

Abstract
Aging is accompanied by progressive declines in skeletal muscle mass and strength and impaired regenerative capacity, predisposing older adults to debilitating age-related muscle deteriorations and severe morbidity. Muscle stem cells (muSCs) that proliferate, differentiate to fusion-competent myoblasts, and facilitate muscle regeneration are increasingly dysfunctional upon aging, impairing muscle recovery after injury. While regulators of muSC activity can offer novel therapeutics to improve recovery and reduce morbidity among aged adults, there are no known muSC regenerative small molecule therapeutics. We recently developed small molecule inhibitors of nicotinamide N-methyltransferase (NNMT), an enzyme overexpressed with aging in skeletal muscles and linked to impairment of the NAD+ salvage pathway, dysregulated sirtuin 1 activity, and increased muSC senescence. We hypothesized that NNMT inhibitor (NNMTi) treatment will rescue age-related deficits in muSC activity to promote superior regeneration post-injury in aging muscle. 24-month old mice were treated with saline (control), and low and high dose NNMTi (5 and 10 mg/kg) for 1-week post-injury, or control and high dose NNMTi for 3-weeks post-injury. All mice underwent an acute muscle injury (barium chloride injection) locally to the tibialis anterior (TA) muscle, and received 5-ethynyl-2’-deoxyuridine systemically to analyze muSC activity. In vivo contractile function measurements were conducted on the injured TA muscle and tissues collected for ex-vivo analyses, including myofiber cross-sectional area (CSA) measurements to assess muscle recovery. Results revealed that muscle stem cell proliferation and subsequent fusion were elevated in NNMTi-treated mice, supporting nearly 2-fold greater CSA and shifts in fiber size distribution to greater proportions of larger sized myofibers and fewer smaller sized fibers in NNMTi-treated mice compared to controls. Prolonged NNMTi treatment post-injury further augmented myofiber regeneration evinced by increasingly larger fiber CSA. Importantly, improved muSC activity translated not only to larger myofibers after injury but also to greater contractile function, with the peak torque of the TA increased by ~70% in NNMTi-treated mice compared to controls. Similar results were recapitulated in vitro with C2C12 myoblasts, where NNMTi treatment promoted and enhanced myoblast differentiation with supporting changes in the cellular NAD+/NADH redox states. Taken together, these results provide the first clear evidence that NNMT inhibitors constitute a viable pharmacological approach to enhance aged muscle regeneration by rescuing muSC function, supporting the development of NNMTi as novel mechanism-of-action therapeutic to improve skeletal muscle regenerative capacity and functional recovery after musculoskeletal injury in older adults.
Keywords: muscle regeneration, aged muscle, therapeutics, nicotinamide N-methyltransferase, inhibitor, muscle stem cells, satellite cells
Graphical Abstract
1. INTRODUCTION
The population of older (60+ years of age) adults is rapidly expanding in the United States and throughout the world, placing ever-increasing strains on health care resources and an urgent need for improved approaches to elder care [1]. One of the most significant impacts of aging is the progressive decline in skeletal muscle mass and strength [2, 3], with concomitant deteriorations in physical function and mobility that are strongly associated with numerous chronic diseases and increased mortality [4, 5]. While all older individuals experience muscle degeneration, approximately 30% of adults over 60 years of age and 50% of adults over 80 years of age develop sarcopenia, a geriatric disease characterized by significant and objective defects in muscle mass, strength, and function [6–8]. Sarcopenic elderly individuals are at a 2-to 5-fold increased risk for permanent disability and greatly diminished quality of life arising from progressive muscle degeneration, decreased muscle function, and poor muscle quality that predispose them to debilitating falls and substantial disease burden [4]. Furthermore, as muscle regenerative capacity of older adults becomes increasingly compromised, it leads to delayed and impaired recovery following muscle injury [9–11], decreased mobility and independence, increased hospitalization costs [12], and higher mortality rates [13].
Muscle stem cells (muSCs; also termed satellite cells) are responsible for mediating skeletal muscle repair by proliferating and differentiating into fusion-competent myoblasts and facilitating myofiber regeneration following injury [14–16]. The major driving factor for delayed and impaired recovery of aged muscle following injury appears to be a significant decrease in muSC regenerative capacity and function [10, 11, 17]. These events occur independent of sarcopenia [18], resulting in the formation of dysfunctional senescent muSCs that are no longer activated by muscle injury or turnover stimuli and present compromised ability to proliferate, differentiate, fuse, and promote the repair and replacement of myofibers [19, 20]. Additionally, muSC abundance has been observed to decline in some aged skeletal muscle tissues [21] as muSCs develop diminished intrinsic capacity for reversible quiescence, which further reduces the population of muSCs supporting the post-injury repair cascade [19]. Applications of muSCs in promoting muscle repair have been preclinically investigated [16]; however, there are no approved pharmacological therapeutics that can safely and effectively enhance muSC activation and regenerative capacity to promote recovery from injury among aging adults.
Nicotinamide adenine dinucleotide (NAD+) is a fundamental cellular regulator of energy metabolism, mitochondrial function, and bioenergetics [22, 23] that declines with age [24–26]. Age-related diminished NAD+ contributes to altered skeletal muscle metabolism and mitochondrial function [25, 27], progressive muscle degeneration and loss of function [28], which further link to increased muSC senescence, reduced muSC function, dysfunctional muscle regeneration, and impaired muscle repair [29, 30]. Recently, several studies have observed that nutraceutical supplements such as nicotinamide mononucleotide (NMN), nicotinamide riboside (NR), and nicotinamide analogues (e.g., acipimox), which increase intracellular NAD+ levels and stimulate muscle NAD+ biosynthetic pathways (e.g., NAD+ salvage pathway), can improve muscle metabolic, mitochondrial, and muSC function, and accelerate muscle regeneration in aged mice [29, 31, 32]. Thus, pharmacologically enhancing NAD+ levels in aging skeletal muscle tissues may provide a viable approach to improve the regenerative function of aged muSCs.
The cytosolic enzyme nicotinamide N-methyltransferase (NNMT) has been newly discovered to modulate the levels of nicotinamide precursors required for NAD+ biosynthesis, and thus plays a crucial role in regulating the NAD+ salvage pathway and cellular metabolism [33]. NNMT is expressed in skeletal muscle, with progressively greater expression associated with aging in muscle tissues [34, 35]. Importantly, NNMT is a dominant component of the gene expression signature for sarcopenia [36], overexpression of which could lead to significantly reduced levels of NAD+ and associated declines in muscle mass, strength, and function that accompany aging [37, 38]. We recently discovered and developed first-in-class small molecule inhibitors of NNMT that have amenable drug-like properties and selectively inhibit NNMT activity in vivo [38, 39]. In the present study, we generated proof-of-concept data demonstrating the efficacy of a novel NNMT small molecule inhibitor to accelerate muscle regeneration in an aged mouse muscle injury model. Our results indicated that treatment of aged mice with a small molecule NNMT inhibitor (NNMTi) enhanced proliferation, fusion, and regenerative capacity of muSCs, subsequently increasing functional performance of skeletal muscle. Similar results were demonstrated in vitro with C2C12 cells that closely mimic muSCs, where NNMTi treatment promoted myoblast differentiation and supporting metabolic changes in NAD+ salvage pathway-related cellular metabolites. To the best of our knowledge, this is the first study to convincingly demonstrate that NNMT inhibition rescues age-related muSC deficits and serves as a suitable therapeutic approach to reactivate skeletal muSCs in aging skeletal muscle, thus mitigating age-associated impaired muscle regeneration and improving skeletal muscle remodeling and function following injury.
2. MATERIALS AND METHODS
2.1. Chemicals.
NNMT inhibitor (NNMTi), 5-amino-1-methylquinolium was synthesized by a previously established synthetic method [39]. Internal standard (IS) for LC/MS/MS studies was a deuterated 5-amino-1-methylquinolinium analogue that was produced using a modified synthetic scheme [39]. Other standards for LC/MS/MS studies were purchased from established commercial suppliers; S-(5′-Adenosyl)-L-methionine (SAM), NAD+, nicotinamide (NA), and NADH (reduced form of NAD+) were obtained from Sigma-Aldrich (St. Louis, MO, USA). 1-methylnicotinamide (1-MNA) and S-Adenosyl-L-homocysteine (SAH) were obtained from Cayman Chemical Company (Ann Arbor, MI, USA).
