

Chaperone regulation of individual microscopic events in Aβ42 aggregation reveals the nature of active sites on amyloid fibrils.
Abstract
The aggregates of the Aβ peptide associated with Alzheimer’s disease are able to both grow in size as well as generate, through secondary nucleation, new small oligomeric species, that are major cytotoxins associated with neuronal death. Despite the importance of these amyloid fibril-dependent processes, their structural and molecular underpinnings have remained challenging to elucidate. Here, we consider two molecular chaperones: the Brichos domain, which suppresses specifically secondary nucleation processes, and clusterin which our results show is capable of inhibiting, specifically, the elongation of Aβ fibrils at remarkably low substoichiometric ratios. Microfluidic diffusional sizing measurements demonstrate that this inhibition originates from interactions of clusterin with fibril ends with high affinity. Kinetic experiments in the presence of both molecular chaperones reveal that their inhibitory effects are additive and noncooperative, thereby indicating that the reactive sites associated with the formation of new aggregates and the growth of existing aggregates are distinct.
INTRODUCTION
A large number of observations, including genetic and epidemiological studies, indicate that the aberrant aggregation of normally soluble peptides and proteins into insoluble amyloid fibrils is associated with the onset and the progression of a wide range of neurodegenerative disorders (1–6). Biophysical studies are currently revealing important molecular details underlying the formation of the fibrillar aggregates from initially supersaturated monomer solutions (7–9). From these studies, a picture is emerging in which the aggregation process is seen to be the result of a complex cascade of individual microscopic steps of nucleation and growth (10). Secondary nucleation, in particular, increases the rate of formation of low–molecular weight oligomers that accompany the formation of mature fibrils and are currently thought to represent the most toxic species (3, 10–13). Inhibition of these key aggregation processes therefore represents a potentially attractive therapeutic opportunity in the search for strategies to combat amyloid-associated diseases (14).
Understanding the molecular determinants of these different mechanistic processes is therefore of fundamental importance to design strategies that target specifically the reactions responsible for the formation of the most highly toxic species. In the context of developing drugs to combat protein aggregation disorders, targeted interventions, rather than nonspecific suppression of the aggregation process, are required (14). The inhibition of different microscopic steps can lead to similar effects on the overall formation of amyloid fibrils, yet can have marked differences in the generation of toxic oligomeric species (14–16). An intervention strategy targeting the specific microscopic steps that represent the major sources of the formation of these oligomers is, therefore, likely to be an optimal approach to suppress the most detrimental effects of protein aggregation.
Recent developments in the analysis of kinetic profiles of aggregation reactions represent a powerful tool in the analysis of the complex mechanistic details at the molecular level, generating information from experimental measurements that is not otherwise achievable (9, 17). An important consequence of these advances is the ability to investigate the ways in which nature suppresses amyloid formation in normally functioning organisms by analyzing the mechanism of action of molecular chaperones (18). These species are key components of the protein homeostasis network whose mode of action ranges from assisting protein folding following biosynthesis to facilitating the degradation of these systems that have failed to fold or to remain completely folded (19, 20). By investigating a series of molecular chaperones that can inhibit selectively one or more specific steps in the aggregation process of different amyloidogenic proteins, we have shown that these vital molecules can influence aggregation processes through a variety of different microscopic events and reveal the complex mechanism that nature has evolved to maintain protein homoeostasis (18, 21, 22). With this information, we can exploit molecular chaperones to perturb, in a selective way, a given aggregation network and thereby investigate the effects of individual steps in the reaction on the formation of key intermediate species. For example, a molecular chaperone belonging to the Brichos family (proSP-C Brichos) has been shown to inhibit the aggregation of Aβ42, a 42-residue protein fragment of the amyloid-β peptide that is the most abundant isoform in amyloid deposits closely associated with Alzheimer’s disease (AD), by suppressing specifically the secondary nucleation reaction and resulting in a marked reduction in the generation of toxic oligomeric species (11).
In the present study, we have built on this approach to reveal the effects of two well-characterized molecular chaperones on different microscopic steps of the aggregation of Aβ(M1-42). In particular, we first investigate the inhibitory mechanism of clusterin. Clusterin is an important member of a family of largely extracellular chaperones, which have been observed to interact with different aggregation states of amyloidogenic proteins, including oligomers, prefibrillar species, and mature fibrils, and hence to affect events leading to the formation of amyloid fibrils (23–28). In particular, experiments with synthetic Aβ40 and Aβ42 have shown that clusterin can bind to oligomers and prefibrillar species, while our experiments on recombinant Aβ(M1-42) demonstrate that clusterin is highly effective in retarding the aggregation process of Aβ(M1-42) at low substoichiometric ratios and physiological concentrations by inhibiting specifically the elongation process (23, 26, 27, 29). We then examine the combined ability of clusterin and another type of molecular chaperone, a Brichos domain, to influence specific microscopic events in the aggregation process and analyze the cooperative nature of the inhibition process to extract information about the location of specific sites on the fibrils that are involved with secondary nucleation and fibril elongation.
RESULTS
Clusterin suppresses specifically the elongation step of Aβ(M1-42) aggregation
To investigate the capability of clusterin to inhibit the aggregation process of Aβ(M1-42), we first incubated the peptide in the absence and presence of different concentrations of the molecular chaperone and monitored the time evolution of fibril formation by recording the fluorescence signal of the dye thioflavin T (ThT), which is strongly enhanced by its interaction with amyloid structures (Fig. 1). We observed a delay in the fibril formation process that is proportional to the concentration of clusterin present in the system even at very low substoichiometric molar ratios (including 1:500) of clusterin. We then carried out a series of kinetic experiments at different concentrations of Aβ(M1-42) (see fig. S1) to explore the dependence of the various microscopic steps in the aggregation cascade and the presence of the molecular chaperone by means of kinetic analysis (14). According to this strategy, the experimental kinetic profiles determined at a range of Aβ(M1-42) concentrations are compared to simulations in which individual events are specifically suppressed (Fig. 1, A to C). The least-squares error function, defined in Materials and Methods, at a clusterin concentration of 135 nM is 1.5, 0.2, and 1.5 for the specific inhibition of kn, k+, and k2, respectively. This comparison suggests that clusterin inhibits the overall aggregation process by suppressing specifically the rate of elongation of the Aβ(M1-42) fibrils while having no detectable effect on primary and secondary nucleation events.
Fig. 1. Analysis of the effects of clusterin on the aggregation kinetics of Aβ(M1-42).
