

Abstract
A major risk for patients having estrogen receptor α (ERα)-positive breast cancer is the recurrence of drug-resistant metastases after initial successful treatment with endocrine therapies. Recent studies have implicated a number of activating mutations in the ligand-binding domain of ERα that stabilize the agonist conformation as a prominent mechanism for this acquired resistance. There are several critical gaps in our knowledge regarding the specific pharmacophore requirements of an antagonist that could effectively inhibit all or most of the different mutant ERs. To address this, we screened various chemotypes for blocking mutant ER-mediated transcriptional signaling and identified RU58668 as a model compound that contains structural elements that support potent ligand-induced inhibition of mutant ERs. We designed and synthesized a focused library of novel antagonists and probed how small and large perturbations in different ligand structural regions influenced inhibitory activity on individual mutant ERs in breast cancer cells. Effective inhibition derives from both non-polar and moderately polar motifs in a multifunctional side chain of the antagonists, with the nature of the ligand core making important contributions by increasing the potency of ligands possessing similar types of side chains. Some of our new antagonists potently blocked the transcriptional activity of the three most common mutant ERs (L536R, Y537S, D538G) and inhibited mutant ER-mediated cell proliferation. Supported by our molecular modeling, these studies provide new insights into the role of specific components, involving both the ligand core and multifunctional side chain, in suppressing wild-type and mutant ER-mediated transcription and breast cancer cell proliferation.
Graphical Abstract:

Introduction
Estrogen receptor-alpha (ERα), a ligand-regulated transcription factor that controls many important physiological and disease states,1 is the target of endocrine therapies for breast cancers, as more than 70% of these cancers are ERα-positive.2 Endocrine therapy agents such as the aromatase inhibitor (AI) letrozole are effective in the initial treatment of breast cancer; however, cancer often returns as metastases that are no longer sensitive to these therapies.3 This acquired drug resistance is one of the primary reasons behind the high mortality rate of metastatic breast cancer patients.
Recently, we and others reported on the molecular basis of much of the acquired endocrine-therapy resistance by identifying somatic mutations in the gene (ESR1) coding for ERα in metastatic breast cancers of patients who had earlier received AI treatment.4–7 The most common mutations lead to the substitution of amino acids in the ERα ligand-binding domain (LBD): tyrosine 537 with serine (Y537S), and aspartic acid 538 with glycine (D538G), as well as several others.8 Significantly, these mutations produce an ERα having much higher transcriptional activity than wild-type ERα in the absence of an agonist (estradiol), rendering it resistant to AIs.4 Our structural, biochemical, and molecular modeling studies suggest that these ERα LBD mutations stabilize an agonist conformation of ERα, thereby favoring recruitment of nuclear receptor (NR) co-activator proteins, including members of p160 steroid receptor coactivator (SRC) family, without the need for agonist binding.9 High doses of current antiestrogen therapeutics (tamoxifen and the selective ER downregulator (SERD), fulvestrant) are needed to inhibit these mutant ERs, thereby seriously impacting their clinical utility.4
Recent reports have highlighted the important role of oral bioavailability of ERα antagonists for controlling their internal exposure and thus inhibitory activity.10, 11 However, there are limits to the efficacy of these improved ERα antagonists in the absence of a pharmacophore that is designed and specifically optimized to counter the unique agonist conformations adopted by these mutant ERs. This is evident from recent findings, which show that some new orally available antiestrogens, exemplified by GDC081011 and AZD9496,10 are less effective against some mutants (Y537S) than others (D538G),8 a pattern of selectivity reflected as well by some other new antiestrogens.12 These results indicate the need for a deeper understanding of the pharmacophore requirements for an ER ligand that might be effective in blocking the multiple mutant forms of ER.
In view of the critical role of the above mutant ERα’s in endocrine therapy-resistant metastatic breast cancer and the significant gaps in our knowledge of ERα inhibitory pharmacophore requirements, we sought to elucidate the underlying rules that govern ligand-controlled inhibition of these mutant ERs. In particular, we wanted to probe the following questions: (a) How do alterations in different structural regions of the ligands influence their ability to inhibit mutant ER signaling? and (b) Is it possible to develop a pan-antagonist that would uniformly and effectively inhibit the activity of wild-type and mutant ERs, and do the different mutant ERs respond in a uniform—or distinct manner—to a series of antagonists?
In this study, we have first searched broadly for compounds that have particularly potent inhibitory activity against the mutant forms of ERα. We then used the best candidate molecule as a template, defining and then interrogating different segments of its structure to see how they contribute to effective inhibition of the ERα mutants; we used both screening and dose-response assays of ERα transcriptional activity, ERα-driven cell proliferation, and ERα downregulation in breast cancer cells, and we developed structure-based molecular models that illustrate some key ligand-receptor interactions that appear to be central to explaining considerable inhibition. In this way, we were able to identify features of these segments that are important for inhibitory activity and define the respective role of the side-chain vs. ligand-core elements as contributors. Our studies also make clear that endemic, structural features of the ERα mutants appear to be the principal elements that control their response to a series of structurally diverse antiestrogens.
Results
Initial Screening of Different Chemotypes for Inhibiting Mutant ERα Signaling.
To identify a suitable antagonist scaffold for probing the pharmacophore properties of mutant ER response to antiestrogens, we initially screened a 33-member in-house library of structurally varied ligands for their ability to block ERα signaling in luciferase assays in MCF-7 cells transfected with three major mutant genotypes, Y537S, D538G, and L536R. This panel of ligands represented a highly diverse array of chemotypes that incorporated several variations in two key structural elements: ligand cores and their side chains known to degrade or induce an antagonistic conformation of ERα. A single dose of 100 nM was chosen to assess a combination of potency and efficacy. The result of these studies (SI, Table S1) revealed that a majority of the ligands inhibited one or more of the mutants to varying degrees. Many of the better compounds had basic side chains as found in tamoxifen,13 bazedoxifene, and the recently described RAD190114 or acrylic acids reminiscent of the Glaxo SmithKline GW563815 and the more recently described AZD949610 and GDC081011 antiestrogens.