2.2. Quantitative measurement of NNMT expression in young and aged muscle tissue.
20–40 mg of tibialis anterior (TA) muscle tissue were collected from young (4-month-old) and aged (24-month-old) C57Bl/6 mice, pulverized on ice, added to RIPA buffer (Cell Signaling Technology, Danvers, MA, USA; product #9806) containing protein phosphatase inhibitor cocktail (P-5726, Sigma Aldrich, St. Louis, MO) (1:10 w/v), and homogenized using a hand-held tissue homogenizer (Tissue-Tearor™, Model 985370, BioSpec Products, Inc., Bartlesville, OK, USA). Homogenized samples were centrifuged at 10,000 g for 10 min at 4°C. Supernatants were collected and stored at - 20°C until further processing. Protein concentration in samples were determined using the bicinchoninic acid (BCA) protein assay (Pierce™ BCA Protein Assay Kit, Thomas Scientific, Swedesboro, NJ, USA). Briefly, 40 μg of tissue homogenates from young and aged TA muscle samples were separated using sodium dodecyl sulfate polyacrylamide gel electerophoresis (SDS-PAGE). Separated proteins were transferred onto a polyvinylidene fluoride (PVDF) membrane and probed for NNMT protein expression using anti-NNMT primary antibody (ab58743, rabbit polyclonal; Abcam, Cambridge, MA, USA) and HRP-conjugated polyclonal goat anti-rabbit secondary antibody (ab205718; Abcam) via western blotting. Membrane was striped and re-probed for β-actin using anti-β-actin primary antibody (ab101173, monoclonal mouse; Abcam) as the loading control. Western blots were analyzed for NNMT and β-actin expression using ImageJ software (NIH). NNMT protein expression (bands detected at ~27 kDa) was normalized to β-actin expression (bands detected at ~42 kDa) and compared between young and aged TA muscle tissues. Samples were run and analyzed in technical duplicates.
2.3. Efficacy of NNMTi to activate and improve regenerative capacity of muSC in an aged mouse muscle injury model.
Aged, 24-month-old (N=48), male C57Bl/6 mice were obtained from the National Institute of Aging (NIA). Upon arrival, mice were single-housed and allowed to acclimate to the controlled environment vivarium maintained at a constant temperature (21–23°C) and humidity (40–50%) on a 12-hour light-dark cycle (lights on 0600–1800 h) for at least 7 days before initiation of experiments. Food and water were available ad libitum. All experiments were carried out in accordance with the Guide for the Care and Use of Laboratory Animals and with approval from the Institutional Animal Care and Use Committee at the University of Texas Medical Branch.
2.3.1. Short-term treatment effects of NNMTi on muscle regeneration after injury.
Following acclimation, mice were randomized into control (saline; n=13), low dose NNMTi (5 mg/kg; n=10), and high dose NNMTi (10 mg/kg; n=13) treatment groups, ensuring similar mean average body weights across the three cohorts. Mice received twice daily (BID) subcutaneous (SC) injections of saline or NNMTi for 2 weeks (1 week pre-injury and 1 week post-injury). Mice were weighed and the body weights were recorded every other day. On day 8 (following 1 week of treatment), mice were briefly anesthetized using isoflurane and 30 µL of barium chloride (BaCl2) toxin (1.2% concentration) was injected locally into the TA muscle of one hindlimb to induce muscle injury, followed by carprofen for analgesia. Starting on the day of the injury (day 8) and on days 10, 12, and 14, mice received a single intraperitoneal (IP) injection of 5-ethynyl-2´-deoxyuridine (EdU) (150 ug in saline) to track muSC proliferation and fusion into regenerating TA muscle fibers. All groups of mice continued to receive BID, SC injections of saline or NNMTi on days 8 through 14. At the terminal end of the study, a sub-cohort of mice from the 3 treatment groups (n=6/group) were deeply anesthetized using ketamine/xylazine cocktail anesthetic (IP injection) and the blood was drawn by cardiac puncture procedure, following which muscle tissues were harvested for additional processing. Remaining mice (n=7/group in control and high dose groups) were subject to TA contractile function measurements (measures described in detail below) under isoflurane-induced anesthesia to assess muscle strength. Terminal bleeds were performed on all remaining mice and the tissues harvested and flash frozen for ex-vivo analyses as described below.
2.3.2. Long-term treatment effect of NNMTi on complete muscle recovery after injury.
A separate cohort of 24-month-old, aged mice were randomized into control (saline; n=6) and high dose NNMTi (10 mg/kg; n=6) treatment groups, ensuring similar mean average body weights across the two cohorts. In contrast to the short-term treatment regimen (1-week post-injury treatment) described above, mice received twice daily (BID) subcutaneous (SC) injections of saline or NNMTi for 4 weeks; pretreatment for 7 days prior to BaCl2 injury to one TA muscle, followed by 3 weeks of post-injury treatment to examine a more complete muscle recovery. Muscle tissues were harvested and flash frozen for ex-vivo analyses as described below.
2.4. Effect of NNMTi on in vivo contractile physiology determined by dorsiflexor torque measurement.
This protocol was adapted and modified from previously published literature [40, 41]. Briefly, mice were anesthetized with isoflurane (3–4% for induction and ~2.5% for maintenance of anesthesia via a nose cone; oxygen maintained at ~1.5 l/min with a VetEquip vaporizer), the BaCl2 injured leg trimmed (Wahl Bravmini; Sterling, IL, USA), and set on a platform (Aurora Scientific 809c in situ testing apparatus; Aurora, ON, Canada) heated to 37°C (with an Anova Industries Model 10 water circulator; Stafford, TX, USA). The leg was then mounted into a clamp to hold it static at the knee, with the tibia perpendicular to the femur and the foot perpendicular to the tibia. The foot was secured in a mount connected to the force transducer, with the ankle wedged in place and with a piece of tape holding the foot to the mount. Platinum needle electrodes were set percutaneously, approximately near the top of the knee and halfway down TA, between the TA and the gastrocnemius (needles were adjusted to find the optimal placing for maximum force production with minimum current and minimal antagonistic muscle response). With an Aurora Physiology system (Model 6650LR Force Transducer, Dual Mode lever System, Hi power Bi-Phase Stimulator, Signal Interface, and software: Dynamic Muscle Control v5.500 and Dynamic muscle Analysis version 5.300), the proper maximal current used to stimulate muscle contraction was determined by eliciting a twitch (pulse duration 0.2 s) with the stimulus starting at 5 mA and increased incrementally to ~50 mA until the maximum twitch torque at the minimum amperage needed to stimulate this torque was recorded. This minimum current was then maintained throughout a torque-frequency curve to find the maximum torque output (twitch1, 10 Hz, 40 Hz, 80 Hz, 100 Hz, 120 Hz, 150 Hz, 180 Hz, 200 Hz, 250 Hz and a final twitch2). A twitch1/twitch2 minimum ratio of 90% determined muscle integrity post-procedure (<90% may indicate extreme muscle fatigue or damage). No mice failed the twitch1/twitch2 test. Peak torque per gram of body mass and peak torque normalized to CSA (average cross-sectional area of fibers) per animal were recorded as outcomes, group averaged, and compared between control and high dose NNMTi (10 mg/kg)-treated mice. Data from one treated mouse was not included in analysis since torque could not be reliably measured above background levels.
2.5. Immunohistochemistry.
For all immunohistochemical analyses, dissected muscles from mice were mounted in Tissue Tek (O.C.T. Compound, Sakura Finetek, Torrance, CA, USA) at resting length and frozen in liquid nitrogen-cooled 2-methylbutane and stored at −80°C until analysis. 7 µm-thick sections were cut with a cryostat (HM525-NX, Thermo Fisher Scientific, Waltham, MA, USA) and allowed to air dry for 1 h prior to storage at −20 °C.
2.5.1. NNMT expression in 4-month old young and 24-month old aged tibialis anterior (TA) muscle.
For NNMT immunohistochemical staining, sections were fixed in 4% paraformaldehyde (PFA). Slides were blocked for 1 h in 1% TSA blocking reagent and incubated overnight at 4 °C in primary antibody against NNMT (ab58743, Abcam, Cambridge, MA; 1:100). The following day, slides were incubated for 1 h at room temperature with IgG anti-rabbit secondary conjugated to AF555 (no. A21428, 1:500; Life Technologies/Thermo Fisher Scientific, Waltham, MA USA) and AF488-cojugated wheat germ agglutinin (WGA) (no. W11261, Life Technologies/Thermo Fisher Scientific) to denote myofiber borders. Slides were incubated in 4′,6-diamidino-2-phenylindole (DAPI; 10 nM, LifeTechnologies/Thermo Fisher Scientific) for 10 min and washed and mounted with Vectashield fluorescence mounting media (Vector Laboratories, Burlingame, CA, USA).
2.5.2. muSC immunohistochemical analyses.
Pax7 is a transcription factor unique to muSCs and a routinely used marker to identify muSCs; laminin is routinely used to denote muscle fiber borders. For Pax7-EdU-laminin staining, sections were fixed in 4% PFA followed by antigen retrieval using sodium citrate (10mM, pH 6.5) at 92°C for 20 min. Slides were then placed in 3% hydrogen peroxide in PBS for 7 min to block endogenous peroxidase activity followed by a second blocking step in Mouse-on-Mouse Blocking Reagent (Vector Laboratories, Burlingame, CA, USA). Sections were incubated overnight at 4 °C in primary antibodies against Pax7 (DSHB, Iowa City, IA, USA; 1:100) and laminin (L9393, Sigma-Aldrich, St Louis, MO, USA; 1:100). Next, slides were incubated with biotinylated anti-mouse IgG secondary antibody for Pax7 (no. 115–065-205, Jackson ImmunoResearch, West Grove, PA, USA; 1:500) and anti-rabbit IgG secondary antibody for laminin (Alexa Fluor 647, no. A11034, Life Technologies/Thermo Fisher Scientific; 1:500) followed by incubation with streptavidin-horseradish peroxidase (HRP) included with a commercially available tyramide signal amplification kit (TSA, Life Technologies/Thermo Fisher Scientific). TSA-Alexa Fluor 488 was used to visualize Pax7 antibody binding. Following all Pax7-laminin steps, slides were permeabilized in 0.1% Triton-X100 in phosphate-buffered saline and incubated with the Life Technologies Click-iT Kit per manufacturer’s instructions, with EdU visualized with Alexa Fluor 555. Slides were incubated in 4′,6-diamidino-2-phenylindole (DAPI; 10 nM, LifeTechnologies/Thermo Fisher Scientific) for 10 min, washed, and mounted using Vectashield fluorescence mounting media (Vector Laboratories).