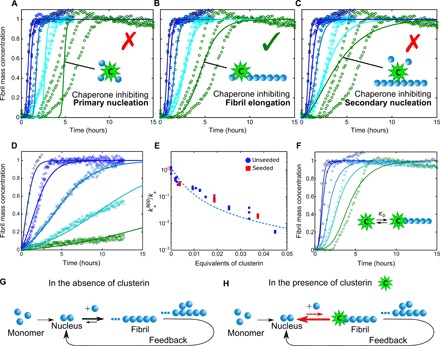
(A to C) Kinetic reaction profiles for the aggregation of 4 μM Aβ(M1-42) solutions are shown in each panel from left (blue) to right (green) in the absence and presence of 7.5, 37, 75, and 135 nM clusterin, with each color representing repetitions at the same concentration. The integrated rate law for Aβ(M1-42) aggregation in the absence of clusterin using the rate constants, previously determined by a least-squares error function, is shown as a dark blue line in each case (10). Predicted profiles of the specific inhibition processes of (A) primary nucleation, (B) fibril elongation, and (C) secondary nucleation generated by clusterin are shown as continuous lines. Note the characteristic differences in the changes in the shape of the reaction profiles in each case. The prediction for the case where the molecular chaperone suppresses only elongation events matches closely the experimental data in the presence of different concentrations of clusterin. (D) Kinetic reaction profiles for the aggregation reaction of a 2 μM Aβ(M1-42) solution seeded with 100 nM preformed fibrils in the absence and presence of 7.5, 37, 75, and 135 nM clusterin. The lines represent the integrated rate laws for Aβ(M1-42) aggregation, where the elongation rate has been selectively reduced. The apparent elongation reaction rates as a function of the molecular chaperone concentration evaluated from the fitting in (B) and (D) are reported in (E) for both unseeded and seeded reactions. The continuous line in (E) represents a simplified correlation between the elongation rate and the binding affinity constant (see Materials and Methods), from which KD,37°C = 8 nM is determined. (F) Comparison between the experimental data reported in (B) and theoretical predictions of the reaction profiles calculated from a kinetic model, which considers the association and dissociation rate constants in the reaction scheme with KD,37°C = 2.5 nM. (G and H) Schematic diagrams showing the molecular pathways involved in Aβ(M1-42) aggregation (G) and the mechanism by which clusterin perturbs the aggregation process (H).
To test this idea experimentally, we performed kinetic experiments where a solution of 5% (w/w) of preformed seed fibrils was added to solutions of monomeric Aβ(M1-42) (Fig. 1D); under these conditions, primary and secondary nucleation events are negligible, and fibril elongation is the major contributor to the increase in fibril mass (30). By increasing the concentration of clusterin, we observed a progressive reduction in the rate of aggregation, supporting the conclusion that the molecular chaperone inhibits this specific microscopic process. In addition, the elongation rate constants calculated from simulations of the experimental data under seeded conditions are in excellent agreement with the values determined under unseeded conditions, confirming the robustness of our analysis (Fig. 1E).
Previous kinetic analysis of Aβ(M1-42) profiles indicates that monomeric species participate directly in elongation and primary/secondary nucleation rates (10). In particular, the elongation reaction involves the addition of monomers to fibril ends and smaller propagons, such as oligomers and other prefibrillar species, and therefore, the inhibition effect of clusterin could result from its interactions with either of the three species (9, 31). Substantial binding of the molecular chaperone to monomeric Aβ(M1-42) would, however, also affect primary and secondary nucleation reactions, because these processes also depend on the concentration of monomers. As the kinetic analysis reveals that clusterin inhibits only the elongation rate, we can conclude that it interacts preferentially with the fibril ends and all species capable of elongation. The fact that inhibition is observed at very low substoichiometric ratios is completely associated with the interaction of clusterin with the ends of fibrils or prefibrillar species, as interactions with monomeric Aβ(M1-42) would require a much higher stoichiometric concentration of clusterin to affect the rates to such a substantial extent.
To interpret the kinetic effects in a more quantitative manner, we compared the experimental data with the prediction of a kinetic model that considers explicitly binary interactions between clusterin and the ends of Aβ(M1-42) fibrils (see Materials and Methods). The theoretical predictions account well for the concentration dependence of the inhibition at 37°C by using the fitting parameters kon,37°C = 4 × 105 M−1 s−1 and koff,37°C = 1 × 10− 3 s−1 (Fig. 1F), corresponding to KD,37°C = 2.5 nM. From an analysis of the dependence of the apparent elongation rate constant on the molecular chaperone concentration (Fig. 1E) using a simplified kinetic model, in which the binding reaction process is assumed to be under equilibrium conditions (see Materials and Methods), we obtain a similar value of KD,37°C = 8 nM. We repeated the experiment at a temperature of 21°C and obtained a value of KD,21°C = 1 nM (fig. S2), indicating that the binding is exothermic. The exergonic binding and the high interaction affinity are consistent with the fact that clusterin interacts with Aβ(M1-42) fibrils in an adenosine 5′-triphosphate (ATP)–independent manner (32).
The interactions between clusterin and fibrils of Aβ(M1-42) were probed in additional kinetic experiments. To this effect, Aβ(M1-42) fibrils were generated in the presence or absence of clusterin and then added to freshly prepared monomer solutions in the presence or absence of clusterin (fig. S3). Even in the absence of clusterin in the monomer solution (fig. S3B), the preformed fibrils that had been produced in the presence of clusterin accelerated aggregation to a smaller extent than did fibrils produced in the absence of the chaperone (fig. S3A). Application of the kinetic analysis reveals a reduction of ca. 40% in the elongation rate in this latter case (fig. S3B), indicating that a substantial fraction of the molecular chaperone must have remained bound to the aggregates during the time course of the reaction, and immunogold transmission electron microscopy (TEM) analysis (Fig. 2, B and D) indicates that clusterin is bound to these Aβ(M1-42) fibrils. Furthermore, the ability of fibril ends to grow was also observed to be substantially reduced when preformed aggregates generated in the absence of the chaperone were added to a mixture of clusterin and monomeric Aβ(M1-42) (fig. S3C), demonstrating the ability of clusterin to inhibit even ongoing reactions. By contrast, in this set of experiments, the fibrils that had not been exposed to the molecular chaperone at any stage maintained their full ability to elongate (fig. S3A).
Fig. 2. Analysis of clusterin interactions with Aβ(M1-42) fibrils using immunogold TEM.
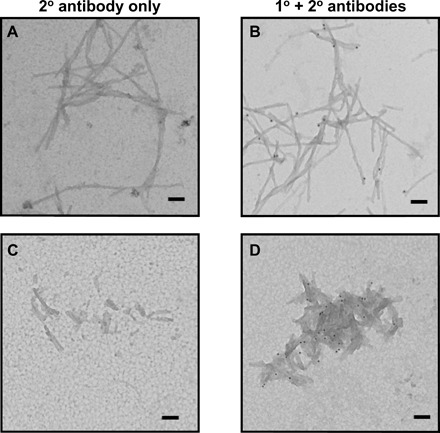
Aβ(M1-42) fibrils formed under quiescent condition imaged as is (A and B) and after sonication (C and D) were incubated with BSA and clusterin and stringently washed. Incubation with an anti-mouse secondary antibody conjugated to a gold particle showed no nonspecific labeling (A and C), whereas incubation with an anti-clusterin monoclonal antibody followed by an anti-mouse secondary antibody conjugated to a gold particle shows the presence of clusterin interacting with the Aβ(M1-42) fibrils (black dots). Scale bars, 100 nm.
