu X, Huse M, Hone JC, Kam LC, Proc. Natl. Acad. Sci. U. S. A 2014, 111, 2241; [DOI] [PMC free article] [PubMed] [Google Scholar]; Hui KL, Balagopalan L, Samelson LE, Upadhyaya A, Mol Biol Cell 2015, 26, 685; [DOI] [PMC free article] [PubMed] [Google Scholar]; Hui KL, Upadhyaya A, Proc. Natl. Acad. Sci. U. S. A 2017, 114, 4175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Grakoui A, Bromley SK, Sumen C, Davis MM, Shaw AS, Allen PM, Dustin ML, Science 1999, 285, 221;10398592 [Google Scholar]; Judokusumo E, Tabdanov E, Kumari S, Dustin Michael L., Kam Lance C., Biophys. J 2012, 102, L5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Bashour KT, Tsai J, Shen K, Lee JH, Sun E, Milone MC, Dustin ML, Kam LC, Mol. Cell Biol 2014, 34, 955; [DOI] [PMC free article] [PubMed] [Google Scholar]; Hu J, Gondarenko AA, Dang AP, Bashour KT, O’Connor RS, Lee S, Liapis A, Ghassemi S, Milone MC, Sheetz MP, Dustin ML, Kam LC, Hone JC, Nano Lett 2016, 16, 2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.