2.5.3. Image acquisition and quantification.
Immunohistochemical images were captured at 200X total magnification room temperature with a Zeiss upright microscope (AxioImager M1; Zeiss, Göttingen, Germany). muSC abundance was evaluated by Pax7 staining along with laminin and DAPI; only cells that were Pax7+ /DAPI+ within the laminin border were counted. Proliferating muSC included only those that were Pax7+/EdU+/DAPI+ within the laminin border. muSC fusion to myofibers was determined by newly acquired myonuclei classified as Pax7−/EdU+/DAPI+. Mean myofiber cross-sectional area (CSA) was measured by manually tracing myofiber area using the laminin border. Only injured myofibers (fibers with central myonucleation) were included in CSA analysis.
Image analysis was performed in a blinded manner using AxioVision Rel software (v4.9). To assess the reliability, accuracy, and reproducibility of the image quantification procedures, three blinded raters assessed images from six mice; two mice were randomly selected from each treatment group for validation of counts of muSC abundance, proliferating muSCs, and muSC fusion (saline: n=2, low dose NNMTi: n=2, high dose NNMTi: n=2). Intraclass correlation coefficients (ICCs) were calculated for each variable to determine reliability of the data. ICC estimates and 95% confidence intervals were calculated using SPSS statistical software (IBM Corp. IBM SPSS Statistics for Windows, Version 25. Armonk, NY, USA) based on a mean-rating, consistency, 2-way mixed-effects model with 3 raters across 6 mice. ICC values below 0.5 indicate poor reliability, 0.5 to 0.75 indicate moderate reliability, 0.75 to 0.9 indicate good reliability, and values greater than 0.90 are indicative of excellent reliability and reproducibility [42, 43].
2.6. Plasma chemistry panel.
Blood samples were collected from each animal immediately upon sacrifice, processed to isolate plasma, frozen in liquid nitrogen, and shipped on dry ice to Texas A&M Veterinary Medical Diagnostic Laboratory (TVMDL; College Station, TX, USA) for small animal serum chemistry analysis via standard protocols. TVMDL is accredited by the American Association of Veterinary Laboratory Diagnosticians and all results were confirmed with positive and negative controls. Complete plasma measures included hepatic panel (AST, ALT, ALP, GGT, GLDH, total bilirubin, albumin, globulin, AG ratio, total protein), renal panel (amylase, BUN, creatine kinase, calcium, phosphorus), amylase, electrolytes (sodium, potassium, and chloride), glucose, and cholesterol to validate systemic effects of repeated NNMTi treatment on major organ functions.
2.7. Differentiation of C2C12 myoblasts.
C2C12 myoblast cells (CRL-1772, American Type Culture Collection; Manassas, VA, USA) were cultured with standard growth media (DMEM, 4.5 g/L glucose, L-glutamine, sodium pyruvate, 10% FBS, 1% penicillin/streptomycin) and grown to 80% confluency before initiating differentiation. To begin differentiation of myoblasts, growth media was replaced by differentiation media (DMEM, 4.5 g/L glucose, L-glutamine, sodium pyruvate, 2% Horse serum, 1% penicillin/streptomycin); low nutrient condition and increased cell-to-cell contact stimulated differentiation of myoblasts by inhibiting proliferation and promoting the formation of multi-nucleated myotubes. Differentiation media was replaced every other day to maintain myotubes in culture for 3–4 days, during which time experiments as described below were performed.
2.8.1. Immunocytochemical analysis of NNMT expression in C2C12 myoblasts and differentiated myotubes.
C2C12 myoblasts and myotubes grown on glass cover slips were fixed in 4% PFA warmed to 37°C for 15 min, and then washed 3 times with PBS. Myoblasts and myotubes were permeabilized with a 10 min wash in PBS + 0.2% Triton-X100, and then blocked in 1% BSA made in PBS + 0.2% Triton-X100 for 1 hr. For myoblasts, cells were incubated in primary antibody (1:100, NMMT cat #ab119758, mouse IgG2b, Abcam) for 2 hr at room temp in 1% BSA, followed by PBS washing, and then secondary antibody (Gt anti-Ms IgG2b AF488, cat # A-21141, ThermoFisher) for 1 hr followed by a co-stain with DAPI and mounting. For myotubes, cells were incubated in primary antibody (NMMT, 1:100, cat #ab119758, mouse IgG2b, Abcam and myosin heavy chain, 1:200, cat #M4276, mouse IgG1, Sigma) for 2 hr at room temp in 1% BSA, followed by PBS washing, and then secondary antibody (Gt anti-Ms IgG2b AF488, cat # A-21141 and Gt anti-Ms IgG1 AF555, cat # A-21127, ThermoFisher) for 1 hr followed by a co-stain with DAPI and mounting. Images were captured at 200X total magnification.
2.8.2. Effect of NNMTi on C2C12 myoblast differentiation.
To determine the effect of NNMTi on C2C12 myoblast differentiation, myoblasts were cultured to confluency as described above and stimulated to differentiate in the absence or presence of NNMTi (10 and 30 µM concentrations) in differentiation media for 96 h. Concentrations of NNMTi were chosen based on our previous published work in adipocytes, where significant phenotypic and metabolic changes were observed in the presence of the inhibitor relative to untreated conditions [38]. After 96 h of differentiation, media was removed and cells were fixed in 4% PFA pre-warmed to 37°C for 15 min. PFA was then removed and the cells were washed three times (3 min each wash) with PBS containing 0.2% Triton X-100. Cells were blocked for 1 h in 1% TSA (fTSA kit – compound D, Invitrogen ThermoFisher Scientific #T-30955) in PBS/Triton. Following blocking, cells were incubated for 2 h at room temperature with anti-myosin heavy chain (MHC) primary antibody (Sigma #M4276 – from MY-32 clone) at 1:200 concentration made in TSA/PBS/Triton diluent. Cells were washed in PBS/Triton and incubated for 1 h at room temperature with goat anti-mouse secondary antibody (IgG1 AF488 conjugated; Invitrogen #A21121) at 1:500 concentration made in PBS/Triton. Cells were washed, incubated with DAPI (1:10,000 concentration; Invitrogen #D35471 in PBS) for 10 min to stain nuclei, and washed finally with PBS/Trition before imaging. Four random field of views were captured per well; total DAPI stained nuclei and MHC positive nuclei were counted to calculate %MHC positive nuclei. Control and NNMTi-treated conditions were run in duplicates with results averaged for each condition. %MHC positive nuclei analyzed were averaged and compared between control and NNMTi-treated myotubes with experiments repeated three times.
2.8.3. Effect of NNMTi treatment on NAD+ and NADH levels in C2C12 myoblasts and myotubes.
C212 myoblasts were differentiated to myotubes using the protocol described above (see section 2.7) and maintained in culture for 4 days before NNMTi treatment. On day 3 of differentiation, media was removed and replaced with fresh differentiation media (control) or NNMTi (10 and 30 µM concentrations) made in differentiation media for 24 h. Similarly, C2C12 myoblasts were cultured to ~40% confluency and treated for 24 h with growth media alone (control) or in the presence of NNMTi (10 and 30 µM concentrations) in growth media for 24 h. Following 24 h of treatment with NNMTi, cells were treated with trypsin, centrifuged, and washed once in ice cold PBS. Cells were re-suspended in 300 µL of 75% ethanol / 25% 10 mM HEPES solution pre-heated to 80°C and extracted using a previously described protocol to obtain soluble NAD+ and NADH metabolites [44]. Levels of NAD+ and NADH in the extracted samples were assessed using a spectrophotometric enzymatic assay. Briefly, samples were re-suspended in distilled water and treated with acid (0.1 M hydrochloric acid) or base (0.1 M sodium hydroxide) and heated at 75°C for 30 min to remove all the NADH and NAD+ in samples, respectively. Samples were then neutralized by appropriately adding acid (0.1 M HCl/0.5 M bicene) or base (0.1 M NaOH/0.5 M bicene) and treated with alcohol dehydrogenase (1x final; A3263–7, Sigma-Aldrich, St. Louis, MO, USA) for 15 min. Following incubation, 10 µL of WST-1 reagent (Roche Applied Science, Mannheim, Germany) to initiate the enzymatic reaction and the pink colored product formed over time was spectrophotometrically measured via absorbance detected at 450 nm wavelength and compared between control and treated samples.