Quantification of interactions between clusterin and Aβ(M1-42) fibrils by microfluidic diffusional sizing
Together, the kinetic data described in the previous sections suggest that clusterin is highly effective at inhibiting the aggregation of Aβ(M1-42) at low substoichiometric ratios by specifically reducing the rate of elongation, and provide strong evidence that this process is mediated by the noncovalent association of clusterin with Aβ(M1-42) fibril ends and prefibrillar species. The interactions between the molecular chaperone and the fibrils, therefore, represent the key molecular process underlying the inhibition of fibril elongation.
Evidence of these interactions between clusterin and the fibril ends is provided by immunogold (TEM) (Fig. 2) when we specifically probe for the presence of clusterin on the Aβ(M1-42) fibrils; under a variety of conditions, which implement stringent washing steps and the use of bovine serum albumin (BSA) incubation to reduce nonspecific binding, it is apparent that clusterin binds to the Aβ(M1-42) fibrils. Although it appears that gold labeling is occurring mainly at the fibril ends, it is not possible to conclude that there are no interactions of clusterin with the surfaces of the fibrils. The quantity of bound clusterin can be seen to be increased when the molecular chaperone was incubated with fibrils that were exposed to mechanical breakage by sonication. These shorter fibrils have a larger number of ends at constant surface area compared to the unsonicated fibrils (Fig. 2D). Although this imaging analysis cannot be carried out rigorously in a quantitative manner, this result suggests that the molecular chaperone interacts preferentially with fibril ends, in agreement with the conclusion from the specific inhibition of the elongation rate observed in this study by the kinetic analysis and discussed earlier.
To characterize the interactions between clusterin and Aβ(M1-42) fibrils in a more quantitative manner, we made use of microfluidic diffusional sizing that has recently been developed to measure the interactions between biomolecules directly in solution under native conditions (33, 34). Briefly, the principle of this technique consists of acquiring, in both space and time, the longitudinal diffusion profiles of molecules in a solution flowing in a microfluidic channel (Fig. 3A). The diffusion profiles acquired in these experiments can then be analyzed by considering advection-diffusion processes to extract the distribution of diffusion coefficients and therefore the distributions of the hydrodynamic radii of the individual species present in solution (33). With this technique, interactions between macromolecules can be detected by monitoring the increase in size associated with these events.
Fig. 3. Microfluidic analysis of clusterin binding to Aβ(M1-42) fibrils.
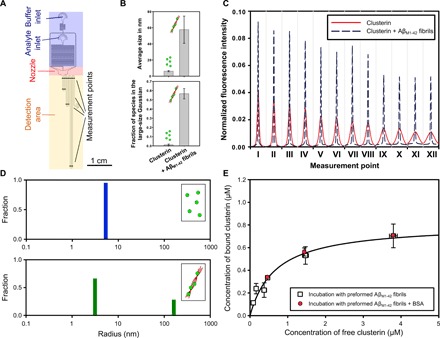
(A) Schematic diagram of the microfluidic diffusional sizing device used in this work indicating its most relevant components (33). (B) The bar charts show the average size and fraction of the species in the large-size range in the absence and presence of Aβ(M1-42) fibrils. The average sizes and the fraction of species in the large-size range reported are the means and SDs of at least three independent repetitions. (C) Diffusion profiles acquired at 12 different positions along the microfluidic channel for a 0.8 μM clusterin solution in the absence (red curves) and presence (black dashed curves) of 17.5 μM preformed Aβ(M1-42) fibrils in 20 mM sodium phosphate buffer at pH 8.0. (D) The size distributions in the absence (blue) and presence (green) of 2 μM Aβ(M1-42) fibrils were evaluated by fitting model simulations on the basis of advection-diffusion equations to the experimental diffusion profiles reported in (C) (see the Supplementary Materials). (E) Binding curve of clusterin to 17.5 μM Aβ(M1-42) fibrils in 20 mM sodium phosphate at pH 8.0 and 21°C measured by the microfluidic diffusion technique. In a first set of experiments (squares), different concentrations of clusterin were incubated with previously generated Aβ(M1-42) fibrils and size distributions were measured after 48 hours of incubation to ensure equilibrium conditions. In a second set of experiments (circles), different concentrations of clusterin were incubated with 17.5 μM Aβ(M1-42) fibrils and BSA at equimolar concentrations to clusterin. Each point represents the mean and SD of at least two independent repetitions. The regression line represents the best fit to the nonlinear Langmuir binding isotherm with KD = 0.67 ± 0.19 μM and M = 0.80 ± 0.08 μM [corresponding to one clusterin molecule per 21 Aβ(M1-42) monomers], with R2 = 0.97.
We therefore monitored the binding of clusterin to Aβ(M1-42) fibrils by measuring the diffusion of clusterin labeled with the fluorescent dye Alexa Fluor 488 in the absence and presence of Aβ(M1-42) fibrils (Fig. 3C and fig. S4). Clusterin bound to a fibril will diffuse substantially slower than the unbound species because of the much greater size of the fibrils related to the molecular chaperones and hence exhibit a distinct diffusion profile (Fig. 3C). The size distributions extracted from the diffusion profiles are shown in Fig. 3D; in the absence of Aβ(M1-42) fibrils, clusterin exhibits a monomodal distribution centered at a hydrodynamic radius of about 8 nm. In the presence of the fibrils, however, a bimodal distribution is observed, with a population similar to the hydrodynamic radius of the monomodal distribution corresponding to unbound molecular chaperone, and a peak in the larger size range corresponding to complexes formed by clusterin and amyloid fibrils. The integral of the area under the two subpopulations is proportional to the concentration of free and bound clusterin. From the titration curve, obtained by performing experiments at different clusterin concentrations between 0 and 4.5 μM with a fibril concentration of 17.5 μM (Fig. 3E), we obtained a KD,21°C value of 670 nM. This value is two orders of magnitude larger than the KD estimated from the kinetic analysis at the same temperature (KD,21°C = 1 nM); the difference may arise from the specific types of information provided by the two approaches. As can be seen by TEM images, clusterin binds both to the surfaces and to the ends of the fibrils, and the microfluidic diffusion technique detects the total quantity of bound clusterin resulting from these two interactions. By contrast, the kinetic analysis is sensitive only to interactions with the growing fibril ends and the smaller propagons, which determine the modulation of the elongation rate, and therefore reports a higher affinity. These findings indicate that clusterin is capable of suppressing Aβ(M1-42) aggregation under physiological concentration, but higher concentrations of molecular chaperone are needed to reduce substantially the effect of toxic species, in agreement with previously reported results (35, 36). As a control experiment, we studied the interaction and the inhibition effects of clusterin when a nonchaperone protein, BSA, is added into the system (Fig. 3E and fig. S5) and detected no effect on binding.
