

Abstract
Radical hydrofunctionalization occurs with ease using metal-hydride atom transfer (MHAT) catalysis to couple alkenes and competent radicalophilic electrophiles. Traditional two-electron electrophiles have remained unreactive. Herein we report the reductive coupling of electronically-unbiased olefins with imines and aldehydes. Iron-catalysis allows addition of alkyl-substituted olefins into imines through the intermediacy of free-radicals, whereas a combination of catalytic Co(Salt-Bu,t-Bu) and chromium salts enable a branch-selective coupling of olefins and aldehydes through the formation of a putative alkyl chromium intermediate.
Graphical Abstract
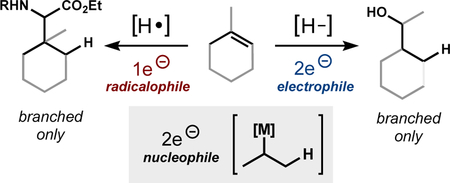
Hyfrometallation of unbiased alkenes with high branched selectivity by radical-anion crossover
Branch-selective reactions of alkyl-substituted olefins via carbocationic1 or radical intermediates2 benefits from an abundance of methods, but the analogous transformation into branched-carbanion equivalents remains underdeveloped (Figure 1). A common way to transform an olefin into a carbanion equivalent is via hydrometallation of a double bond. However, such branch-selective hydrometallation of alkenes is generally limited to styrenes, allenes, and dienes—all electronically biased systems that stabilize a developing carbon-metal bond.3 In the absence of electronic bias, canonical metal hydrides favor linear selectivity.4 To obtain branch-selectivity with electronically-unbiased alkenes, we have investigated M−H hydrogen atom transfer (MHAT) catalysis and subsequent capture of the nascent intermediates by a second metal complex.5,6,7 For example, we recently established that nickel complexes intercept Co(Salt-Bu,t-Bu)-catalyzed MHAT cycles in a direct organocobalt to organonickel transmetallation.6 Similar alkyl transmetallations have been reported in non-catalytic systems between alkyl−Co(dmgBF2)2Py and inorganic nickel,8 and proposed for bioorganometallic9 and catalytic processes.10 This alkyl transfer does not appear limited to nickel: vitamin B12-mimetics (such as Co(salen) derivatives) can undergo alkyl transfer to palladium,11 rhodium,12 other cobalt,13 platinum,14 gold,15 chromium,16 and zinc17 salts and organometallic species. Yet despite the apparent generality of this transformation, there is a paucity of preparative cross-coupling methods that leverage this reactivity.
Figure 1:
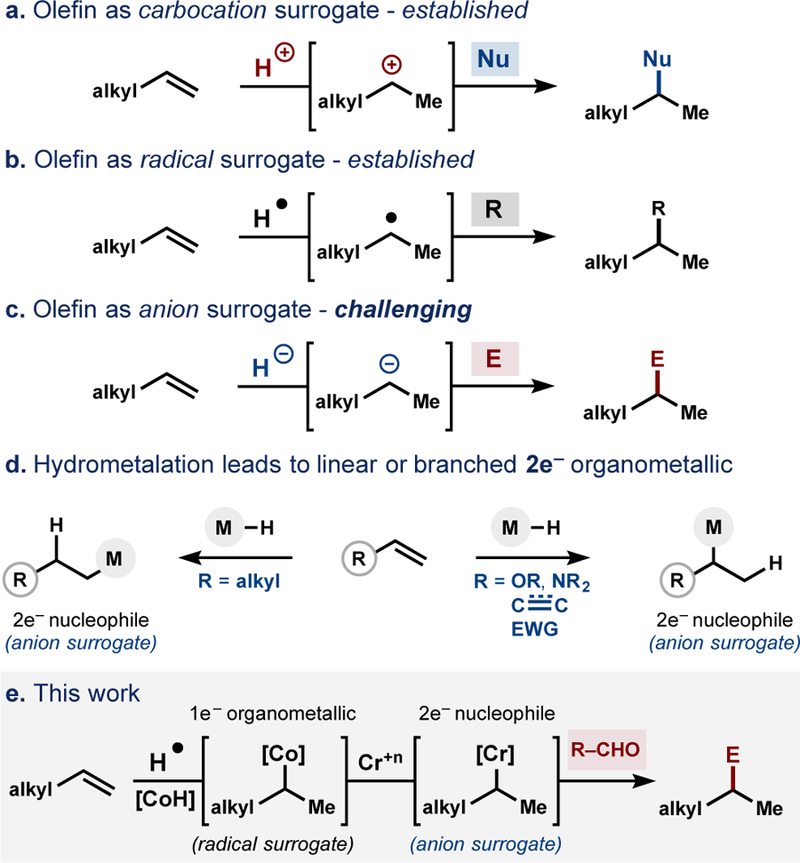
Transformation of olefins into carbanion equivalents by a radical/polar crossover.
