

Abstract
Break induced replication (BIR) is a pathway that repairs one-ended double strand breaks (DSBs). For decades, yeast model systems offered the only opportunities to study eukaryotic BIR. These studies described an unusual mode of BIR synthesis that is carried out by a migrating bubble and shows conservative inheritance of newly synthesized DNA, leading to genomic instabilities like those associated with cancer in humans. Yet, evidence of BIR functioning in mammals or during repair of other DNA breaks has been missing. Recent studies have uncovered multiple examples of BIR working in replication restart and repair of eroded telomeres in yeast and mammals as well as some unexpected findings, including the RAD51 independence of BIR. Strong interest remains in determining the variations in molecular mechanisms that drive and regulate BIR in different genetic backgrounds, across organisms, and particularly in the context of human disease.
Keywords: break-induced replication, alternative lengthening of telomeres, Rad51-dependent BIR, RAD51-independent BIR, MMBIR, mutation cluster
Why study BIR?
The integrity of DNA in living organisms is challenged by various factors that inflict damage upon DNA, which then requires repair to maintain normal cell function (see Box 1 for details on DNA damage). Double-strand DNA breaks (DSBs), the most deleterious type of DNA lesions, can be repaired by either non-homologous end-joining (NHEJ) or homologous recombination (HR) (reviewed in [1, 2]). Historically, HR was regarded as the “safer” repair option, as DSB end processing during NHEJ introduces small insertions or deletions that frequently manifest as missense or frameshift mutations. Conversely, HR pathways utilize an intact homologous DNA molecule as a template for repair (see Box 2 for details of DSB repair pathways). However, recent findings suggest that HR promotes genome destabilization through mutagenesis and chromosomal rearrangements, with variable frequency of these events associated with the different HR pathways ([3–6], reviewed in [7]).
Box 1. DNA damage.
DNA Damage is an alteration in the chemical structure of DNA. Common types of DNA damage include DNA base modifications, e.g. base oxidation, deamination, alkylation, etc., DNA inter- and intra-strand crosslinks, and DNA single- and double strand breaks (SSBs and DSBs, respectively). Unrepaired DNA lesions compromise genomic integrity by impeding DNA replication and promoting mutations and chromosomal rearrangements.
Endogenous Sources of DNA damage (see [95] for more details) include reactive oxygen species or some other products of cell and DNA metabolism. Endogenous sources of DNA damage can lead to DNA base oxidation, alkylation, hydrolysis (producing deamination, depurination, and depyrimidination), and to formation of bulky adducts. Also, problems in DNA metabolism including problems in DNA replication and chromosomal segregation can lead to DNA breaks including both SSBs and DSBs. In addition, members of the apolipoprotein B mRNA editing enzymes catalytic polypeptide-like (APOBEC) family of cytidine deaminases have been recently identified as tumor-specific endogenous source of DNA damage (reviewed in [96, 97]). The main goal of APOBECs is to restrict retrovirus propagation and retrotransposon mobility by introducing multiple DNA lesions. However, APOBECs can also attack ssDNA regions formed in chromosomal DNA leading to formation of mutation clusters that are frequently associated with cancer in humans [96, 97].
Exogenous Sources of DNA damage (see [95] for more details) are external (environmental) agents including ionizing radiation (IR), ultraviolet radiation (UV) and a variety of chemical agents. IR, including alpha, beta, gamma, and X-ray radiation, can oxidize DNA bases by producing reactive oxygen species (ROSs), and also generate SSBs and DSBs threatening genomic integrity. UV irradiation promotes formation of photodimers, including cyclobutane pyrimidine dimers and 6,4-photoproducts that can impede DNA replication and promote mutagenesis. DNA intercalating agents (e.g. proflavine, acridine, etc.) include substances that insert themselves into DNA structure. DNA crosslinking agents, including cisplatin (which is often used in chemotherapy treatment), psoralen and Mitomycin C form covalent linkage between two DNA bases. DNA crosslinks impede DNA replication promoting replication collapse. DNA alkylating agents, including nitrogen mustards, dacarbazine used for chemotherapy, and also ethyl- and methyl methanesulfonate (EMS and MMS, respectively) that are frequently used in research experiments attach alkyl groups to DNA bases. For example, methyl methanesulfonate (MMS) is an alkylating agent that is often used in experiments and which can produce various alkylated bases, including N7-methylguanine, N3-methyladenine, N1-methyladenine, and N3-methylcytosine. The latter modification is most often responsible for the mutagenesis promoted by MMS in ssDNA [97, 98].
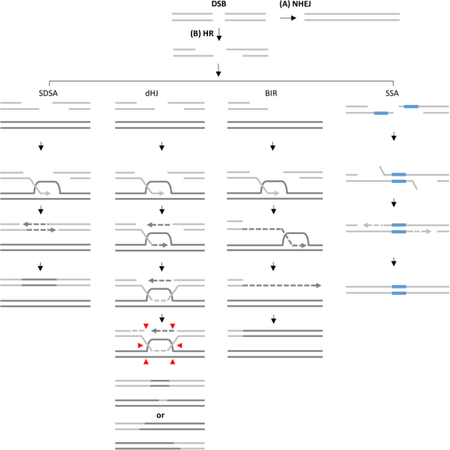
Box 2. DSB repair pathways (see the Box 2 figure for details).
A DSB can be repaired by either (A) Non Homologous End Joining (NHEJ) that ligates broken ends or (B) Homologous Recombination (HR) where following 5’−3’ resection, the DSB ends can be channeled into following four sub-pathways: (i) SDSA is a mechanism leading to gene conversion (GC), and is initiated by invasion of 3’ ssDNA end into a homologous template forming a displacement loop (D-loop). The 3’ end of the invading strand is then used as a primer to initiate new DNA synthesis. The other resected DSB end anneals to the newly synthesized strand once displaced from its template, and initiates a second round of DNA synthesis. (ii) The double Holliday junction (dHJ) pathway represents another GC mechanism that copies only a short DNA patch and involves strand invasion followed by stabilization of a D-loop via annealing to the second broken DSB end. The second round of DNA synthesis promotes double dHJ formation, and resolution (vertical or horizontal, red arrows) produces non-crossover or crossover outcomes. (iii) BIR is initiated when only one broken end is available for strand invasion. The invasion triggers DNA repair synthesis proceeding via a migrating bubble with asynchronous leading and lagging strand synthesis, and leads to conservative inheritance of newly-synthesized DNA. (iv) In SSA 5’−3’ resection continues until flanking homologous sequences are exposed and anneal to each other. The protruding non-homologous 3’ tails are clipped off and the 3’ ends are used as primers to fill in the gaps.
During most HR repair, the two broken DNA ends work in concert to invade a single, homologous DNA sequence, synthesize new DNA, and ligate to each other to repair the ruptured molecule. Break-induced replication (BIR) is a unique HR mechanism employed in situations wherein a single end of the DSB acts independently, which may occur when one side of the break fails to engage with a homologous sequence, or when the two ends find different homologous templates ( reviewed in [8]). A third scenario that promotes repair via BIR arises when the DSB manifests as a “one-ended break” , which can occur due to replication through a DNA lesion that results in fork stalling and collapse, or telomere erosion that exposes a single DNA end (reviewed in [8–10]).