Modulation of Aβ(M1-42) aggregation by Brichos reveals that the reactive sites of secondary nucleation and elongation are distinct
Although there is evidence from the immunogold labeling and the diffusion experiments that clusterin may be able to bind along the surface of the fibrils, as well as to their ends, the kinetic experiments show that clusterin does not inhibit detectable surface-catalyzed secondary nucleation. By contrast, a molecular chaperone belonging to the Brichos family (proSP-C Brichos) has been found to inhibit the secondary nucleation rate associated with Aβ(M1-42) aggregation but not the rate of elongation (11). This observation suggests that inhibition of secondary nucleation processes requires specific interactions with defined reactive sites on the fibrils rather than nonspecific binding along their surfaces. It also suggests that the sites involved in the nucleation and elongation processes are likely to be distinct. To address this question, as well as to conclude whether or not the effect of the two molecular chaperones is additive, we added 18 nM clusterin and 2 μM proSP-C Brichos, both individually and together, to a solution of 2.5 μM Aβ(M1-42) and monitored the aggregation over time. We observed a larger inhibition of the aggregation kinetics when the two molecular chaperones are present together in the solution (Fig. 4). Moreover, the kinetic profiles can be described very closely by simulation using the rate constants determined in the experiments where the two molecular chaperones were added individually. In particular, the results show that inhibition by proSP-C Brichos on the secondary nucleation rate is essentially the same in the presence or absence of clusterin, and reduction of the elongation rate constant is not detectably affected by the presence of proSP-C Brichos (corresponding to the values reported in Fig. 1E).
Fig. 4. Brichos and clusterin exhibit modular and additive behavior of their specific inhibition processes.
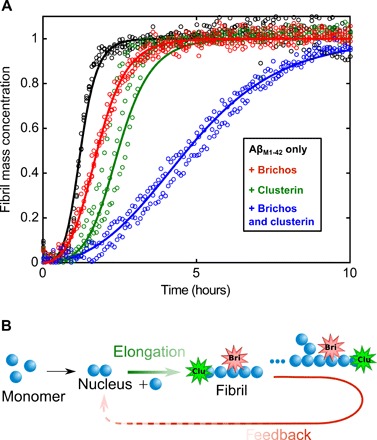
(A) Kinetic profiles of 3 μM Aβ(M1-42) solutions in 20 mM sodium phosphate buffer at pH 8.0 in the absence and presence of 18 nM clusterin and 2 μM proSP-C Brichos added either individually or together as indicated at 37°C. (B) The additivity of the inhibition effects reveals that the sites associated with the two different microscopic steps of elongation and secondary nucleation are distinct. Continuous lines represent model simulations where either the elongation rate constant (green line), secondary nucleation rate constant (red line), or both (blue line) have been selectively inhibited.
The finding that the inhibition effects of the two molecular chaperones studied here are additive is extremely interesting and has a number of important implications: First, it indicates that the sites on the amyloid fibrils involved with the two different microscopic processes of elongation and secondary nucleation pathways are distinct, a result consistent with a recent study showing the development of antibodies able to target specifically distinct steps (15, 37). Moreover, biologically relevant molecular chaperones can interact with these different sites in an additive noncooperative way. Experimental repetitions (fig. S7) at different inhibitor concentrations confirm the additive and noncooperative inhibition effect of the two individual chaperones.
DISCUSSION AND CONCLUSION
Increasing evidence indicates that the aggregation networks leading to the formation of amyloid fibrils are composed of a series of distinct microscopic reactions (9, 38, 39). For Aβ(M1-42), secondary nucleation under the in vitro conditions studied has emerged as the most important source of toxic oligomeric species (10, 40). Understanding the molecular details of this process, which is now feasible through the development of chemical kinetic analysis, is therefore of vital importance for understanding the fundamental processes that are likely to be involved in AD. In this work, we have built on the ability of this kinetic platform to identify the different microscopic mechanisms of the inhibitory effects of two molecular chaperones on amyloid formation by Aβ(M1-42). We have observed that clusterin induces a similar reduction of the parameters k+k2 and k+kn, in agreement with previous findings (26). Moreover, our analysis of seeded aggregation profiles demonstrates that, for our recombinant peptide, this observation can be explained at the microscopic level by a reduction of the elongation rate constant and not by an equal reduction of primary and secondary nucleation rates. In particular, we have shown that the additive nature of the inhibition by the different molecular chaperones implies a selective inhibition of two different microscopic reactions, namely, elongation and surface-induced secondary nucleation. This result suggests that the natural protective mechanisms that have evolved to maintain the protein homoeostasis network are highly sophisticated and work together to suppress different steps in the series of events that give rise to protein aggregation and amyloid formation. Specific inhibition of elongation reactions alone would inhibit the formation of surface available for secondary nucleation but could lead to an accumulation of soluble intermediates and an increase in toxicity. For a complete inhibition of the aggregation process, therefore, additional interactions with the surfaces of the fibrils are required to suppress secondary nucleation events, a finding in good agreement with previous reports (36). Future developments of the analysis described in this work may also clarify the behavior observed in the more complex environment of in vivo systems, for example, where overexpression of clusterin has been shown to accelerate amyloid deposition in mice but to decrease the risk of AD in humans (41–43). More generally, understanding how the aggregation process is altered by specific molecules is crucial to evaluate the consequences for the generation of potential toxic oligomeric species. These molecular details are particularly relevant in the context of the rational design of drug molecules that could, potentially in combination, target multiple specific aggregation steps in a selective manner.
MATERIALS AND METHODS
Materials
The expression and purification procedure for the peptide Aβ(M1-42) (MDAEFRHDSGYEVHHQKLVFFAEDVGSNKGAIIGLMVGGVVIA) and the molecular chaperones clusterin and proSP-C Brichos were carried out as described in previous papers (44–47). In short, Aβ(M1-42) inclusion bodies were extracted from Escherichia coli cells by sonication and dissolved in 8 M urea. Further purification was performed by ion exchange in batch mode on DEAE cellulose resin with additional lyophilization and gel filtration on a 3.4 cm × 200 cm gel-filtration column at 4°C (44).
To obtain the proSP-C Brichos domain, E. coli cells were lysed by lysozyme (1 mg/ml) for 30 min and incubated with deoxyribonuclease and 2 mM MgCl2 for another 30 min on ice. The centrifuged cell pellet was dissolved in 2 M urea in 20 mM tris and 0.1 M NaCl (pH 8) and sonicated for another 5 min. After another centrifugation step, the supernatant was filtered through a 5-μm filter and purified with a 2.5-ml nickel-agarose column. The thioredoxin and His6 tag were removed by adding thrombin for 16 hours at 4°C, followed by another run through a nickel column. The protein was further purified by ion exchange chromatography (45, 46).