The capacity to form cobalt organometallics via MHAT followed by cage-collapse6,18,19,20 prompted us to explore transmetallation partners that might lead to otherwise inaccessible branched products. Here we show that olefins can be added to imines and aldehydes to form sp3−sp3 bonds. The former reaction with an activated electrophile occurs under standard MHAT catalysis, whereas the latter reaction requires interception of MHAT intermediates with chromium salts (Figure 1e). This transformation expands the current scope of olefins as carbanion surrogates21 which has heretofore required the use of electronically-activated olefins, such as styrene, allenyl, or dienes. Alkyl-substituted olefins, in contrast, react with carbonyls at the least-substituted position through a Prins mechanism,22 or undergo iron-catalyzed hydromagnesiation reactions to form linear nucleophiles.23,24,25
We initially investigated the Markovnikov addition of alkenes into carbonyl derivatives by utilizing the intermediacy of the free radicals and noticed that productive reactions were only obtained with standard radicalophiles, such as radical-stabilizing imines. Glyoxylimines are precedented as radical acceptors,26,27 and chiral sulfinyl auxiliaries can be used to impart stereocontrol. Although the competitive reduction of these electrophiles by the metal hydrides or the stoichiometric silane was observed, this could be minimized by using a slight excess of the olefin and Fe+3 salts as the catalyst.28 Several feedstock alkenes served as competent nucleophilic components and delivered unnatural amino acids derivatives with good to excellent diastereoselectivities. (Figure 2). The early transition state of radical reactions allows facile formation of sterically hindered unnatural α-amino acids bearing β-quaternary carbons, and reactive groups like free-hydroxyls (13) or two-electron electrophiles such as esters, epoxides, or aldehydes (8, 15, and 17) are tolerated. Complex feedstock terpenes can engage the sulfinylimines to deliver adducts 12, 14, and 15, and even glycans deliver amino esters with good diastereocontrol (16). Comparison of the optical rotation obtained from our reaction to that of t-butyl glycine derivatives shows that sulfinimes with the (S)-configuration affords the (S)-amine whereas the (R)-sulfinime affords the (R)-amine.28 Better diastereoselectivity is obtained with the more hindered mesitylene or tri-isopropyl arene-derived sulfinamide, whereas the use of Ellman’s tert-butyl sulfinamide was not compatible with these radical conditions.28 Given the ease with which these compounds are made, requiring no prefunctionalization prior to radical formation, we anticipate that this method will find application in the synthesis of unnatural amino acids.29
Figure 2.
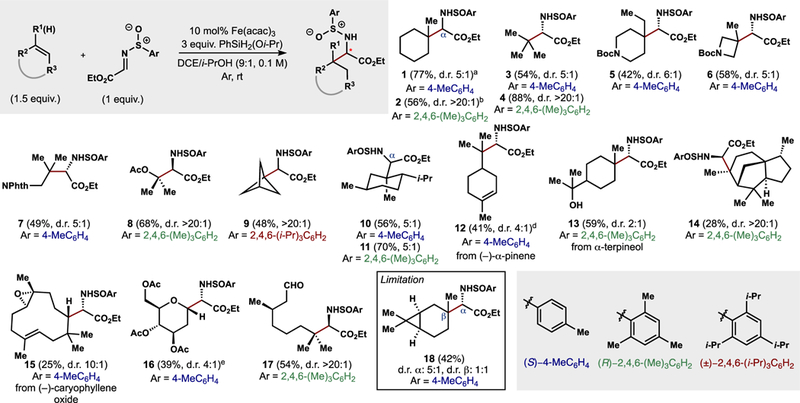
Alkyl radicals generated by MHAT add to chiral sulfinimines. d.r. of two major diastereomers reported. astereochemistry at the α-carbon is (S). bstereochemistry in the α-carbon is (R). cMn(dpm)3 instead of Fe(acac)3 was used. dcontains 15% unrearranged pinene product and its diastereomer. e3 equiv. of olefin used.
Addition of the free radical to aldehydes, however, proved challenging (see Table 1), as may be expected due to the lower stability of an O-centered radical relative to a C-centered radical, which is reflected by the more facile C–C bond scission than C–C bond formation.30 Strategies to drive this energetically disfavorable addition include sequestering the unstable O-centered radical as an alkoxide (which cannot undergo homolytic β-scission) in an intramolecular setting31 or accessing excited-states via photochemistry.32 However, neither strategy may be used for intermolecular addition with alkyl-substituted olefins.33 In light of the precedence for alkyl-cobalt complexes to transmetallate other metal species and the apparent facility with which organocobalt species can form from olefins,6,20 we wondered if a two-electron nucleophile equivalent could arise from unactivated olefins via sequential one-electron reductions via interception of organocobalt with chromium species.
Table 1.