Over the last 30 years, BIR has been reported in various model organisms in several different contexts. In bacteriophage T4 and Escherichia coli, BIR (termed recombination-dependent replication (RDR)) is required to repair stalled replication forks (reviewed in [11–13]). In eukaryotes, BIR is implicated in telomere maintenance in the absence of telomerase, as well as in replication fork restart [8–10]. As our understanding of the BIR mechanism has improved, it has become apparent that BIR is associated with elevated levels of mutagenesis and chromosomal rearrangements, many of which mimic genome lesions annotated in cancer cells [6, 14–19]. Nevertheless, BIR remained studied almost exclusively in the context of site-specific DSBs in yeast models until very recently. This review focuses on the recurring aspects of BIR with an emphasis on recent studies using yeast and human cells, including those addressing the role of BIR in replication fork restart and telomere maintenance.
The molecular mechanism of BIR: what we have learned from yeast
Eukaryotic BIR has been studied most extensively in budding yeast in experimental systems with controlled induction of DSBs. Most frequently, a site-specific HO endonuclease expressed under a galactose-regulated promoter was used to induce one-ended DSBs with homology located on another chromosome [20–23]. Similar to other HR pathways, BIR is preceded by extensive 5’-to-3’ end resection to generate a 3’ ssDNA end that is first bound by Replication Protein A (RPA), followed by Rad52-mediated formation of a Rad51 nucleoprotein filament that searches for a homologous template to invade, forming a D- loop (Figure 1) [21–26]. The beginning of DNA synthesis is often delayed by dissociation and reinvasion of the 3’ end [17], though the exact kinetics vary across experimental systems [22, 23, 25, 27–29]. However, the delay in BIR initiation is often sufficient to trigger G2/M cell cycle arrest, and, consequently, data from currently available experimental systems support that a majority of BIR events complete during G2/M [21–23, 27, 28]. Once synthesis begins, it often proceeds through the end of the template chromosome.
Figure 1. Model of RAD51-dependent BIR.

(A) Following DSB resection, the 3’ ssDNA end invades a homologous DNA molecule to form a D-loop and DNA synthesis is initiated (possibly with participation of Polα) at the 3’ invaded end. (B) DNA leading strand synthesis (dotted line) is carried out by Polδ or Polε. Progression of the D-loop is driven by a helicase, potentially Pif1 or MCM. (C) The newly synthesized leading strand persists as a long tract of ssDNA behind the BIR bubble and accumulates unrepaired DNA lesions (asterisks). (D) Polδ or Polε, and also Polα carry out lagging strand synthesis which might proceed through formation of Okazaki fragments. The translesion bypass of DNA damage in the template during lagging strand synthesis results in mutations and mutation clusters (rectangles). The remaining questions regarding the mechanism and participating proteins are indicated by question marks.
As noted above, the canonical BIR pathway is RAD51-dependent, although BIR was first described in the context of a less-efficient, RAD51-independent pathway. While both pathways require Rad52 [20, 23], a number of the other enzymes differ between the two. As specific examples, RAD51-independent BIR requires Rad59, Rdh54, and the Mre11/Rad50/Xrs2 (MRX) complex [30], whereas the RAD51-dependent pathway is dependent upon Rad54, Rad55 and Rad57 [23]. It has been proposed that Rad51 suppresses the Rad52-mediated annealing of ssDNA strands formed during extensive DSB resection that initiates RAD51-independent BIR [31, 32]. . Interestingly, RAD51-independent BIR described in yeast frequently involves interactions between repeated DNA elements that are often homeologous to each other, which can initiate genomic rearrangements that are suppressed by Rad51-mediated strand invasion [31, 32]. Although its incidence is well documented, many mechanistic details of RAD51-independent BIR remain mysterious. Our continuing discussion will focus on RAD51-dependent BIR, for which the molecular mechanism has been studied in detail.
The most unusual feature of RAD51-dependent BIR is its mode of DNA synthesis (Figure 1). BIR progresses via a migrating D-loop, or bubble, driven by branch migration of an unresolved Holliday junction that displaces the newly synthesized strand. In contrast with S-phase DNA replication, BIR synthesis is asynchronous; thus, the leading strand primed at the 3’-OH end accumulates as ssDNA while the D-loop migrates [14, 33]. To complete the repair, the lagging strand utilizes the leading strand as the template, resulting in conservative inheritance of the newly synthesized DNA [14, 28]. The identity of the main helicase that drives BIR progression remains unclear. MCM2–7 was originally proposed as the main replicative helicase in one yeast BIR experimental system [34], but was deemed dispensable in a different model [33]. Also, Pif1 helicase is essential for BIR to complete long-tract DNA synthesis, though its specific role in the process remains unknown [14, 33]. Hypotheses being considered position Pif1 ahead of the progressing D-loop to unwind the DNA duplex, or, alternatively, posit that Pif1 may function behind the D-loop to extract the newly synthesized DNA strand from the replication machinery [33]. Srs2 helicase activity is also relevant during BIR, but within the context of preventing formation of toxic structures resulting from invasion of the newly synthesized leading strand into the template duplex DNA [35].
Polε, Polα, and Polδ each participate in BIR DNA synthesis, though when and how each of these polymerases is used is not well understood. Evidence suggests that Polδ is the main polymerase executing leading-strand synthesis [22, 36, 37]. It has been demonstrated that the Pol32 subunit of Polδ, which is non-essential during S-phase DNA synthesis, is required for BIR, possibly by enabling strand displacement during bubble migration [22]. Studies demonstrate that Polα-primase participates in lagging-strand synthesis, and it is also believed to participate in initiation of leading-strand synthesis [22], although it is currently unclear what role it may play in this context. Also, data suggests that Polε is not required to initiate DNA synthesis during BIR; rather, it appears to promote efficient synthesis after BIR has been established [22]. Though our understanding of polymerase activity during BIR is improving, the requirement for these polymerases during S-phase replication and the difficulty to efficiently deplete them specifically during BIR synthesis has certainly affected our ability to address unanswered questions regarding the process of DNA synthesis during BIR with currently available systems.
The alternate, conservative mechanism of DNA synthesis executed during BIR confers an elevated risk of genetic instabilities compared with S phase DNA synthesis, including both increased frequencies of mutagenesis and chromosomal rearrangements [6,15–18, 27, 36–38]. At least two factors contribute to the increased level of frameshift mutations during BIR: (i) frequent dissociations of Polδ from the template due to the DNA synthesis within the D-loop (see in [8, 17, 27]), and (ii) reduced efficiency, versus its activity during S phase, of mismatch repair (MMR) in correcting DNA synthesis errors introduced during BIR [6]. The increased level of base substitutions result from unrepaired DNA damage accumulated in the long ssDNA tracts formed by leading strand BIR synthesis ([14], Figure 1C; D). Further, exposure of cells to ssDNA-specific damaging agents (for example, methyl methanesulfonate (MMS)[14]), specifically during BIR amplifies the accumulation of base substitutions and results in clusters of mutations within the newly synthesized DNA [16]. It remains unknown whether endogenous sources of DNA damage, such as APOBEC enzymes that mediate formation of mutation clusters in human cancer cells (See Box 1), may similarly induce mutagenesis during BIR. At the level of chromosomal rearrangements, premature resolution of BIR intermediates have been demonstrated to lead to formation of so-called half-crossovers, which resemble chromosome fusions annotated in human cancer cells ([36, 37], see also Figure 2 for details).