2.9. Statistical analysis.
Effects of NNMTi on muSC regeneration were expressed as incidence of activated muSCs and fibers with myonuclei and analyzed using R statistical software (R Core Team, 2018, version 3.5.1) by mixed effect logistic regression as a function of dose group, adjusting for cohort (given that the experiments were conducted across separate cohorts), and controlling for repeated measures effect. Differences between dose groups were assessed by Tukey adjusted contrasts posthoc test. Fiber CSA data were log-transformed (for 1-week post-injury treatment data to better approximate normal distribution) or analyzed without transformation (3-week post-injury treatment data) by mixed effect analysis of variance (ANOVA) followed by Tukey multiple contrasts posthoc as a function of dose, adjusting for cohort (given that the experiments were conducted across separate cohorts), and controlling for repeated measures effect. Where applicable, statistical analysis for two-group comparisons was conducted using unpaired Student’s t-test (e.g., serum chemistry results) or an appropriate non-parametric analysis (where datasets did not pass normality test) using Graphpad Prism (version 7.0). Two-way ANOVA with Sidak’s multiple comparison posthoc test was used to compare frequency of fibers as a function of binned CSA/fiber size in controls versus NNMTi treated groups using Graphpad Prism (version 7.0). One-way ANOVA with Dunnett’s posthoc test was used to compare %MHC positive nuclei in NNMTi-treated myotubes versus control using Graphpad Prism (version 7.0). Absorbance data obtained at various timepoints for NAD+ and NADH extracted control and NNMTi-treated C2C12 cell samples were transferred to Graphpad Prism (version 7.0) to perform linear regression analysis and obtain slopes from the linear reaction progress curves. Slopes for NAD+ and NADH extracted samples and the ratio of NAD+/NADH slopes were compared using one-way ANOVA or Kruskal-Wallis test (for datasets not passing normality) followed by Dunnett’s posthoc to compare NNMTi treatments to controls. All statistical analyses were performed with an experiment-wise error rate of α = 0.05, for a 95% level of confidence.
3. RESULTS
3.1. NNMT expression increases in aged TA muscle.
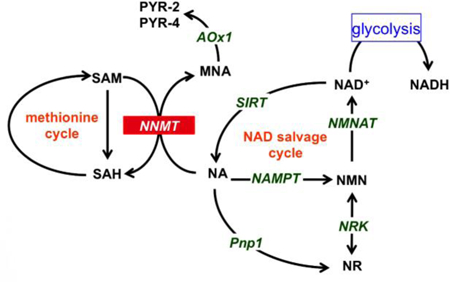
Immunolabeling of NNMT revealed NNMT-specific staining both in the TA muscle obtained from 24-month-old and 4-month-old mice (Fig. 1A, 1B). Expression of NNMT was qualitatively demonstrated to be relatively greater in aged muscle based on the immunohistochemical evaluations of the aged TA compared to young TA muscle tissues (Fig. 1A, 1B). To validate the qualitative findings using a semi-quantitative approach, NNMT immunoblots were established using aged and young mouse TA tissues (Fig. 1C). Consistent with the differential immunohistochemical NNMT results in aged versus young TA tissue, the expression level of NNMT protein in aged TA muscle was ~3-fold higher compared to the level of NNMT protein in young TA tissue (Fig. 1D; p < 0.05 vs. NNMT expression in young TA tissue). Taken together, these results support increased NNMT protein expression in the skeletal muscle as a function of age, suggesting likely enhanced NNMT activity in aged muscle tissue.
Figure 1.
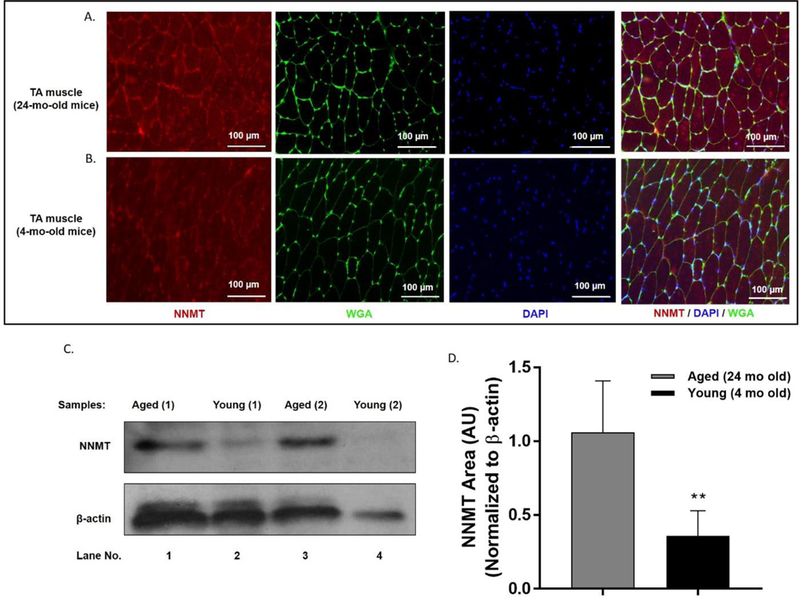
NNMT expression in aged (24 mo) and young (4 mo) TA muscle. Representative images of (A) aged (top panels) and (B) young (bottom panels) TA muscle tissues immunolabeled for detection of NNMT protein (red), myofiber membrane border (marked by wheat germ agglutinin [WGA] in green), and nuclei (marked by DAPI in blue). (C) Representative immunoblots obtained from replicate aged and young TA mouse tissue samples. (D) NNMT protein expression normalized to the loading control β-actin in aged and young TA muscle tissues as analyzed using western blotting. **, p < 0.01 vs. aged TA muscle NNMT expression.
3.2. In vivo NNMT inhibition activates muSCs and promotes aged muscle regeneration.
Fig. 2A and 2B present representative images that highlight muSC proliferation and fusion into damaged myofibers of aged TA muscle in control and NNMTi (10 mg/kg, bid)-treated mice. As indicated by white arrowheads (rightmost panels, Fig. 2A, 2B), NNMT inhibition increased the number of proliferating muSCs (measured by counting Pax7+/EdU+/DAPI+ cells per unit area); however, the total number of muSCs (i.e., Pax7+/DAPI+ cell counts per unit area) were unchanged in the control, low NNMTi dose, and high NNMTi dose groups, and averaged 209 ± 23, 213 ± 44, and 205 ± 9 muSC/mm2, respectively (Fig. 2A, 2B, representative panels showing Pax7+ positive cells indicated in green). Similarly, as indicated by yellow arrowheads (rightmost panel, Fig. 2A and 2B), NNMT inhibition promoted greater fusion of muSCs into damaged myofibers (measured by newly acquired myonuclei; Pax7−/EdU+/DAPI+ within laminin border), suggesting improved muscle regeneration following injury.
Figure 2.
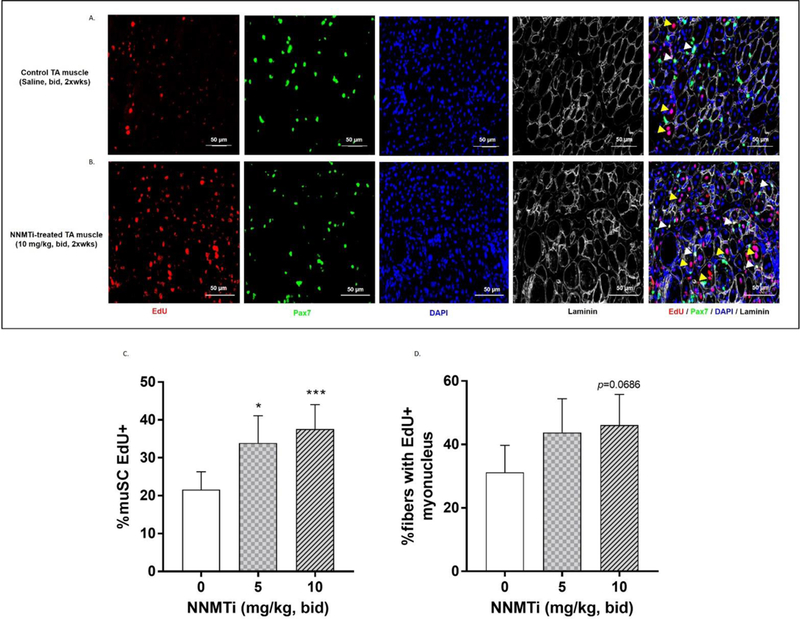
Effects of NNMTi on proliferation and fusion of muSCs into myofibers following injury of aged TA muscle. (A) Representative image of control aged and injured TA muscle tissue immunolabeled to detect proliferation and fusion of muSCs using EdU (red), Pax7 (green), DAPI (blue), and laminin (white/gray) to trace myofibers.. (B) Representative image of aged and injured TA muscle tissue following 1-week post-injury treatment with NNMTi immunolabeled as in panel (A). White arrowheads denote muSCs that have proliferated post-injury (EdU+/Pax7+/DAPI+). Yellow arrowheads indicate myonuclei fused into damaged myofibers post-injury (EdU+/Pax7−/DAPI+). (C) Relative number of activated muSCs compared to total population of muSCs (% muSC EdU+) in NNMTi-treated and control aged TA muscle tissues following injury (n=10–12 per group). Data represent adjusted mean % muSC EdU+ ± 95% CI). *, p < 0.05 and ***, p < 0.001 vs. control as determined by Tukey-adjusted posthoc comparisons. (D) Percentage of myofibers containing EdU+ myonucleus (% fibers with EdU+ myonucleus) in NNMTi-treated and control aged TA muscle tissues following injury (n=10–12 per group). Data represent adjusted mean % fibers with EdU+ myonucleus ± 95% CI. Consistent with % muSC EdU+ data (C), an increase in % fibers with EdU+ myonucleus trend was observed with NNMTi treatment relative to control, with the high dose of 10 mg/kg treatment showing a near statistically significant increase (p = 0.069) vs. control.