In this initial screen, RU58668 (Fig. 1A), a pure antiestrogen prepared some years ago by the Roussel Company,16 stood out from all the other ligands we examined, including fulvestrant, by its remarkable ability to block much of the activity of all three mutants at this 100 nM concentration.8 Therefore, we selected RU58668 as an archetype compound for the design of a more focused library of candidates to investigate the structural parameters that are required for ligand-induced mutant ERα inhibition.
Figure 1. Chemical Structures.

(A) Structure of initial lead compound, RU58668. (B) Segmented elements for the design of a focused library of novel antagonists based on RU58668: Ligand core, Linker, Central Polar group, and Terminal Component.
Design of a Focused Library of New Antagonists.
We identified four key regions or segments in an RU58668-based structure for diversification into a library of antagonists to be evaluated for their inhibitory activity on mutant forms of ERα (Fig. 1B).
Ligand Core.
We replaced the steroid core of RU58668 with structurally simpler bisphenolic adamantyl, bicyclo[3.3.1]nonyl, cyclohexyl, and heterocyclic cores (Fig. 1B). This “core switch” gave us much more synthetically accessible systems for chemical diversification;12 ligands having these bulky and rigid cores also possess very high affinities for ERα.17, 18 These ligand-core scaffolds anchor one of the phenols (A) inside the LBD (hydrogen bonded to E353 and R394) and thereby orient the other phenol (B) to project its side chain outside the LBD, thus mimicking the phenyl ether at the 11β position in RU58668.16, 19
Linker.
We designated the region that extended from the phenol (B) to the central polar functional group as the “linker,” and within this linker substructure, the length (n) and polarity of the alkyl chain were varied (Fig. 1).
Central Polar Functional Group.
Two different moieties were used to probe the importance of this substructure: sulfur in different oxidation states (sulfone vs. sulfoxide), exemplified in RU58668 and in fulvestrant, and a carboxamide found in the AstraZeneca antiestrogen, ICI 164,384.20 A compound lacking any polar functional group was also designed.
Terminal Component.
This region appeared to be critical for conferring a repressive function to the antagonists. We incorporated partially fluorinated and non-fluorinated alkyl, branched alkyl, and heterocyclic moieties into this terminal region.
The synthesis and optimization of antagonists possessing different cores and side chains were done sequentially. In particular, the optimal side chain was identified by screening antagonists possessing adamantyl, bicyclononyl, and cyclohexyl cores, and this side chain was later installed on a heterocyclic core.
Synthesis.
The bisphenolic adamantyl, bicyclononyl, and cyclohexyl cores were synthesized through McMurray coupling18 of commercially available 2-adamantanone, bicyclo[3.3.1]nonan-9-one, and cyclohexanone, respectively, with 4,4’-dihydroxybenzophenone.17 Thereafter, the alkyl linkers were attached to one of the phenols by SN2 displacement with appropriate bromo-chloro alkanes (Scheme 1A and SI, Scheme S-1). The remaining terminal chloride group was replaced with iodide to allow substitution with a thioacetate group, which was then hydrolyzed and converted into the sulfides by treatment with pentafluoropentyl iodide. Chemoselective oxidation of these sulfides with different equivalents of oxone furnished the desired sulfone and sulfoxide derivatives (1–10, Table 1).21 The above method was also followed for the synthesis of 16 (Table 1), except that a polyethylene glycol chain substituted with a tosylate was used instead of the bromo-chloro alkane for alkylation of the phenolic core (SI, Scheme S-5). The synthesis of 11 started with demethylation of commercially available 6-methoxy-2-(4-methoxyphenyl)benzo[b]thiophene and protection of the two phenols with MOM groups (Scheme 1B and see SI, Scheme S-2 for details). The MOM-protected benzothiophene derivative was brominated at the C-2 position, followed by oxidation of the thiophene ring with oxaziridine.22 This oxidation activated the resulting bromo-thiophene-S-oxide ring to participate in an SNAr reaction with a phenol that had a sulfide side chain at its para positon. Reduction of the thiophene-S-oxide with LAH followed by deprotection of the two MOM groups and chemoselective oxidation of sulfide side chain gave the sulfoxide (11, Table 1). Compounds 12–13 were prepared by alkylation of 2-thiomethyl pyridine with an appropriate chloro derivative described above (SI, Scheme S-3). Compounds 14–15 were prepared by alkylation of corresponding phenolic core (described above) with 1-iododecane or 1-bromopentadecane (SI, Scheme S-4). Compounds 17–18 were synthesized by treating the corresponding phenol cores with Boc-protected 4-amino-butyl methanesulfonate followed by Boc-deprotection and coupling with the NHS ester of the appropriate carboxylic acid (SI, Scheme S-6).
Scheme 1.

General scheme for synthesis of novel antagonists (1–11). See SI for further details of the synthetic procedures for all the antagonists mentioned in Table 1.
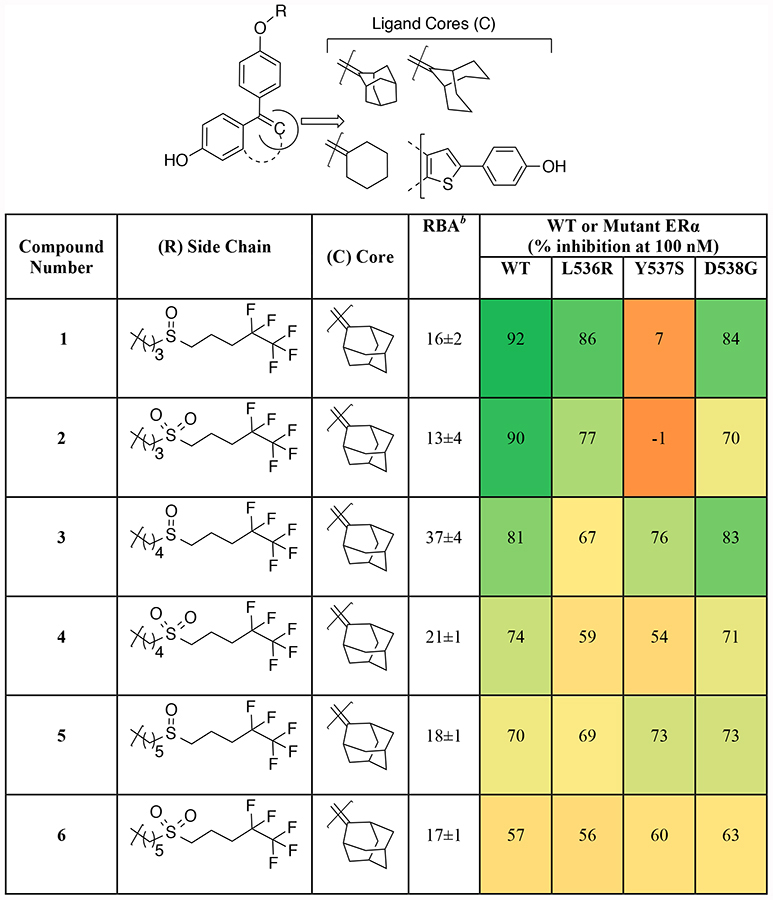
Table 1.