Figure 2. Mechanisms of chromosomal rearrangements associated with BIR.

Interruption of BIR synthesis can lead to a variety of possible outcomes: (A) Microhomology-mediated BIR (MMBIR, leading to complex genomic rearrangements similar to those underlying cancer and developmental disorders in humans (reviewed in [103]) occurs when the 3’-end of newly-synthesized DNA dissociates from its template and anneals in the region of microhomology (purple block) in the ssDNA accumulated behind the progressing BIR bubble [15]. Synthesis primed by a translesion polymerase at the re-annealed 3’ end proceeds (red dashes), but shortly disrupts, which leads to a second template switch by re-annealing back to the BIR track at a second microhomology (green block). (B) Half-crossovers are produced when the BIR is interrupted and the migrating bubble is resolved, leading to fusion of the recipient and donor chromosomes (shown in the blue and black, respectively) [36, 37]. The remaining portion of the broken donor chromosome can either be lost leading to formation of a half-crossover outcome (shown at right), or it can invade another donor or ectopic position leading to half-crossover cascades [38] resulting in chromosomal rearrangements (shown at left).
BIR in replication restart
BIR plays a crucial role in replication fork restart in bacteriophage T4, as well as in bacteria (see [12, 13, 40, 41]). In Escherichia coli, where replication is initiated from a single origin, BIR (termed RDR) is the primary pathway to ensure completion of replication following fork collapse (see in [13, 42]). Eukaryotes are less dependent on BIR to rescue replication fork collapse due to their multiple origins of replication: extensive repair synthesis from collapsed replication forks is avoided by merging with other forks. In an elegant experiment, Mayle et al observed [43] that BIR was initiated but quickly terminated via Mus81-mediated resolution of the D-loop that formed following collapse of an S-phase replication fork upon encountering a single DNA nick created by a modified FLP nuclease; the D-loop subsequently merged with a replication fork approaching from the opposite direction [43]. Long-tract BIR DNA synthesis was induced only when the DNA nick was generated in regions of the genome devoid of replication origins and within a MUS81-null background [43]. Overall, the literature supports that BIR is suppressed in S-phase, but precisely how this is accomplished remains unclear. One possibility is that Mus81 activity suppresses extensive BIR DNA synthesis during S-phase. Deletion of MUS81 only marginally effects BIR initiated during G2 [25], but this could be related to modifications in Mus81 interacting protein Mms4 that occur during G2 [44], which could potentially alter its substrate specificity and/or eliminate its suppressor function versus its activity during S phase, though these questions have not yet been tested experimentally. Other, currently unappreciated suppressors and regulators of BIR may also exist in eukaryotic cells.
Even if suppressed, BIR initiated by replication collapse does occur in eukaryotes, and studying some of these exceptional cases that have been documented provides an opportunity to hone in on factors that may serve to regulate (suppress) the process. One example is the frequent induction of BIR following profound replication stress (Figure 3, Key Figure, Box 3). In particular, overexpression of cyclin E in mammalian cells leads to massive collapse of replication forks and, consequently, DSBs (see in [45, 46]) that are repaired by BIR and yield chromosomal rearrangements [47, 48]. BIR has also been reported in cancer cells with deregulated origin licensing, which induces over-replication and massive replication fork collapse [49]. When re-replication occurs, DSBs form late in the cell cycle, at the end of S phase and possibly within G2, making these events similar to BIR studied extensively in yeast systems during G2/M arrest. It remains elusive why re-replication triggers BIR. Several factors may be at play, and additional clues on how BIR initiates and progresses following re-replication may eventually be revealed by studies in other eukaryotic models where re-replication has also been reported. For example, it was recently observed that re-replication initiated at yeast centromeres led to DNA breakage followed by RAD52-dependent repair similar to BIR [50]. This repair event duplicated an entire sister chromatid, suggesting that BIR could produce aneuploidy. Also, in Drosophila, deletion of POL32 and PIF1, homologs of genes required for BIR in yeast, attenuated fork progression following re-replication [51]. Thus, BIR is implicated in replication fork recovery following re-replication in various eukaryotes, including yeast, flies and humans.
Figure 3, Key Figure. Initiation of BIR by collapse of a replication fork and telomere erosion.

Replication fork collapse due to replication stress resulting from over-expression of oncogenes, decreased dNTP pools, re-replication, transcription-replication conflicts, and telomere erosion lead to formation of “one-ended” DNA breaks that can be repaired by RAD51-dependent or RAD51-independent BIR.
Box 3. DNA locations and special conditions promoting BIR-like events.
BIR is often initiated in situations leading to excessive DNA breakage and formation of one ended DSBs. Such BIR-permissive conditions might arise at eroded telomeres, common fragile sites or under increased replication stress load.
Eroded Telomeres. Telomeres are nucleoprotein structures protecting the chromosomal ends from degradation. Telomeres are maintained by telomerase, a reverse transcriptase enzyme that extends telomeres. In cells lacking telomerase, telomeres gradually erode, and upon becoming critically short, they trigger the DNA damage response and cell senescence [99]. Alternative lengthening of telomeres (ALT) is a mechanism of telomere maintenance that elongates telomeres in the absence of telomerase (reviewed in [74]). BIR is proposed to underlie ALT proceeding by copying telomeric repeats from other telomeres or ECTRs to the one-ended telomeric ends.
Common Fragile Sites (CFS) represent locations within the human genome that frequently undergo breakage under conditions of perturbed DNA synthesis, for example upon partial inhibition of replicative polymerases by aphidicolin (reviewed in [52, 53]). Several features of CFS loci, including their late replication, paucity of replication origins, enrichment for large actively transcribed genes, and their propensity to form DNA secondary structures were proposed to contribute to their breakage. CFS loci frequently include AT microsatellite sequences, inverted DNA repeats, trinucleotide repeats and other motifs prone to adopting non-B DNA structures that can impede replication (see Box 4). In addition, large transcription units promote replication collapse resulting from collision of replication with ongoing transcription and with stabilized R-loops. Interestingly, telomeres show replication-dependent defects that resemble those of aphidicolin induced CFS [100], which most likely results from the proneness of telomeric sequences to adopt specific secondary structures.
Collapse of replication forks due to re-replication or oncogene overexpression. Overexpression of oncogenes promotes massive collapse of replication forks. Possible reasons for this include, but are not limited to, perturbed licensing/firing of replication origins, decreased level of dNTPs, and elevated transcription that might interfere with fork progression ([46], reviewed in [101]). Re-replication is the repeated firing of an origin within the same cell cycle that leads to frequent collapse of replication forks (see for example in [49]).
