

Abstract
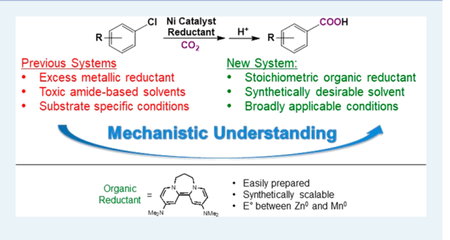
The nickel-catalyzed carboxylation of organic halides or pseudohalides using carbon dioxide is an emerging method to prepare synthetically valuable carboxylic acids. Here, we report a detailed mechanistic investigation of these reactions using the carboxylation of aryl halides with (PPh3)2NiIICl2 as a model reaction. Our studies allow us to understand several general features of nickel-catalyzed carboxylation reactions. For example, we demonstrate that both a Lewis acid and halide source are beneficial for catalysis. To this end, we establish that heterogeneous Mn(0) and Zn(0) reductants are multifaceted reagents that generate noninnocent Mn(II) or Zn(II) Lewis acids upon oxidation. In a key result, a rare example of a well-defined nickel(I) aryl complex is isolated, and it is demonstrated that its reaction with carbon dioxide results in the formation of a carboxylic acid in high yield (after workup). The carbon dioxide insertion product undergoes rapid decomposition, which ca These three oxidation states correspond to the onbe circumvented by a ligand metathesis reaction with a halide source. Our studies have led to both a revised mechanism and the development of a broadly applicable strategy to improve reductive carboxylation reactions. A critical component of this strategy is that we have replaced the heterogeneous Mn(0) reductant typically used in catalysis with a well-defined homogeneous organic reductant. Through its use, we have increased the range of ancillary ligands, additives, and substrates that are compatible with the reaction. This has enabled us to perform reductive carboxylations at low catalyst loadings. Additionally, we demonstrate that reductive carboxylations of organic (pseudo)halides can be achieved in high yields in more practically useful, non-amide solvents. Our results describe a mechanistically guided strategy to improve reductive carboxylations through the use of a homogeneous organic reductant, which may be broadly translatable to a wide range of cross-electrophile coupling reactions.
Keywords: carbon dioxide, nickel, cross-electrophile coupling, homogeneous organic reductant, catalysis, mechanism
Graphical Abstract
INTRODUCTION
Carbon dioxide (CO2) is an attractive C1 source because it is renewable, inexpensive, abundant, and nontoxic.1 The catalytic formation of C−C bonds from CO2 is a particularly interesting class of reactions due to the prevalence of C−C bonds in fuels, commodity chemicals, and pharmaceuticals.1 For instance, in synthetic chemistry, it would be valuable to use CO2 as a feedstock for the preparation of carboxylic acids,2 which are commonly found in bioactive molecules and are important intermediates in complex molecule synthesis due to their synthetic versatility and facile purification.3 Additionally, current methods to generate carboxylic acids, such as the stoichiometric reaction of CO2 with highly reactive organometallic reagents,4 the oxidation of alcohols and aldehydes,5 and nitrile hydrolysis,6 all have poor functional group tolerance and atom economy. Therefore, the development of efficient catalytic methods for the preparation of carboxylic acids from CO2 that are functional group tolerant could have widespread applications in organic synthesis.
Over the past decade, it has been demonstrated that the catalytic reductive coupling of CO2 with unsaturated hydrocarbons7 and organic halides or pseudohalides8–12 is a highly chemoselective strategy to generate carboxylic acids. For example, in 2012, Tsuji and co-workers demonstrated that carboxylic acids can be generated from aryl chlorides and CO2 using a well-defined (PPh3)2NiIICl2 precatalyst (5 mol %), a heterogeneous Mn0 reductant (300 mol %), tetraethylammonium iodide (Et4NI) (10 mol %), and PPh3 (10 mol %) in the amide-based solvent 1,3-dimethylimidazolidinone (DMI) (Figure 1a).8a Furthermore, in a series of seminal reports, Martin demonstrated that related Ni-catalyzed systems can be used to couple CO2 with a variety of electrophiles, including aryl, alkyl, and vinyl halides or pseudohalides with high functional group tolerance (Figure 1b).8b−e,h−l,h However, despite the impressive progress in Ni-catalyzed carboxylation reactions, there are a number of general problems that are preventing both further development and practical application of this method.2g These include (1) the need for super-stoichiometric amounts of heterogeneous Zn0 or Mn0 metallic reductants in most reactions,13 which give rise to poorly reproducible kinetic profiles, limiting industrial applications and complicating mechanistic studies, (2) the need for synthetically undesirable, highly toxic amide-based solvents,14 which are increasingly subject to strict regulation and reduce the number of substrates that are compatible with this method, (3) the need for excess ligand and other inscrutable additives, which complicate mechanistic analysis and make it difficult to predict the outcome of reactions with different substrates, (4) the need for high catalyst loadings (10 mol % Ni is frequently used), (5) the need for different ancillary ligands for even closely related substrates, and (6) limitations in the substrate scope for each particular class of electrophile. For instance, in Tsuji’s system, the reaction is restricted to substrates that are not ortho, amino, or hydroxy substituted.8a,15 It is also noteworthy that many of these limitations in Ni-catalyzed carboxylation reactions also apply to Ni-catalyzed cross-electrophile couplings involving substrates other than CO2, such as reductive couplings between alkyl and aryl halides, which are also currently attracting significant attention in synthetic chemistry.16
Figure 1.

(a) Generic scheme showing Tsuji’s carboxylation of aryl halides. (b) Range of electrophiles that can be used in Ni-catalyzed carboxylation reactions.
One of the main reasons why the design of improved systems for Ni-catalyzed carboxylation reactions has proven challenging is our relative lack of understanding of the mechanism of these transformations.2g Nevertheless, a general mechanism is commonly proposed for all Ni-catalyzed reductive carboxylation reactions. This is summarized in Figure 2 for the Tsuji system.8a In the proposed mechanism, precatalyst activation generates a Ni0 active species, which undergoes oxidative addition with an organic halide (or pseudohalide) to form an organometallic NiII halide (or pseudohalide) complex. Subsequent one electron reduction removes the halide (or pseudohalide) and forms an organometallic NiI complex, which is proposed to insert CO2 and generate a NiI carboxylate. Reduction of the NiI carboxylate removes the carboxylate product from the metal and regenerates the catalytically active Ni0 species. However, with the exception of oxidative addition,17 there is little experimental precedent for the other elementary steps in the proposed catalytic cycle. In particular, the reactivity and speciation of the proposed NiI intermediates remain largely uninvestigated, and there are no characterized examples of CO2 insertion into well-defined NiI complexes.18 Additionally, the role of the additives, such as Et4NI in the Tsuji system, which are typically required for productive catalysis, are not accounted for in the proposed mechanism.
Figure 2.

Proposed catalytic cycle for the carboxylation of aryl chlorides using (PPh3)2NiIICl2.
Here, we perform a detailed mechanistic study of the Tsuji carboxylation system. We explain the roles of all of the required additives and propose a modified catalytic cycle. In a key result, we isolate a well-defined NiI aryl complex and demonstrate that reaction with CO2 results in the formation of a carboxylic acid in high yield (after workup). Lewis acids increase the rate of the proposed CO2 insertion, and we show that a Lewis acid, as well as a halide source are beneficial for catalysis. Our mechanistic studies have enabled us to develop a general strategy to improve catalytic systems for aryl halide carboxylation. A central feature of this strategy is that we can replace the superstoichiometric Mn0 reductant with a well-defined homogeneous organic reductant and an alkali metal halide additive. This has enabled us to decrease the catalyst loading and reaction time, carboxylate more sterically bulky substrates, and perform reactions in more synthetically practical, non-amide solvents. Overall, our results provide fundamental understanding about Ni-catalyzed carboxylation reactions, which may be broadly applicable to improving a variety of cross-electrophile coupling reactions.
RESULTS AND DISCUSSION
Empirical Investigation into the Role of Different Reagents in the Tsuji System for Carboxylation.
Precatalyst Screen.
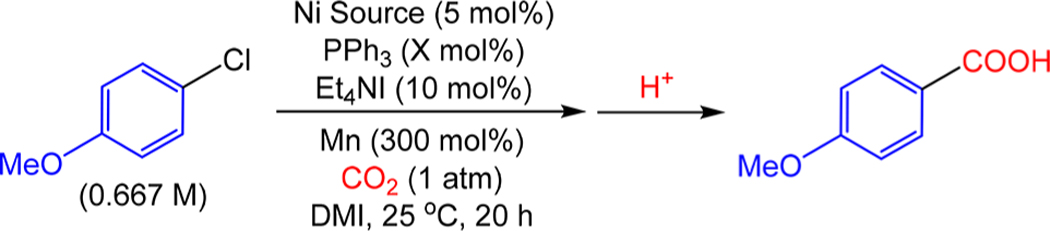
To begin our investigation, we compared the catalytic performance of precatalysts in the Ni0, NiI, and NiII oxidation states for the carboxylation of 4-chloroanisole to 4-anisic acid using the conditions reported by Tsuji et al. (Table 1).8a These three oxidation states correspond to the oxidation states of the Ni intermediates in the mechanism proposed in Figure 2. The amount of free PPh3 ligand added was varied so the overall ratio of PPh3 to NiI was always 4:1. The Ni precatalyst, (PPh3)3Ni Cl, gave comparable activity to Tsuji’s NiII precatalyst, (PPh3)2NiIICl2, with yields of approximately 75% observed in both cases. In contrast, the Ni0 precatalyst, (PPh3)4Ni0, gave a lower yield (51%) of 4-anisic yield. This result is surprising as our control experiments suggest that when (PPh3)2NiIICl2 is used as the precatalyst, (PPh3 ) 4Ni0 is the catalyst resting state, which suggests that PPh3 dissociation is likely turnover limiting in catalysis (see SI). When either (PPh3)3NiICl or (PPh3)2NiIICl2 is used as the precatalyst, MnCl2 is presumably generated as a byproduct of precatalyst activation to form the Ni0 active species; however,this step is not required when (PPh3)4Ni0 is used as the precatalyst. The addition of 5 mol % MnCl2 to a catalytic reaction using (PPh3)4Ni0 as the precatalyst resulted in an increase in product yield from 51% to 81%, which is comparable to results obtained with the NiI and NiII precatalysts. This increase in yield strongly suggests that the Mn0 reductant plays a dual role in catalysis—it not only provides electrons but is also a source of MnCl2, which is beneficial for catalysis. These results may also explain why Mn0 (or Zn0, which presumably generates ZnCl2) has been the reductant of choice for Ni-catalyzed carboxylation reactions and may guide the development of reductive carboxylation systems that employ alternative reductants. This hypothesis is explored further in our reductant screen.
Table 1.
Carboxylation of 4-Chloroanisole with CO2 Using PPh3-Supported Nickel Precatalysts in Different Oxidation Statesa
 | ||
|---|---|---|
| Ni source | PPh3b (mol %) | yieldc (%) |
| (PPh3)2NiIICl2 | 10 | 76 |
| (PPh3)3NiICl | 5 | 73 |
| (PPh3)4Ni0 | 0 | 51 |
| (PPh3)4Ni0 with 5 mol % MnCl2 | 0 | 81 |
Reaction conditions: 4-chloroanisole (0.25 mmol), Ni source (0.0125 mmol), PPh3 (NiII: 0.025 mmol; NiI: 0.0125 mmol; Ni0: 0 mmol), Et4NI (0.025 mmol), Mn (0.75 mmol), CO2 (1 atm) in DMI (0.375 mL) at 25 °C for 20 h.
Added PPh3 was varied to maintain a 4:1 ratio of PPh3 /Ni across reactions.
Yields are reported as the average of two trials and were determined by integration of 1H NMR spectra against a hexamethylbenzene external standard.
Reductant Screen.