Binding affinity and effect of new antagonists on inhibition of transcriptional activity of WT and mutant ERs in MCF-7 cells.a
|

|
MCF-7 cells were transfected with plasmids for ERE-Firefly luciferase, ERE-Renilla luciferase, as well as WT or mutant ERα, as indicated. Cells were grown in full medium, which contains sufficient residual estrogens to stimulate luciferase activity. The percent inhibition of Firefly luciferase activity by compound at 100 nM was measured after 24 hours. The values are color-coded, with the dark green corresponding to the highest level of inhibition, and light green, yellow, dark yellow, and orange corresponding to progressively less inhibition, and even to stimulation (dark orange, negative numbers).
Relative Binding Affinity (RBA) of compounds for WT ERα.
Biological Results.
Most Ligands Have High ERα Binding Affinity.
The binding affinity of the new compounds for wild-type (WT) ERα (Table 1) was determined by a competitive radiometric binding assay using full-length WT ERα and [3H]estradiol as the tracer.23 The affinity, relative to the tracer, is given as relative binding affinity (RBA) values, with the affinity for estradiol considered to be 100. The compounds had high binding affinities for ERα, ranging from 15–60% that of estradiol, except for 14 and 15, which had affinities below 1%.
Inhibition of Transcriptional Activity.
Side Chain Steric and Polar Factors Needed for Effective ERα Antagonism.
The novel ligands were tested for their ability to inhibit WT ERα activity, and several of the best were also tested for their ability to downregulate ERα. Inhibition of mutant ERα-mediated transcription in MCF-7 cells was evaluated using the same protocol as in the broad screen. The results (Table 1) are color-coded on a gradient scale, with high inhibition in green, low inhibition in orange, or even slight stimulation in dark orange.
This designed compound set provided several insights into the role of different structural elements of each ligand. In general, bulky rings such as adamantane and bicyclononane (3–4, 7–8) afforded better inhibitory activity than the simple cyclohexane (9–10). Moreover, the bisphenolic heterocyclic benzothiophene core moiety (11), an analog of the core found in the Lilly antiestrogen Arzoxifene,24–26 was also effective in suppressing mutant ER signaling.
The length of the linker region (C3 vs. C4 vs. C5 chain lengths) did not have a major effect on the inhibitory activity of the corresponding ligands (1–8) on mutant ERs (except Y537S), but the C4 linker (3–4, 7–8) provided somewhat better and/or more uniform activity. The type of central polar group was also important, with sulfoxides displaying better antagonistic behavior than sulfones (1 vs. 2; 3 vs. 4; 5 vs. 6; 7 vs. 8; 9 vs. 10). On the other hand, replacement of the alkyl chain with a much more polar triethylene glycol chain (16) attenuated inhibitory activity. Certain amide groups (17–18) had some, though mostly modest activity in this position.
Together with the central polar group, the terminal component had a profound effect on inhibition of transcription, evident from the absence of antagonistic activity of 14–15, which retained the initial alkyl linker but lacked any other polar/functional group in the side chain. Interestingly, the substitution of the pentafluoropentyl region with the trifluoroethyl group (17) or isohexyl group (18) maintained modest inhibitory activity, while attachment of heterocycles in this region, such as the α-picoline sulfide that was explored by the Roussel group27 (12–13), essentially eliminated antagonistic activity. In a number of cases (1, 2, and 8) some ERα mutants, particularly Y537S, were less fully inhibited than WT ERα.
To understand how these antiestrogens might be working, we evaluated their ability to downregulate ERα. MCF-7 cells were treated with fulvestrant and our compounds (3, 7, and 11), and ER protein was analyzed by in-cell Western blot analysis (Figure 2). All three compounds effected marked (70%) degradation of WT ERα to levels nearly that observed with fulvestrant. The IC50 value for ERα downregulation by fulvestrant is 0.21 nM, and the three compounds had IC50 values between 0.55 and 1.2 nM.
Figure 2.

Downregulation of ERα levels in MCF-7 cells. Cells were treated with the indicated concentration of compounds for 24 hours, and ERα levels were determined by cell Western blot analysis.
Ligand Core Modulates Inhibitory Potency Against Different Mutant ERs.
To probe the inhibitory pattern of these new antagonists more deeply, we selected four of the most promising new compounds (3, 5, 7, 11, Table 1) for dose-dependent inhibition of mutant and WT ERα-mediated transcription in both MCF-7 and MDA-MB-231 cells. The MCF-7 cells contain endogenous WT plus transfected WT or mutant ERα, whereas the ERα-negative MDA-MB-231 cells contain solely transfected WT or transfected mutant ERα.4, 8 The compounds were selected for their inhibitory activity across the three mutants, and comparisons were made with fulvestrant.