BIR-like synthesis triggered by replication fork collapse at so-called common fragile sites (CFS, reviewed in [52, 53]), which represent locations within the human genome that frequently undergo breakage under conditions of perturbed DNA synthesis, have been documented in human cells during mitosis [54, 55]. A combination of CFS features (see Box 3) appear to contribute to formation and persistence of DNA breaks resulting from replication fork collapse until mitosis, when BIR is induced to mend the lesion, seemingly as a last resort. Although the specific features of CFS sites that initiate mitotic DNA synthesis (MiDAS) remain unclear, inverted repeats and R-loop formation have been implicated ([56], see below for further discussion). MiDAS involves MUS81-mediated cleavage of the stalled replication fork followed by DNA synthesis in which the newly synthesized strands are conservatively inherited [55], as is the case in BIR described during G2/M in yeast. The use of MiDAS to finish replication late in cell division suggests that even the genomic instabilities conferred during this process are likely less consequential to the daughter cells than the consequences of unfinished replication [56]; however, the mechanism of MiDAS and the precise types and frequencies of mutations and rearrangements associated with this synthesis remain unclear.
While it is difficult to dissect the propensity of individual structural features of CFSs to promote BIR in mammals, modeling some of the known elements of CFSs in yeast, including R-loops and DNA motifs that form secondary structures (inverted DNA repeats and trinucleotide repeats) has enabled important studies on this topic that led to conclusion that all these structures promote replication collapse leading to BIR ([57–63], see Box 4 for specific examples). Notably, BIR can also promote expansions of trinucleotide repeats. However, despite the evidence obtained in yeast, it is still uncertain whether these structural features of CFSs are indeed responsible for replication collapse leading to MiDAS in humans, and whether they may play any additional roles in promoting BIR-like events at CFS sites within the human genome. For example, it is plausible that, in addition to initiating replication collapse, they may also block replication forks approaching from another direction from merging with the collapsed fork, which could yield DSBs that persist into G2/M, when BIR is presumably de-repressed.
Box 4. DNA motifs and structures inducing BIR.
Inverted DNA repeats (IRs) are characterized by a mirror symmetry allowing them to switch between inter- and intra-strand base pairing (reviewed in [61]) to form secondary DNA structures, such as cruciforms, hairpins, or fold-backs when included into single-strand DNA (ssDNA) regions. Such structures can promote stalling and collapse of a replication fork, which leads to double-strand breaks (DSBs) and genome instability, including ectopic recombination, deletions, and chromosome rearrangements [58, 59, 61]. Some of the rearrangements might stem from BIR. For example, yeast mutants with reduced activity of Polα or Polδ (mimicking replication stress) promote chromosome rearrangements by ectopic BIR initiated at positions of inverted DNA repeats, mimicking human CFSs [57, 58]. It was proposed that collapse of replication at hairpin structures formed by inverted DNA repeats triggered ectopic BIR involving Ty and delta repeated elements, which led to translocations. Similarly, chromosome breakage at the quasipalindromic human Alu-repeats inserted in the yeast genome caused replication collapse followed by BIR resulting in translocations and amplifications [59].
Trinucleotide repeats. Expansions of trinucleotide DNA repeats, including (CGG)n·(CCG)n, (CAG)n·(CTG)n, (GAA)n·(TTC)n and (GCN)n·(NGC)n are responsible for severe human pathologies, such as Huntington’s disease, myotonic dystrophy type 1, Friedreich’s ataxia, and fragile X syndrome. It has been proposed that expansions and contractions of trinucleotide repeats result from their propensity to adopt non-B-DNA structures that hinder and collapse DNA replication (reviewed in [62]). Recent results obtained in yeast suggested that large expansions of (CAG)n and (GAA)n repeats are mediated by BIR initiated by collapse of replication at hairpins or triplex structures (respectively) formed by these repeats [60, 63]. However, it remains unclear which aspect of BIR leads to trinucleotide repeat expansion.
R-loops are three-stranded hybrid DNA-RNA structures that form by invasion of single strand RNA into the DNA duplex. As R-loops induce replication stalling and collapse, they have also been implicated in BIR initiation. It was demonstrated in yeast that disrupting activity of two RNaseH enzymes that prevent R-loop formation leads to DNA breaks that are frequently repaired by BIR [102]. This BIR was PIF1-dependent, while depletion of topoisomerase I in the RNase H mutants led to cell death, supposedly resulting from accumulation of unresolved BIR intermediates. Importantly, the authors proposed that R-loops promote BIR not only by increasing the frequency of DNA breakage, but also by precluding processing of one of the two DSB ends, making the breaks effectively “one-ended”.
In each scenarios of BIR initiated by replication collapse described above, including oncogene overexpression, re-replication, and CFSs, BIR is dependent on POLD3 (a mammalian homologue of POL32, which essential for BIR in yeast) and RAD52, but RAD51 is not required [48, 49, 55]. It remains unclear how BIR is initiated without Rad51 in mammalian cells and whether this process is similar to RAD51-independent BIR described in yeast [20, 32] (Figure 4). As mammalian systems to study HR continue to improve, we anticipate our understanding of the genetics of BIR throughout the mammalian cell cycle will improve markedly.
Figure 4. RAD51- independent BIR.

Left: A model of RAD51-independent BIR in yeast mediated by Rad52, Rad59, (and also Rdh54, and MRX complex, not shown) in the absence of Rad51, where Rad52 promotes annealing of the 3’ end of a one-ended broken chromosome to regions of transient ssDNA. Right: Possible DNA structures providing ssDNA, for RAD52-dependent RAD51-independent ssDNA annealing for initiating RAD51-independent BIR.
BIR and Alternative Lengthening of Telomeres (ALT)
Eroded telomeres formed in the absence of telomerase mimic one-ended DSBs (reviewed in [64]) and represent a substrate for BIR. The BIR-mediated lengthening of eroded telomeres, termed Alternative Lengthening of Telomeres (ALT, see Box 3), has been investigated in yeast and human cells. In yeast, ALT is responsible for the rare survivors that emerge from cell cultures lacking telomerase [65]. Two ALT pathways each generating phenotypically different telomeres have been described in yeast. In a notable parallel with the genetics of G2/M BIR described in yeast models, both Type I and Type II ALT rely on RAD52 [65] and POL32 [22]. However, only Type I requires RAD51, while Type II does not [65–67]. Telomeres resulting from Type I ALT consist of Y’ element arrays with very short terminal tracts of a telomeric sequence [65–68], which are the result of strand invasion of the eroded chromosome end into large regions of homology (Y’ elements). In contrast, Type II ALT generates long, heterogeneous telomeres consisting of the classical telomeric repeats [66, 67]. Type II ALT requires the Mre11/Rad50/Xrs2 complex, as well as Rad59, which assists Rad52 with annealing between single-strand DNA regions [66, 67, 69], see also [64]. Thus, based on our current knowledge of the basic genetics of these two pathways, the phenotypically different telomeres are possibly the result of different mechanisms of ALT initiation: Rad51-mediated invasion of a homologous sequence (Type I) versus Rad52/Rad59-mediated single-strand annealing between shorter homologous or homeologous sequences (Type II).
BIR has also been implicated in ALT in mammalian cells (see in [70, 71]). Approximately 15% of all human cancers induce ALT to maintain telomeres versus re-activating telomerase ([72, 73], see also [74]). Similarly to BIR in yeast, ALT represents a conservative type of DNA synthesis [71]. The genetic requirements of ALT were investigated recently in human cells where ALT was initiated using telomere-specific breakage induced by a TRF1-Fok1 fusion protein [70, 75]. This breakage triggered DNA repair synthesis that was up to 70 kilobases long and, similar to BIR in yeast required POLD3, but was RAD51-independent [70].