Conversion of C-centered radicals to 2-electron nucleophiles.
 | |||
| 19 (1 equiv.) | 20 (2 equiv.) | “standard conditions” | 21 70%a (72%)b |
| Entry | Deviations from above |
Yield (%)a |
|
| 1 | Fe(dpm)3, Fe(acac)3, Co(acac)2 or Mn(dpm)3 instead of Co(Salt-Bu,t-Bu) |
< 10% | |
| 2 | CrCl2 instead of CrCl3 | trace | |
| 3 | with Zn° or Mn° | 11% | |
| 4 | Co(salen)Cl and no [O] | 45%c | |
| 5 | no [O] | − | |
| 6 | 0.2 equiv. of CrCl3 instead of 1 equiv. | 22% | |
| 7 | 0.2 equiv. of CrCl3 and TMSCl (1 equiv.) | − | |
| 8 | in DMF instead of THF/CH3CN | − | |
| 9 | without CH3CN | 35% | |
| 10 | under air | 46% | |
| 11 | with 1 equiv. of H2O | − | |
| 12 | No Co(Salt-Bu,t-Bu) | − | |
| 13 | No CrCl3 | −d | |
yield determined by GC/FID using a calibrated internal standard
isolated yield with 20 mol % of Co(salen)Cl and CrCl3(THF)3
1 equiv. of NaBF4 added
isomerization and hydrogenation was observed; d.r. 1:1 in all cases.
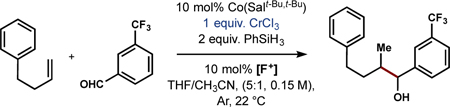
We were drawn to chromium chemistry for several reasons: 1) organochromium reagents are known to add into carbonyls in a 1,2-fashion 2) Cr+2 salts are proposed to intercept alkyl-radicals to form organochromium species with bimolecular rate constants on the order of 107 M−1s−1,34 3) alkyl-cobalamines and -cobaloximes can also be intercepted by Cr+2,16,35,36 and 4) chromium salts are inexpensive and of low toxicity in the +2 and +3 oxidation states.37 Furthermore, the weak Brønsted acidity of organochromium complexes allows for a high functional group tolerance and for their use in late-stage functionalization for complex molecule synthesis.38
Initially, attempts to merge MHAT catalysis and chromium chemistry met with poor results. β-diketonate complexes of Co and Mn were not productive, although iron salts afforded the product in low yield (Table 1, Entry 1).39 We discovered, however, that use of Co(Salt-Bu,t-Bu) and equimolar amounts of 1-fluoro-2,4,6-trimethylpyridinium tetrafluoroborate in the presence of phenylsilane and CrCl3 could couple the terminal olefin in 19 to 3-trifluoromethyl benzaldehyde in good yields. Given that Cr+2 is typically the active species in the addition of alkyl halides into carbonyls, we initially explored the reaction using CrCl2 or CrCl3 alongside an external metal reductant only to discover that these conditions led to less product formed than the amount of [Co] pre-catalyst added (Entries 2 and 3). One explanation is that the external reductants impede the Co-cycle by unproductive reduction of Co+3 intermediates.40 Our optimized conditions appear to circumvent this problem by reductive formation of Cr+2 in situ (see below). Although it is possible to perform this reaction with the pre-oxidized Co(Salt-Bu,t-Bu)Cl, use of Co+2 and an external oxidant generally afforded higher yields (Entry 4), thereby allowing use of the more convenient +3 and +2 oxidation states of Cr and Co, respectively. Control experiments indicate that both metals are necessary for product formation (Entries 12–13)41
Evaluation of the scope (Figure 3) revealed that both aromatic and alkyl aldehydes are competent electrophiles, and a wide range of electronic variation is tolerated. In general, electron withdrawn substrates afford higher yields than electron-rich electrophiles, yet even vanillin-derived aldehydes such as 33 and 34 react in high yield. Various heteroaromatic aldehydes may be employed (37 − 39), as well as terpene-derived substrates (41). Esters (45), tosylates (44), and chlorides (46) are orthogonal electrophiles, but competitive reduction of bromides, and iodides was observed. A switch in solvent from THF to DME allows 1,2-disubstituted olefins to be engaged (54 − 57), although trisubstituted olefins are not yet competent. Modest diastereocontrol is imparted by a chiral directing group (49), and sterically bulky substrates (35, 43, 50 and 52).42
Figure 3.
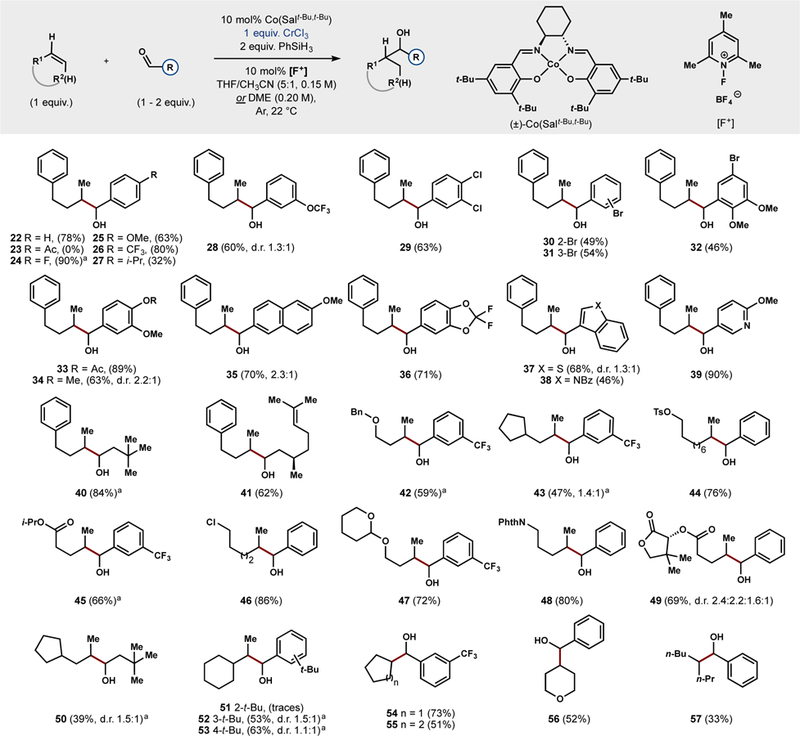
Conversion of alkenes into carbanion surrogates with branched-selectivity. d.r. at the formed bond is close to 1:1 unless otherwise noted. See Supporting Information for the d.r. of the isolated compounds. a20 mol% of [Co] and 20 mol% of [F+] were used.