The results, expressed as IC50 or Ki values, are summarized in Table 2. The selected antagonists, in most cases, showed potent inhibitory effects on WT and mutant ERs (IC50 ≤ 5 nM), and some compounds (in particular 11) resembled fulvestrant in potency. In comparison with WT ERα, the mutant ERs consistently required higher concentrations of the antagonists to suppress transcriptional activity. These assays also revealed that the sensitivity of WT and different mutant ERα’s for antagonism decreased in the order of WT > L536R > D538G > Y537S, a trend that also reflected their binding affinities for these four receptors. Mutant ERα activity in MDA-MB-231 cells was somewhat more resistant to inhibition than in MCF-7 cells, as also observed by others,12, 28 indicating cell-type dependence of antagonist activity. This difference could reflect the fact that the MDA-MB-231 cells have only the more active, mutant forms of ERα, whereas MCF-7 cells also contain some of the more easily inhibited WT ERα, or it could be rooted in other differences between the cell lines (coregulator levels, etc.). Again, the length of the linker region (C4 vs. C5 chain lengths, compound 3 vs. 5, Table 2) had little effect on the biological activity. Compound 3, an adamantyl core antagonist with a C4 linker, was more effective than its analog possessing bicyclononane core (7). A closer comparison of two antagonists (3 vs. 11) highlights the dependence of potency and inhibitory activity on the type of ligand core in each antagonist. Compound 11 differs from compound 3 in possessing a heterocyclic core in place of the adamantane, while both have the same linker, central functional group and terminal components. Compound 11, however, not only had better activity than compound 3, but it required lower concentrations for inhibiting all the three mutants in both MCF-7 and MDA-MB-231 cells, differences that cannot be explained by binding affinities alone. In summary, from these studies, an adamantane core is better than a bicyclononane, and the heterocyclic benzothiophene is the best of all, being nearly as effective as fulvestrant.
Table 2.
Inhibition of transcriptional activity of mutant and WT ERs by compounds in two different breast cancer cell lines.

|

|
Relative binding affinity (RBA) values were determined with a radiometric ligand-binding assay, using tritiated estradiol as the tracer and ligand-binding domains of WT ERα and the three mutants.23 RBA = [IC50 (estradiol)/IC50 (compound)] × 100.23
The Ki values can be calculated from the formula: Ki = (Kd[estradiol]/RBA) × 100. The Kd[estradiol] values were determined by Scatchard analysis.23 Kd: wild type 0.22 nM, L536R 1.35 nM, Y537S 1.40 nM, D538G 1.77 nM. Because the IC50 values from the competitive binding assay are affected by the estradiol binding affinities, the Ki values are a better representation of the absolute affinity of the compounds for WT and mutant ERs, equivalent to correcting binding values using the Cheng-Prusoff relationship.29
Both cell types were transfected with plasmids for ERE-Firefly luciferase, ERE-Renilla luciferase, and the indicated WT or ERα mutant. Cells were grown in full medium, which contains sufficient estrogen to stimulate luciferase activity. The percent inhibition of luciferase activity was measured after 24 hours over a range of compound concentrations, and IC50 values were determined.
Inhibition of Proliferative Activity.
Antagonists Show Qualitative Similarities but Quantitative Differences Across Different Mutant ERα’s.
To further characterize the ER antagonist activity of the most promising compounds, we evaluated the same four compounds plus fulvestrant in dose-response studies of potency in suppressing the proliferation of MCF-7 cells containing WT ERα or WT ERα plus mutant ERα (Figure 3 and Table 3). Regardless of whether inhibition was by fulvestrant or any of the four new antiestrogens, the overall rank order of mutant sensitivity to inhibition was the same and paralleled that found in the ERE-luciferase assays (Table 2): WT-ERα was the most sensitive, Y537S was least sensitive, and D538G and L536R were intermediate. Proliferation of cells with the Y537S mutation, in particular, proved to be very difficult to fully inhibit, as we and others have noted with different antiestrogens.8, 12, 30 This is particularly evident in Table 3, showing both potencies as IC50 values and fold-reduction in potency on mutant ERα compared to WT ERα. Fulvestrant was 50-fold less potent on Y537S ERα than on WT ERα, with the factor for the new compounds being even greater (100- to 200-fold) or difficult to determine. Hence, the mutant ERα genotype appeared to be a dominant determinant of sensitivity to inhibition of proliferation by these ER antagonists.
Figure 3.

Inhibition of MCF-7 cell proliferation driven by WT or mutant ERα. MCF-7 cells that were expressing doxycycline-inducible WT or mutant ER were treated with various concentrations of the indicated compound in regular medium and proliferation was measured after 5 days using the MTT assay. Graphs were plotted with the mean ±SD of 6 biological replicates.
Table 3.
Potencies for inhibition of proliferation of MCF-7 cells driven by WT or mutant forms of ERα.
| Genotype | Inhibition of Proliferation IC50 (nM ± SD)a Fold-reduction in potency relative to wild-type ERa | ||||
|---|---|---|---|---|---|
| 3 | 5 | 7 | 11 | fulvestrant | |
| Wild type | 4.4 ± 0.98 [1x] |
5.1 ± 0.72 [1x] |
3.8 ± 1.1 [1x] |
0.65 ± 0.08 [1x] |
0.31 ± 0.03 [1x] |
| L536R | 8.8 ± 3.6 [2.0x] |
12 ± 2.1 [2.4x] |
9.7 ± 3.5 [2.6x] |
2.2 ± 0.45 [3.4] |
2.6 ± 0.40 [8.4x] |
| Y537S | NCb | NCb | NCb | 191 ± 25 [294x] |
16 ± 1.8 [52x] |
| D538G | 20 ± 2.5 [4.5x] |
46 ± 4.8 [9.0x] |
18 ± 4.8 [4.7x] |
17 ± 1.8 [26x] |
5.9 ± 0.60 [19x] |
IC50 values ± standard deviations were obtained from the dose-response experiments presented in Figure 3. Potency values for the mutants relative to WT ERα (set at 1x) were determined by IC50 mutant/IC50WT and are indicated in square brackets.
Not calculated: only 50–60% inhibition at 1 μM.
Spectrum of Compound Potencies.
The antiproliferative activity of our compounds varied roughly in the order: 11 > 7 > 3 > 5, although not all differences were large. New ligand potencies were rather modestly reduced on the L536R mutant, with 11 rivaling fulvestrant on this genotype. Compound 3 was also potent on D538G and L536R, being nearly as good as fulvestrant, but far less potent on Y537S. Perhaps most intriguing, the dose-response curves for WT, L536R, and D538G were quite similar, more so with compounds 3 and 7 than with 5 and 11, whereas the curves for fulvestrant were different, with inhibition of all of the mutant ERs being significantly right-shifted relative to WT-ERα. In terms of overall potency, the best new compound was 11, having the heterocyclic core.
Molecular Modeling.