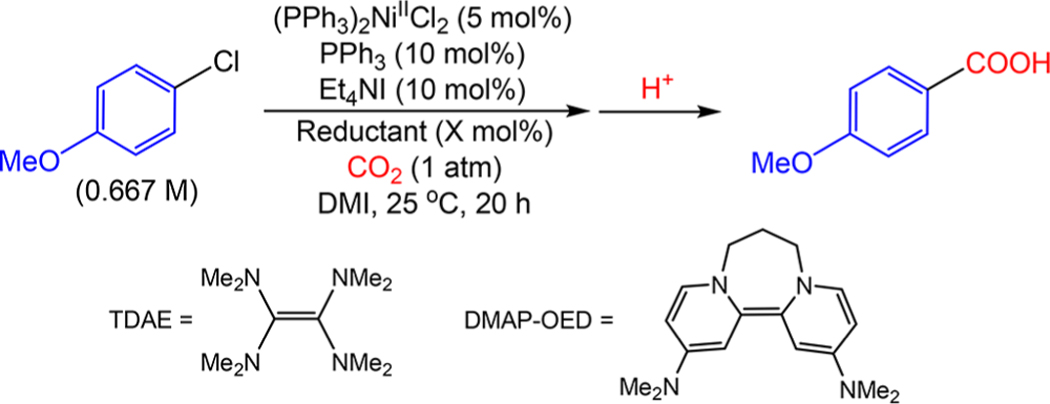
Despite the rising interest in reductive coupling reactions, there are limited comparative studies exploring the impact of the nature of the reductant on catalysis.19 To this end, a diverse series of reductants spanning a range of reduction potentials was evaluated in the carboxylation of 4-chloroanisole with (PPh3)2NiIICl2 as the precatalyst to explore the effect of reductant speciation and potential on catalyst performance (Table 2). Specifically, we tested the one electron organometallic, homogeneous reductants decamethylcobaltocene (Cp*2Co)20 and cobaltocene (Cp2Co),21 as well as the organic, homogeneous two-electron reductants tetrakis(dimethylamino)ethylene (TDAE)22 and DMAP-OED,23 which is an organic electron donor derived from 4-(dimethylamino)pyridine that was first reported by Murphy and co-workers. These reductants were compared to the standard metallic, heterogeneous two electron reductants Mn0 and Zn0 in catalysis.24 Under the standard Tsuji conditions, only the two strongest reductants gave significant yields of product (Table 2, column 1). The heterogeneous reductant Mn0 gave a yield of 76%, while the homogeneous reductant Cp*2Co generated 4-anisic acid in 40% yield. No weaker reductants formed product in a yield of greater than 12%, demonstrating that strong reductants are critical for catalysis under the Tsuji conditions.
Table 2.
Carboxylation of 4-Chloroanisole with CO2 Using a Variety of Reductants of Differing Strength under Various Reaction Conditionsa
 | ||||
|---|---|---|---|---|
| homogeneous reductant | E°d (V) | yieldf (%) | yield without Et4NIf (%) | yield without Et4NI and with100 mol % MnCl2f (%) |
| Cp*2Co | −1.1618 | 40 | 36 | 83g |
| DMAP-OED | −1.0019 | <1 | 7 | 62h |
| Cp2Co | −0.6720 | 1 | 1 | 34 |
| TDAE | −0.5721 | 0 | 1 | <1 |
| heterogeneous reductant | E°e (V) | yield (%)f | yield without Et4NIf (%) | yield with Et4NI and 100 mol % MnCl2f (%) |
| Mn0 | −1.1922 | 76 | <1 | 86 |
| Zn0 | −0.7622 | 12 | 3 | 35 |
Reaction conditions: 4-chloroanisole (0.25 mmol), (PPh3)2NiIICl2 (0.0125 mmol), PPh3 (0.025 mmol), Et4NI (0 or 0.025 mmol), reductant (see fnts b and c), CO2 (1 atm) in DMI (0.375 mL) at 25 °C for 20 h.
When using homogeneous reductants a stoichiometric number of electron equivalents was added relative to 4-chloroanisole and (PPh3) NiIICl2 (which presumably needs to be reduced as part of the activation process): Cp*2Co and Cp2Co (0.525 mmol, 210 mol %), DMAP-OED and TDAE (0.2625 mmol, 105 mol %).
Three equivalents of electrons relative to 4-chloroanisole was added when using heterogeneous reductants: Zn0 and Mn0 (0.75 mmol, 300 mol %).
Values reported in DMF vs NHE.
Values reported as potentials at the metal surface vs NHE.
Yields are reported as the average of two trials and were determined by integration of 1H NMR spectra against a hexamethylbenzene external standard.
Control experiments showed that Cp*2Co could not reduce MnCl2 in DMI, as determined by 1H NMR spectroscopy.
Control experiments showed no catalysis in the absence of a Ni catalyst.
One of the additives that is present in the Tsuji conditions is Et4NI, which is proposed to assist with electron transfer between the heterogeneous Mn0 reductant and the solution-state Ni catalyst.25 To explore this hypothesis, catalytic reactions were performed with our full series of reductants in the absence of Et4NI (Table 2, column 2). No significant changes in product yields were observed with homogeneous reductants in the absence of Et4NI. For example, the yield of product using Cp*2Co as the reductant was 36% in the absence of Et4NI, which is essentially the same as the yield in the presence of Et4NI. In contrast, when Et4NI was removed from a reaction with the heterogeneous reductant Mn0 the yield diminished entirely to <1%. These results are consistent with the hypothesis that Et4NI is required to facilitate electron transfer when the reaction is performed using heterogeneous reductants. For that reason, in the remainder of this work, unless otherwise stated, Et4NI was not present in catalytic reactions using a homogeneous reductant but was added in catalytic reactions using a heterogeneous reductant.
Our previous results showed that MnCl2, which is generated in situ upon oxidation of Mn0, is beneficial in catalysis. To further investigate its role, 100 mol % MnCl2 was added to a series of reactions with different reductants (Table 2, column 3). Upon addition of MnCl2, an increase in yield was observed with all reductants tested except for TDAE, which is incapable of producing catalytically active (PPh3)4Ni0, as reaction of (PPh3)3NiIICl2 with an excess of TDAE only generates (PPh3)3NiICl in the presence of 2 equiv of PPh3 (see the SI). Using both homogeneous and heterogeneous reductants, product yields increased with increasing reductant strength. Interestingly, a significant decrease in the reductant strength required for productive catalysis was observed upon addition of MnCl2. This is best illustrated by the fact that the yield obtained using Cp*2Co as the reductant in the absence of MnCl2 (36%) was the same as the yield obtained using Cp2Co as the reductant in the presence of MnCl2 (34%). Essentially, the addition of MnCl2 allows for comparable catalytic performance between structurally similar reductants with a 490 mV (13.6 kcal/mol) difference in reduction potential in DMF. This suggests that the addition of MnCl2 changes either the speciation of the catalyst in solution or the reaction mechanism, a topic that is explored further in a subsequent section.
From a practical perspective, the most noteworthy result is that the addition of MnCl2 leads to a significant yield (62%) of product when a stoichiometric amount of DMAP-OED is used as the reductant. This is the first example of an organic reductant being utilized in a reductive carboxylation reaction without concomitant photoredox catalysis.8n,9d Additionally, DMAP-OED can be prepared in two steps from relatively inexpensive starting materials (each less than $100/kg) and is a solid at room temperaure,23 which makes it easier to work with than volatile liquid organic reductants such as TDAE.19,26 From a mechanistic perspective, no catalysis was observed with DMAP-OED as the reductant in the absence of MnCl2. This indicates that when DMAP-OED is used in catalysis, the source of the electrons is decoupled from the production of MnCl2, unlike when Mn0 is used the reductant. This observation is important because it means that when DMAP-OED is used as the reductant, we can discretely study the role of MnCl2 (and related additives) in catalysis in the absence of in situ generated MnCl2 from the reductant, which provides an opportunity to improve the reaction.
Additive Screen.
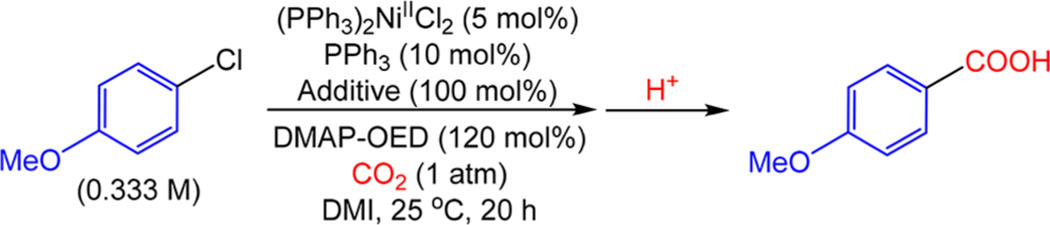
Metal halide type additives are commonly employed in Ni-catalyzed reductive carboxylation reactions, but the role of such additives has remained unclear.2g,i To empirically investigate the role of MnCl2 in catalysis, the metal halide-type additive was systematically varied across catalytic reactions using (PPh3)2NiIICl2 as the catalyst and a slight excess of DMAP-OED (120%) as the reductant and catalyst performance was monitored (Table 3). When the reaction was performed with MnCl2 as the additive, a 68% yield was obtained. Simple alkali halide salts with high solubility in DMI, such as LiCl and LiBr, performed comparably to MnCl2, giving yields of 68% and 67%, respectively (see SI for a full list of additives that were evaluated). This result is significant in understanding the mechanism of these reactions because it indicates that organometallic Mn (or Zn when it is used as the reductant) species do not play a crucial role in Ni-catalyzed carboxylation reactions as the reaction proceeds in the absence of any Mn-containing species. Previous stoichiometric studies have demonstrated that Ni-mediated carboxylation can occur in significant quantities in the absence of Mn or Zn species;7f,8a,e,j,k however, they had not been able to rigorously exclude this hypothesis in catalysis.2g As a result, mechanisms in which carboxylation requires species derived from Mn0 could not be excluded.13 It is also important from a practical perspective because Li+ salts are both available in greater variety and tend to be significantly cheaper than Mn2+ salts. For this reason, LiBr (or LiCl) was used in place of MnCl2 in many of our further studies.
Table 3.
 | |
|---|---|
| additive | yieldc (%) |
| MnCl2 | 68 |
| LiCl | 68 |
| LiBr | 67 |
| LiOTf | 15 |
| LiPF6 | 14 |
| LiBF4 | 36 (23d) |
| nBuBr | 3 |
| B(OPh)3 | 17 |
| B(OPh)3 with 100 mol % nBu4Br | 44 |
Reaction conditions: 4-chloroanisole (0.25 mmol), (PPh3)2NiIICl2 (0.0125 mmol), PPh3 (0.025 mmol), DMAP-OED (0.30 mmol), CO2 (1 atm) in DMI (0.750 mL) at 25 °C for 20 h.
Reactions were performed at more dilute concentrations to ensure additive solubility and with a slight excess of reductant to ensure it was not the limiting reagent.
Yields are reported as the average of two trials and were determined by integration of 1H NMR spectra against a hexamethylbenzene external standard.
Reaction performed with 5 mol % (PPh3)4Ni0 (0.0125 mmol) in place of (PPh3)2NiIICl2 and PPh3.