While Fok1-induced telomere breakage models have given rise to breakthroughs in our understanding of ALT mechanisms and genetics, details of endogenous events that lead to ALT initiation remain obscure. Most findings converge on two main factors that contribute to ALT induction: changes in chromatin structure and replication stress within telomere regions. In particular, it was demonstrated that loss of chromatin remodeling factors, including ATRX and ASF1, and changes in histone modification create permissive conditions for the initiation of homologous recombination that is otherwise suppressed at telomeres [76–80]. Thus, it is possible that the restructured chromatin facilitates the strand invasion and/or annealing that is required for the initiation of repair synthesis. Replication stress results from several sources, including aforementioned chromatin changes [76–78], but it is also induced by RNA transcribed from telomeric regions, termed telomeric RNA or TERRA [81, 82]. This RNA specifically binds to telomeric DNA creating R-loop structures, which promotes replication fork collapse and ultimately contributes to ALT initiation. Also, TERRA facilitates loading of RPA onto telomeres, making telomeres more permissive to HR [81]. In addition, ALT is associated with a conservative mitotic DNA synthesis [83, 84], similar to MiDAS induced by replication collapse at CFSs [54, 55]. Various sources of replication stress, including oncogene overexpression and stabilization of G4 quadruplex structures at telomeres, have been implicated in the initiation of telomeric MiDAS [83].
Another important question concerns the role of RAD51 during ALT. On one hand, a growing amount of data supports RAD51 independency of ALT. This includes the already mentioned Fok1-induced ALT [70], and ALT-associated mitotic synthesis detected on naturally eroded telomeres that requires RAD52 and POLD3, but is RAD51-independent [83]. Conversely, several observations are indicative of RAD51 involvement in ALT, specifically: (i) the presence of Rad51 in PML bodies that represent special subnuclear structures where ALT occurs [85]; (ii) the dependence of telomere movement and clustering on RAD51 [75]; (iii) the dependence of BLM promoted telomere extension detected in telomerase-deficient cells on RAD51 [84]. In total, it appears that two separate ALT pathways, RAD51-dependent and RAD51-independent are at work in mammals, in parallel with Type I and Type II pathways of ALT in yeast. While RAD51-dependent ALT is likely to operate via the well understood RAD51-dependent BIR, the mechanism of RAD51-independent ALT and BIR remain unclear. It was proposed that RAD51-independent ALT proceeds via annealing of the ssDNA from eroded telomeres to telomeric C-circles (ECTRs) that are partially single-stranded (Figure 4), and that this annealing is facilitated by Rad52 where rolling-circle-like synthesis is initiated, leading to formation of ALT survivors (see in [72, 86, 87]). It is also possible that RAD51-independent ALT results from intratelomeric strand invasion facilitated by shelterin proteins, see in [88].
Concluding Remarks.
BIR remains a very interesting but poorly understood pathway. Recent findings provide new examples of BIR in higher eukaryotes and allowed to compare molecular mechanisms that execute BIR across different organisms and at different stages of the cell cycle. For example, the requirement of POL32 or its mammalian homolog, POLD3, has been consistently reported for a majority of BIR events described, which underscored similarity between BIR in yeast and humans. However, since POLD3 is essential in mammals [89], it is important for future studies to characterize the involvement of other proteins in POLD3-dependent BIR mechanisms before making direct comparisons with pathways characterized in yeast. Another interesting, and possibly unexpected, scenario to emerge has been the identification of multiple RAD51-independent BIR pathways in mammalian cells. The mechanism of RAD51-independent BIR, including proteins involved and the genome-destabilizing consequences across organisms, remains elusive, including in yeast, and is an important area of focus for future research.
The variety of DNA structures that initiate BIR are also likely to contribute to the diversity among BIR mechanisms. For example, the structure of one-ended breaks produced by HO endonuclease may be sufficiently different from a collapsed replication fork to influence the pathway of BIR. Moreover, it is conceivable that the context within the replication fork fails, e.g., due to oncogene overexpression, within a CFS, or while traversing trinucleotide repeats, may create variable molecular structures that dictate the choice of BIR mechanism. In addition, though beyond the scope of this review, reports have emerged of other contexts wherein BIR has occurred, including BIR resulting from Mus81-mediated cleavage of reversed mammalian replication forks in the absence of BRCA2 and replication restart [90], and it is not yet known whether this new case of BIR is similar to those described for other situations. Another case that we could not describe here in detail includes restart of replication collapsed at fission yeast RTS sequence (reviewed in [91]). Similarly to BIR, this restart often leads to genetic instabilities (reviewed in [91]). However, a semi-conservative mode of DNA synthesis during this restart and also no involvement of a DSB intermediate [92], also reviewed in [91], underscore other deviations from what has been described in S. cerevisiae during G2/M repair of site-specific DSBs. As a final point, investigation of long-tract gene conversion (LTGC), which was described in yeast [27] and human cells [93, 94] and involves long D-loop repair DNA synthesis suggests that this pathway, which has been classified as an alteration of gene conversion (GC) , may be more appropriately considered a variation of BIR.
In sum, BIR represents an unusual type of DNA synthesis that is best described in yeast and within the G2/M stage of the cell cycle. Recent progress in the development of new models has enabled investigations of BIR in different cellular contexts, e.g., in telomere maintenance, and in replication restart. Though we are only beginning to understand the myriad ways in which cellular context can initiate and orchestrate apparently multiple different BIR pathways, the progress made in the field has only heightened interest in the topic, as it is increasingly apparent that BIR plays a critical role in maintaining genomic stability during numerous critical – and common – events in eukaryotic life cycle. Through an improved understanding of these molecular mechanisms, we are hopeful that we may reach new horizons in our understanding of how dysregulated BIR may promote human pathologies, including cancer, and potentially identify novel therapeutic opportunities (See Outstanding Questions).
How is the initiation of BIR regulated? Why does replication stress lead to BIR while the collapse of a single replication fork often does not?
How is BIR regulated through the cell cycle? Is it promoted at G2/M, but restricted in S-phase? If so, how does the regulation work?
Does initiation of leading strand synthesis require primase, and if yes, why?
How is lagging strand synthesis carried out during RAD51-dependent BIR? Are there Okazaki fragments formed?
How is the labor distributed between DNA polymerases in a course of BIR? Is Pol δ synthesizing both leading and lagging strands?
Why is BIR DNA synthesis in the context of a D-loop often interrupted? Why does this interruption lead to template switching and frameshift mutations?
Which protein(s) serve as a replicative helicase during BIR?
Why BIR is more efficient when initiated by a DNA break near the end of the chromosome than in the middle? Is it an unusual telomere effect?
What is the mechanism of RAD51-independent BIR? How does the synthesis proceed? Which proteins are involved?
How is RAD51-independent BIR initiated? What are the structural characteristics of chromosomal regions that serve as templates (donors) for RAD51-independent BIR?
What is the mechanistic role of Rad52 in RAD51-independent BIR?
What are the homology requirements for initiation of RAD51-independent BIR?
How is the choice between RAD51-dependent and RAD51-independent BIR made?
How is the choice between Type I and Type II ALT regulated in yeast and human cells? What are the preferred templates/donors for each of these pathways?
What events trigger BIR-driven ALT in cells lacking telomerase?
Which polymerase(s) can mediate MMBIR in humans and other eukaryotes?