Clusterin was extracted from human blood plasma obtained from Wollongong Hospital (Wollongong, New South Wales, Australia). Complete protease inhibitor was added, and the mixture was filtered through (i) a GF/C glass fiber filter and then (ii) a 0.45-mm cellulose nitrate filter. The filtrate was further purified on a 5-ml G7 anti-CLU monoclonal antibody column. After severe washing steps, the specifically bound material was eluted using 2 M GdnHCl in phosphate-buffered saline (PBS). The fraction was dialyzed against 20 mM MES (pH 6.0) and loaded on a 1-ml HiTrap SP XL cation exchange column, collecting the flow through. Last, the pure protein was obtained by size exclusion chromatography on a Superose 6 10/300 column (47). For the microfluidic experiments, clusterin was covalently labeled with Alexa Fluor 488 N-hydroxysuccinimide ester (Thermo Fisher Scientific, Waltham, USA). To achieve this, the protein (2 mg/ml) was incubated with a 10-fold molar excess of the fluorophore (added from a 10 mM stock in dimethyl sulfoxide) [1 hour, room temperature (RT)]. Unconjugated dye was removed by buffer exchange into PBS using a PD-10 column (GE Healthcare, Chicago, USA). The final protein concentration and labeling efficiency was measured according to the manufacturer’s instructions. We carried out kinetic experiments to show that the labeling with the Alexa dye, required for the fluorescence detection, does not modify the inhibition and the binding properties of clusterin (fig. S6). BSA, obtained from Sigma-Aldrich (St. Louis, USA), was used. All aggregation assays and binding reactions were carried out in 20 mM sodium phosphate buffer at pH 8.0. All chemicals were of analytical grade and purchased from Sigma-Aldrich, unless otherwise stated.
Kinetic aggregation assay
Aggregation reactions in the presence and absence of clusterin and proSP-C Brichos were followed by recording ThT fluorescence emission at 480 nm after excitation at 440 nm. Samples (100 μl) were incubated in the Corning (Corning, USA) 96-Well (COSTAR) Half-Area Black with Clear Flat Bottom Polystyrene NBS Microplate, and measurements were recorded in a FLUOstar OPTIMA plate reader (BMG Labtech, Ortenberg, Germany) at 21° or 37°C under quiescent conditions (10). For seeded reactions, fibrils were freshly prepared before each experiment and mixed with freshly purified monomeric Aβ(M1-42) at the desired concentrations. The concentrations of clusterin, monomeric, and fibrillar Aβ(M1-42) for the different experiments are reported in the main text and Supplementary Materials as well as in the captions of the figures. The concentration of ThT was 20 μM in all experiments that we carried out at 37°C, unless otherwise stated.
Kinetic analysis and simulations
The aggregation profiles in the absence and presence of different concentrations of molecular chaperones were simulated individually according to the following equation
| (1) |
where the kinetic parameters B±, C±, κ, κ∞, and are functions of the mass and number concentrations of seeds as well as of the two combinations of the microscopic rate constants k+k2 and knk2, where kn, k+, and k2 are the rate constants of primary nucleation, elongation, and secondary nucleation, respectively (38, 39).
The microscopic rate constants in the absence of the molecular chaperones were considered equal to the values estimated in previous work (10). The rate constants in the presence of different concentrations of clusterin were determined by fitting the individual reaction profiles by minimizing a least-squares error function defined as , where Msim(ti) and Mexp(ti) are the simulated and experimental total fibril mass fractions at time ti, respectively. In the simulations shown in Fig. 1, kn, k+, and k2 were varied individually, but in the simulations shown in the other figures, only k+ has been modified.
The experimental data in the absence and presence of clusterin were described by a second kinetic model, which considers binary interactions between clusterin and fibril ends. The detailed description of the corresponding equations can be found in (18). In this model, the rate constants kn, k+, and k2 were assumed to be equal to the values estimated in the absence of the chaperone and considered to be independent of the concentration of clusterin, the effect of which is included in the association and dissociation rates. The association and dissociation rate constants were estimated by the global analysis of the reaction profiles at different concentrations of clusterin by minimizing a least-squares error function similar to that described above (18). The simulation reported in Fig. 1E is based on a simplified expression, which can be derived from this second kinetic model, and relates the apparent elongation rate constant to the total concentration of chaperone and the equilibrium constant of the association reaction (18)
| (2) |
In this study, we report the dissociation constant KD = 1/KeqEnd.
Immunogold-labeling TEM
Aβ(M1-42) fibrils [1.5 μM Aβ(M1-42) monomer equivalents] were incubated with BSA (1 μM; New England Biolabs) in 20 mM phosphate buffer (pH 8.0) (10 min, RT). Clusterin (0.15 μM) was added to the solution and further incubated (10 min, RT). The fibril sample was centrifuged (15,000 rpm, 20 min, 4°C), and the supernatant was removed. The pellet was resuspended in PBS containing 0.01% (v/v) Triton and 0.01% (v/v) Tween 20 (15 μl), and then the sample was centrifuged (15,000 rpm, 20 min, 4°C). The pellet was resuspended in PBS (10 μl). For the sonicated samples, Aβ(M1-42) fibrils (1.5 μM) were sonicated using a probe sonicator (Bandelin, SONOPULS HD 2070) for 1 min with 10% maximum power and 30% cycles before incubation with BSA and clusterin. The prepared fibril samples (5 μl) were applied to a carbon support film, 400 mesh, 3-mm nickel grid (EM Resolutions Ltd., Saffron Walden, UK) and incubated (5 min, RT). The grid was blocked with BSA (1 mg/ml) in PBS for 15 min and incubated with 1:100 G7 mouse anti-human clusterin monoclonal antibody [stock solution (2 mg/ml)] in PBS for 30 min. For the secondary antibody–only negative controls, samples were incubated with PBS only at this step. The grid was washed (three times for 5 min), first, in PBS/0.01% Triton/0.01% Tween 20 and then twice with PBS only, followed by incubation with 1:500 gold-labeled anti-mouse secondary antibody (BBI Solutions, Cardiff, UK) in PBS for 30 min. Last, the grid was washed three times as described above, twice with water, and then incubated for 2 min with 2% uranyl acetate (w/v). To remove excess uranyl acetate, the grid was washed twice with water and dried completely before imaging. The fibrils were imaged on a FEI Tecnai G2 TEM (Cambridge Advanced Imaging Centre, University of Cambridge, UK). Images were analyzed using the SIS MegaView II Image Capture System (Olympus, Tokyo, Japan).