It was not clear from our preliminary work if the cation, anion, or both components of LiBr or MnCl2 were crucial for enhancing catalysis. To explore this, we used either Li+ or Br− additives with generally innocent counterions such as trifluoromethanesulfonate (OTf−), hexafluorophosphate (PF6−), and tetrafluoroborate (BF4−) or tetrabutylammonium (nBu4+) in catalysis. Reactions using LiOTf, LiPF6, and LiBF4 gave product yields of 15%, 14%, and 36%, respectively, while the reaction using nBu4Br gave a yield of 3%. The difference in the yields with the different lithium salts may be related to ion-pairing effects between the cation and anion in DMI, which changes the effective concentration of free Li+.27 The reaction with LiBF4 was repeated using (PPh3)4Ni0 as the precatalyst in place of (PPh3)2NiIICl2 (with 2 equiv of PPh3) to limit the number of halide sources present in the reaction. This decreased the yield to 23%. We note that it is not possible to completely eliminate halide sources from the reaction when 4-chloroanisole is used as the substrate, but catalytic data collected using phenyl triflate as the substrate demonstrated that while a Lewis acid is essential for catalysis, a halide source only increases catalyst performance and is not essential (see the SI for further details). Overall, our results demonstrate that neither Li+ nor Br− sources with innocent counterions are sufficient to provide catalytic results comparable to those obtained with LiBr, which indicates that both the cation and anion are important in catalysis.
One potential role of Li+ in catalysis is to act as a Lewis acid. To investigate the potential role of Lewis acids, a nonionic Lewis acid, triphenoxyborane (B(OPh)3), was employed as an additive in catalysis, giving a product yield of 17%, which is significantly above the baseline reaction with no Lewis acid. Furthermore, when B(OPh)3 was used as an additive in catalysis with nBu4Br, an increase in yield to 44% was observed. This result provides evidence that the role of Li+ or Mn2+ in catalysis is that of a Lewis acid and further exemplifies the benefits of both a Lewis acid and halide source in catalysis. In fact, a review of the literature indicates that every system for the reductive carboxylation of organic halides and pseudohalides reported to date has both a Lewis acid and halide source present in catalysis, demonstrating the critical nature of these reagents to productive catalysis.8–11 In most cases, these species have not been added deliberately but form in situ, and there is presumably significant scope for optimizing these reagents to enhance catalysis, especially if the reductant is decoupled from the Lewis acid source. Additionally, despite their ubiquity in carboxylation reactions, only one computational study has proposed any sort of role for Lewis acids and halides (vide infra),28 and it is noteworthy that in the proposed mechanism for the carboxylation of aryl chlorides (Figure 2), neither a Lewis acid nor an external halide source is involved as a reagent in any elementary reaction. Therefore, we sought to investigate the elementary steps of the proposed catalytic cycle through stoichiometric reactions in order to explore their validity and elucidate the role of Lewis acids and halides in catalysis.
Investigation of Proposed Elementary Steps.
LnNiII(Ar)(Cl) Reduction.
In all Ni-catalyzed reductive carboxylation reactions involving alkyl or aryl halide or pseudohalide substrates, the reduction of an organometallic NiII halide or pseudohalide is proposed to be an elementary step (Figure 2).8 For example, in the Tsuji system the reduction of (PPh3)2NiII(Ar)(Cl) is proposed to occur as opposed to CO2 insertion into (PPh3)2NiII(Ar)(Cl).8a Two observations from the literature provide support for this step: (i) complexes of the type LnNiII(Ar)(X) (X = (pseudo)halide) do not react directly with CO2,29 an observation which is consistent with our own control experiments using complexes of the form (PPh3)2NiII(Ar)(Cl); (ii) Tsuji et al. reported that when (PPh3)2Ni In agreement with our hypothesis,II(C6H5)(Cl) was treated with a Mn0/Et4NI reductant pair iFollowing this precedent, we preparn DMI under an atmosphere of CO2, 47% yield of methylbenzoate was produced after esterification of the product.8a Given this precedent, we have not studied the reduction of NiII to NiI in detail, although we have performed some experiments exploring the effect of the aryl group on reduction. Specifically, in direct contrast to Tsuji’s results with (PPh3)2NiII(C6H5)(Cl), when we stirred (PPh3)2NiII(o-tol)-(Cl) with a Mn0/Et4NI reductant pair in DMI under both an N2 and CO2 atmosphere for 1 day, no reaction was observed. However, when (PPh3)2NiII(o-tol)(Cl) was treated with 1 equiv of DMAP-OED in DMI under an N2 atmosphere, starting material was consumed rapidly and a black precipitate was formed (see the SI). When the same reaction was performed under a CO2 atmosphere, 2-toluic acid was produced in 42% yield (see the SI for details). Since Mn0 is a stronger reductant than DMAP-OED, these results indicate that kinetic factors, presumably related to the increased steric bulk around the metal center, prevent electron transfer from the Mn0/Et4NI reductant pair to (PPh3)2NiII(o-tol)(Cl). Our stoichiometric observations are consistent with the inability of the Tsuji system to carboxylate ortho-substituted aryl halides and suggest that when DMAP-OED is used as the reductant it may be possible to carboxylate these substrates.8a In agreement with our hypothesis, when 2-chlorotoluene was employed as a substrate in catalysis using DMAP-OED as the reductant, MnCl2 as an additive, and (PPh3)2NiIICl2 as the catalyst, 2-toluic acid was produced in 53% yield (Table 4). This result demonstrates that in the Tsuji system, the restriction in substrate scope is not intrinsic to the reaction, but is a limitation imposed by the choice of reductant and suggests that improved and distinct reactivity can be achieved with homogeneous reductants compared to heterogeneous reductants in reductive coupling reactions. Additionally, the NiII complex (PPh3)2NiII(o-tol)(Cl) performed comparably as a catalyst to the literature (PPh3)2NiIICl2 precatalyst for the carboxylation of 2-chlorotoluene using DMAP-OED (see the SI for details). This result provides further evidence that complexes of the type (PPh3)2NiII(Ar)(Cl) are intermediates in catalysis.
Table 4.
Carboxylation of 2-Chlorotoluene with CO2 Using Mn0/Et4NI and DMAP-OED Reductantsa
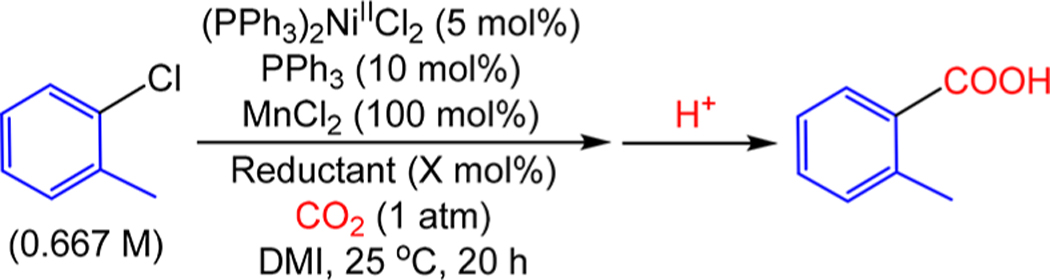
Reaction conditions: 2-chlorotoluene (0.25 mmol), (PPh3)2NiIICl2 (0.0125 mmol), PPh3 (0.025 mmol), reductant (see fnts b and c), CO2 (1 atm) in DMI (0.375 mL) at 25 °C for 20 h.
300 mol % Mn0 (0.75 mmol) and 10 mol % Et4NI (0.025 mmol) were utilized.
105 mol % DMAP-OED (0.2625 mmol) was utilized.
Yields are reported as the average of two trials and were determined by integration of 1H NMR spectra against a hexamethylbenzene external standard.
Preparation of a NiI Aryl Complex and Reaction with CO2.
One-electron reduction of compounds of the type LnNiII(R)-(X) is proposed to generate highly reactive organometallic LnNiI(R) intermediates (in this work (PPh3)nNiI(Ar)) in catalysis, which are proposed to react with CO2; however, studying this class of complexes is difficult due to their instability.30 Indeed, the direct reduction of (PPh3)2NiII(o-tolyl)(Cl) with DMAP-OED did not lead to the formation of any organometallic complexes that could be spectroscopically characterized. Recently, it was demonstrated that metastable NiI aryl species supported by the bidentate phosphine ligand dppf (dppf = 1,1′-bis(diphenylphosphino)ferrocene) can be synthesized using sterically bulky aryl ligands.30e Following this precedent, we prepared (PPh3)2NiI(2,4,6-iPr3C6H2), which contains a bulky aryl group, through the treatment of (PPh3)3NiI(Cl) with 2,4,6-triisopropylmagnesium bromide (Scheme 1). This compound, which is a model for the proposed NiI aryl intermediates in the carboxylation of aryl chlorides (Figure 2), is a rare example of a well-defined NiI aryl species.30b,c,e,f Although (PPh3)2NiI(2,4,6-iPr3C6H2) decomposes over 5 h in THF at room temperature to 1,3,5-triispropylbenzene and a mixture of unidentifiable products, we were able to grow single crystals suitable for X-ray diffraction (Scheme 1). The Ni(1)−C(1) bond length of 1.9369(16) Å is similar to those observed in the few previous examples of NiI aryl complexes.30b,c,e,f The geometry around the Ni center is highly distorted trigonal planar with the P(2)−Ni(1)−C(1) bond angle of 140.72(5)° being significantly larger than either the P(1)−Ni(1)−C(1) or P(1)−Ni(1)−P(2) bond angles, which are 109.22(5) and 109.278(11)°, respectively. The 1H NMR spectrum of (PPh ) NiI(2,4,6-iPr C H ) is consistent with a paramagnetic complex.31 There is a broad diagnostic resonance integrating to 12 protons at 10.86 ppm in toluene-d8, which enabled us to use NMR spectroscopy to determine the stability of the complex and check if it is present in a reaction mixture (see the SI for details). The rate of decomposition of (PPh3)2NiI(2,4,6-iPr3C6H2) is unaffected by the presence of a strong reductant such as Cp*2Co, indicating that the NiI aryl complex is likely not reduced during catalysis. The decomposition of (PPh3)2NiI(2,4,6-iPr3C6H2) is significantly slower in the presence of 2 equiv of PPh3 (2 days compared to 5 h at room temperature in THF), suggesting that ligand dissociation provides a decomposition pathway and providing a rationale for the need for excess PPh3 in catalysis.8a Owing to the instability of (PPh3)2NiI(2,4,6-iPr3C6H2), we were not able to directly evaluate the competence of an isolated sample of the NiI aryl complex as a (pre)catalyst in a catalytic reaction. The E PR spectrum of (PPh3)2NiI(2,4,6-iPr3C6H2) is consistent with the presence of S = 1/2 species and is dependent upon the concentration of PPh3. Similar to its dppf congener,30e the EPR spectrum shows metal-centered radical character, in agreement with the proposed NiI oxidation state (see the SI for details).
Scheme 1.
Synthesis and ORTEP of (PPh3)2NiI(2,4,6-iPr3C6H2)a

aThermal ellipsoids are shown at 30% probability and hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and angles (deg): Ni(1)−C(1) 1.9369(16), P(1)−Ni(1)−P(2) 109.278(11), P(2)−Ni(1)−C(1) 140.72(5), C(1)−Ni(1)−P(1) 109.22(5).
In catalysis, the formation of a new C−C bond and the incorporation of CO2 into the catalytic cycle is proposed to occur via CO2 insertion into a (PPh3)nNiI(Ar) species.8a Due to the instability of NiI aryl species, the only evidence to support this elementary step is from DFT calculations29a and there are no examples of reactions of CO2 with NiI aryl species resulting in the formation of carboxylic acids.2g To this end, a THF solution of (PPh3)2NiI(2,4,6-iPr3C6H2) was placed under 1 atm of CO2 in the presence of 2 equiv of PPh3 (Table 5). The excess PPh3 was added to slow down the background decomposition of (PPh3)2NiI(2,4,6-iPr3C6H2) (vide supra). After 5 h, 1H NMR spectroscopy indicated that all of the (PPh3)2NiI(2,4,6-iPr3C6H2) had been consumed, and treatment of the reaction mixture with acid gave a 78% yield of 2,4,6-triisopropylbenzoic acid. Efforts to identify the metal-containing product from the reaction of CO2 with (PPh3)2NiI(2,4,6-iPr3C6H2) prior to treatment with acid are described in a subsequent section. The carboxylic acid product we obtain after an acid workup clearly establishes for the f irst time that LnNiI(R) complexes are capable of activating CO2, supporting the commonly proposed elementary step in reductive carboxylation reactions. It also provides evidence against the proposal that Mn or Zn are required for the activation of CO2 and confirms that NiI aryl species are highly nucleophilic. This suggests that they may also be able to insert other molecules with polar double bonds such as carbonyls, which could be relevant to cross-electrophile coupling reactions between aryl halides and aryl aldehydes or cyclic anhydrides.32
Table 5.