What is the role of BIR and MMBIR in onset of various pathologies such as neurological diseases and cancer?
TIGS quiz questions.
-
1What features of canonical BIR in yeast make it distinct from S-phase DNA replication?
- Conservative Inheritance
- Asynchronous leading and lagging strand synthesis
- Requirement of Pol32
- Increased mutagenesis
- All of the above
Answer: E. All of these factors make canonical BIR as described in yeast a distinct and unusual pathway when compared to S-phase DNA replication.
-
2Which statement concerning alternative lengthening of telomeres (ALT) is TRUE:
- Approximately 15% of human cancers have reactivated telomerase to elongate telomeres
- Approximately 15% of human cancers use ALT to elongate telomeres
- Rad51 is always required for ALT in mammalian and yeast cells.
- Rad52 is not required for ALT in mammalian and yeast cells.
Answer: B. Approximately 15% of all human cancers induce ALT to maintain telomeres versus re-activating telomerase. There are Rad51-independent and Rad52-dependent pathways for both yeast and mammalian cells.
-
3Which of the following processes does not require recombination?
- RDR
- BIR
- NHEJ
- GC
Answer: C. Non-homologous end joining (NHEJ) is a pathway to repair DSBs, but does not require recombination to complete.
- Which of the following DNA damaging agents can be produced both endogenously and as a result of exogenous exposures?
-
DMitomycin C
-
EReactive oxygen species
-
FGamma Radiation
-
GMethyl methanesulfonate
-
HAPOBEC
-
D
Answer: B. Reactive oxygen species can be produced as byproducts of cellular metabolism (endogenous source), and are produced by exposure to ionizing radiation (exogenous source)
Highlights.
Under physiological, non-stressed conditions, eukaryotes seem to be reluctant to use BIR for repair of collapsed replication forks. However, this changes under conditions of replication stress when BIR is employed to handle replication fork collapse.
Mammalian BIR involves RAD52, but is often independent from RAD51, similar to yeast RAD51-independent BIR.
Similar to yeast, mammalian BIR often leads to genomic rearrangements, e.g. tandem duplications and copy number variations (CNVs), which can contribute to development of cancer and other diseases in humans.
Similar to BIR in yeast, mammalian break-induced telomere synthesis underlies alternative lengthening of telomeres (ALT) and proceeds with conservative inheritance of newly synthesized DNA.
Acknowledgments
We thank Zachary Kockler for helpful comments on the manuscript. The research in AM laboratory is funded by NIH grant GM084242.
Glossary.
- Double strand break (DSB)
a DNA lesion where both phosphodiester bonds in double stranded DNA are interrupted (see Box 1, Box 2)
- Non-homologous end joining (NHEJ)
a repair pathway that ligates two DNA ends of a double strand break back together (see Box 2)
- Homologous recombination (HR)
a DNA repair pathway where a resected DNA end invades an identical (or similar) sequence, which is then used as template for repair (see Box 2)
- Displacement loop (D-loop)
a DNA structure where one strand of a double-stranded DNA molecule is displaced by invasion of a single-stranded DNA molecule. A similar structure termed
- R-loop
may be formed by invasion of an RNA strand into double-stranded DNA
- Conservative/Semiconservative DNA synthesis
most of the DNA in living organisms is replicated in a semiconservative fashion i.e. the daughter DNA molecules contain one of the original template strands paired to a newly synthesized strand. Alternatively, conservative synthesis yields daughter DNA molecules consisting of either two original strands or the two newly replicated strands
- Holliday junction
a four-way branched DNA structure that forms at a junction between two recombining DNA molecules. The Holliday junction is a key structure in DNA recombination processes because its resolution allows for the formation of crossover products that are necessary for the success of homologous chromosome segregation in meiosis
- Rad51 filament
formed when Rad51 recombination protein monomers bind to the single-strand DNA forming a nucleoprotein filament. The Rad51 filament facilitates homology search and strand invasion into the homologous double stranded DNA molecule during recombination
- Extrachromosomal telomeric repeats (ECTR)
DNA molecules other than chromosomes that carry telomeric repeats. ECTR most often exist in the form of telomeric circles, but linear and branched species may also occur
- Telomeric repeat-containing RNA (TERRA)
a non-coding RNA transcribed from telomeric repeats
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest
Authors declare no conflict of interest.
References.
- 1.Rodgers K and McVey M (2016) Error-Prone Repair of DNA Double-Strand Breaks. J Cell Physiol 231 (1), 15–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Heyer WD (2015) Regulation of recombination and genomic maintenance. Cold Spring Harb Perspect Biol 7 (8), a016501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Strathern JN et al. (1995) DNA synthesis errors associated with double-strand-break repair. Genetics 140 (3), 965–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hicks WM et al. (2010) Increased mutagenesis and unique mutation signature associated with mitotic gene conversion. Science 329 (5987), 82–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang Y et al. (2008) Hypermutability of damaged single-strand DNA formed at double-strand breaks and uncapped telomeres in yeast Saccharomyces cerevisiae. PLoS Genet 4 (11), e1000264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Deem A et al. (2011) Break-induced replication is highly inaccurate. PLoS Biol 9 (2), e1000594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Malkova A and Haber JE (2012) Mutations arising during repair of chromosome breaks. Annu Rev Genet 46, 455–73. [DOI] [PubMed] [Google Scholar]
- 8.Sakofsky CJ and Malkova A (2017) Break induced replication in eukaryotes: mechanisms, functions, and consequences. Crit Rev Biochem Mol Biol 52 (4), 395–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Llorente B et al. (2008) Break-induced replication: what is it and what is it for? Cell Cycle 7 (7), 859–64. [DOI] [PubMed] [Google Scholar]
- 10.Anand RP et al. (2013) Break-induced DNA replication. Cold Spring Harb Perspect Biol 5 (12), a010397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Heller RC and Marians KJ (2006) Replisome assembly and the direct restart of stalled replication forks. Nat Rev Mol Cell Biol 7 (12), 932–43. [DOI] [PubMed] [Google Scholar]
- 12.Mosig G (1998) Recombination and recombination-dependent DNA replication in bacteriophage T4. Annu Rev Genet 32, 379–413. [DOI] [PubMed] [Google Scholar]
- 13.Kogoma T (1997) Stable DNA replication: interplay between DNA replication, homologous recombination, and transcription. Microbiol Mol Biol Rev 61 (2), 212–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Saini N et al. (2013) Migrating bubble during break-induced replication drives conservative DNA synthesis. Nature 502 (7471), 389–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sakofsky CJ et al. (2015) Translesion Polymerases Drive Microhomology-Mediated Break-Induced Replication Leading to Complex Chromosomal Rearrangements. Mol Cell 60 (6), 860–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sakofsky CJ et al. (2014) Break-induced replication is a source of mutation clusters underlying kataegis. Cell Rep 7 (5), 1640–1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smith CE et al. (2007) Template switching during break-induced replication. Nature 447 (7140), 102–5. [DOI] [PubMed] [Google Scholar]