Using molecular modeling, we explored how the ligand core and side chain of some of our representative compounds (1, 3, 5, and 11) might be interacting with the ERα LBD (Fig. 4; see SI for more detail). All of the antagonists we have modeled have one phenol that mimics the A-ring of estradiol and forms hydrogen bonds to E353 and R394 within the ERα LBD, allowing another phenyl group to project outside the ligand-binding and act as a side-chain attachment site. For guidance in the manual docking of the studied ligands, there are two relevant and useful X-ray complexes: ICI 164,384 in rERβ (PDB:1HJ1), which is the only available crystal structure of an ICI-type antiestrogen, and raloxifene in wild-type hERα (PDB:1ERR). The orientation of the Y526 side chain varies in other hERα X-rays structures, and we selected 1ERR because the Y526 side chain is close to the binding site and is within hydrogen-bond distance of a water molecule. Also, in other hERα X-rays structures, a water molecule appears in approximately the same point in space. The binding models for compounds 5, 3, and 1 superimpose well on ICI 164,384 in the rERβ (PDB:1HJ1) complex (Fig. 4 (iii)); for clarity, we do not show the other water molecules in PDB:1ERR. The binding models of compounds 5 and 3 are similar, in that their perfluorinated tail traces the N-butyl tail of ICI 164,384 in the X-ray structure; in contrast, the perfluorinated tail of compound 1 superimposes on the N-methyl unit of the ligand in the X-ray structure.
Figure 4.

i) Superimposed Protein Data Bank (PDB)34 entries of the Y537S mutant in the agonist conformation (with and without protein ribbon representations in panels A and B, respectively) exhibit a channel, which is defined by H11, H12, and the H11–H12 loop and which is stabilized by a hydrogen bond between S537 and D351.
ii) Binding models for compounds 5, 3, and 1 (depicted in panels A, B, and C, respectively) were developed in a wild-type hERα protein structure (PDB:1ERR). Compounds 5 and 3 benefit from a water-mediated hydrogen bond involving Y526 and the sulfoxide oxygen atom; compound 1 cannot achieve this interaction without the loss of important interactions with the protein’s hydrophobic surface.
iii) The binding models, which also are presented in the previous figure section, for compounds 5, 3, and 1 in wild-type hERα (PDB:1ERR), as depicted in panels A, B, and C, respectively, are superimposed on the rERβ (PDB:1HJ1) X-ray complex (yellow carbon atoms) with its ligand, ICI 164,384. Hydrogen bonds are indicated by dashed, yellow lines.
iv) A water-mediated hydrogen bond, which involves Y526 and the sulfoxide oxygen atom, is observed in the wild-type hERα (PDB:1ERR) binding models for compounds 3, 5, and 11 (panels A and B). For compound 11, the linker adopts a lower-energy linear conformation (panel B), and the ligand’s core is further stabilized by a hydrogen bond between His524 and the oxygen atom of the rotatable phenol group of the ligand. The origin of the linker conformational difference is the rotation of the phenoxy group at the base of the linker (in compound 11 vs. compounds 3 and 5), as shown in the superimposition (panel C).
v) In the wild-type hERα (PDB:1ERR) binding models for compound 11 (panel A) and fulvestrant (panel B), the perfluorinated end of the flexible chain binds more deeply in a surface pocket when fulvestrant is compared to compound 11. The protein model in each case is represented by a color-coded surface, with green indicating a mainly hydrophobic region. The water of the hydrogen bond involving Y526 is also shown. Hydrogen bonds are indicated by dashed, magenta lines.
Discussion
Prospecting for Effective Inhibitors of ERα Bearing Activating Mutations.
Through our initial screen, we identified RU58668 as the most active inhibitor of the transcriptional activity of WT ERα and three major constitutively active ERα mutants found in endocrine therapy-resistant recurrent breast cancer. Using RU58668 as a template, we extracted four distinct sub-structural components that might be contributing to its antagonistic activity (Fig. 1). These were then systematically installed in simpler, more synthetically accessible non-steroidal model cores with high ER-binding affinity and evaluated for their ability to block WT and mutant ERα ligand binding, transcriptional activity, and proliferation in breast cancer cells.
Contributions to Antagonism from the Side Chain.
Through side-chain variations, we found a strict requirement for a non-polar linker region, as installation of a polyethylene glycol abrogated inhibitory activity (Table 1, 16). On the other hand, the remaining regions of the side chain need to embody some mildly polar and steric elements to induce ER antagonism. Specifically, replacing the pentafluoropentyl fragment of 3 with shorter alkyl (trifluoroethyl, 17, Table 1) or branched alkyl (isoamyl, 18) chains maintained or decreased the inhibitory activity of antagonists, while planar heterocycles (Table 1, 12–13) abolished the inhibitory activity. The terminal region of the side chain alone is not responsible for inducing antagonism, because increasing the polarity of the linker region (Table 1, 16) or decreasing the polarity of the linker and the central polar group (compounds 14–15) lowered the antagonistic activity. Hence, non-polarity of the linker might be essential for it to make favorable hydrophobic contacts along the surface of ERα, the central polar functional group perhaps interacting with a polar residue or solvent, providing an important type of chain flexibility (see modeling discussion below).
Despite the widespread use of fulvestrant in endocrine therapy, there is little information on how its side chain induces ER antagonism and degradation. There is only one crystal structure (PDB code: 1HJ1) of a fulvestrant-type compound (ICI 164,384), which is in a complex with ERβ, the other ER subtype.19 In this structure, the steroidal core of ICI 164,384 is inverted, allowing the long 7α side chain to exit in the same 11β-direction as the chain in RU58668; this enables the end of the ICI side chain to dock into a hydrophobic cleft within the AF2 surface, thereby disordering the position of helix 12 and abrogating its interaction with the ERα LBD. These molecular interactions are thought to expose hydrophobic surfaces on ERα and thus promote ERα degradation. We too found that several of our new antagonists also effectively downregulated ERα. Some studies have shown that the ER-fulvestrant complex has stronger interaction with corepressors than those with tamoxifen or raloxifene.31, 32 Hence, the terminal components of these multifunctional side chains may not be just occupying the AF-2 cleft as in the crystal structure19 but rather actively facilitating the binding of corepressors to the AF-2 coregulator interaction site.