Time required for Complete Consumption of (PPh3)2NiI(2,4,6-iPr3C6H2) under 1 atm of CO2 in the Presence of Different Additivesa
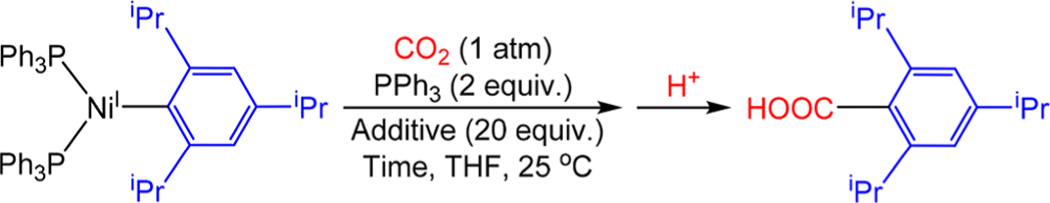
Reaction conditions: (PPh3)2 NiI(2,4,6-iPr3C6H2) (0.0032 mmol), PPh3 (0.0064 mmol), additive (0.064 mmol), CO2 (1 atm) in THF (0.50 mL) at 25 °C.
Time until consumption of starting material.
Yields were determined by integration of 1H NMR spectra against a hexamethylbenzene external standard.
It has previously been demonstrated that Lewis acids, such as Li+, can promote CO2 insertion reactions into transition-metal hydride and methyl bonds.33 In catalytic carboxylation reactions, this provides a possible role for Lewis acid additives (vide supra). When a T HF so lu tio n o f (PPh3)2NiI(2,4,6-iPr3C6H2) was placed under 1 atm of CO2 in the presence of 2 equiv of PPh3 and 20 equiv of LiPF6, the rate of CO2 insertion increased significantly. All of the (PPh3)2NiI(2,4,6-iPr3C6H2) was consumed in 20 min, and after the reaction mixture was exposed to acid, the yield of 2,4,6-triisopropylbenzoic acid was 79% (Table 5). To check if the increase in rate was due to a change in the ionic strength of the solution, the reaction was repeated with 20 equiv of nBu4PF6 in place of LiPF6. In this case, no rate enhancement was observed. These results demonstrate that CO2 insertion into (PPh3)2NiI(2,4,6-iPr3C2H6) is promoted by the presence of Li+, which presumably stabilizes the negative charge which builds up on the incipient carboxylate group in the transition state.29b,33d Given the similarities between the calculated transition states for CO2 insertion into NiI aryl and alkyl bonds,28,29 we suggest that this Lewis acid rate enhancement is likely also relevant to carboxylation reactions involving alkyl substrates.
Formation of a Putative NiI Carboxylate Species.
Monomeric NiI carboxylate complexes, formed via the insertion of CO2 into NiI organometallic complexes, are proposed as intermediates in all Ni-catalyzed reductive carboxylation reactions of organic halides or pseudohalides, even though there are no structurally characterized examples.8,34 In an effort to spectroscopically observe a NiI carboxylate species, we monitored the reaction of (PPh3)2NiI(2,4,6-iPr3C6H2) with CO2 in the presence of excess PPh3 (vide supra) using NMR and EPR spectroscopy. Upon consumption of (PPh3)2NiI(2,4,6-iPr3C6H2), no new paramagnetic species were present according to 1H NMR (in THF) or EPR spectroscopy (in 2-MeTHF) in the presence of 20 equiv of LiOTf (Scheme 2, step 1). This suggests that if CO2 insertion is resulting in a monomeric NiI carboxylate it is unstable. Upon complete consumption of (PPh3)2NiI(2,4,6-iPr3C6H2), the only signals observed in the 31P NMR spectrum were consistent with the generation of (PPh3)4Ni0 and (PPh3)2Ni0(κ2-OCO), which is known to form reversibly through the reaction of (PR3)2Ni0 complexes with CO2 (see the SI).35 The observation of these Ni0 products suggests that decomposition of the putative NiI carboxylate occurs alongside an electron-transfer process. We hypothesized that the NiI carboxylate could undergo a disproportionation reaction to generate 1 equiv of Ni0 and 1 equiv of a NiII species containing two carboxylate ligands, which was not detected by either NMR or EPR spectroscopy (Scheme 2, step 2). It is possible that the proposed NiII species was not observed because it is paramagnetic with an integer spin state. This could be caused by the formation of dimeric or higher order species. For example, dimeric NiII paddlewheel complexes featuring four bridging carboxylate units and a capping phosphine ligand are known.34
Scheme 2.
Proposed Decomposition of a Putative NiI Carboxylate Formed from the Reaction of (PPh3)2NiI(2,4,6-iPr3C6H2) with CO2 and Trapping Experiments of the Proposed Decomposition Productsa

aCompounds in brackets were unobserved during the reaction. *74% yield for formation of 2 equiv of (PPh3)2NiIBr of which only 37% of the Ni is presumably from the starting (PPh3)2NiI(2,4,6-iPr3C6H2).
To explore if the putative NiI carboxylate was undergoing disproportionation, we sought to quantify the products. However, since (PPh3)4Ni0 exhibits a broad signal by 31P NMR spectroscopy that is difficult to integrate accurately, especially in the presence of free PPh3, and we could not spectroscopically observe the proposed NiII biscarboxylate complex, trapping experiments were designed to quantify the products of disproportionation (Scheme 2). After the reaction between (PPh3)2NiI(2,4,6-iPr3C6H2) and CO2 had reached full conversion as determined by 1H NMR spectroscopy, (PPh3)2NiIIBr2 was added to selectively react with the observed Ni0 products. This resulted in a comproportionation reaction,36 which generated (PPh3)3NiIBr in a 74% yield. Two equivalents of (PPh3)3NiIBr are formed in the comproportionation reaction: 1 equiv from the Ni0 products formed after decomposition of the NiI carboxylate and 1 equiv from the (PPh3)2NiIIBr2 trapping reagent (Scheme 2, step 3). Therefore, if the trapping reaction is quantitative, 37% of the NiI carboxylate species generates Ni0 products after decomposition. Next, LiBr was added to the reaction mixture to react selectively with the proposed NiII product of disproportionation, the NiII biscarboxylate complex, through a ligand metathesis reaction. This generated (PPh3)2NiIIBr2 in a 41% yield, as well as presumably Li{OC(O)(2,4,6-iPr3C6H2)}, which, when treated with acid, generated 2,4,6-triisopropylbenzoic acid in 83% yield (Scheme 2, step 4). The high mass balance and nearly 1:1 ratio of the trapped Ni0 and NiII species formed after the reaction of (PPh3)2NiI(2,4,6-iPr3C6H2) with CO2 provide strong evidence for disproportionation as the major decomposition pathway for a putative NiI carboxylate species. At this time, the speciation of the proposed NiII species with two carboxylate ligands remains unclear, although when it is treated with acid it presumably generates 2,4,6-triisopropylbenzoic acid (vide supra). It is possible that this species forms under our catalytic conditions, and if it does form, it is probably an off-cycle species.
Our catalytic results indicate that a halide source is beneficial for high catalytic activity. One potential role of a halide source is to trap the NiI carboxylate before it undergoes disproportionation. To probe this hypothesis, (PPh3)2NiI(2,4,6-iPr3C6H2) was treated with CO2 in THF in the presence of 2 equiv of PPh3 and 20 equiv of LiCl. We expected that the LiCl might allow us to trap the NiI carboxylate as the known, stable complex (PPh3)3NiICl through a ligand metathesis reaction (Scheme 3). Complete consumption o f (PPh3)2NiI(2,4,6-iPr3C6H2) was observed in less than 10 min, and (PPh3)3NiICl was formed in 83% yield, alongside a colorless precipitate, which is presumably Li{OC(O)-(2,4,6-iPr3C6H2)}. Subsequent treatment of the reaction mixture with acid generated 2,4,6-triisopropylbenzoic acid in 75% yield, indicating that the Ni that underwent the CO2 insertion was trapped as (PPh3)3NiICl. These results are consistent with (1) an unstable NiI carboxylate complex being a catalytic intermediate; (2) ligand metathesis of a NiI carboxylate complex with a metal halide salt to generate (PPh3)3NiIX and a metal carboxylate salt being a plausible elementary reaction in catalysis, especially given that halide salts are beneficial in catalysis; and (3) ligand metathesis with a halide salt being faster than the decomposition of the putative NiI carboxylate via disproportionation (Scheme 2).
Scheme 3.

Reaction of (PPh3)2NiI(2,4,6-iPr3C6H2) with CO2 in the Presence of LiCl and Proposed Reaction Pathway
Given the apparent instability of NiI carboxylate complexes, it was not possible to investigate the ligand exchange reaction of a NiI carboxylate with a halide in stoichiometric reactions. We note that several alternative synthetic routes, which did not involve CO2 insertion into a NiI aryl complex, were pursued in order to prepare NiI carboxylates and did not give tractable products (see SI). However, our catalytic data provide some insight into the ligand substitution reaction. When DMAP-OED is used as the reductant in the presence of a halide source but in the absence of a Lewis acid, no catalytic activity was observed (Table 3). Although the Lewis acid assists with CO2 insertion, we have shown that CO2 insertion can still occur in the absence of a Lewis acid, albeit at a slower rate (vide supra). It is, therefore, surprising that almost no product is generated in catalysis without a Lewis acid. In fact, these results suggest that the Lewis acid also helps with another step in catalysis. Previous computational studies have suggested a strong Lewis acid−base interaction between the Mg2+ cation of MgCl2 with the noncoordinated oxygen atom of a κ1-carboxylate ligand on a NiI center supported by two tricyclopentylphosphine ligands.28 These calculations suggest that a cationic Lewis acid may play a role in catalysis by stabilizing the NiI carboxylate intermediate toward disproportionation. Additionally, Lewis acids have been shown to interact with κ1-carboxylate ligands and to induce a change in binding mode of carboxylate ligands from κ2 to κ1, both of which can lead to changes in reactivity or reduction potential (see the reductant screen section in the SI for further discussion).37 At this stage, we do not have enough evidence to unequivocally determine whether Lewis acids are playing a role in altering the reactivity of the putative NiI carboxylate in our systems but in light of our results this hypothesis seems plausible.
In Tsuji’s mechanism for the carboxylation of aryl halides, it is proposed that a NiI carboxylate is directly reduced during catalysis (Figure 2). Our catalytic results indicate that when a strong reductant such as Cp*2Co is used, turnover is observed in the absence of a Lewis acid. This suggests that direct reduction of a NiI carboxylate can occur under strong reducing conditions. However, when a weaker reductant such as DMAP-OED is used, catalytic activity is only observed in the presence of a Lewis acid and higher yields are obtained when a halide source is present. Therefore, we suggest that DMAP-OED is not a strong enough reductant to reduce the NiI carboxylate intermediate by itself. As a consequence, with DMAP-OED and weaker reductants, reduction only occurs if the NiI carboxylate is primed for reduction by a Lewis acid (vide supra). In agreement with this hypothesis, weaker reductants can be utilized in catalysis in the presence of a Lewis acid (see the SI). Additionally, if a halide source is present the NiI carboxylate can be converted into a NiI halide, which is presumably easier to reduce, and leads to the best catalytic performance. Our studies provide insight into the role of a putative NiI carboxylate intermediate in catalysis; however, a well-defined system, which will likely be difficult to synthesize, is required to rigorously investigate the structure and reactivity of NiI carboxylate complexes.
Revised Mechanistic Proposal.