Although we currently do not have a complete mechanistic model, several observations are worth noting. First, the yield of the product formed does not vary as a function of delayed Cr+3 addition, which is consistent with intermediate formation of a stable organocobalt species that is engaged by the Cr, and inconsistent with an alternative hypothesis that the cobalt cycle continuously generates a C-centered radical whose reactivity would favor formation of side products prior to addition of the CrCl3.43 These observations draw analogy to our previously reported Ni/Co hydroarylation6 and the mechanistic studies of Espenson and coworkers.35 Stoichiometric experiments support a transmetallation and suggest reaction with Cr+2 rather than the Cr+3 species. In these experiments, a sec-alkyl cobalt was formed in situ by displacement of 2-bromopropane by CoI(Salt-Bu, t-Bu)(py)2; addition of CrCl3 and aldehyde 20 yields no product, whereas CrCl2 produces around 50% of adduct 58 based on the yield of the alkyl-cobalt.28 Control experiments with the alkyl-halide during the same time period yields no product under these conditions. We suspect reduction of Cr+3 to Cr+2 occurs via the stoichiometric silane reductant necessary for the MHAT catalytic cycle.44 By analogy to the proposal of Espenson and coworkers in similar systems,35 a possible mechanism for the alkyl transfer could involve electron transfer from a Cr+2 to an alkyl–Co+3 intermediate to form an unstable alkyl–Co+2 species which is known to homolyze to afford an alkyl radical that could escape the solvent cage and capture a second Cr+2 species, a kinetically facile process (k = 107 M−1s−1).35,45,46
In summary, we have discovered divergent reactivity available to alkenes that enables branch-selective (Markovnikov) addition to radicalophilic and non-radicalophilic electrophiles. First, carbon-centered radicals generated by MHAT are competent to add to chiral sulfinimines, which stabilize the incipient N-centered radical and impart stereocontrol. The products of these reactions are valuable, unnatural amino acid derivatives. Second and complementarily, although these same radicals do not productively add into aldehydes, the addition of Cr+3 salts allows coupling to occur. This latter method circumvents the poor reactivity of free radicals towards carbonyl intermediates while maintaining the Markovnikov reactivity and chemoselectivity of MHAT. Overall, this work enables cross-coupling of abundant chemical feedstocks (aldehydes and olefins) without the need for pre-functionalization. Mechanistic experiments and analogy to the literature is consistent with alkyl–Co+3 transmetallation to alkyl–Cr+3, mediated by Cr+2. This second example5,6 of catalytic MHAT organocobalt transmetallation calls attention to the potentially general use of these alkyl-cobalt complexes as catalytically-generated organometallic species capable of transferring their alkyl ligands to various other transition metals (including Ni and Cr) for previously inaccessible branch-selective bond-forming processes from olefins. This reactivity complements catalytically-generated organocuprate species which can also engage in hydrometallation/transmetallation, but with linear selectivity.47
Supplementary Material
Figure 4.