Consistent with these results, extensive quantification of numerous images revealed a dose-related increase in the relative abundance of proliferating muSCs (Fig. 2C) and fibers with integrated myonuclei via muSC fusion (Fig. 2D) following 1-week post-injury NNMTi treatment. The 5 mg/kg and 10 mg/kg doses of NNMTi tested resulted in 60% and 75% higher incidence of proliferating/active muSCs, respectively, relative to control (Tukey adjusted P values: p = 0.013, 5 mg/kg dose vs. control; p = 0.0007, 10 mg/kg vs. control). The odds ratio of active muSCs for control was 0.54 and 0.46 times the odds at the low and high doses, respectively. Although the higher treatment dose produced ~11% higher incidence of activated muSC compared to the lower treatment dose, this observed difference did not rise to the level of being statistically significant (p > 0.05, n.s.) (Fig. 2C). ICC for muSC abundance was 0.944 [95% CI (0.765,0.992)] and for proliferating muSCs 0.754 [95% CI (−0.43,0.963)] indicating excellent reliability of the data recorded by independent experimenters in a blinded fashion.
Similarly, dose-dependent trends were observed with NNMTi treatment in the incidence of newly acquired myonuclei via muSC fusion into damaged myofibers (Fig. 2D). The relative numbers of fibers with an EdU+ myonucleus increased 40% and 48% with NNMTi treatment at 5 mg/kg and 10 mg/kg, respectively, relative to control. The odds ratio of fused myonuclei for control were 0.58 and 0.53 times the odds at the low and high NNMTi dose, respectively, and the Tukey adjusted P values bordered on the cutoff generally considered statistically significant (p = 0.0686, 10 mg/kg dose vs. control). ICC for muSC fusion was 0.971 [95% CI (0.878,0.996)] indicating excellent reliability of data scored independently by blinded experimenters.
3.3. In vivo NNMT inhibition promotes muscle fiber growth after aged muscle injury.
Damaged TA muscle tissues were immunolabeled with laminin to trace fibers, and the cross-sectional area (CSA) of the fibers was analyzed and compared between control and NNMTi-treated tissues. Representative laminin labeled, muscle fiber-traced images obtained from control (left panel) and NNMTi 10 mg/kg dose (right panel) treatment groups are shown in Fig. 3A. One-week post-injury treatment with NNMTi at the high dose (10 mg/kg) produced a significant 1.8-fold increase in the mean CSA of damaged TA muscle fibers relative to control (Tukey adjusted P values: p = 0.0052, 10 mg/kg dose vs. control) (Fig. 3B). A similar (1.6-fold) dose-dependent increase was observed at the high dose of NNMTi treatment compared to the low dose tested (Tukey adjusted P values: p = 0.023, 10 mg/kg dose vs. 5 mg/kg), while the low dose of NNMTi treatment did not significantly alter the muscle fiber mean CSA compared to control (Tukey adjusted p > 0.05) (Fig. 3B). These results demonstrate that small molecule-mediated in vivo NNMT inhibition improves muscle regeneration and growth, consistent with the enhanced muSC activation and fusion observed following injury of the aged muscle (Fig. 2).
Figure 3.
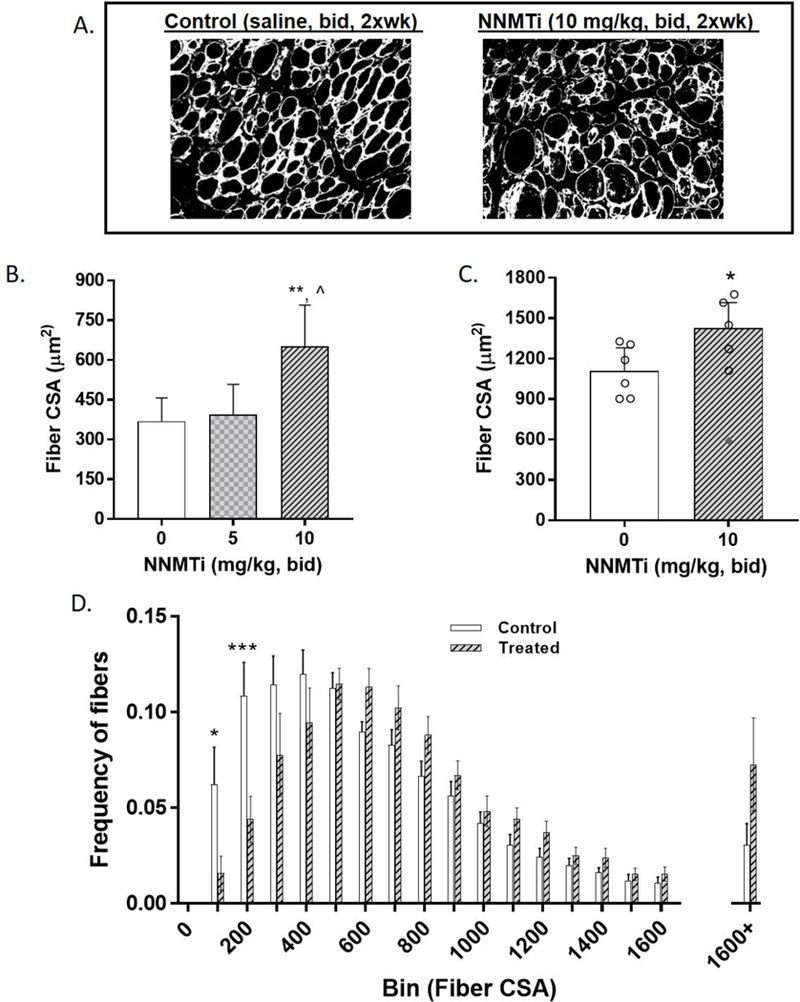
Effect of NNMTi on damaged TA muscle fiber cross-sectional area (CSA). (A) Representative images of aged and injured TA muscle tissue following 1-week post-injury in control (left panel) NNMTi-treated (right panel); immunolabeled with laminin (indicated in white) to quantify CSA. (B) Mean CSA of damaged TA muscle fibers analyzed in aged control and NNMTi-treated mice (n=10–12 per group). Data represent adjusted mean CSA (µm2) ± 95% CI. **, p < 0.01 vs. control; ^, p < 0.05 vs. 5 mg/kg. (C) Mean CSA of damaged TA muscle fibers analyzed in aged control and treated (10 mg/kg) mice following longer-term (3-week post-injury) treatment to assess more complete recovery (n=6 per group). Data represent adjusted mean CSA (µM2) ± 95% CI with individual datapoints represented as a scatter plot. *, p < 0.05 vs. control, with one datapoint (indicated in filled diamond) in the treated group eliminated from analysis on the basis of an unusually low (i.e., below threshold) CSA value for complete recovery analysis (p > 0.05, n.s. with this data point included). (D) Frequency of fibers binned as a function of CSA area in aged control and NNMTi (high dose, 10 mg/kg)-treated mice (n=12/group). Data represent mean frequency of fibers per bin ± SEM. *, p < 0.05 vs. control and ***, p < 0.001 vs. control.
The dramatic increase in muscle fiber size following a brief treatment with NNMTi supported the testing of high dose NNMTi in a separate cohort of aged mice to assess the impact of longer-term treatment on complete muscle recovery following injury. As shown in Fig. 3C, the mean CSA of damaged muscle fibers from aged mice was significantly (2-to 3-fold) larger at 3-weeks post-injury compared to 1-week post-injury (Fig. 3C vs. 3B), indicating progressive longitudinal recovery of damaged muscle in aged mice. Consistent with CSA results observed with 1-week post-injury treatment, three-week post-injury treatment with 10 mg/kg NNMTi produced a statistically significant and physiologically relevant 1.5-fold increase in the mean CSA of damaged TA muscle fibers relative to control (Tukey adjusted p = 0.039). As noted in the Methods section, a single aged mouse that showed unusually low CSA (included for completeness and indicated by filled diamond scatter point in Fig. 3C) due to unaccountable factors (i.e., lower CSA than control CSA in even the 1-week post-injury data) was excluded from analysis. Taken together, these results suggest that in vivo NNMT inhibition not only increases the rate of recovery by early activation of muSCs, but also increases the overall magnitude of recovery following injury to aged muscle (Tukey adjusted p > 0.05).