Based on our catalytic and stoichiometric studies, we propose a modified catalytic cycle for the nickel catalyzed reductive carboxylation of aryl chlorides compared to that typically proposed in the literature (Figure 3). Initially, precatalyst activation of (PPh3)2NiIICl2 occurs through reduction with Mn0, which requires Et4NI, to generate a catalytically active Ni0 species and MnCl2. The formation of MnCl2 is crucial because it subsequently acts as a catalytically beneficial Lewis acid and halide source in catalysis. The catalytically active Ni0 species is in equilibrium with (PPh3)4Ni0 due to the presence of excess PPh3. We propose that (PPh3)4Ni0 is the catalyst resting state, which must lose one or more PPh3 ligands to generate the catalytically active Ni0 species. This Ni0 species undergoes oxidative addition with the aryl chloride electrophile to generate a (PPh3)2NiII(Ar)-(Cl) intermediate, which is reduced by one electron by Mn0 to generate a highly reactive (PPh3)nNiI(Ar) species. The NiI aryl species is unstable but the presence of free PPh3 decreases the rate of its decomposition. In the key step, CO2 inserts into (PPh3)nNiI(Ar) to generate a new C−C bond. This process is assisted by the presence of a Lewis acid, which increases the rate of insertion. The product of CO2 insertion is presumably a NiI carboxylate, which is highly unstable. At this point the mechanism diverges and the NiI carboxylate can undergo two different processes. First, in the presence of a strong reductant, such as Mn0, direct reduction of the NiI carboxylate regenerates the catalytically active Ni0 species and releases the carboxylated product from the Ni center. It is likely that in the presence of a Lewis acid there is a decrease in required reducing power for this elementary reaction, so weaker reductants can be used with a Lewis acid. Alternatively, the NiI carboxylate can undergo a ligand metathesis reaction with a halide source to generate a more stable (PPh3)3NiICl intermediate and release the carboxylated product from the Ni center. The NiI halide complex, (PPh3)3NiICl, is readily reduced by Mn0 to regenerate the active Ni0 species. We suggest that our revised mechanism is likely to be general to nickel-catalyzed reductive carboxylations of other organic halides and pseudohalides and possibly to the broader field of cross-electrophile coupling, where Lewis acids and halide sources are also commonly used.16,32 Additionally, our revised mechanism provides guidance on how to improve reductive carboxylation reactions, a topic which is addressed in the following section.
Figure 3.

Revised catalytic cycle for the carboxylation of aryl chlorides with (PPh3)2NiIICl2 and Mn0 as the reductant.
Development of an Improved System for the Carboxylation of Aryl Halides and Pseudohalides.
Optimization of Reaction Conditions in DMI.
As described in the Introduction, there are several major limitations associated with the conditions and reagents typically used in Ni-catalyzed reductive carboxylation reactions. Our discovery that DMAP-OED can be used as the reductant instead of the combination of Mn0/Et4NI has several advantages beyond the fact that it is a homogeneous reductant. Specifically, when DMAP-OED is used as the reductant instead of Mn0/Et4NI: (1) only a slight excess of the reductant is required, (2) the substrate scope is expanded to include ortho substituted aryl chloride substrates, and (3) there is a greater range of additives and ancillary ligands that can promote the carboxylation reaction, which provides more opportunities to improve the reaction conditions (see the SI for more details). For example, when (dppf)NiIICl2, a complex featuring a bidentate ancillary ligand, was used as the precatalyst for the carboxylation of 4-chloroanisole with Mn0/Et4NI as the reductant under Tsuji’s conditions, no product was observed (see SI). In contrast, when (dppf)NiIICl2 was used as the precatalyst for the same reaction using DMAP-OED as the reductant and MnCl2 as an additive it outperformed the literature precatalyst (PPh3)2NiIICl2. In light of these findings, a system for carboxylation was optimized in DMI using (dppf)NiIICl2 as the precatalyst, near-stoichiometric equivalents of DMAP-OED as the reductant, and a slight excess of LiCl as a cost-effective additive for the carboxylation of 4-chloroanisole (see the SI for the full optimization). Notably, the reaction proceeded with high yields in the absence of excess ligand, which the Tsuji system required for high selectivity.8a In fact, under our optimized conditions, high yields could be obtained with catalyst loadings as low as 1 mol %, which is the lowest catalyst loading reported for a reductive carboxylation reaction of any organic halide or pseudohalide (Figure 4a). Addition-ally, at 5 mol % catalyst loading reaction times could be reduced to 30 min, which is the shortest reported time for any catalytic reductive carboxylation reaction (Figure 4b). These results demonstrate that despite the higher cost of DMAP-OED compared to heterogeneous reductants such as Mn0 or Zn0, there is a clear improvement in the catalyst loading and reaction time when using DMAP-OED in combination with ancillary ligands and additives that are incompatible with Mn0 or Zn0. As a result, our findings should translate well to the synthesis of high value products such as the incorporation of 13C- and 14C-labeled carbon atoms into pharmaceutically relevant molecules via reductive carboxylation recently described by Baran et al.38
Figure 4.

Carboxylation of 4-chloroanisole (0.25 mmol) with CO2 (1 atm) using DMAP-OED (105 mol %) as the reductant and LiCl (110 mol %) as an additive in DMI (0.375 mL) at 25 °C under optimized reaction conditions for (a) low catalyst loadings (1 mol % (dppf)NiIICl2 for 20 h) and (b) short reaction times (5 mol % (dppf)NiIICl2 for 0.5 h). Yields are reported as the average of two trials and were determined by integration of 1H NMR spectra against a hexamethylbenzene external standard.
Role of Amide-Containing Solvents in Catalysis.
The most significant synthetic challenge associated with reductive carboxylation reactions is the requirement of an amide-based solvent. In fact, every reductive carboxylation reaction of an organic halide or pseudohalide reported to date has been performed in a highly undesirable amide-based solvent.8–11 Therefore, we sought to understand the critical nature of amide-based solvents in catalysis. As part of our mechanistic work, we established that the MnCl2 generated in situ upon oxidation of Mn0 plays a critical role in catalysis. Similarly, when DMAP-OED was used as the reductant, addition of MnCl2 (or a related soluble alkali metal salt) was necessary for appreciable amounts of product to be formed. However, while MnCl2 is highly soluble in amide-based solvents such as DMI, it is only sparingly soluble in more synthetically desirable solvents such as THF. On this basis, we hypothesized that the reason catalytic activity is not observed in non-amide-based solvents is because the MnCl2, which is both the required Lewis acid and halide source in carboxylation, is not soluble in these solvents. Our previous results showed that simple alkali metal salts such as LiCl, which are soluble in THF, can be used instead of MnCl2 to promote carboxylation in DMI. Given these results, we performed a carboxylation reaction in THF using 4-chloroanisole as the substrate, (dppf)NiIICl2 as the precatalyst, DMAP-OED as the reductant, and LiCl as the additive. Consistent with our hypothesis the reaction was successful and an 86% yield of 4-anisic was obtained (Figure 5). This reaction is the f irst reported example of a successf ul reductive carboxylation of any organic halide or pseudohalide performed in a non-amide-containing solvent. Interestingly, with the proper choice of additive (LiI), catalysis can also be performed in THF using Mn0 as the electron source (see SI). It is likely that our observation that carboxylation reactions involving aryl halides can be performed in non-amide-containing solvent if the Lewis acid and halide sources are soluble extends to other reductive carboxylation reactions. As a result, our method of employing DMAP-OED as a reductant and LiCl as an additive may be a broadly applicable strategy for performing this class of reactions in more synthetically desirable solvents. Additionally, since reductive carboxylation reactions involve related intermediates to those invoked in other reductive coupling reactions, such as sp3−sp2 couplings, DMAP-OED may also find utility as a reductant in the broader class of cross-electrophile coupling.16
Figure 5.

Carboxylation of 4-chloroanisole (0.25 mmol) with CO2 (1 atm) using (dppf)NiIICl2 (2.5 mol %) as the precatalyst, DMAP-OED (105 mol %) as the reductant, and LiCl (110 mol %) as an additive in THF (0.75 mL) at 25 °C for 20 h. Volume of THF was increased to accommodate LiCl solubility. Yields are reported as the average of two trials and were determined by integration of 1H NMR spectra against a hexamethylbenzene external standard.
Substrate Scope.
Using (dppf)NiIICl2 as the precatalyst, DMAP-OED as the reductant, and LiCl as an additive, the scope of the reaction was explored in THF (Figure 6). Aryl chlorides containing both electron-donating and -withdrawing groups were carboxylated in high yields (1a−c). Aryl chlorides bearing simple electron-donating groups, such as 4-chloroanisole, could be carboxylated at 2.5 mol % catalyst loadings at 25°C, whereas elevated temperatures and catalyst loadings were required for substrates with electron-withdrawing groups. The carboxylation of ortho-substituted aryl bromides,8n,9a,d iodides,11 and triflates8g have been reported; however, carboxylation of less expensive and more commercially available aryl chlorides bearing ortho substitution had not been successful. To this end, in contrast to Tsuji’s carboxylation system,8a our system is able to carboxylate ortho-substituted aryl chloride electrophiles for the first time owing to the use of the homogeneous reductant DMAP-OED (vide supra). In fact, both mono-ortho-substituted aryl chlorides and di-ortho-substituted aryl bromides were carboxylated in good yields (1d−f). A reaction with a di-ortho-substituted aryl chloride was unsuccessful. Pseudohalide electrophiles, including synthetically valuable phenol derivatives, were readily carboxylated under our reaction conditions. For example, similar to Tsuji’s initial report,8a phenyl triflate and tosylate could be carboxylated to benzoic acid in good yields (1g, 1h), with successful reactions of phenyl triflate occurring at 25 °C with a catalyst loading of 2.5 mol %. Additionally, aryl sulfamates and pivalates could be carboxylated in moderate yields (1i, 1j). Martin also demonstrated carboxylation of 1j;8c however, this is the first report of a sulfamate electrophile, which can be used as a directing group for C−H activation of aryl substrates,36c being utilized in any cross-electrophile coupling reaction, demonstrating the broad applicability of our system to various pseudohalide substrates.
Figure 6.

Isolated yields of products for the carboxylation of aryl halides and pseudohalides. (a) Yields are reported as the average of two trials. (b) Reaction conditions: substrate (0.25 mmol), (dppf)NiIICl2 (0.0125 mmol), LiCl (0.375 mmol), DMAP-OED (0.2625 mmol), CO2 (1 atm) in THF (0.750 mL) at 50 °C for 20 h. (c) (dppf)NiIICl2 (0.00625 mmol) at 25 °C. (d) (dppf)NiIICl2 (0.025 mmol) for 40 h. (e) (PPh3)2NiIICl2 (0.0125 mmol) and PPh3 (0.025 mmol) instead of (dppf)NiIICl2. (f) (PCy3)2NiIICl2 (0.025 mmol) instead of (dppf)NiIICl2.
Although our (dppf)NiIICl2 precatalyst is effective for the carboxylation of a range of sterically congested aryl halide electrophiles, low selectivity is observed when using a substrate with a functional group, such as an ester. In this reaction, a significant amount of biaryl homocoupling product is formed from the electrophile. We observed that, in this case, using Tsuji’s precatalyst and 2 equiv of PPh3 in place of (dppf)NiIICl2 provides higher selectivity for the desired carboxylated product over the biaryl product. We suggest that this difference in selectivity arises because of the stabilizing effect that excess PPh3 has on (PPh3)2NiII(Ar)(Cl) and (PPh3)nNiI(Ar) intermediates in catalysis (vide supra). This effect is not observed when using an excess of the bidentate ligand dppf and the (dppf)NiIICl2 precatalyst. Using Tsuji’s precatalyst, we are able to carboxylate an aryl chloride bearing an ester functional group (1l), which suggests that the high chemoselectivity that has been observed in reductive carboxylation reactions in amide-based solvents with metallic reductants is preserved when performing the same reactions in THF with an organic reductant.