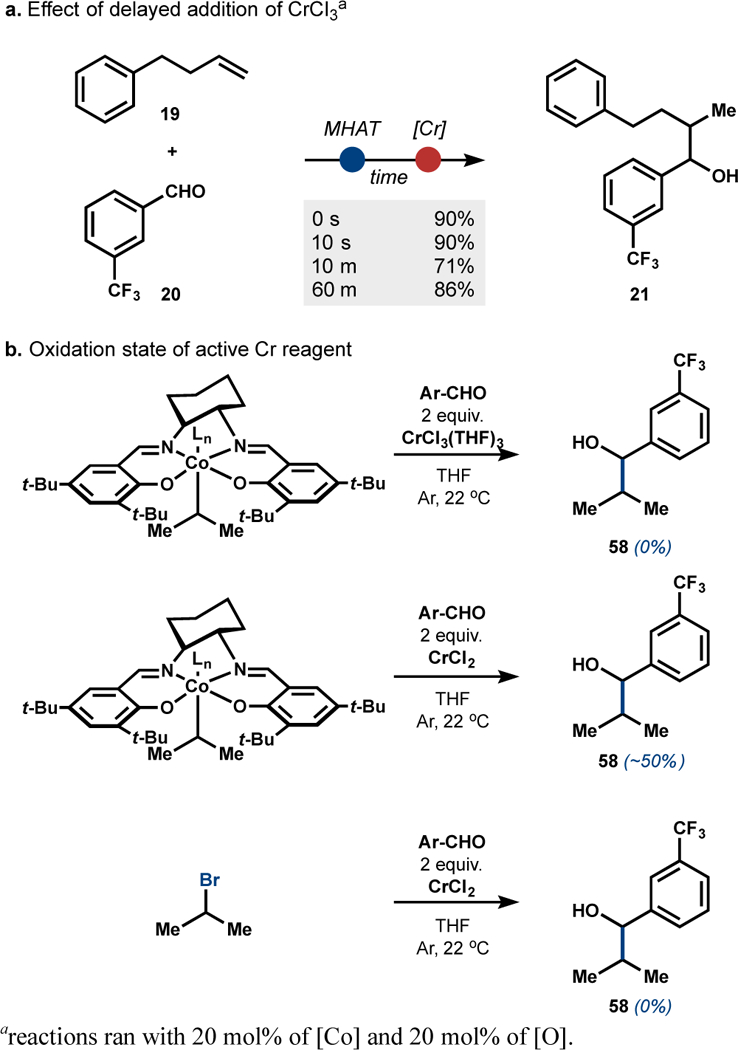
Delayed addition and stoichiometric reactions suggest transmetallation of alkyl−Co+3 to alkyl−Cr+3.
Acknowledgments
Funding Sources
Generous support was provided by the National Institutes of Health (R35 GM122606), the NSF (GRFP to J. L. M. M.), and Shionogi & Co., Ltd. (funding for T.O.). We thank Dr. L. Pasternack and Dr. D.-H. Huang for NMR assistance, Dr. J. Chen and Britanny Sánchez for HRMS measurements, Dr. R. Gianatassio for providing the starting material for 9, and Dr. S. Crossley, B. Huffman, R. Demoret, S. Shevick, and M. Baker for constructive feedback during preparation of the manuscript.
Footnotes
ASSOCIATED CONTENT
Supporting Information.
The Supporting Information is available free of charge on the ACS Publications website.
Detailed experimental procedures, compound characteriza-tion and spectral data.
REFERENCES
- 1.Schevenels FT; Shen M; Snyder SA Isolable and Readily Handled Halophosphonium Pre-reagents for Hydro- and Deuteriohalogenation. J. Am. Chem. Soc 2017, 139, 6329. [DOI] [PubMed] [Google Scholar]
- 2.Crossley SWM; Martinez RM; Obradors C; Shenvi RA Mn-, Fe-, and Co-Catalyzed Radical Hydrofunctionalizations of Olefins. Chem. Rev 2016, 116, 8912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Coombs JR; Morken JP Catalytic Enantioselective Functionalization of Unactivated Terminal Alkenes. Angew. Chem. Int. Ed 2016, 55, 2636. [DOI] [PMC free article] [PubMed] [Google Scholar]; The use of directing groups can also lead to branch-selective hydrometallation. See: Murphy SK, Coulter MM, & Dong VM β-hydroxy ketones prepared by regioselective hydroacylation. Chem. Sci, 2012, 3, 355. [Google Scholar]
- 4.Crabtree RH; The Organometallic Chemistry of the Transition Metals, Sixth Edition. John Wiley & Sons: New Jersey, 2014. [Google Scholar]
- 5.Green SA; Matos JLM; Yagi A; Shenvi RA Branch-Selective Hydroarylation: Iodoarene-Olefin Cross-Coupling. J. Am. Chem. Soc 2016, 138, 12779. [DOI] [PubMed] [Google Scholar]
- 6.Shevick SL; Obradors C; Shenvi RA Mechanistic Interrogation of Co/Ni-Dual Catalyzed Hydroarylation. J. Am. Chem. Soc 2018, 140, 12056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Green SA; Vásquez-Céspedes S; Shenvi RA Iron-Nickel Dual-Catalysis: A New Engine for Olefin Functionalization. J. Am. Chem. Soc 2018, 140, 11317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Ram MS; Yap GPA; Liable-Sands L; Rheingold AL; Marchaj A; Norton JR Kinetics and Mechanism of Alkyl Transfer from Organocobalt(III) to Nickel(I): Implications for the Synthesis of Acetyl Coenzyme A by CO Dehydrogenase. J. Am. Chem. Soc 1997, 119, 1648. [Google Scholar]; (b) Ram MS; Riordan CG Methyl Transfer From a Cobalt Complex to Ni(tmc)+ Yielding Ni(tmc)Me+: A model for Methylcobalamin Alkylation of CO Dehydrogenase. J. Am. Chem. Soc 1995, 117, 2365. [Google Scholar]
- 9.Can M, Armstrong FA; Ragsdale SW Structure, Function, and Mechanism of the Nickel Metalloenzymes, CO Dehydrogenase, and Acetyl-CoA Synthase. Chem. Rev, 2014, 114, 4149–4174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Komeyama K; Yamahata Y; Osaka I Nickel and Nucleophilic Cobalt-Catalyzed Trideuteriomethylation of Aryl Halides Using Trideuteriomethyl p-Toluenesulfonate. Org. Lett 2018, 20, 4375. [DOI] [PubMed] [Google Scholar]
- 11.Scovell WM Kinetics and Mechanism of Methyl Transfer from Methylcobalamin to Palladium(II). J. Am. Chem. Soc 1974, 96, 3451. [Google Scholar]
- 12.Dodd D; Johnson MD Bimolecular Nucleophilic Displacement as a Mechanism of Alkyl Transfer from Cobalt. J. Chem. Soc. D, 1971, 1371.