Preparation of samples for binding measurements
Aβ(M1-42) fibrils were generated by incubating 17 μM monomeric Aβ(M1-42) in 20 mM sodium phosphate buffer at pH 8.0 in a 96-well plate in a FLUOstar OPTIMA plate reader (BMG Labtech) at 37°C with double orbital rotation (400 rpm). Aggregation was monitored by following the increase in the fluorescence emission of a similar sample implemented with 20 μM ThT dye initiated upon its binding to amyloid fibrils. After completion of the aggregation reaction, the fibrils were collected, supplemented with Alexa Fluor 488–labeled clusterin in the concentration range between 0 and 4.5 μM, and incubated for at least 2 days at 21°C to ensure that equilibrium was obtained. Diffusion sizing measurements were then performed at 21°C, as described below. In a second set of experiments, three more points were evaluated with the addition of BSA (at the same concentration as used in the clusterin experiments, i.e., 0.8, 2, and 4.5 μM) added to a mixture of clusterin and fibrils, as described above, to examine the specificity of the binding with 17.5 μM Aβ(M1-42) fibrils. The samples were again analyzed by microfluidic diffusion methods.
Microfluidic diffusional sizing
The fabrication and the operation of the microfluidic diffusion device used in the present studies have been described in previous papers (33, 48). Briefly, the microfluidic chips were fabricated by using standard soft lithography. The sample to be analyzed and the buffer were introduced into the system through reservoirs connected to the inlets, and the flow rate in the channel was controlled by applying a negative pressure at the outlet by a syringe pump (CETONI neMESYS, Korbussen, Germany) at typical flow rates in the range from 60 to 90 μl/hour. Lateral diffusion profiles were recorded at 12 different positions (3.5, 5.3, 8.6, 10.3, 18.6, 20.3, 28.6, 30.4, 58.7, 60.4, 88.7, and 90.5 mm) by standard epifluorescence microscopy using a cooled charge-coupled device camera (Photometrics Evolve 512, Tucson, USA). The diffusion profiles were fitted to model simulations on the basis of advection-diffusion equations assuming a bimodal Gaussian distribution (48). From the area under the curves of the two Gaussian populations, the concentrations of the bound and free molecular chaperones were evaluated. The dissociation constant KD was calculated by direct nonlinear regression on the basis of the Langmuir binding isotherm (49, 50)
| (3) |
where M is the total concentration of binding sites available on the Aβ(M1-42) fibrils, representing the maximum concentration of bound molecular chaperone, while [clusterinbound] and [clusterinfree] are the concentrations of bound and free molecular chaperone, respectively.
Supplementary Material
Acknowledgments
Funding: The research leading to these results has received funding from the European Research Council under the European Union’s Seventh Framework Programme (FP7/2007-2013) through the ERC grant PhysProt (agreement no. 337969) (to T.S., T.P.J.K., and S.L.). Furthermore, we acknowledge financial support from the Marie Curie fellowship scheme for career development (to P.A.), EPSRC, BBSRC, the Frances and Augustus Newman Foundation (to T.P.J.K.), Swedish Research Council (to S.L.), and the Wellcome Trust (094425/Z/10/Z) (to C.M.D., M.V., and T.P.J.K.). Author contributions: P.A., J.R.K., and T.P.J.K. designed the study. P.A., T.S., U.L., J.R.K., and D.R.W. performed the experiments. S.L. and M.R.W. provided new materials. T.S., U.L., P.A., J.R.K., S.L., C.M.D., M.V., and T.P.J.K. analyzed the data. T.S., P.A., C.M.D., M.V., and T.P.J.K wrote the paper. All authors discussed the results and commented on the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from T.P.J.K. (tpjk2@cam.ac.uk) or P.A. (paolo.arosio@chem.ethz.ch).
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/4/eaau3112/DC1
Fig. S1. Analysis of the effects of clusterin on the aggregation kinetics of Aβ(M1-42) at 37°C.
Fig. S2. Analysis of the effects of clusterin on the aggregation kinetics of Aβ(M1-42) at 21°C.
Fig. S3. Seeding experiments of Aβ(M1-42) in the presence and absence of clusterin.
Fig. S4. Diffusion profiles of specific samples at 12 different positions in a microfluidic device.
Fig. S5. Analysis of interfering effects of transient proteins on clusterin inhibition activity.
Fig. S6. Effects of the fluorescent label of clusterin on the inhibition process on Aβ(M1-42) aggregation.
Fig. S7. Kinetic analysis on the aggregation kinetics of Aβ(M1-42) in the presence of Brichos and clusterin separately and combined.
REFERENCES AND NOTES
- 1.Knowles T. P. J., Vendruscolo M., Dobson C. M., The amyloid state and its association with protein misfolding diseases. Nat. Rev. Mol. Cell Biol. 15, 384–396 (2014). [DOI] [PubMed] [Google Scholar]
- 2.Eisenberg D., Jucker M., The amyloid state of proteins in human diseases. Cell 148, 1188–1203 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hardy J., Selkoe D. J., The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 297, 353–356 (2002). [DOI] [PubMed] [Google Scholar]
- 4.Hardy J., Expression of normal sequence pathogenic proteins for neurodegenerative disease contributes to disease risk: ‘Permissive templating’ as a general mechanism underlying neurodegeneration. Biochem. Soc. Trans. 33, 578–581 (2005). [DOI] [PubMed] [Google Scholar]
- 5.Karran E., Mercken M., De Strooper B., The amyloid cascade hypothesis for Alzheimer’s disease: An appraisal for the development of therapeutics. Nat. Rev. Drug Discov. 10, 698–712 (2011). [DOI] [PubMed] [Google Scholar]
- 6.Chiti F., Dobson C. M., Protein misfolding, amyloid formation, and human disease: A summary of progress over the last decade. Annu. Rev. Biochem. 86, 27–68 (2017). [DOI] [PubMed] [Google Scholar]
- 7.F. Ferrone, Amyloid, Prions, and Other Protein Aggregates (Academic Press Inc., 1999), vol. 309, pp. 256–274. [Google Scholar]
- 8.Tanaka M., Collins S. R., Toyama B. H., Weissman J. S., The physical basis of how prion conformations determine strain phenotypes. Nature 442, 585–589 (2006). [DOI] [PubMed] [Google Scholar]
- 9.Knowles T. P. J., Waudby C. A., Devlin G. L., Cohen S. I. A., Aguzzi A., Vendruscolo M., Terentjev E. M., Welland M. E., Dobson C. M., An analytical solution to the kinetics of breakable filament assembly. Science 326, 1533–1537 (2009). [DOI] [PubMed] [Google Scholar]