Contributions to Antagonism from the Core.
Dose-response studies of ER transcriptional activity of our best compounds indicate that the type of side chain does not alone determine the pattern of inhibitory activity, as compounds bearing similar side chains but different ligand cores (3 vs. 11) showed differences in potency and selectivity for mutant ERs. This suggests that for inhibition of mutant ER, there is a strict requirement for a specific multifunctional side chain and the nature of the core also contributes to inhibition. In fact, we found that changing the nature of ligand core can be a good strategy for increasing antiestrogen potency towards mutant ERs. Nevertheless, the mutant ERα genotype appeared to be a dominant determinant of sensitivity to inhibition of proliferation by our ER antagonists, with Y537S ERα showing the greatest requirement for high concentrations of antiestrogens to suppress its constitutive activity, as noted also for other antagonists.8, 12
Potential Mode of Interaction of Our Best New Antiestrogens with the ERα LBD.
Our modeling analysis suggests that the formation of a water-mediated hydrogen bond involving the sulfoxide oxygen atom of our compounds and Y526 is critical for their antagonistic activities, as the water-mediated hydrogen bond helps drive the equilibrium away from the constitutively active agonist conformation of hERα(Y537S) (Fig. 4 (i)), which is stabilized by a hydrogen-bond involving S537 and D351.9,33 The transformation of hERα(Y537S) to adopt an antagonist conformation with a mobile, dislodged helix-12 (H12) is accomplished by this compensating water-mediated hydrogen bond, which is evident in models for compounds 3, 5, and 11 (Fig. 4 (ii and iv)) but is absent in the model for compound 1 (Fig. 4 (ii)). This hypothesis is further corroborated by two experimental observations: 1) Compound 1 shows unusually low antagonistic activity against Y537S (Table 1, entry 1) and 2) Compounds 14 and 15 did not show any activity against mutant ERs. These two results can be rationalized by taking into account the water-mediated hydrogen-bond interaction between the sulfoxide oxygen and Y526. This crucial hydrogen-bond interaction is inaccessible to the sulfoxide group in 1 (Fig. 4 (ii)) unless there is a loss of important interactions with the hydrophobic surface of the protein. In the case of 14 and 15, there is no sulfoxide or sulfone group in the side chain, and thus these two compounds have no possibility for interaction with Y526.
Further, this hypothesis, which is supported by our modeling, also provides an explanation for the other observed trends in biological activity. Examining Tables 2 and 3 for Y537S-related data (specifically, MCF-7 IC50 values for inhibition of transcription and proliferation, respectively), the following composite rank-ordering (from less to more potent) emerges: [3 ≈ 5] < 11 < fulvestrant. In the binding models for compounds 3 and 5, a portion of the linking alkane chain adopts a somewhat strained gauche conformation (Fig. 4 (iv)) to enable formation of the water-mediated hydrogen bond involving Y526 and the sulfoxide oxygen atom. In contrast, the corresponding chain segment in compound 11 is linear (i.e., a lower-energy conformational state), and the same hydrogen bond is achieved, while preserving a reasonable superimposition of the terminal portion of the hydrophobic chain in ligands 3, 5, and 11. Also, compound 11 has an additional hydrogen bond involving H524, which helps stabilize the buried core of the ligand. The ability of compound 11 to adopt a (lower-energy) linear conformation is due to the orientation of the chain from the ligand core; in compound 11, the phenyl ring at the start of the chain is rotated relative to the phenyl ring in compounds 3 and 5. Thus, compound 11’s linear-chain conformation and additional (H524) hydrogen bond lead to increased potency for compound 11 compared to compounds 3 and 5.
Fulvestrant, like compound 11, benefits from an additional hydrogen bond involving H524, but fulvestrant, compared to compound 11, has a bulkier and less flexible (mainly hydrophobic) core, which likely enhances affinity. Also, the perfluorinated end of the flexible chain appears to bind more deeply in a surface pocket for fulvestrant than for compound 11 (Fig. 4 (v)). These structural and interaction attributes likely contribute to the increased potency of fulvestrant in comparison to compounds 11, 5, and 3.
Collectively, the above analysis suggests that increasing the strength of a hydrogen-bond interaction of antagonists with Y526 might further increase their antagonistic activity on mutant ERs such as Y537S.
Conclusion
In summary, we have probed how structural perturbations in different regions of a series of novel ligands influenced their ability to effectively block the activity of WT ERα and the constitutive activity of three mutant ERs responsible for much of endocrine therapy-resistant breast cancer. Both the ligand core and the multifunctional side chain make distinct contributions, the core being a determinant of potency, while different hydrophobic and moderately polar regions located on a multifunctional side chain play more detailed roles in ligand-receptor interactions responsible for inducing antagonism. Our studies led to the identification of several new antiestrogens that inhibit the transcriptional activity of the three ER mutants and also suppress mutant ER-mediated breast cancer cell proliferation. Nevertheless, with all of our new antiestrogens, the ease of inhibiting ERα activity follows the same progression: WT > L536R > D538G > Y537S, thus highlighting ERα genotype as the major determinant of pharmacological efficacy. The results of our studies also allowed us to build a structure-based binding model through which we could identify some novel interactions (e.g., a hydrogen bond of sulfoxide with Y526) that could be targeted to further improve the potency of mutant ER antagonists. These structure-function relationships provide new insights in the search for antiestrogens providing effective suppression of mutant ERα-driven drug-resistant breast cancers, and they also highlight the challenge of developing a truly pan-antagonist that will uniformly and effectively inhibit the activity of WT and all activating-mutant ERs.
Supplementary Material
Acknowledgments.
AS gratefully acknowledges Stevens Institute of Technology (SIT) for financial support. Support for this research through grants from the National Institutes of Health (R01DK015556 to JAK, R01CA220284 to JAK and BSK, predoctoral fellowship T32GM070421 to VSG, P41GM104601 to CGM and the University of Illinois, R01CA204999 to SC and P30CA008748 to SC, P30CA14599 to GLG and University of Chicago Cancer Center), the Breast Cancer Research Foundation (BCRF-17–083 to BSK and JAK), Department of Defense Breast Cancer Research Program (W81XWH-14-1-0360 to GLG), and the Virginia and D.K. Ludwig Fund for Cancer Research (to GLG) is gratefully acknowledged. The Center for Healthcare Innovation at SIT funded the computational chemistry component of this work.