- 13.(a) Stolzenberg AM; Cao Y Alkyl Exchange Reactions of Organocobalt Porphyrins. A Bimolecular Homolytic Substitution Reaction. J. Am. Chem. Soc 2001, 123, 9078–9090. [DOI] [PubMed] [Google Scholar]; (b) Dodd D; Johnson MD; Lockman BL Kinetics and Mechanism of Apparent Alkyl Transfer from Alklcobaloximes to Cobaloxime(I), Cobaloxime(II), and Cobaloxime(III) reagents. J. Am. Chem. Soc 1977, 99, 3664. [Google Scholar]
- 14.Fanchiang YT; Ridley WP; Wood JM Methylation of Platinum Complexes by Methylcobalamin. J. Am. Chem. Soc 1979, 101, 1442. [Google Scholar]
- 15.Agnes G; Bendle S; Hill HAO; Williams FR; Williams RJP Methylation by Methyl Vitamin B12. J. Chem. Soc., Chem. Commun 1971, 850.
- 16.Espenson JH; Shveima JS Alkyl Transfer from Cobalt to Chromium. J. Am. Chem. Soc 1973, 95, 4468. [Google Scholar]
- 17.Witman MW; Weber JH Methylation of zinc(II), cadmium(II), and lead(II) by a trans-dimethylcobalt complex. Inorg. Chem, 1976, 15, 2375. [Google Scholar]
- 18.Crossley SWM; Barabe F; Shenvi RA Simple, Chemoselective, Catalytic Olefin Isomerization. J. Am. Chem. Soc 2014, 136, 16788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eisenberg DC; Norton JR Hydrogen-Atom Transfer Reactions of Transition-Metal Hydrides. Isr. J. Chem 1991, 31, 55. [Google Scholar]
- 20.Formation of an organocobalt from olefins is also postulated on: (a) Shigehisa H; Aoki T; Yamaguchi S; Shimizu N; Hiroya K Hydroalkoxylation of Unactivated Olefins with Carbon Radicals and Carbocation Species as Key Intermediates. J. Am. Chem. Soc 2013, 135, 10306. [DOI] [PubMed] [Google Scholar]; (b) Shigehisa H; Nishi E; Fujisawa M; Hiroya K Cobalt-Catalyzed Hydrofluorination of Unactivated Olefins: A Radical Approach of Fluorine Transfer. Org. Lett, 2013, 15, 5158. [DOI] [PubMed] [Google Scholar]; (c) Tokuyasu T; Kunikawa S; Masuyama A; Nojima M Co(III)−Alkyl Complex- and Co(III)−Alkylperoxo Complex-Catalyzed Triethylsilylperoxidation of Alkenes with Molecular Oxygen and Triethylsilane. Org. Lett, 2002, 4, 3595. [DOI] [PubMed] [Google Scholar]; (d) Waser J; Gaspar B; Nambu H; Carreira EM Hydrazines and Azides via the Metal-Catalyzed Hydrohydrazination and Hydroazidation of Olefins. J. Am. Chem. Soc, 2006, 128, 11693. [DOI] [PubMed] [Google Scholar]; (e) Gaspar B; Carreira EM Catalytic Hydrochlorination of Unactivated Olefins with para-Toluenesulfonyl Chloride. Angew. Chem. Int. Ed 2008, 47, 5758. [DOI] [PubMed] [Google Scholar]
- 21.(a) Nguyen KD; Park BY; Luong T; Sato H; Garza VJ; Krische MJ Metal-catalyzed reductive coupling of olefin-derived nucleophiles: Reinventing carbonyl addition. Science, 2016, 354, 6310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zheng Y-L; Liu Y-Y; Wu Y-M; Wang Y-X; Lin Y-T; Ye M Iron‐Catalyzed Regioselective Transfer Hydrogenative Couplings of Unactivated Aldehydes with Simple Alkenes. Angew. Chem. Int. Ed 2016, 55, 6315. [DOI] [PubMed] [Google Scholar]
- 23.Shirakawa E; Ikeda D; Masui S; Yoshida M; Hayashi T Iron-Copper Cooperative Catalysis in the Reactions of Alkyl Grignard Reagents: Exchange Reaction with Alkenes and Carbometalation of Alkynes. J. Am. Chem. Soc 2012, 134, 272. [DOI] [PubMed] [Google Scholar]