- 10.Cohen S. I. A., Linse S., Luheshi L. M., Hellstrand E., White D. A., Rajah L., Otzen D. E., Vendruscolo M., Dobson C. M., Knowles T. P. J., Proliferation of amyloid-β42 aggregates occurs through a secondary nucleation mechanism. Proc. Natl. Acad. Sci. U.S.A. 110, 9758–9763 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cohen S. I. A., Arosio P., Presto J., Kurudenkandy F. R., Biverstal H., Dolfe L., Dunning C., Yang X., Frohm B., Vendruscolo M., Johansson J., Dobson C. M., Fisahn A., Knowles T. P. J., Linse S., A molecular chaperone breaks the catalytic cycle that generates toxic Aβ oligomers. Nat. Struct. Mol. Biol. 22, 207–213 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lambert M. P., Barlow A. K., Chromy B. A., Edwards C., Freed R., Liosatos M., Morgan T. E., Rozovsky I., Trommer B., Viola K. L., Wals P., Zhang C., Finch C. E., Krafft G. A., Klein W. L., Diffusible, nonfibrillar ligands derived from Aβ1–42 are potent central nervous system neurotoxins. Proc. Natl. Acad. Sci. U.S.A. 95, 6448–6453 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Benilova I., Karran E., De Strooper B., The toxic Aβ oligomer and Alzheimer’s disease: An emperor in need of clothes. Nat. Neurosci. 15, 349–357 (2012). [DOI] [PubMed] [Google Scholar]
- 14.Arosio P., Vendruscolo M., Dobson C. M., Knowles T. P. J., Chemical kinetics for drug discovery to combat protein aggregation diseases. Trends Pharmacol. Sci. 35, 127–135 (2014). [DOI] [PubMed] [Google Scholar]
- 15.Aprile F. A., Sormanni P., Perni M., Arosio P., Linse S., Knowles T. P. J., Dobson C. M., Vendruscolo M., Selective targeting of primary and secondary nucleation pathways in Aβ42 aggregation using a rational antibody scanning method. Sci. Adv. 3, e1700488 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Habchi J., Arosio P., Perni M., Costa A. R., Yagi-Utsumi M., Joshi P., Chia S., Cohen S. I. A., Müller M. B. D., Linse S., Nollen E. A. A., Dobson C. M., Knowles T. P. J., Vendruscolo M., An anticancer drug suppresses the primary nucleation reaction that initiates the production of the toxic Aβ42 aggregates linked with Alzheimer’s disease. Sci. Adv. 2, e1501244 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Michaels T. C. T., Šarić A., Habchi J., Chia S., Meisl G., Vendruscolo M., Dobson C. M., Knowles T. P. J., Chemical kinetics for bridging molecular mechanisms and macroscopic measurements of amyloid fibril formation. Annu. Rev. Phys. Chem. 69, 273–298 (2018). [DOI] [PubMed] [Google Scholar]
- 18.Arosio P., Michaels T. C. T., Linse S., Mansson C., Emanuelsson C., Presto J., Johansson J., Vendruscolo M., Dobson C. M., Knowles T. P. J., Kinetic analysis reveals the diversity of microscopic mechanisms through which molecular chaperones suppress amyloid formation. Nat. Commun. 7, 10948 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hartl F. U., Hayer-Hartl M., Molecular chaperones in the cytosol: From nascent chain to folded protein. Science 295, 1852–1858 (2002). [DOI] [PubMed] [Google Scholar]
- 20.Hartl F. U., Bracher A., Hayer-Hartl M., Molecular chaperones in protein folding and proteostasis. Nature 475, 324–332 (2011). [DOI] [PubMed] [Google Scholar]
- 21.Aprile F. A., Arosio P., Fusco G., Chen S. W., Kumita J. R., Dhulesia A., Tortora P., Knowles T. P. J., Vendruscolo M., Dobson C. M., Cremades N., Inhibition of α-synuclein fibril elongation by Hsp70 is governed by a kinetic binding competition between α-synuclein species. Biochemistry 56, 1177–1180 (2017). [DOI] [PubMed] [Google Scholar]
- 22.Månsson C., Arosio P., Hussein R., Kampinga H. H., Hashem R. M., Boelens W. C., Dobson C. M., Knowles T. P. J., Linse S., Emanuelsson C., Interaction of the molecular chaperone DNAJB6 with growing amyloid-beta 42 (Aβ42) aggregates leads to sub-stoichiometric inhibition of amyloid formation. J. Biol. Chem. 289, 31066–31076 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Narayan P., Orte A., Clarke R. W., Bolognesi B., Hook S., Ganzinger K. A., Meehan S., Wilson M. R., Dobson C. M., Klenerman D., The extracellular chaperone clusterin sequesters oligomeric forms of the amyloid-β(1-40) peptide. Nat. Struct. Mol. Biol. 19, 79–83 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wyatt A. R., Yerbury J. J., Ecroyd H., Wilson M. R., Extracellular chaperones and proteostasis. Annu. Rev. Biochem. 82, 295–322 (2013). [DOI] [PubMed] [Google Scholar]
- 25.Kumita J. R., Poon S., Caddy G. L., Hagan C. L., Dumoulin M., Yerbury J. J., Stewart E. M., Robinson C. V., Wilson M. R., Dobson C. M., The extracellular chaperone clusterin potently inhibits human lysozyme amyloid formation by interacting with prefibrillar species. J. Mol. Biol. 369, 157–167 (2007). [DOI] [PubMed] [Google Scholar]
- 26.Beeg M., Stravalaci M., Romeo M., Carrá A. D., Cagnotto A., Rossi A., Diomede L., Salmona M., Gobbi M., Clusterin binds to Aβ1–42 oligomers with high affinity and interferes with peptide aggregation by inhibiting primary and secondary nucleation. J. Biol. Chem. 291, 6958–6966 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yerbury J. J., Poon S., Meehan S., Thompson B., Kumita J. R., Dobson C. M., Wilson M. R., The extracellular chaperone clusterin influences amyloid formation and toxicity by interacting with prefibrillar structures. FASEB J. 21, 2312–2322 (2007). [DOI] [PubMed] [Google Scholar]
- 28.Yerbury J. J., Kumita J. R., Meehan S., Dobson C. M., Wilson M. R., α2-Macroglobulin and Haptoglobin suppress amyloid formation by interacting with prefibrillar protein species. J. Biol. Chem. 284, 4246–4254 (2009). [DOI] [PubMed] [Google Scholar]
- 29.M. R. Wilson, J. J. Yerbury, S. Poon, Extracellular chaperones and amyloids, in Heat Shock Proteins and the Brain: Implications for Neurodegenerative Diseases and Neuroprotection (Heat Shock Proteins, vol. 3), A. A. A. Asea, I. R. Brown, Eds. (Springer Science & Business Media, 2008), pp. 283–315. [Google Scholar]
- 30.Arosio P., Cukalevski R., Frohm B., Knowles T. P. J., Linse S., Quantification of the concentration of Aβ42 propagons during the lag phase by an amyloid chain reaction assay. J. Am. Chem. Soc. 136, 219–225 (2014). [DOI] [PubMed] [Google Scholar]