The observation that Tsuji’s precatalyst is effective under our newly developed conditions, with an organic reductant and non-amide-containing solvent, is significant, as it raises the possibility that our conditions may be directly translatable to carboxylation reactions with other substrates that are facilitated by different catalysts. This would enable us to carboxylate a range of substrates without needing to fully reoptimize the system to accommodate the organic reductant, LiCl additive, and change in solvent. To explore this hypothesis, we performed catalysis with the sp3-hybridized substrate 9-bromofluorene (1m), using the literature precatalyst ((PCy3)2NiIICl2) under our conditions.8b The desired carboxylic acid was isolated in 51% yield, which is comparable to the yield previously reported in the literature (60%). This result suggests that previous work identifying the optimal metal−ligand combination for a specific carboxylation reaction using metallic reductants and amide containing solvents can simply be translated to our new conditions, which are potentially broadly applicable for performing reductive carboxylation reactions under more synthetically desirable conditions.
CONCLUSIONS
In this work, we have used a model system to study the mechanism of nickel-catalyzed carboxylation reactions of organic halides and pseudohalides. Our results explain why certain reagents and additives are required for catalysis. For instance, we show that in previous examples of nickel-catalyzed carboxylation reactions involving aryl halides, heterogeneous reductants, such as Mn0 or Zn0, were required because they generate MnCl2 or ZnCl2 salts upon oxidation, which act as necessary sources of a Lewis acid and halide in catalysis. The Lewis acid assists with CO2 insertion, which we demonstrate by establishing that Lewis acids increase the rate of CO2 insertion into a NiI aryl complex to generate a carboxylic acid (after workup). The halide source is proposed to undergo a ligand-exchange reaction and facilitate reduction of the proposed NiI carboxylate to Ni0, which regenerates the active catalyst. On the basis of these experiments, we propose a revised mechanism for carboxylation in which we have strong evidence for most of the elementary reactions and an understanding of the factors that are important for promoting catalysis. In the case of nickel-catalyzed carboxylation of aryl halides, we used our mechanistic insight into provide strategies to address many of the current challenges associated with these reactions. For example, by using LiCl as a cost-effective additive, which provides both a Lewis acid and halide source, we can, for the first time, perform catalysis using a stoichiometric amount of an easily prepared solid organic reductant instead of a vast excess of a heterogeneous metallic reductant. The use of a homogeneous organic reductant should allow carboxylation reactions to be performed on scale in situations where the use of heterogeneous reductants is problematic. Additionally, by using LiCl as an additive, we can perform catalytic reactions in non-amide-based solvents, such as THF, because the required Lewis acid and halide source is now soluble under the reaction conditions. The fact that previous systems for carboxylation could only operate in non-amide-based solvents was a major limitation and our advance should also assist in making carboxylation reactions more practical. Our mechanistic studies have also enabled us to lower the catalyst loadings required and increase the substrate scope. Furthermore, in preliminary studies we have demonstrated that our use of an organic reductant in a non-amide-based solvent is generalizable to carboxylation reactions involving alkyl halides. This suggests that it may be possible to apply our findings to many other carboxylation reactions. Finally, our results are likely also relevant to cross-electrophile coupling reactions that do not involve CO2, as the use of superstoichiometric amounts of metallic reductants has also been a problem in these reactions. In the future, our laboratory intends to more fully evaluate the scope of cross-electrophile coupling reactions that can be facilitated through the combination of a solid organic reductant and soluble Lewis acid source.
Supplementary Material
ACKNOWLEDGMENTS
N.H. acknowledges support from the NIHGMS under Award No. R01GM120162. The EPR spectroscopy work was supported by the Department of Energy, Office of Basic Energy Sciences, Division of Chemical Sciences Grant No. DE-FG02–05ER15646 (H.M.C.L. and G.W.B.). N.H. is a Camille and Henry Dreyfus Foundation Teacher−Scholar. We are grateful to Rubén Martín, John Murphy, Megan Mohadjer Beromi, and Rosemarie Somerville for valuable discussions and Brandon Mercado and Nicholas Smith for assistance with X-ray crystallography.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscatal.9b00566.
Full characterization data, experimental procedures, and details about EPR spectra (PDF)
X-ray data for NiIAryl complex (CIF)
Notes
The authors declare no competing financial interest.
REFERENCES
- (1).(a) Darensbourg DJ Chemistry of Carbon Dioxide Relevant to Its Utilization: A Personal Perspective. Inorg. Chem 2010, 49, 10765–10780. [DOI] [PubMed] [Google Scholar]; (b) Riduan SN; Zhang Y Recent Developments in Carbon Dioxide Utilization Under Mild Conditions. Dalton Trans 2010, 39, 3347–3357. [DOI] [PubMed] [Google Scholar]; (c) Cokoja M; Bruckmeier C; Rieger B; Herrmann WA; Kühn FE Transformation of Carbon Dioxide with Homogeneous Transition-Metal Catalysts: A Molecular Solution to a Global Challenge? Angew. Chem., Int. Ed 2011, 50, 8510–8537. [DOI] [PubMed] [Google Scholar]; (d) Peters M; Köhler B; Kuckshinrichs W; Leitner W; Markewitz P; Müller TE Chemical Technologies for Exploiting and Recycling Carbon Dioxide into the Value Chain. ChemSusChem 2011, 4, 1216–1240. [DOI] [PubMed] [Google Scholar]; (e) Appel AM; Bercaw JE; Bocarsly AB; Dobbek H; DuBois DL; Dupuis M; Ferry JG; Fujita E; Hille R; Kenis PJA; Kerfeld CA; Morris RH; Peden CHF; Portis AR; Ragsdale SW; Rauchfuss TB; Reek JNH; Seefeldt LC; Thauer RK; Waldrop GL Frontiers, Opportunities, and Challenges in Biochemical and Chemical Catalysis of CO2 Fixation. Chem. Rev 2013, 113, 6621–6658. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Centi G; Quadrelli EA; Perathoner S Catalysis for CO2 Conversion: A Key Technology for Rapid Introduction of Renewable Energy in the Value Chain of Chemical Industries. Energy Environ. Sci 2013, 6, 1711–1731. [Google Scholar]; (g) Aresta M; Dibenedetto A; Angelini A Catalysis for the Valorization of Exhaust Carbon: from CO2 to Chemicals, Materials, and Fuels. Technological Use of CO2. Chem. Rev 2014, 114, 1709–1742. [DOI] [PubMed] [Google Scholar]; (h) Wang W-H; Himeda Y; Muckerman JT; Manbeck GF; Fujita E CO2 Hydrogenation to Formate and Methanol as an Alternative to Photo- and Electrochemical CO2 Reduction. Chem. Rev 2015, 115, 12936–12973. [DOI] [PubMed] [Google Scholar]; (i) Klankermayer J; Wesselbaum S; Beydoun K; Leitner W Selective Catalytic Synthesis Using the Combination of Carbon Dioxide and Hydrogen: Catalytic Chess at the Interface of Energy and Chemistry. Angew. Chem., Int. Ed 2016, 55, 7296–7343. [DOI] [PubMed] [Google Scholar]; (j) Bernskoetter WH; Hazari N Reversible Hydrogenation of Carbon Dioxide to Formic Acid and Methanol: Lewis Acid Enhancement of Base Metal Catalysts. Acc. Chem. Res 2017, 50, 1049–1058. [DOI] [PubMed] [Google Scholar]; (k) Artz J; Müller TE; Thenert K; Kleinekorte J; Meys R; Sternberg A; Bardow A; Leitner W Sustainable Conversion of Carbon Dioxide: An Integrated Review of Catalysis and Life Cycle Assessment. Chem. Rev 2018, 118, 434–504. [DOI] [PubMed] [Google Scholar]
- (2).(a) Martín R; Kleij AW Myth or Reality? Fixation of Carbon Dioxide into Complex Organic Matter under Mild Conditions. ChemSusChem 2011, 4, 1259–1263. [DOI] [PubMed] [Google Scholar]; (b) Huang K; Sun C-L; Shi Z-J Transition-Metal-Catalyzed C−C Bond Formation Through the Fixation of Carbon Dioxide. Chem. Soc. Rev 2011, 40, 2435–2452. [DOI] [PubMed] [Google Scholar]; (c) Omae I Recent Developments in Carbon Dioxide Utilization for the Production of Organic Chemicals. Coord. Chem. Rev 2012, 256, 1384–1405. [Google Scholar]; (d) Maeda C; Miyazaki Y; Ema T Recent Progress in Catalytic Conversions of Carbon Dioxide. Catal. Sci. Technol 2014, 4, 1482–1497. [Google Scholar]; (e) Liu Q; Wu L; Jackstell R; Beller M Using Carbon Dioxide as a Building Block in Organic Synthesis. Nat. Commun 2015, 6, 5933. [DOI] [PubMed] [Google Scholar]; (f) Yu D; Teong SP; Zhang Y Transition Metal Complex Catalyzed Carboxylation Reactions with CO2. Coord. Chem. Rev 2015, 293–294, 279–291.; (g) Börjesson M; Moragas T; Gallego D; Martin R Metal-Catalyzed Carboxylation of Organic (Pseudo)halides with CO2. ACS Catal 2016, 6, 6739–6749. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Luo J; Larrosa I C−H Carboxylation of Aromatic Compounds through CO2 Fixation. ChemSusChem 2017, 10, 3317–3332. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Tortajada A; Juliá-Hernández F; Börjesson M; Moragas T; Martin R Transition-Metal-Catalyzed Carboxylation Reactions with Carbon Dioxide. Angew. Chem., Int. Ed 2018, 57, 15948–15982. [DOI] [PubMed] [Google Scholar]
- (3).(a) Parai S The Chemistry of Acid Derivatives; Wiley: New York, 1992. [Google Scholar]; (b) Goossen LJ; Rodríguez N; Goossen K Carboxylic Acids as Substrates in Homogeneous Catalysis. Angew. Chem., Int. Ed 2008, 47, 3100. [DOI] [PubMed] [Google Scholar]