- 18.Anand RP et al. (2014) Chromosome rearrangements via template switching between diverged repeated sequences. Genes Dev 28 (21), 2394–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pardo B and Aguilera A (2012) Complex chromosomal rearrangements mediated by break-induced replication involve structure-selective endonucleases. PLoS Genet 8 (9), e1002979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Malkova A et al. (1996) Double-strand break repair in the absence of RAD51 in yeast: a possible role for break-induced DNA replication. Proc Natl Acad Sci U S A 93 (14), 7131–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Davis AP and Symington LS (2004) RAD51-dependent break-induced replication in yeast. Mol Cell Biol 24 (6), 2344–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lydeard JR et al. (2007) Break-induced replication and telomerase-independent telomere maintenance require Pol32. Nature 448 (7155), 820–3. [DOI] [PubMed] [Google Scholar]
- 23.Malkova A et al. (2005) RAD51-dependent break-induced replication differs in kinetics and checkpoint responses from RAD51-mediated gene conversion. Mol Cell Biol 25 (3), 933–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chung WH et al. (2010) Defective resection at DNA double-strand breaks leads to de novo telomere formation and enhances gene targeting. PLoS Genet 6 (5), e1000948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Anand R et al. (2017) Rad51-mediated double-strand break repair and mismatch correction of divergent substrates. Nature 544 (7650), 377–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ruff P et al. (2016) RPA Stabilization of Single-Stranded DNA Is Critical for Break-Induced Replication. Cell Rep 17 (12), 3359–3368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jain S et al. (2009) A recombination execution checkpoint regulates the choice of homologous recombination pathway during DNA double-strand break repair. Genes Dev 23 (3), 291–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Donnianni RA and Symington LS (2013) Break-induced replication occurs by conservative DNA synthesis. Proc Natl Acad Sci U S A 110 (33), 13475–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mehta A et al. (2017) Homology Requirements and Competition between Gene Conversion and Break-Induced Replication during Double-Strand Break Repair. Mol Cell 65 (3), 515–526 e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Signon L et al. (2001) Genetic requirements for RAD51- and RAD54-independent break-induced replication repair of a chromosomal double-strand break. Mol Cell Biol 21 (6), 2048–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.VanHulle K et al. (2007) Inverted DNA repeats channel repair of distant double-strand breaks into chromatid fusions and chromosomal rearrangements. Mol Cell Biol 27 (7), 2601–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Downing B et al. (2008) Large inverted repeats in the vicinity of a single double-strand break strongly affect repair in yeast diploids lacking Rad51. Mutat Res 645 (1–2), 9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wilson MA et al. (2013) Pif1 helicase and Poldelta promote recombination-coupled DNA synthesis via bubble migration. Nature 502 (7471), 393–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lydeard JR et al. (2010) Break-induced replication requires all essential DNA replication factors except those specific for pre-RC assembly. Genes Dev 24 (11), 1133–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Elango R et al. (2017) Break-induced replication promotes formation of lethal joint molecules dissolved by Srs2. Nat Commun 8 (1), 1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Smith CE et al. (2009) Aberrant double-strand break repair resulting in half crossovers in mutants defective for Rad51 or the DNA polymerase delta complex. Mol Cell Biol 29 (6), 1432–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Deem A et al. (2008) Defective break-induced replication leads to half-crossovers in Saccharomyces cerevisiae. Genetics 179 (4), 1845–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vasan S et al. (2014) Cascades of genetic instability resulting from compromised break-induced replication. PLoS Genet 10 (2), e1004119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jain S et al. (2016) Sgs1 and Mph1 Helicases Enforce the Recombination Execution Checkpoint During DNA Double-Strand Break Repair in Saccharomyces cerevisiae. Genetics 203 (2), 667–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Formosa T and Alberts BM (1986) DNA synthesis dependent on genetic recombination: characterization of a reaction catalyzed by purified bacteriophage T4 proteins. Cell 47 (5), 793–806. [DOI] [PubMed] [Google Scholar]
- 41.Kreuzer KN and Brister JR (2010) Initiation of bacteriophage T4 DNA replication and replication fork dynamics: a review in the Virology Journal series on bacteriophage T4 and its relatives. Virol J 7, 358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Marians KJ (2000) PriA-directed replication fork restart in Escherichia coli. Trends Biochem Sci 25 (4), 185–9. [DOI] [PubMed] [Google Scholar]
- 43.Mayle R et al. (2015) Mus81 and converging forks limit the mutagenicity of replication fork breakage. Science 349 (6249), 742–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Matos J et al. (2011) Regulatory control of the resolution of DNA recombination intermediates during meiosis and mitosis. Cell 147 (1), 158–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Halazonetis TD et al. (2008) An oncogene-induced DNA damage model for cancer development. Science 319 (5868), 1352–5. [DOI] [PubMed] [Google Scholar]
- 46.Macheret M and Halazonetis TD (2018) Intragenic origins due to short G1 phases underlie oncogene-induced DNA replication stress. Nature, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Costantino L et al. (2014) Break-induced replication repair of damaged forks induces genomic duplications in human cells. Science 343 (6166), 88–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sotiriou SK et al. (2016) Mammalian RAD52 Functions in Break-Induced Replication Repair of Collapsed DNA Replication Forks. Mol Cell 64 (6), 1127–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Galanos P et al. (2016) Chronic p53-independent p21 expression causes genomic instability by deregulating replication licensing. Nat Cell Biol 18 (7), 777–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hanlon SL and Li JJ (2015) Re-replication of a centromere induces chromosomal instability and aneuploidy. PLoS Genet 11 (4), e1005039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Alexander JL et al. (2016) Multiple mechanisms contribute to double-strand break repair at rereplication forks in Drosophila follicle cells. Proc Natl Acad Sci U S A 113 (48), 13809–13814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Durkin SG and Glover TW (2007) Chromosome fragile sites. Annu Rev Genet 41, 169–92. [DOI] [PubMed] [Google Scholar]
- 53.Glover TW et al. (2017) Fragile sites in cancer: more than meets the eye. Nat Rev Cancer 17 (8), 489–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Minocherhomji S et al. (2015) Replication stress activates DNA repair synthesis in mitosis. Nature 528 (7581), 286–90. [DOI] [PubMed] [Google Scholar]
- 55.Bhowmick R et al. (2016) RAD52 Facilitates Mitotic DNA Synthesis Following Replication Stress. Mol Cell 64 (6), 1117–1126. [DOI] [PubMed] [Google Scholar]
- 56.Bhowmick R and Hickson ID (2017) The “enemies within”: regions of the genome that are inherently difficult to replicate. F1000Res 6, 666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lemoine FJ et al. (2008) Reduced levels of DNA polymerase delta induce chromosome fragile site instability in yeast. Mol Cell Biol 28 (17), 5359–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lemoine FJ et al. (2005) Chromosomal translocations in yeast induced by low levels of DNA polymerase a model for chromosome fragile sites. Cell 120 (5), 587–98. [DOI] [PubMed] [Google Scholar]
- 59.Narayanan V et al. (2006) The pattern of gene amplification is determined by the chromosomal location of hairpin-capped breaks. Cell 125 (7), 1283–96. [DOI] [PubMed] [Google Scholar]