Abbreviations:
- AI
aromatase inhibitor
- ER
estrogen receptor
- ESR1
gene name for ER
- LBD
ligand-binding domain
- NR
nuclear receptor
- SERD
selective ER downregulator
- SERM
selective ER modulator
- SRC
steroid receptor coactivator
- WT
wild type
- RBA
relative binding affinity
Footnotes
Supporting Information Available:
The Supporting Information is available free of charge via the internet on the ACS Publications website. Detailed experimental procedures for synthesis of compounds, characterization data of compounds, biological data from screening of local compound library, biological assay protocols, and molecular modeling data/method.
References
- 1.Heldring N, Pike A, Andersson S, Matthews J, Cheng G, Hartman J, Tujague M, Strom A, Treuter E, Warner M, and Gustafsson JA (2007) Estrogen receptors: how do they signal and what are their targets, Physiol. Rev 87, 905–931. [DOI] [PubMed] [Google Scholar]
- 2.Ariazi EA, Ariazi JL, Cordera F, and Jordan VC (2006) Estrogen receptors as therapeutic targets in breast cancer, Curr. Top. Med. Chem 6, 181–202. [PubMed] [Google Scholar]
- 3.Forbes JF, Cuzick J, Buzdar A, Howell A, Tobias JS, and Baum M (2008) Effect of anastrozole and tamoxifen as adjuvant treatment for early-stage breast cancer: 100-month analysis of the ATAC trial, Lancet Oncol. 9, 45–53. [DOI] [PubMed] [Google Scholar]
- 4.Toy W, Shen Y, Won H, Green B, Sakr RA, Will M, Li Z, Gala K, Fanning S, King TA, Hudis C, Chen D, Taran T, Hortobagyi G, Greene G, Berger M, Baselga J, and Chandarlapaty S (2013) ESR1 ligand-binding domain mutations in hormone-resistant breast cancer, Nat. Genet 45, 1439–1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Merenbakh-Lamin K, Ben-Baruch N, Yeheskel A, Dvir A, Soussan-Gutman L, Jeselsohn R, Yelensky R, Brown M, Miller VA, Sarid D, Rizel S, Klein B, Rubinek T, and Wolf I (2013) D538G mutation in estrogen receptor-alpha: A novel mechanism for acquired endocrine resistance in breast cancer, Cancer Res. 73, 6856–6864. [DOI] [PubMed] [Google Scholar]
- 6.Li S, Shen D, Shao J, Crowder R, Liu W, Prat A, He X, Liu S, Hoog J, Lu C, Ding, et al. (2013) Endocrine-therapy-resistant ESR1 variants revealed by genomic characterization of breast-cancer-derived xenografts, Cell Rep. 4, 1116–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jeselsohn R, Yelensky R, Buchwalter G, Frampton G, Meric-Bernstam F, Gonzalez-Angulo AM, Ferrer-Lozano J, Perez-Fidalgo JA, Cristofanilli M, Gomez H, et al. (2014) Emergence of constitutively active estrogen receptor-alpha mutations in pretreated advanced estrogen receptor-positive breast cancer, Clin. Cancer Res 20, 1757–1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Toy W, Weir H, Razavi P, Lawson M, Goeppert AU, Mazzola AM, Smith A, Wilson J, Morrow C, Wong, et al. (2017) Activating ESR1 Mutations Differentially Affect the Efficacy of ER Antagonists, Cancer Discov. 7, 277–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fanning SW, Mayne CG, Dharmarajan V, Carlson KE, Martin TA, Novick SJ, Toy W, Green B, Panchamukhi S, Katzenellenbogen BS, Tajkhorshid E, Griffin PR, Shen Y, Chandarlapaty S, Katzenellenbogen JA, and Greene GL (2016) Estrogen receptor alpha somatic mutations Y537S and D538G confer breast cancer endocrine resistance by stabilizing the activating function-2 binding conformation, eLife 5, e12792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.De Savi C, Bradbury RH, Rabow AA, Norman RA, de Almeida C, Andrews DM, Ballard P, Buttar D, Callis RJ, Currie GS et al. (2015) Optimization of a Novel Binding Motif to (E)-3-(3,5-Difluoro-4-((1R,3R)-2-(2-fluoro-2-methylpropyl)-3-methyl-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indol-1-yl)phenyl)acrylic Acid (AZD9496), a Potent and Orally Bioavailable Selective Estrogen Receptor Downregulator and Antagonist, J. Med. Chem 58, 8128–8140. [DOI] [PubMed] [Google Scholar]
- 11.Lai A, Kahraman M, Govek S, Nagasawa J, Bonnefous C, Julien J, Douglas K, Sensintaffar J, Lu N, Lee K. j., Aparicio A, Kaufman J, Qian J, Shao G, Prudente R, Moon MJ, Joseph JD, Darimont B, Brigham D, Grillot K, Heyman R, Rix PJ, Hager JH, and Smith ND (2015) Identification of GDC-0810 (ARN-810), an Orally Bioavailable Selective Estrogen Receptor Degrader (SERD) that Demonstrates Robust Activity in Tamoxifen-Resistant Breast Cancer Xenografts, J. Med. Chem 58, 4888–4904. [DOI] [PubMed] [Google Scholar]
- 12.Zhao Y, Laws MJ, Guillen VS, Ziegler Y, Min J, Sharma A, Kim SH, Chu D, Park BH, Oesterreich S, Mao C, Shapiro DJ, Nettles KW, Katzenellenbogen JA, and Katzenellenbogen BS (2017) Structurally Novel Antiestrogens Elicit Differential Responses from Constitutively Active Mutant Estrogen Receptors in Breast Cancer Cells and Tumors, Cancer Res. 77, 5602–5613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shiau AK, Barstad D, Loria PM, Cheng L, Kushner PJ, Agard DA, and Greene GL (1998) The Structural Basis of Estrogen Receptor/Coactivator Recognition and the Antagonism of This Interaction by Tamoxifen, Cell 95, 927–937. [DOI] [PubMed] [Google Scholar]