- (4).Sakakura T; Choi J-C; Yasuda H Transformation of Carbon Dioxide. Chem. Rev 2007, 107, 2365–2387. [DOI] [PubMed] [Google Scholar]
- (5).Caron S; Dugger RW; Ruggeri SG; Ragan JA; Ripin DHB Large-Scale Oxidations in the Pharmaceutical Industry. Chem. Rev 2006, 106, 2943–2989. [DOI] [PubMed] [Google Scholar]
- (6).Debabov VG; Yanenko AS Biocatalytic Hydrolysis of Nitriles. Rev. J. Chem 2011, 1, 385–402. [Google Scholar]
- (7).(a) Takimoto M; Kawamura M; Mori M; Sato Y Nickel-Catalyzed Regio- and Stereoselective Double Carboxylation of Trimethylsilylallene Under an Atmosphere of Carbon Dioxide and its Application to the Synthesis of Chaetomellic Acid A Anhydride. Synlett 2005, 2019–2022.; (b) Williams CM; Johnson JB; Rovis T Nickel-Catalyzed Reductive Carboxylation of Styrenes Using CO2. J. Am. Chem. Soc 2008, 130, 14936–14937. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Li S; Yuan W; Ma S Highly Regio- and Stereoselective Three-Component Nickel-Catalyzed syn-Hydrocarboxylation of Alkynes with Diethyl Zinc and Carbon Dioxide. Angew. Chem., Int. Ed 2011, 50, 2578–2582. [DOI] [PubMed] [Google Scholar]; (d) Mizuno T; Oonishi Y; Takimoto M; Sato Y Total Synthesis of (−)-Corynantheidine by Nickel-Catalyzed Carboxylative Cyclization of Enynes. Eur. J. Org. Chem 2011, 2011, 2606–2609. [Google Scholar]; (e) Fujihara T; Horimoto Y; Mizoe T; Sayyed FB; Tani Y; Terao J; Sakaki S; Tsuji Y Nickel-Catalyzed Double Carboxylation of Alkynes Employing Carbon Dioxide. Org. Lett 2014, 16, 4960–4963. [DOI] [PubMed] [Google Scholar]; (f) Wang X; Nakajima M; Martin R Ni-Catalyzed Regioselective Hydrocarboxylation of Alkynes with CO2 by Using Simple Alcohols as Proton Sources. J. Am. Chem. Soc 2015, 137, 8924–8927. [DOI] [PubMed] [Google Scholar]; (g) Kirillov E; Carpentier JF; Bunel E Carboxylic Acid Derivatives via Catalytic Carboxylation of Unsaturated Hydrocarbons: Whether the Nature of a Reductant may Determine the Mechanism of CO2 Incorporation? Dalton Trans 2015, 44, 16212–16223. [DOI] [PubMed] [Google Scholar]; (h) Cao T; Ma S Highly Stereo- and Regioselective Hydrocarboxylation of Diynes with Carbon Dioxide. Org. Lett 2016, 18, 1510–1513. [DOI] [PubMed] [Google Scholar]; (i) Nogi K; Fujihara T; Terao J; Tsuji Y Carboxyzincation Employing Carbon Dioxide and Zinc Powder: Cobalt-Catalyzed Multicomponent Coupling Reactions with Alkynes. J. Am. Chem. Soc 2016, 138, 5547–5550. [DOI] [PubMed] [Google Scholar]; (j) Juliá-Hernández F; Gaydou M; Serrano E; van Gemmeren M; Martin R Ni- and Fecatalyzed Carboxylation of Unsaturated Hydrocarbons with CO2. Top. Curr. Chem 2016, 374, 45. [DOI] [PubMed] [Google Scholar]; (k) Cao T; Yang Z; Ma S Selectivities in Nickel-Catalyzed Hydrocarboxylation of Enynes with Carbon Dioxide. ACS Catal 2017, 7, 4504–4508. [Google Scholar]; (l) Diccianni JB; Heitmann T; Diao T Nickel-Catalyzed Reductive Cycloisomerization of Enynes with CO2. J. Org. Chem 2017, 82, 6895–6903. [DOI] [PubMed] [Google Scholar]; (m) Doi R; Abdullah I; Taniguchi T; Saito N; Sato Y Nickel-Catalyzed Hydrocarboxylation of Ynamides with CO2 and H2O: Observation of Unexpected Regioselectivity. Chem. Commun 2017, 53, 7720–7723. [DOI] [PubMed] [Google Scholar]; (n) Gaydou M; Moragas T; Juliá-Hernández F; Martin R Site-Selective Catalytic Carboxylation of Unsaturated Hydrocarbons with CO2 and Water. J. Am. Chem. Soc 2017, 139, 12161–12164. [DOI] [PubMed] [Google Scholar]; (o) Higuchi Y; Mita T; Sato Y Palladium-Catalyzed Intramolecular Arylative Carboxylation of Allenes with CO2 for the Construction of 3-Substituted Indole-2-Carboxylic Acids. Org. Lett 2017, 19, 2710–2713. [DOI] [PubMed] [Google Scholar]; (p) Murata K; Numasawa N; Shimomaki K; Takaya J; Iwasawa N Construction of a Visible Light-Driven Hydrocarboxylation Cycle of Alkenes by the Combined use of Rh(I) and Photoredox Catalysts. Chem. Commun 2017, 53, 3098–3101. [DOI] [PubMed] [Google Scholar]; (q) Meng Q-Y; Wang S; Huff GS; König B Ligand-Controlled Regioselective Hydrocarboxylation of Styrenes with CO2 by Combining Visible Light and Nickel Catalysis. J. Am. Chem. Soc 2018, 140, 3198–3201. [DOI] [PubMed] [Google Scholar]; (r) Tortajada A; Ninokata R; Martin R Ni-Catalyzed Site-Selective Dicarboxylation of 1,3-Dienes with CO2. J. Am. Chem. Soc 2018, 140, 2050–2053. [DOI] [PubMed] [Google Scholar]
- (8).(a) For Ni-catalyzed systems, see:Fujihara T; Nogi K; Xu T; Terao J; Tsuji Y Nickel-Catalyzed Carboxylation of Aryl and Vinyl Chlorides Employing Carbon Dioxide. J. Am. Chem. Soc 2012, 134, 9106–9109. [DOI] [PubMed] [Google Scholar]; (b) León T; Correa A; Martin R Ni-Catalyzed Direct Carboxylation of Benzyl Halides with CO2. J. Am. Chem. Soc 2013, 135, 1221–1224. [DOI] [PubMed] [Google Scholar]; (c) Correa A; León T; Martin R Ni-Catalyzed Carboxylation of C(sp2)− and C(sp3)−O Bonds with CO2. J. Am. Chem. Soc 2014, 136, 1062–1069. [DOI] [PubMed] [Google Scholar]; (d) Liu Y; Cornella J; Martin R Ni-Catalyzed Carboxylation of Unactivated Primary Alkyl Bromides and Sulfonates with CO2. J. Am. Chem. Soc 2014, 136, 11212–11215. [DOI] [PubMed] [Google Scholar]; (e) Moragas T; Cornella J; Martin R Ligand-Controlled Regiodivergent Ni-Catalyzed Reductive Carboxylation of Allyl Esters with CO2. J. Am. Chem. Soc 2014, 136, 17702–17705. [DOI] [PubMed] [Google Scholar]; (f) Rebih F; Andreini M; Moncomble A; Harrison-Marchand A; Maddaluno J; Durandetti M Direct Carboxylation of Aryl Tosylates by CO2 Catalyzed by In situ-Generated Ni0. Chem. - Eur. J 2016, 22, 3758–3763. [DOI] [PubMed] [Google Scholar]; (g) Nogi K; Fujihara T; Terao J; Tsuji Y Cobalt- and Nickel-Catalyzed Carboxylation of Alkenyl and Sterically Hindered Aryl Triflates Utilizing CO2. J. Org. Chem 2015, 80, 11618–11623. [DOI] [PubMed] [Google Scholar]; (h) Wang X; Liu Y; Martin R Ni-Catalyzed Divergent Cyclization/Carboxylation of Unactivated Primary and Secondary Alkyl Halides with CO2. J. Am. Chem. Soc 2015, 137, 6476–6479. [DOI] [PubMed] [Google Scholar]; (i) Moragas T; Martin R Nickel-Catalyzed Reductive Carboxylation of Cyclopropyl Motifs with Carbon Dioxide. Synthesis 2016, 48, 2816–2822. [Google Scholar]; (j) Moragas T; Gaydou M; Martin R Nickel-Catalyzed Carboxylation of Benzylic C−N Bonds with CO2. Angew. Chem., Int. Ed 2016, 55, 5053–5057. [DOI] [PubMed] [Google Scholar]; (k) Börjesson M; Moragas T; Martin R Ni-Catalyzed Carboxylation of Unactivated Alkyl Chlorides with CO2. J. Am. Chem. Soc 2016, 138, 7504–7507. [DOI] [PubMed] [Google Scholar]; (l) van Gemmeren M; Börjesson M; Tortajada A; Sun S-Z; Okura K; Martin R Switchable Site-Selective Catalytic Carboxylation of Allylic Alcohols with CO2. Angew. Chem., Int. Ed 2017, 56, 6558–6562. [DOI] [PubMed] [Google Scholar]; (m) Juliá-Hernández F; Moragas T; Cornella J; Martin R Remote Carboxylation of Halogenated Aliphatic Hydrocarbons with Carbon Dioxide. Nature 2017, 545, 84. [DOI] [PubMed] [Google Scholar]; (n) Meng Q-Y; Wang S; König B Carboxylation of Aromatic and Aliphatic Bromides and Triflates with CO2 by Dual Visible-Light−Nickel Catalysis. Angew. Chem., Int. Ed 2017, 56, 13426–13430. [DOI] [PubMed] [Google Scholar]; (o) Chen Y-G; Shuai B; Ma C; Zhang X-J; Fang P; Mei T-S Regioselective Ni-Catalyzed Carboxylation of Allylic and Propargylic Alcohols with Carbon Dioxide. Org. Lett 2017, 19, 2969–2972. [DOI] [PubMed] [Google Scholar]
- (9).For Pd-catalyzed systems, see: (a) Correa A; Martín R Palladium-Catalyzed Direct Carboxylation of Aryl Bromides with Carbon Dioxide. J. Am. Chem. Soc 2009, 131, 15974–15975. [DOI] [PubMed] [Google Scholar]; (b) Mita T; Higuchi Y; Sato Y Highly Regioselective Palladium-Catalyzed Carboxylation of Allylic Alcohols with CO2. Chem. - Eur. J 2015, 21, 16391–16394. [DOI] [PubMed] [Google Scholar]; (c) Zhang S; Chen W-Q; Yu A; He L-N Palladium-Catalyzed Carboxylation of Benzyl Chlorides with Atmospheric Carbon Dioxide in Combination with Manganese/Magnesium Chloride. ChemCatChem 2015, 7, 3972–3977. [Google Scholar]; (d) Shimomaki K; Murata K; Martin R; Iwasawa N Visible-Light-Driven Carboxylation of Aryl Halides by the Combined Use of Palladium and Photoredox Catalysts. J. Am. Chem. Soc 2017, 139, 9467–9470. [DOI] [PubMed] [Google Scholar]
- (10).For Co-catalyzed systems, see ref 8g and: Nogi K; Fujihara T; Terao J; Tsuji Y Cobalt-Catalyzed Carboxylation of Propargyl Acetates with Carbon Dioxide. Chem. Commun 2014, 50, 13052–13055. [DOI] [PubMed] [Google Scholar]
- (11).For a Cu-catalyzed system, see: Tran-Vu H; Daugulis O Copper-Catalyzed Carboxylation of Aryl Iodides with Carbon Dioxide. ACS Catal 2013, 3, 2417–2420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Liao L-L; Cao G-M; Ye J-H; Sun G-Q; Zhou W-J; Gui Y-Y; Yan S-S; Shen G; Yu D-G Visible-Light-Driven External-Reductant-Free Cross-Electrophile Couplings of Tetraalkyl Ammonium Salts. J. Am. Chem. Soc 2018, 140, 17338–17342. [DOI] [PubMed] [Google Scholar]
- (13). Systems involving photoredox catalysis have been described for the carboxylation of aryl bromides and triflates; see refs 8n, 9d, and 12. These do not require metallic reductants, but a range of other challenges need to be solved to make these reactions practical.
- (14).Diorazio LJ; Hose DRJ; Adlington NK Toward a More Holistic Framework for Solvent Selection. Org. Process Res. Dev 2016, 20, 760–773. [Google Scholar]
- (15). By changing the ancillary ligand, Tsuji was able to carboxylate ortho-substituted aryl triflates; see ref 8g. However, to the best of our knowledge, there are no examples of the carboxylation of ortho-substituted aryl chlorides.