- 24.α-olefins have also been added to activated α-hydroxy ketones or diols via cyclometallation (a) Yamaguchi E; Mowat J; Luong T; Krische MJ Regio‐ and Diastereoselective C-C Coupling of α‐Olefins and Styrenes to 3‐Hydroxy‐2‐oxindoles by Ru‐Catalyzed Hydrohydroxyalkylation. Angew. Chem. Int. Ed 2013, 52, 8428. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Park BY; Luong T; Sato H; Krische MJ Osmium(0)-Catalyzed C–C Coupling of Ethylene and α-Olefins with Diols, Ketols, or Hydroxy Esters via Transfer Hydrogenation. J. Org. Chem, 2016, 81, 8585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Branch-selective hydroformylations of alkyl olefins have been reported in a number of cases, but at generally low ratios, with the exception of idiosyncratic substrates. See: (a) Noonan GN; Fuentes JA; Cobley CJ; Clarke ML “An Asymmetric Hydroformylation Catalyst that Delivers Branched Aldehydes from Alkyl Alkenes Angew. Chem., Int. Ed 2012, 51, 2477; [DOI] [PubMed] [Google Scholar]; (b) Pittaway R; Fuentes JA; Clarke ML Diastereoselective and Branched-Aldehyde-Selective Tandem Hydroformylation−Hemiaminal Formation: Synthesis of Functionalized Piperidines and Amino Alcohols. Org. Lett 2017, 19, 2845. [DOI] [PubMed] [Google Scholar]; (c) Kuil M; Soltner T; van Leeuwen PWNM; Reek JNH High-Precision Catalysts: Regioselective Hydroformylation of Internal Alkenes by Encapsulated Rhodium Complexes. J. Am. Chem. Soc 2006, 128, 11344. [DOI] [PubMed] [Google Scholar]
- 26.a) Garrido-Castro AF; Choubane H; Daaou M; Maestro MC Alemán J Asymmetric radical alkylation of N-sulfinimines under visible light photocatalytic conditions. Chem. Commun 2017, 53, 7764. [DOI] [PubMed] [Google Scholar]; (b) Fernández-Salas JA; Maestro MC; Rodríguez-Fernández MM; García-Ruano JL; Alonso I Intermolecular Alkyl Radical Additions to Enantiopure N-tert-Butanesulfinyl Aldimines. Org. Lett 2013, 15, 1658. [DOI] [PubMed] [Google Scholar]; (c) Zhong Y-W; Dong Y-Z; Fang K; Izumi K; Xu M-H; Lin G-Q A Highly Efficient and Direct Approach for Synthesis of Enantiopure β-Amino Alcohols by Reductive Cross-Coupling of Chiral N-tert-Butanesulfinyl Imines with Aldehydes. J. Am. Chem. Soc, 2005, 127, 11956–11957. [DOI] [PubMed] [Google Scholar]; (d) Robak MT; Herbage MA; Ellman JA Synthesis and Applications of tert-Butanesulfinamide. Chem. Rev, 2010, 110, 3600–3740. [DOI] [PubMed] [Google Scholar]
- 27.Intermolecular addition of olefins into hydrazones and oxime ethers via radical intermediates have also been reported: (a) Dao HT; Li C; Michaudel Q; Maxwell BD; Baran PS Hydromethylation of Unactivated Olefins. J. Am. Chem. Soc, 2015, 137, 8046. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Gaspar B; Carreira EM Cobalt Catalyzed Functionalization of Unactivated Alkenes: Regioselective Reductive C−C Bond Forming Reactions. J. Am. Chem. Soc, 2009, 131, 13214. [DOI] [PubMed] [Google Scholar]
- 28. See Supporting Information.
- 29.For research performed concurrent with this work: Ni S; Garrido-Castro AF; Merchant RR; deGruyter JN; Schmitt DC; Mousseau JJ; Gallego GM; Yang S; Collins MR; Qiao JX; Yeung K; Langley DR; Poss MA; Scola PM; Qin T; Baran PS A General Amino Acid Synthesis Enabled by Innate Radical Cross-Coupling. Angew. Chem. Int. Ed 2018, 10.1002/anie.201809310 [DOI] [PMC free article] [PubMed]
- 30.Beckwith ALJ; Hay BP Kinetics of the reversible .beta.-scission of the cyclopentyloxy radical. J. Am. Chem. Soc, 1989, 111, 230 [Google Scholar]
- 31.Saladrigas M; Bosch C; Saborit GV; Bonjoch J; Bradshaw B Radical Cyclization of Alkene-Tethered Ketones Initiated by Hydrogen-Atom Transfer. Angew. Chem. Int. Ed 2018, 57, 182. [DOI] [PubMed] [Google Scholar]
- 32.Pitzer L; Sandfort F; Strieth-Kalthoff F; Glorius F Intermolecular Radical Addition to Carbonyls Enabled by Visible Light Photoredox Initiated Hole Catalysis. J. Am. Chem. Soc, 2017, 139, 13652. [DOI] [PubMed] [Google Scholar]
- 33.Reference 32 has a single example.