Further, binned analysis of the muscle fiber CSA size distribution demonstrated a significant interaction between fiber size and NNMTi treatment (F(17, 396) = 3.315; p < 0.0001) as analyzed by a two-way ANOVA (Fig. 3D). Particularly, a significant downward shift in the frequency of smaller sized myofibers (Fig. 3D; p = 0.03, NNMTi treatment vs. control in the frequency of fibers up to 100 µm2 CSA; p = 0.0002, NNMTi treatment vs. control for 200 µm2 CSA bin) and prominent rightward and upward shifts toward larger sized fibers (p = 0.07, NNMTi treatment vs. control in the frequency of fibers at sizes greater than 1600 µm2 CSA) were noted in the damaged myofibers of aged mice treated with the high dose NNMTi (Fig. 3D).
3.4. In vivo NNMT inhibition improves aged TA muscle contractile force.
In vivo dorsiflexor torque measurements were conducted 1-week post-injury on the damaged TA muscle of control and NNMTi treated aged mice to assess muscle contractile function properties at maximum applied force conditions. Peak torque output of the damaged TA muscle, when normalized to body weight of each animal, was 67% higher in NNMTi-treated group compared to control group. This increase was functionally and statistically significant (p = 0.033 vs. control; Fig. 4A), and consistent with the increased fiber size observed in the treated cohort. In contrast, peak torque output when normalized to the muscle fiber size (i.e., normalized to regenerating TA myofiber CSA and presented as torque per unit area, Fig. 4B) showed no difference between the treated and control groups (p > 0.05; Fig. 4B); this lack of difference was largely driven by the significantly larger muscle fiber mean CSA in the NNMTi-treated mice relative to controls. These data clearly provide support for greatly improved functional strength in aged muscle with in vivo NNMTi treatment, in addition to the improved muscle regeneration response observed following injury.
Figure 4.
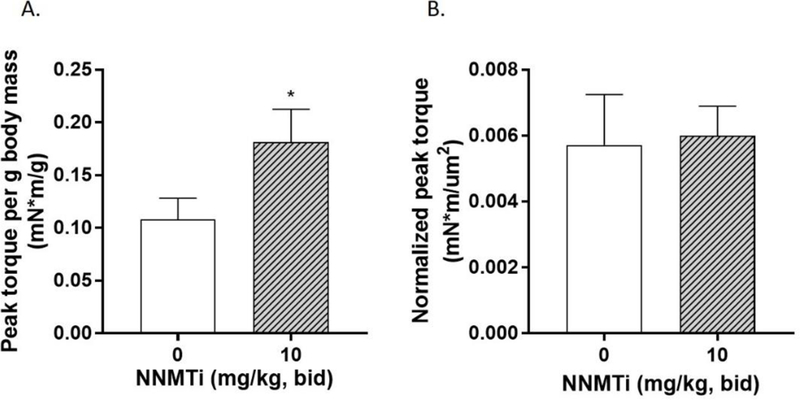
Effect of NNMTi (1-week treatment post-injury) on TA muscle contractile force in aged mice following TA muscle injury. (A) Maximum torque measured per g of body mass in vivo in response to maximum force delivered via minimal electric stimulation of the TA muscle in aged control and NNMT-treated (10 mg/kg, bid) mice (n=6–7 per group). Data represent mean peak torque per g of body mass (mN*m/g) ± SEM. *, p < 0.05 vs. control. (B) Maximum torque measured normalized to fiber CSA analyzed per animal in aged control and NNMT-treated mice (n=6–7 per group). Data represent mean normalized peak torque (mN*m/µM2) ± SEM.
3.5. Chronic administration of NNMTi produces no systemic toxicity.
As shown in Table 1, a complete plasma chemistry panel including major organ enzymes (liver, pancreas, gastrointestinal, renal, endocrine), total lipid, plasma proteins, and major electrolytes were profiled and compared between control and high dose (10 mg/kg) NNMTi-treated plasma samples. No significant differences were noted in the levels of the tested enzymes, glucose, cholesterol, plasma proteins, and electrolytes between control and NNMTi-treated samples (Table 1). Amylase levels were noted to be higher in all aged animals (regardless of control/treatment conditions) relative to young animals, likely indicative of general age-related dysfunctions (Table 1). Moreover, control and treated cohorts had similar body weight changes in all studies, suggesting similar feeding patterns (data not shown). Taken together, 1-week post-injury daily repeat-dosing of NNMTi was well tolerated with no untoward systemic toxicity or behavioral implications in aged mice.
Table 1.
Serum chemistry panel from control and NNMTi-treated (10 mg/kg) aged mice. p-value calculated using 2-tailed Student’s t-test with Welch’s correction (populations not assumed to have same standard deviations).
| Serum marker | Control | Treated | p-value* |
|---|---|---|---|
| Creatine kinase (U/L) | 648 ± 229 | 419 ±88 | 0.39 |
| AST(U/L) | 280 ± 84 | 248 ± 81 | 0.79 |
| ALT(U/L) | 142 ± 61 | 110 ± 44 | 0.69 |
| ALP (U/L) | 85 ± 15 | 75 ± 8 | 0.55 |
| GGT (U/L) | < 3 | < 3 | |
| GLDH (U/L) | 73 ± 46 | 103 ± 43 | 0.66 |
| Amylase(U/L)^ | 1365 ± 748 | 1344 ± 554 | 0.77 |
| Total bilirubin (mg/dL) | 0.18 ± 0.03 | 0.18 ± 0.03 | >0.99 |
| BUN (mg/dL) | 23 ± 2 | 43 ± 12 | 0.17 |
| Glucose (mg/dL) | 194 ± 23 | 173 ± 10 | 0.43 |
| Cholesterol (mg/dL) | 81 ± 3 | 94 ± 12 | 0.36 |
| Total protein (g/dL) | 4.4 ± 0.2 | 4.4 ± 0.1 | 0.91 |
| Albumin (g/dL) | 2.4 ± 0.063 | 2.4 ± 0.048 | 0.25 |
| Globulin (g/dL) | 1.9 ± 0.2 | 2.2 ± 0.075 | 0.29 |
| Calcium (mg/dL) | 8.7 ± 0.3 | 8.7 ± 0.2 | 0.94 |
| Phosphorous (mg/dL) | 6.7 ± 0.8 | 5.6 ± 0.5 | 0.29 |
| Sodium (mEq/L) | 147.5 ± 1.7 | 153.8 ± 2.8 | 0.10 |
| Potassium (mEq/L) | 5.6 ± 0.5 | 5.0 ± 0.2 | 0.30 |
| Chloride (mEq/L) | 114.5 ± 1.1 | 119.5 ± 2.4 | 0.10 |
Data represent mean ± SEM; n=4/group
, p > 0.05; no significant difference
, elevated levels in both cohorts suggesting age-related dysfunctions
3.6. NNMTi treatment promotes differentiation of C2C12 myoblasts and alters NAD+/NADH redox cofactors in differentiated C2C12 myotubes.
In vitro experiments were conducted to probe phenotypic and metabolic implications of NNMT inhibition in C2C12 cells that capture many characteristics of muSCs. Prior to determining the effects of NNMTi treatment in C2C12 cells, presence of NNMT target protein was qualitatively confirmed both in C2C12 myoblasts and differentiated myotubes using immunocytochemistry (Fig. 5A, 5B). NNMT expression was observed in C2C12 myoblast cells (Fig. 5A), differentiating myoblasts that were myogenic committed precursor cells (Fig. 5B, elongated, MHC-expressing cells indicated by yellow arrowheads), as well as in fully developed myotubes, which was confirmed by co-expression of NNMT with MHC (marker for myotubes/myofibers; Fig. 5B). Addition of NNMTi to confluent myoblasts under differentiation conditions produced a concentration-related increase in myoblast differentiation (Fig. 5C; F(2, 21) = 10.27, p = 0.0008 for NNMTi treatment factor). 30 µM NNMTi resulted in 18 ± 0.03% MHC-positive myotube nuclei, representing a 45% increase in the extent of myoblast differentiation (expressed as %MHC positive myotube nuclei) compared to untreated differentiating myoblasts that only produced 12 ± 0.4 % MHC-positive myotube nuclei (Fig. 5C; p = 0.0004 versus control).
Figure 5.