- 60.Kim JC et al. (2017) The role of break-induced replication in large-scale expansions of (CAG)n/(CTG)n repeats. Nat Struct Mol Biol 24 (1), 55–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lobachev KS et al. (2007) Hairpin- and cruciform-mediated chromosome breakage: causes and consequences in eukaryotic cells. Front Biosci 12, 4208–20. [DOI] [PubMed] [Google Scholar]
- 62.Schmidt MH and Pearson CE (2016) Disease-associated repeat instability and mismatch repair. DNA Repair (Amst) 38, 117–26. [DOI] [PubMed] [Google Scholar]
- 63.Neil AJ et al. (2018) RNA-DNA hybrids promote the expansion of Friedreich’s ataxia (GAA)n repeats via break-induced replication. Nucleic Acids Res, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.McEachern MJ and Haber JE (2006) Break-induced replication and recombinational telomere elongation in yeast. Annu Rev Biochem 75, 111–35. [DOI] [PubMed] [Google Scholar]
- 65.Lundblad V and Blackburn EH (1993) An alternative pathway for yeast telomere maintenance rescues est1- senescence. Cell 73 (2), 347–60. [DOI] [PubMed] [Google Scholar]
- 66.Teng SC and Zakian VA (1999) Telomere-telomere recombination is an efficient bypass pathway for telomere maintenance in Saccharomyces cerevisiae. Mol Cell Biol 19 (12), 8083–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Teng SC et al. (2000) Telomerase-independent lengthening of yeast telomeres occurs by an abrupt Rad50p-dependent, Rif-inhibited recombinational process. Mol Cell 6 (4), 947–52. [DOI] [PubMed] [Google Scholar]
- 68.Louis EJ et al. (1994) The chromosome end in yeast: its mosaic nature and influence on recombinational dynamics. Genetics 136 (3), 789–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Le S et al. (1999) RAD50 and RAD51 define two pathways that collaborate to maintain telomeres in the absence of telomerase. Genetics 152 (1), 143–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dilley RL et al. (2016) Break-induced telomere synthesis underlies alternative telomere maintenance. Nature 539 (7627), 54–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Roumelioti FM et al. (2016) Alternative lengthening of human telomeres is a conservative DNA replication process with features of break-induced replication. EMBO Rep 17 (12), 1731–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dilley RL and Greenberg RA (2015) ALTernative Telomere Maintenance and Cancer. Trends Cancer 1 (2), 145–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Neumann AA and Reddel RR (2002) Telomere maintenance and cancer -- look, no telomerase. Nat Rev Cancer 2 (11), 879–84. [DOI] [PubMed] [Google Scholar]
- 74.Conomos D et al. (2013) Alternative lengthening of telomeres: remodeling the telomere architecture. Front Oncol 3, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cho NW et al. (2014) Interchromosomal homology searches drive directional ALT telomere movement and synapsis. Cell 159 (1), 108–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Clynes D et al. (2015) Suppression of the alternative lengthening of telomere pathway by the chromatin remodelling factor ATRX. Nat Commun 6, 7538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Conomos D et al. (2014) NuRD-ZNF827 recruitment to telomeres creates a molecular scaffold for homologous recombination. Nat Struct Mol Biol 21 (9), 760–70. [DOI] [PubMed] [Google Scholar]
- 78.O’Sullivan RJ et al. (2014) Rapid induction of alternative lengthening of telomeres by depletion of the histone chaperone ASF1. Nat Struct Mol Biol 21 (2), 167–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Schwartzentruber J et al. (2012) Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 482 (7384), 226–31. [DOI] [PubMed] [Google Scholar]
- 80.Heaphy CM et al. (2011) Altered telomeres in tumors with ATRX and DAXX mutations. Science 333 (6041), 425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Flynn RL et al. (2015) Alternative lengthening of telomeres renders cancer cells hypersensitive to ATR inhibitors. Science 347 (6219), 273–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Arora R et al. (2014) RNaseH1 regulates TERRA-telomeric DNA hybrids and telomere maintenance in ALT tumour cells. Nat Commun 5, 5220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Min J et al. (2017) Alternative Lengthening of Telomeres Mediated by Mitotic DNA Synthesis Engages Break-Induced Replication Processes. Mol Cell Biol 37 (20). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sobinoff AP et al. (2017) BLM and SLX4 play opposing roles in recombination-dependent replication at human telomeres. EMBO J 36 (19), 2907–2919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Draskovic I et al. (2009) Probing PML body function in ALT cells reveals spatiotemporal requirements for telomere recombination. Proc Natl Acad Sci U S A 106 (37), 15726–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.McEachern MJ and Blackburn EH (1996) Cap-prevented recombination between terminal telomeric repeat arrays (telomere CPR) maintains telomeres in Kluyveromyces lactis lacking telomerase. Genes Dev 10 (14), 1822–34. [DOI] [PubMed] [Google Scholar]
- 87.Tomaska L et al. (2009) Telomeric circles: universal players in telomere maintenance? Nat Struct Mol Biol 16 (10), 1010–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Verma P and Greenberg RA (2016) Noncanonical views of homology-directed DNA repair. Genes Dev 30 (10), 1138–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Murga M et al. (2016) POLD3 Is Haploinsufficient for DNA Replication in Mice. Mol Cell 63 (5), 877–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lemacon D et al. (2017) MRE11 and EXO1 nucleases degrade reversed forks and elicit MUS81-dependent fork rescue in BRCA2-deficient cells. Nat Commun 8 (1), 860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Carr AM and Lambert S (2013) Replication stress-induced genome instability: the dark side of replication maintenance by homologous recombination. J Mol Biol 425 (23), 4733–44. [DOI] [PubMed] [Google Scholar]
- 92.Miyabe I et al. (2015) Polymerase delta replicates both strands after homologous recombination-dependent fork restart. Nat Struct Mol Biol 22 (11), 932–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Chandramouly G et al. (2013) BRCA1 and CtIP suppress long-tract gene conversion between sister chromatids. Nat Commun 4, 2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Willis NA et al. (2014) BRCA1 controls homologous recombination at Tus/Ter-stalled mammalian replication forks. Nature 510 (7506), 556–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Friedberg EC et al. (2005) DNA Repair and Mutagenesis, ASM Press. [Google Scholar]
- 96.Chan K and Gordenin DA (2015) Clusters of Multiple Mutations: Incidence and Molecular Mechanisms. Annu Rev Genet 49, 243–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Roberts SA and Gordenin DA (2014) Hypermutation in human cancer genomes: footprints and mechanisms. Nat Rev Cancer 14 (12), 786–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Yang Y et al. (2010) A single-strand specific lesion drives MMS-induced hyper-mutability at a double-strand break in yeast. DNA Repair (Amst) 9 (8), 914–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Palm W and de Lange T (2008) How shelterin protects mammalian telomeres. Annu Rev Genet 42, 301–34. [DOI] [PubMed] [Google Scholar]
- 100.Sfeir A et al. (2009) Mammalian telomeres resemble fragile sites and require TRF1 for efficient replication. Cell 138 (1), 90–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Macheret M and Halazonetis TD (2015) DNA replication stress as a hallmark of cancer. Annu Rev Pathol 10, 425–48. [DOI] [PubMed] [Google Scholar]
- 102.Amon JD and Koshland D (2016) RNase H enables efficient repair of R-loop induced DNA damage. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Carvalho CM and Lupski JR (2016) Mechanisms underlying structural variant formation in genomic disorders. Nat Rev Genet 17 (4), 224–38. [DOI] [PMC free article] [PubMed] [Google Scholar]