- 34.Kochi JK; Powers JW; Mechanism of reduction of alkyl halides by chromium(II) complexes. Alkylchromium species as intermediates. J. Am. Chem. Soc 1970, 92, 137. [Google Scholar]
- 35.a) Bakac A; Espenson JH Unimolecular and bimolecular homolytic reactions of organochromium and organocobalt complexes. Kinetics and equilibria. J. Am. Chem. Soc 1984, 106, 5197. [Google Scholar]; (b) Espenson JH; Sellers TD Kinetics and mechanism of alkylchromium formation in the reductive cobalt-carbon bond cleavage of alkylcorrins by chromium(II). J. Am. Chem. Soc, 1974, 96, 94. [Google Scholar]
- 36.This reactivity has also been used for the coupling of unactivated alkyl halides and aldehydes using Cr+2: Takai K; Nitta K; Fujimura O; Utimoto K Preparation of alkylchromium reagents by reduction of alkyl halides with chromium(II) chloride under cobalt catalysis. J. Org. Chem, 1989, 54, 4732.; [Google Scholar]; as well as the coupling of dienes with aldehydes: Takai K; Toratsu C B12-Catalyzed Generation of Allylic Chromium Reagents from 1,3-Dienes, CrCl2, and Water. J. Org. Chem 1998, 63, 6450. [Google Scholar]; In more recent examples, combination of Co/Cr have been used in: Xiong Y; Zhang G Enantioselective 1,2-Difunctionalization of 1,3-Butadiene by Sequential Alkylation and Carbonyl Allylation. J. Am. Chem. Soc 2018, 140, 2735. [DOI] [PubMed] [Google Scholar]
- 37.a) Steib AK; Kuzmina OM; Fernandez S; Flubacher D; Knochel P Efficient chromium(II)-catalyzed cross-coupling reactions between Csp2 centers. J. Am. Chem. Soc 2013, 135, 15346. [DOI] [PubMed] [Google Scholar]; (b) Oral-rat LD 50 for Cr2O3 > 2700 mg/kg. Chromium(III) and its Inorganic Compounds. 2014, The MAK‐Collection for Occupational Health and Safety doi: 10.1002/3527600418.mb1606583vee4614 [DOI]
- 38.Gil A; Albericio F and Álvarez M Role of the Nozaki–Hiyama–Takai–Kishi Reaction in the Synthesis of Natural Products. Chem. Rev 2017, 117, 8420. [DOI] [PubMed] [Google Scholar]
- 39.Kurosu M; Lin MH; Kishi Y Fe/Cr- and Co/Cr-mediated catalytic asymmetric 2-haloallylations of aldehydes. J. Am. Chem. Soc 2004, 126, 12248. [DOI] [PubMed] [Google Scholar]
- 40.Co+3 is the oxidation state necessary for formation of the Co−H with the silane. Use of external metal reductants typically lead to formation of an inactive Co+2.
- 41.Different attempts to make the chromium catalytic have been so far unsuccessful; presumably the additives necessary to dissociate the Cr−O bond, as proposed in standard Cr-catalyzed reactions, can also react with the salen ligand from the cobalt (see: Namba K; Kishi Y New catalytic cycle for couplings of aldehydes with organochromium reagents. Org. Lett 2004, 6, 5031.) [DOI] [PubMed] [Google Scholar]; Recent examples of combining Ir-photocatalysis with Cr where dissociation of the putative Cr−O bond seems to occur without an external agent, could in a future be explored for these endeavors (see: Schwarz JL; Schaf̈ers F; Tlahuext-Aca A; Lückemeier L; Glorius F; Diastereoselective Allylation of Aldehydes by Dual Photoredox and Chromium Catalysis. J. Am. Chem. Soc 2018, 140, 12705.) [DOI] [PubMed] [Google Scholar]
- 42.Addition of Lewis basic ligands did not change diastereoselectivity and tended to suppress reactivity.
- 43.Formation of an intermediate alkyl-chloride that leads to formation of product is unlikely, given that we do not see reaction through the alkyl-chloride in 46.
- 44.Aluminum- and borohydrides are known to reduce Cr+3 into Cr+2, perhaps reduction by the silane or a derivative could occur through a similar mechanism. See: Fürstner A Carbon−Carbon Bond Formations Involving Organochromium(III) Reagents. Chem. Rev, 1999, 99, 991. [DOI] [PubMed] [Google Scholar]
- 45.Outer sphere electron transfer from Cr to Co: Candlin JP; Halpern J; Trimm DL Kinetics of the Reduction of Some Cobalt(III) Complexes by Chromium(II), Vanadium (II), and Europium (II). J. Am. Chem. Soc, 1964, 86, 1019. [Google Scholar]
- 46.An alternative mechanism of SH2, as finally proposed in Ref. 35a, is not likely operative given that Co+2 is not catalytically active in our system (Table 1, entry 5). A mechanism similar to our previously reported Co/Ni dual catalysis involving oxidation of the alkyl−Co+3 to an alkyl−Co+4 prior to homolysis and concomitant reduction of the Cr+3 to Cr+2 (see Ref. 6) is also unlikely given that control experiments indicate there is no reactivity with Cr+3 (Figure 4).
- 47.Pyea DR; Mankad NP Bimetallic catalysis for C–C and C–X coupling reactions. Chem. Sci 2017, 8, 1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.