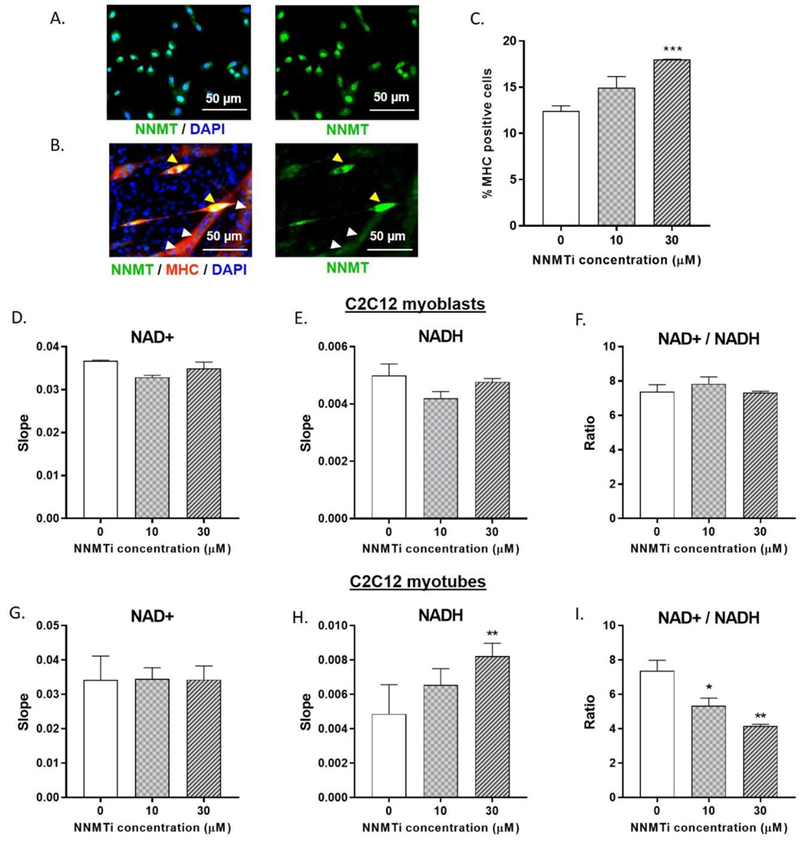
Effects of NNMTi on C2C12 myoblast differentiation and NAD+ salvage pathway metabolites in C2C12 myoblasts and myotubes. (A) Representative image of C2C12 myoblasts immunolabeled to highlight NNMT (green) and nuclei (blue). All myoblast cells appear to express NNMT. (B) Representative image of C2C12 differentiated myotubes immunolabeled to highlight NNMT (green), myosin heavy chain (MHC; red), and nuclei (blue). Yellow arrowheads represent NNMT expression in elongated, differentiation-committed myogenic precursor cells (co-labeled with MHC) and white arrowheads indicate NNMT expression in fully-differentiated myotubes (co-labeled with MHC marking myofibers). (C) Relative frequency of MHC positive cells in control and NNMTi -treated myoblasts at 96 h post-treatment in culture and under differentiation conditions. Data represent mean % MHC positive cells ± SEM measured in duplicates per condition. ***, p < 0.001 vs. control. (D) NAD+ levels, (E) NADH levels, and (F) NAD+/NADH ratio in control and NNMTi-treated (24 hr) C2C12 myoblasts. (G) NAD+levels, (H) NADH levels, and (I) NAD+/NADH ratio in control and NNMTi-treated (24 hr) differentiated C2C12 myotubes. Data represent mean slopes calculated from reaction progress curves (D, E, G, H) or ratios of slopes (F, I) obtained from spectrophotometric enzyme assays, where the slope is proportional to NAD+ and NADH concentrations (performed in duplicates across two experiments). *, p < 0.05 and **, p < 0.01 vs. control.
C2C12 myoblasts and differentiating myotubes were treated with NNMTi for 24 h to determine the effects of NNMTi treatment on NAD+ and NADH levels and the metabolic redox states. NNMTi treatment did not significantly alter NAD+ (Fig. 5D; H(2) = 4.57, p = 0.067) and NADH (Fig. 5E; H(2) = 3.71, p = 0.2) levels or the redox state (NAD+/NADH ratio; Fig. 5F) in C2C12 myoblasts. Similarly, NNMTi treatment produced no significant change in the NAD+ levels (Fig. 5G, F(2, 8) < 1.0, p > 0.05, n.s.) in differentiated C2C12 myotubes. However, NNMTi treatment produced concentration-dependent increases in NADH levels (Fig. 5H; F(2, 8) = 7.059, p = 0.0171), as indicated by 50% higher slopes generated from reaction progress curves for NADH extracted samples with 30 µM NNMTi treatment suggesting 50% higher NADH concentrations compared to control untreated myotubes (Fig. 5H; p = 0.0118 versus control). Given the observed NAD+ and NADH responses to NNMTi treatment, NAD+/NADH ratios decreased in a concentration-related manner with NNMTi treatment of myotubes (Fig. 5I; F(2, 8) = 11.65, p = 0.0043). The NAD+/NADH ratio was 25% lower with 10 µM NNMTi (Fig. 5I; p = 0.0443 versus control) and 40% lower with 30 µM NNMTi (Fig. 5I; p = 0.0024 versus control) compared to control.
4. DISCUSSION
To our knowledge, this is the first study to demonstrate that systemic treatment of aged animals with a small molecule drug-like NNMT inhibitor [38, 39] can rejuvenate muSCs, thereby robustly promoting muSC activation and fusion which are critical to support de novo myofiber regeneration and repair following injury [14–16]. At 1-week post-injury, control aged mice showed poor myofiber growth, minimal muSC activity, and a compromised skeletal muscle repair profile; these observations were consistent with impaired muSC regenerative capacity previously reported in skeletal muscles from aged mice [45, 46]. In contrast, aged mice treated with NNMTi demonstrated dramatically increased frequency of activated muSCs and greatly enhanced muscle repair via muSC fusion and subsequent myonuclear accrual. Both the number of activated muSC and the average size of regenerating myofibers on the injured limb had nearly doubled at 1-week post-injury with treatment facilitating the development of myofibers with much larger cross-sectional areas, similar to youthful myogenic growth response [45, 46]. Consistent with increased muscle fiber growth, NNMTi treatment nearly doubled peak dorsiflexor torque output on the injured limb, suggesting clinically meaningful overall improvements in muscle strength and function. Additionally, at 3-weeks post-injury, when the damaged muscles of the control animals had undergone additional recovery, prolonged NNMTi treatment resulted in further enhancement of myofiber growth compared to controls. These results suggest that prolonged NNMTi treatment enables a more complete recovery of the injured aged muscles, as average myofiber sizes were comparable between injured TA muscles of the treated aged mice and uninjured TA muscles of similar aged (24-mo-old) mice [18].
Consistent with our findings, Zhang and colleagues recently observed that 6–8-week treatment of 22–24-mo-old mice with 400 mg/kg/day of NR (an NAD+ precursor agent) accelerated muscle regeneration following cardiotoxin-induced injury to the TA muscle [29]. While Zhang et al. observed that NR treatment also increased the absolute number of muSCs pooled and analyzed from numerous (e.g., gastrocnemius, soleus, quadriceps, TA) aged muscle tissues [29], NNMTi treatment did not alter the pool of muSCs analyzed from the TA muscle of aged mice. This difference may be due to the shorter (2-week) treatment regimen used in the current study compared to the longer (6–8 weeks) treatment regimen employed with NR [29], or to the different skeletal muscle groups examined in the previous studies since it has been observed that only a few mouse muscle groups (e.g., gastrocnemius, soleus) experience age-related declines in muSC abundance [21].
The molecular mechanisms by which NNMT inhibition promotes muSC activation and myogenic response are currently unknown, but potentially associates with NNMT’s central role in regulating the NAD+ salvage pathway, which in turn affects intracellular NAD+ levels and SIRT1 (NAD-dependent deacetylase sirtuin 1) activity that modulate a cell’s metabolic and transcriptional responses [33, 47]. Since muSCs represent only about 3–5% of adult muscle fiber nuclei and a small proportion of the cells within the whole muscle tissue [29, 48], we performed additional experiments in cultured C2C12 myoblasts undergoing programmed differentiation, a system that recapitulates many of the features of muSC activation and thus can serve as a model to better understand how NNMTi treatment rejuvenates aged muSC and alters metabolic states of cells during differentiation [48, 49]. NNMTi treatment enhanced C2C12 myoblast differentiation and promoted myotube formation, selectivity increasing NADH levels and reducing NAD+/NADH ratio in differentiating myotubes, without impacting the metabolic states of myoblasts. These in vitro results suggest that NNMT inhibition enables muSCs to become increasingly activated and committed to muscle precursor cells with increased potential to differentiate, which is consistent with the increased differentiation and fusion of activated muSCs observed in NNMTi-treated aged mice following muscle injury.
muSC differentiation-dependent metabolic reprogramming has been previously demonstrated by Ryall and colleagues [48]. Specifically, activated and differentiation-committed C2C12 myoblasts were shown to switch to glycolytic metabolism with marked reductions in NAD+/NADH ratio (mediated by increased NADH levels and reduced NAD+ levels) and SIRT-1 histone deacetylase activity, and accompanied by myogenic stimulation and increased MyoD expression (muscle-specific transcription factor routinely used to denote activated muSCs). In contrast, oxidation-dependent metabolism marked by higher NAD+ levels and SIRT-1 activity predominate in C2C12 myoblasts that are more equivalent to quiescent muSCs [29, 48, 50]. Thus, our findings that NNMTi treatment selectively increased NADH levels and reduced NAD+/NADH ratio in differentiating myotubes, without impacting metabolic states in quiescent myoblasts are consistent with results from Ryall et al., favoring a switch towards glycolytic metabolism reprogramming (i.e., increased NAD+ substrate utilization in glycolysis for meeting muSC differentiation energy demands) that supports enhanced myoblast differentiation. Taken together, our working model regarding the molecular mechanisms whereby NNMT inhibition promotes muSC activation and increases myogenic response is illustrated schematically in Figure 6. As muscles age, NNMT expression and activity increases in the skeletal muscles, which impairs NAD+ flux through the salvage pathway and dysregulates SIRT-1 activity, thereby interfering with the capacity of muSC to maintain quiescence as well as the efficiency to proliferate, differentiate, and promote efficient muscle regeneration following injury [29, 48]. Given that NNMT appears to be uniformly expressed in skeletal muscles independent of fiber type distribution (i.e., type I, IIa, IIb) as previously demonstrated [34], suggests its dynamic involvement in the regulation of both oxidative and glycolytic metabolism. Future studies will investigate if similar metabolic changes are observed in muSCs isolated from NNMTi-treated skeletal muscle ex-vivo, and subsequently investigate the effects of NNMTi treatment on mitochondrial biogenesis and epigenetic modulations (both via SIRT-1 and SAM in the methionine pathways) in aged muscles.