- 31.Collins S. R., Douglass A., Vale R. D., Weissman J. S., Mechanism of prion propagation: Amyloid growth occurs by monomer addition. PLOS Biol. 2, e321 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Poon S., Easterbrook-Smith S. B., Rybchyn M. S., Carver J. A., Wilson M. R., Clusterin is an ATP-independent chaperone with very broad substrate specificity that stabilizes stressed proteins in a folding-competent state. Biochemistry 39, 15953–15960 (2000). [DOI] [PubMed] [Google Scholar]
- 33.Arosio P., Müller T., Rajah L., Yates E. V., Aprile F. A., Zhang Y., Cohen S. I. A., White D. A., Herling T. W., De Genst E. J., Linse S., Vendruscolo M., Dobson C. M., Knowles T. P. J., Microfluidic diffusion analysis of the sizes and interactions of proteins under native solution conditions. ACS Nano 10, 333–341 (2016). [DOI] [PubMed] [Google Scholar]
- 34.Yates E. V., Müller T., Rajah L., De Genst E. J., Arosio P., Linse S., Vendruscolo M., Dobson C. M., Knowles T. P. J., Latent analysis of unmodified biomolecules and their complexes in solution with attomole detection sensitivity. Nat. Chem. 7, 802–809 (2015). [DOI] [PubMed] [Google Scholar]
- 35.Yerbury J. J., Wilson M. R., Extracellular chaperones modulate the effects of Alzheimer’s patient cerebrospinal fluid on Aβ1–42 toxicity and uptake. Cell Stress Chaperones 15, 115–121 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Drews A., De S., Flagmeier P., Wirthensohn D. C., Chen W.-H., Whiten D. R., Vincke C., Muyldermans S., Paterson R. W., Slattery C. F., Fox N. C., Schott J. M., Zetterberg H., Dobson C. M., Gandhi S., Klenerman D., Inhibiting the Ca2+ influx induced by human CSF. Cell Rep. 21, 3310–3316 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Munke A., Persson J., Weiffert T., De Genst E., Meisl G., Arosio P., Carnerup A., Dobson C. M., Vendruscolo M., Knowles T. P. J., Linse S., Phage display and kinetic selection of antibodies that specifically inhibit amyloid self-replication. Proc. Natl. Acad. Sci. U.S.A. 114, 6444–6449 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cohen S. I. A., Vendruscolo M., Welland M. E., Dobson C. M., Terentjev E. M., Knowles T. P. J., Nucleated polymerization with secondary pathways. I. Time evolution of the principal moments. J. Chem. Phys. 135, 065105 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cohen S. I. A., Vendruscolo M., Dobson C. M., Knowles T. P. J., Nucleated polymerization with secondary pathways. II. Determination of self-consistent solutions to growth processes described by non-linear master equations. J. Chem. Phys. 135, 065106 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Törnquist M., Michaels T. C. T., Sanagavarapu K., Yang X., Meisl G., Cohen S. I. A., Knowles T. P. J., Linse S., Secondary nucleation in amyloid formation. Chem. Commun. 54, 8667–8684 (2018). [DOI] [PubMed] [Google Scholar]
- 41.DeMattos R. B., O’dell M. A., Parsadanian M., Taylor J.W., Harmony J. A. K., Bales K. R., Paul S. M., Aronow B. J., Holtzman D. M., Clusterin promotes amyloid plaque formation and is critical for neuritic toxicity in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. U.S.A. 99, 10843–10848 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wojtas A. M., Kang S. S., Olley B. M., Gatherer M., Shinohara M., Lozano P. A., Liu C.-C., Kurti A., Baker K. E., Dickson D.W., Yue M., Petrucelli L., Bu G., Carare R. O., Fryer J. D., Loss of clusterin shifts amyloid deposition to the cerebrovasculature via disruption of perivascular drainage pathways. Proc. Natl. Acad. Sci. U.S.A. 114, E6962–E6971 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tan L., Wang H.-F., Tan M.-S., Tan C.-C., Zhu X.-C., Miao D., Yu W.-J., Jiang T., Tan L., Yu J.-T.; Alzheimer’s Disease Neuroimaging Initiative , Effect of CLU genetic variants on cerebrospinal fluid and neuroimaging markers in healthy, mild cognitive impairment and Alzheimer’s disease cohorts. Sci. Rep. 6, 26027 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Walsh D. M., Thulin E., Minogue A. M., Gustavsson N., Pang E., Teplow D. B., Linse S., A facile method for expression and purification of the Alzheimer’s disease-associated amyloid beta-peptide. FASEB J. 276, 1266–1281 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Johansson H., Nordling K., Weaver T. E., Johansson J., The Brichos domain-containing C-terminal part of pro-surfactant protein C binds to an unfolded poly-Val transmembrane segment. J. Biol. Chem. 281, 21032–21039 (2006). [DOI] [PubMed] [Google Scholar]
- 46.Willander H., Presto J., Askarieh G., Biverstal H., Frohm B., Knight S. D., Johansson J., Linse S., BRICHOS domains efficiently delay fibrillation of amyloid β-peptide. J. Biol. Chem. 287, 31608–31617 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dabbs R. A., Wilson M. R., Expression and purification of chaperone-active recombinant clusterin. PLOS ONE 9, e86989 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Müller T., Arosio P., Rajah L., Cohen S. I. A., Yates E. V., Vendruscolo M., Dobson C. M., Knowles T. P. J., Particle-based Monte-Carlo simulations of steady-state mass transport at intermediate Péclet numbers. Int. J. Nonlinear Sci. Numer. 17, 175–183 (2016). [Google Scholar]
- 49.Langmuir I., The constitution and fundamental properties of solids and liquids. Part I. Solids. J. Am. Chem. Soc. 38, 2221–2295 (1916). [Google Scholar]
- 50.Langmuir I., Vapor pressures, evaporation, condensation and adsorption. J. Am. Chem. Soc. 54, 2798–2832 (1932). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/4/eaau3112/DC1
Fig. S1. Analysis of the effects of clusterin on the aggregation kinetics of Aβ(M1-42) at 37°C.
Fig. S2. Analysis of the effects of clusterin on the aggregation kinetics of Aβ(M1-42) at 21°C.
Fig. S3. Seeding experiments of Aβ(M1-42) in the presence and absence of clusterin.
Fig. S4. Diffusion profiles of specific samples at 12 different positions in a microfluidic device.
Fig. S5. Analysis of interfering effects of transient proteins on clusterin inhibition activity.
Fig. S6. Effects of the fluorescent label of clusterin on the inhibition process on Aβ(M1-42) aggregation.
Fig. S7. Kinetic analysis on the aggregation kinetics of Aβ(M1-42) in the presence of Brichos and clusterin separately and combined.