- 14.Garner F, Shomali M, Paquin D, Lyttle CR, and Hattersley G (2015) RAD1901: a novel, orally bioavailable selective estrogen receptor degrader that demonstrates antitumor activity in breast cancer xenograft models, Anti-Cancer Drugs 26, 948–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Willson TM, Henke BR, Momtahen TM, Charifson PS, Batchelor KW, Lubahn DB, Moore LB, Oliver BB, and Sauls HR (1994) 3-[4-(1,2-Diphenylbut-1-enyl)phenyl]acrylic Acid: A Non-Steroidal Estrogen with Functional Selectivity for Bone over Uterus in Rats, J. Med. Chem 37, 1550–1552. [DOI] [PubMed] [Google Scholar]
- 16.Van de Velde P, Nique F, Bouchoux F, Bremaud J, Hameau MC, Lucas D, Moratille C, Viet S, Philibert D, and Teutsch G (1994) RU 58,668, a new pure antiestrogen inducing a regression of human mammary carcinoma implanted in nude mice, J. Steroid Biochem. Mol. Biol 48, 187–196. [DOI] [PubMed] [Google Scholar]
- 17.Min J, Guillen VS, Sharma A, Zhao Y, Ziegler Y, Gong P, Mayne CG, Srinivasan S, Kim SH, Carlson KE, Nettles KW, Katzenellenbogen BS, and Katzenellenbogen JA (2017) Adamantyl Antiestrogens with Novel Side Chains Reveal a Spectrum of Activities in Suppressing Estrogen Receptor Mediated Activities in Breast Cancer Cells, J. Med. Chem 60, 6321–6336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Muthyala RS, Sheng S, Carlson KE, Katzenellenbogen BS, and Katzenellenbogen JA (2003) Bridged bicyclic cores containing a 1,1-diarylethylene motif are high-affinity subtype-selective ligands for the estrogen receptor, J. Med. Chem 46, 1589–1602. [DOI] [PubMed] [Google Scholar]
- 19.Pike AC, Brzozowski AM, Walton J, Hubbard RE, Thorsell AG, Li YL, Gustafsson JA, and Carlquist M (2001) Structural insights into the mode of action of a pure antiestrogen, Structure 9, 145–153. [DOI] [PubMed] [Google Scholar]
- 20.Bowler J, Lilley TJ, Pittam JD, and Wakeling AE (1989) Novel steroidal pure antiestrogens, Steroids 54, 71–99. [DOI] [PubMed] [Google Scholar]
- 21.Trost BM, and Curran DP (1981) Chemoselective oxidation of sulfides to sulfones with potassium hydrogen persulfate, Tetrahedron Lett. 22, 1287–1290. [Google Scholar]
- 22.Davis FA, Lal SG, and Durst HD (1988) Chemistry of oxaziridines. 10. Selective catalytic oxidation of sulfides to sulfoxides using N-sulfonyloxaziridines, J. Org. Chem 53, 5004–5007. [Google Scholar]
- 23.Carlson KE, Choi I, Gee A, Katzenellenbogen BS, and Katzenellenbogen JA (1997) Altered ligand binding properties and enhanced stability of a constitutively active estrogen receptor: evidence that an open pocket conformation is required for ligand interaction, Biochemistry 36, 14897–14905. [DOI] [PubMed] [Google Scholar]
- 24.Chan S (2002) A review of selective estrogen receptor modulators in the treatment of breast and endometrial cancer, Semin. Oncol 29, 129–133. [DOI] [PubMed] [Google Scholar]
- 25.Sato M, Turner CH, Wang T, Adrian MD, Rowley E, and Bryant HU (1998) LY353381.HCl: a novel raloxifene analog with improved SERM potency and efficacy in vivo, J. Pharmacol. Exp. Ther 287, 1–7. [PubMed] [Google Scholar]
- 26.Marchisano-Karpman C, and DeGregorio MW (2003) Arzoxifene Eli Lilly, IDrugs 6, 880–885. [PubMed] [Google Scholar]
- 27.Claussner A, Nique F, Teutsch J, Patrick VDV. 19-nor steroids having a thiocarbonated chain in position 11β, their preparation process and the intermediates of this process, their use as medicaments and compositions, US6281204B1, (2001)
- 28.Bahreini A, Li Z, Wang P, Levine KM, Tasdemir N, Cao L, Weir HM, Puhalla SL, Davidson NE, Stern AM, Chu D, Park BH, Lee AV, and Oesterreich S (2017) Mutation site and context dependent effects of ESR1 mutation in genome-edited breast cancer cell models, Breast Cancer. Res 19, 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cheng Y, and Prusoff WH (1973) Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction, Biochem. Pharmacol 22, 3099–3108. [DOI] [PubMed] [Google Scholar]
- 30.Wardell SE, Ellis MJ, Alley HM, Eisele K, VanArsdale T, Dann SG, Arndt KT, Primeau T, Griffin E, Shao J, Crowder R, Lai JP, Norris JD, McDonnell DP, and Li S (2015) Efficacy of SERD/SERM Hybrid-CDK4/6 Inhibitor Combinations in Models of Endocrine Therapy-Resistant Breast Cancer, Clin. Cancer Res 21, 5121–5130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Webb P, Nguyen P, and Kushner PJ (2003) Differential SERM Effects on Corepressor Binding Dictate ERα Activity in Vivo, J. Biol. Chem 278, 6912–6920. [DOI] [PubMed] [Google Scholar]
- 32.Heldring N, Pawson T, McDonnell D, Treuter E, Gustafsson J-Å, and Pike ACW (2007) Structural Insights into Corepressor Recognition by Antagonist-bound Estrogen Receptors, J. Biol. Chem 282, 10449–10455. [DOI] [PubMed] [Google Scholar]
- 33.Katzenellenbogen JA, Mayne CG, Katzenellenbogen BS, Greene GL, Chandarlapaty S (2018) Structural underpinnings of oestrogen receptor mutations in endocrine therapy resistance, Nat. Rev. Cancer, 18, 377–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Berman HM; Westbrook J; Feng Z; Gilliland G; Bhat TN; Weissig H; Shindyalov IN; Bourne PE (2000) The Protein Data Bank, Nucleic Acids Res. 28, 235–242. The URL of the RCSB PDB is www.rcsb.org. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.