- (16).(a) Everson DA; Shrestha R; Weix DJ Nickel-Catalyzed Reductive Cross-Coupling of Aryl Halides with Alkyl Halides. J. Am. Chem. Soc 2010, 132, 920–921. [DOI] [PubMed] [Google Scholar]; (b) Everson DA; Weix DJ Cross-Electrophile Coupling: Principles of Reactivity and Selectivity. J. Org. Chem 2014, 79, 4793–4798. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Knappke CEI; Grupe S; Gaertner D; Corpet M; Gosmini C; Jacobi von Wangelin A. Reductive Cross-Coupling Reactions Between Two Electrophiles. Chem. - Eur. J 2014, 20, 6828–6842. [DOI] [PubMed] [Google Scholar]; (d) Weix DJ Methods and Mechanisms for Cross-Electrophile Coupling of Csp2 Halides with Alkyl Electrophiles. Acc. Chem. Res 2015, 48, 1767–1775. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Gu J; Wang X; Xue W; Gong H Nickel-Catalyzed Reductive Coupling of Alkyl Halides with Other Electrophiles: Concept and Mechanistic Considerations. Org. Chem. Front 2015, 2, 1411–1421. [Google Scholar]; (f) Richmond E; Moran J Recent Advances in Nickel Catalysis Enabled by Stoichiometric Metallic Reducing Agents. Synthesis 2018, 50, 499–513. [Google Scholar]
- (17).Tsou TT; Kochi JK Mechanism of Oxidative Addition. Reaction of Nickel(0) Complexes with Aromatic Halides. J. Am. Chem. Soc 1979, 101, 6319–6332. [Google Scholar]
- (18).Lin C-Y; Power PP Complexes of Ni(I): A “Rare” Oxidation State of Growing Importance. Chem. Soc. Rev 2017, 46, 5347–5399. [DOI] [PubMed] [Google Scholar]
- (19).Anka-Lufford LL; Huihui KMM; Gower NJ; Ackerman LKG; Weix DJ Nickel-Catalyzed Cross-Electrophile Coupling with Organic Reductants in Non-Amide Solvents. Chem. - Eur. J 2016, 22, 11564–11567. [DOI] [PubMed] [Google Scholar]
- (20).Aranzaes JR; Daniel M-C; Astruc D Metallocenes as References for the Determination of Redox Potentials by Cyclic Voltammetry. Permethylated Iron and Cobalt Sandwich Complexes, Inhibition by Polyamine Dendrimers, and the Role of Hydroxy-Containing Ferrocenes. Can. J. Chem 2006, 84, 288–299. [Google Scholar]
- (21).Liang G; DeYonker NJ; Zhao X; Webster CE Prediction of the Reduction Potential in Transition-Metal Containing Complexes: How expensive? For What Accuracy? J. Comput. Chem 2017, 38, 2430–2438. [DOI] [PubMed] [Google Scholar]
- (22).Eberle B; Hübner O; Ziesak A; Kaifer E; Himmel H-J What Makes a Strong Organic Electron Donor (or Acceptor)? Chem. - Eur. J 2015, 21, 8578–8590. [DOI] [PubMed] [Google Scholar]
- (23).Murphy JA; Garnier J; Park SR; Schoenebeck F; Zhou S.-z.; Turner AT Super-Electron Donors: Bis-pyridinylidene Formation by Base Treatment of Pyridinium Salts. Org. Lett 2008, 10, 1227–1230. [DOI] [PubMed] [Google Scholar]
- (24).Birbilis N; Buchheit RG Electrochemical Characteristics of Intermetallic Phases in Aluminum Alloys: An Experimental Survey and Discussion. J. Electrochem. Soc 2005, 152, B140–B151. [Google Scholar]
- (25).Iyoda M; Sakaitan M; Otsuka H; Oda M Reductive Coupling of Benzyl Halides Using Nickel(0)-Complex Generated In Situ in the Presence of the Tetraethylammonium Iodide, A Simple and Convenient Synthesis of BiBenzyls. Chem. Lett 1985, 14, 127–130. [Google Scholar]
- (26).(a) García-Domínguez A; Li Z; Nevado C Nickel-Catalyzed Reductive Dicarbofunctionalization of Alkenes. J. Am. Chem. Soc 2017, 139, 6835–6838. [DOI] [PubMed] [Google Scholar]; (b) Suzuki N; Hofstra JL; Poremba KE; Reisman SE Nickel-Catalyzed Enantioselective Cross-Coupling of N-Hydroxyphthalimide Esters with Vinyl Bromides. Org. Lett 2017, 19, 2150–2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Marcus Y; Hefter G Ion Pairing. Chem. Rev 2006, 106, 4585–4621. [DOI] [PubMed] [Google Scholar]
- (28).Sayyed FB; Sakaki S The Crucial Roles of MgCl2 as a Non-Innocent Additive in the Ni-Catalyzed Carboxylation of Benzyl Halide with CO2. Chem. Commun 2014, 50, 13026–13029. [DOI] [PubMed] [Google Scholar]
- (29).(a) Sayyed FB; Tsuji Y; Sakaki S The Crucial Role of a Ni(I) Intermediate in Ni-Catalyzed Carboxylation of Aryl Chloride with CO2: A Theoretical Study. Chem. Commun 2013, 49, 10715–10717. [DOI] [PubMed] [Google Scholar]; (b) Hazari N; Heimann JE Carbon Dioxide Insertion into Group 9 and 10 Metal−Element σ Bonds. Inorg. Chem 2017, 56, 13655–13678. [DOI] [PubMed] [Google Scholar]
- (30).(a) Zhang K; Conda-Sheridan M; Cooke SR; Louie J N-Heterocyclic Carbene Bound Nickel(I) Complexes and their Roles in Catalysis. Organometallics 2011, 30, 2546–2552. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Laskowski CA; Bungum DJ; Baldwin SM; Del Ciello SA; Iluc VM; Hillhouse GL Synthesis and Reactivity of Two-Coordinate Ni(I) Alkyl and Aryl Complexes. J. Am. Chem. Soc 2013, 135, 18272–18275. [DOI] [PubMed] [Google Scholar]; (c) Hatnean JA; Shoshani M; Johnson SA Mechanistic Insight into Carbon−Fluorine Cleavage with a (iPr3P)2Ni source: Characterization of (iPr3P)2NiC6F5 as a Significant Ni(I) byproduct in the Activation of C6F6. Inorg. Chim. Acta 2014, 422, 86–94. [Google Scholar]; (d) Menges FS; Craig SM; Tötsch N; Bloomfield A; Ghosh S; Krüger H-J; Johnson MA Capture of CO2 by a Cationic Nickel(I) Complex in the Gas Phase and Characterization of the Bound, Activated CO2 Molecule by Cryogenic Ion Vibrational Predissociation Spectroscopy. Angew. Chem., Int. Ed 2016, 55, 1282–1285. [DOI] [PubMed] [Google Scholar]; (e) Mohadje Beromi M.; Banerjee G; Brudvig GW; Hazari N; Mercado BQ Nickel(I) Aryl Species: Synthesis, Properties, and Catalytic Activity. ACS Catal 2018, 8, 2526–2533. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Diccianni JB; Katigbak J; Hu C; Diao T Mechanistic Characterization of (Xantphos)Ni(I)-Mediated Alkyl Bromide Activation: Oxidative Addition, Electron Transfer, or Halogen-Atom Abstraction. J. Am. Chem. Soc 2019, 141, 1788. [DOI] [PubMed] [Google Scholar]
- (31).Hwang SJ; Powers DC; Maher AG; Nocera DG Tandem Redox Mediator/Ni(II) Trihalide Complex Photocycle for Hydrogen Evolution From HCl. Chem. Sci 2015, 6, 917–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).(a) Moragas T; Correa A; Martin R Metal-Catalyzed Reductive Coupling Reactions of Organic Halides with Carbonyl-Type Compounds. Chem. - Eur. J 2014, 20, 8242–8258. [DOI] [PubMed] [Google Scholar]; (b) Lin T; Mi J; Song L; Gan J; Luo P; Mao J; Walsh PJ Nickel-Catalyzed Desymmetrizing Cross-Electrophile Coupling of Cyclic Meso-Anhydrides. Org. Lett 2018, 20, 1191–1194. [DOI] [PubMed] [Google Scholar]
- (33).(a) Darensbourg DJ; Rokicki A Reduction of Carbon Dioxide and Carbonyl Sulfide by Anionic Group VIB Metal Hydrides and Alkyls. Carbon-Hydrogen and Carbon-Carbon Bond Formation Processes and the Structure of [PNP][Cr(CO)5SC(O)H]. J. Am. Chem. Soc 1982, 104, 349–350. [Google Scholar]; (b) Darensbourg DJ; Pala M Cation-Anion Interaction in the [Na-kryptofix-221][W(CO)5O2CH] Derivative and its Relevance in Carbon Dioxide Reduction Processes. J. Am. Chem. Soc 1985, 107, 5687–5693. [Google Scholar]; (c) Lau K-C; Petro BJ; Bontemps S; Jordan RF Comparative Reactivity of Zr− and Pd−Alkyl Complexes with Carbon Dioxide. Organometallics 2013, 32, 6895–6898. [Google Scholar]; (d) Heimann JE; Bernskoetter WH; Hazari N; Mayer JM Acceleration of CO2 Insertion into Metal Hydrides: Ligand, Lewis Acid, and Solvent Effects on Reaction Kinetics. Chem. Sci 2018, 9, 6629–6638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Recently, a bimetallic nickel(I) complex was reported with a bridging aryl and carboxylate ligand: Somerville RJ; Hale LVA; Gómez-Bengoa E; Burés J; Martin R Intermediacy of Ni−Ni Species in sp2 C−O Bond Cleavage of Aryl Esters: Relevance in Catalytic C−Si Bond Formation. J. Am. Chem. Soc 2018, 140, 8771–8780. [DOI] [PubMed] [Google Scholar]
- (35).Aresta M; Nobile CF; Albano VG; Forni E; Manassero M New Nickel−Carbon Dioxide Complex: Synthesis, Properties, and Crystallographic Characterization of (carbon dioxide)-bis-(tricyclohexylphosphine)nickel. J. Chem. Soc., Chem. Commun 1975, 0, 636–637. [Google Scholar]
- (36).(a) Guard LM; Mohadjer Beromi M.; Brudvig GW; Hazari N; Vinyard DJ Comparison of dppf-Supported Ni Precatalysts for the Suzuki-Miyaura Reaction: The Observation and Activity of Ni(I). Angew. Chem., Int. Ed 2015, 54, 13352–13356. [DOI] [PubMed] [Google Scholar]; (b) Yin G; Kalvet I; Englert U; Schoenebeck F Fundamental Studies and Development of Nickel-Catalyzed Trifluoromethylthiolation of Aryl Chlorides: Active Catalytic Species and Key Roles of Ligand and Traceless MeCN Additive Revealed. J. Am. Chem. Soc 2015, 137, 4164–4172. [DOI] [PubMed] [Google Scholar]; (c) Mohadjer Beromi M.; Nova A; Balcells D; Brasacchio AM; Brudvig GW; Guard LM; Hazari N; Vinyard DJ Mechanistic Study of an Improved Ni Precatalyst for Suzuki-Miyaura Reactions of Aryl Sulfamates: Understanding the Role of Ni(I) Species. J. Am. Chem. Soc 2017, 139, 922–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).(a) Beh DW; Piers WE; del Rosal I; Maron L; Gelfand BS; Gendy C; Lin J-B Scandium Alkyl and Hydride Complexes Supported by a Pentadentate Diborate Ligand: Reactions with CO2 and N2O. Dalton Trans 2018, 47, 13680–13688. [DOI] [PubMed] [Google Scholar]; (b) Jin D; Schmeier TJ; Williard PG; Hazari N; Bernskoetter WH Lewis Acid Induced β-Elimination from a Nickelalactone: Efforts toward Acrylate Production from CO2 and Ethylene. Organometallics 2013, 32, 2152–2159. [Google Scholar]; (c) Jin D; Williard PG; Hazari N; Bernskoetter WH Effect of Sodium Cation on Metallacycle β-Hydride Elimination in CO2−Ethylene Coupling to Acrylates. Chem. - Eur. J 2014, 20, 3205–3211. [DOI] [PubMed] [Google Scholar]; (d) Rauch M; Parkin G Zinc and Magnesium Catalysts for the Hydrosilylation of Carbon Dioxide. J. Am. Chem. Soc 2017, 139, 18162–18165. [DOI] [PubMed] [Google Scholar]
- (38).Kingston C; Wallace MA; Allentoff AJ; deGruyter JN; Chen JS; Gong SX; Bonacorsi S; Baran PS Direct Carbon Isotope Exchange through Decarboxylative Carboxylation. J. Am. Chem. Soc 2019, 141, 774–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.