Figure 6.
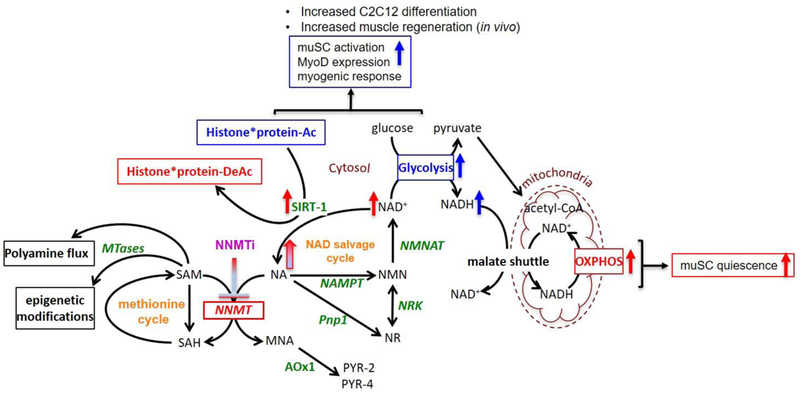
Schematic illustration of pathways regulated by NNMT and proposed working model of mechanisms of action of NNMT inhibition as it relates to muSC activation and metabolic state in aged muscle tissue. Maintenance of flux through the NAD salvage pathway by NNMTi is indicated by gradient blue/red arrow; (i) increase flux of NAD+, activated SIRT-1, and oxidative metabolism that support quiescent muSCs are indicated by red arrows; (ii) increased NADH (i.e., increased NAD+ utilization in glycolysis), enhanced glycolysis, and reduced SIRT-1 mediated histone deacetylation that support muSC activation and differentiation are indicated by blue arrows. Pathway abbreviations include AOx1 (aldehyde oxidase), NA (nicotinamide), MNA (1-methylnicotinmaide), NAMPT (nicotinamide phosphoribosyltransferase), NMN (nicotinamide mononucleoside), NMNAT (nicotinamide adenylyltransferase), NNMT (nicotinamide N-methyltransferase), NR (nicotinamide riboside), NRK (nicotinamide riboside kinase), PNP1 (purine nucleoside phosphorylase), PYR-2/4 (1-methyl-2/4-pyridone-5-carboxamide), SAH (S-adenosyl-L-homocysteine), SAM (S-adenosylmethionine), MTases (methyl transferases), SIRT1 (NAD-dependent deacetylase sirtuin 1), NAD+/NADH (oxidized Nicotinamide adenine dinucleotide/reduced form of NAD+), OXPHOS (oxidative phosphorylation), Histone*protein-Ac/DeAc (Acetylated/deacetylated histones).
In conclusion, the present findings convincingly demonstrate that NNMT inhibition provides a viable, safe, and non-invasive therapeutic approach to rejuvenate dysregulated muSCs and restore their regenerative capacity in aged skeletal muscles, as well as their intrinsic quiescent properties, to support dynamic muscle repair mechanisms after injury. Significantly, NNMT inhibition enhanced myofiber regeneration and growth following injury and dramatically improved the strength of the injured muscle, suggesting overall functional improvements to aged skeletal muscles. These results support NNMT inhibitors as novel therapeutics to restore a healthy myogenic response in injured aging skeletal muscle through rejuvenation of muSC activity, translating into improved muscle function and potentially mitigating the functional decline which sarcopenic older adults typically experience following muscular injury. Evaluations on the longitudinal effects of NNMTi treatment on muscle functional outputs (including measures of strength and endurance), safety/toxicology, and mechanistic implications of NNMT inhibition on aged muscle physiology will be continued to validate the clinical translational benefits of NNMTi therapeutics.
ACKNOWLEDGEMENTS
We thank our biostatistician expert Mr. Clark Anderson (The Office of Biostatistics, Dept. of Preventative Medicine and Community Health, University of Texas Medical Branch) for support on conducting mixed effect logistic regression analysis on the stem cell datasets using R statistical software, and discussions of the results and their interpretations. This work was supported by the Sanofi iAward grant (S.J.W., C.C.F), University of Texas Medical Branch Technology Commercialization Award (S.J.W, H.N), T32 training grant T32AG000270 (C.R.B.), NCATS CTSA TL1TR001440 NSRA Fellowship (T.G.G.), and NIH R01 AR072061 (C.S.F.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The Pax7 (#PAX7) antibody was developed by A. Kawakami and obtained from the Developmental Studies Hybridoma Bank, created by the NICHD of the NIH and maintained at The University of Iowa, Department of Biology, Iowa City, IA 52242.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
5. REFERENCES
- [1].N.C.f.H. Statistics, Health, United States, 2014: With special feature on adults aged 55–64, in: U.D.o.H.a.H. Services (Ed.) US Government Printing Office, Hyattsville, MD, 2015, pp. 1–473. [PubMed] [Google Scholar]
- [2].Abe T, Thiebaud RS, Loenneke JP, Age-related change in handgrip strength in men and women: is muscle quality a contributing factor?, Age (Dordr) 38(1) (2016) 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Goodpaster BH, Park SW, Harris TB, Kritchevsky SB, Nevitt M, Schwartz AV, Simonsick EM, Tylavsky FA, Visser M, Newman AB, The loss of skeletal muscle strength, mass, and quality in older adults: the health, aging and body composition study, J Gerontol A Biol Sci Med Sci 61(10) (2006) 1059–64. [DOI] [PubMed] [Google Scholar]
- [4].Cawthon PM, Fox KM, Gandra SR, Delmonico MJ, Chiou CF, Anthony MS, Sewall A, Goodpaster B, Satterfield S, Cummings SR, Harris TB, A. Health, S. Body Composition, Do muscle mass, muscle density, strength, and physical function similarly influence risk of hospitalization in older adults?, J Am Geriatr Soc 57(8) (2009) 1411–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Liguori I, Russo G, Aran L, Bulli G, Curcio F, Della-Morte D, Gargiulo G, Testa G, Cacciatore F, Bonaduce D, Abete P, Sarcopenia: assessment of disease burden and strategies to improve outcomes, Clin Interv Aging 13 (2018) 913–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Brown JC, Harhay MO, Harhay MN, Sarcopenia and mortality among a population-based sample of community-dwelling older adults, J Cachexia Sarcopenia Muscle 7(3) (2016) 290–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Shafiee G, Keshtkar A, Soltani A, Ahadi Z, Larijani B, Heshmat R, Prevalence of sarcopenia in the world: a systematic review and meta-analysis of general population studies, J Diabetes Metab Disord 16 (2017) 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Studenski SA, Peters KW, Alley DE, Cawthon PM, McLean RR, Harris TB, Ferrucci L, Guralnik JM, Fragala MS, Kenny AM, Kiel DP, Kritchevsky SB, Shardell MD, Dam T-TL, Vassileva MT, The FNIH sarcopenia project: Rationale, study description, conference recommendations, and final estimates, J Gerontol A Biol Sci Med Sci 69 (2014) 547–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Grounds MD, Age-associated changes in the response of skeletal muscle cells to exercise and regeneration, Ann N Y Acad Sci 854 (1998) 78–91. [DOI] [PubMed] [Google Scholar]
- [10].Brack AS, Conboy MJ, Roy S, Lee M, Kuo CJ, Keller C, Rando TA, Increased Wnt signaling during aging alters muscle stem cell fate and increases fibrosis, Science 317(5839) (2007) 807–10. [DOI] [PubMed] [Google Scholar]
- [11].Conboy IM, Rando TA, Aging, stem cells and tissue regeneration: lessons from muscle, Cell Cycle 4(3) (2005) 407–10. [DOI] [PubMed] [Google Scholar]
- [12].Burns ER, Stevens JA, Lee R, The direct costs of fatal and non-fatal falls among older adults - United States, J Safety Res 58 (2016) 99–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Newman AB, Kupelian V, Visser M, Simonsick EM, Goodpaster BH, Kritchevsky SB, Tylavsky FA, Rubin SM, Harris TB, Strength, but not muscle mass, is associated with mortality in the health, aging and body composition study cohort, J Gerontol A Biol Sci Med Sci 61(1) (2006) 72–7. [DOI] [PubMed] [Google Scholar]
- [14].Lepper C, Partridge TA, Fan CM, An absolute requirement for Pax7-positive satellite cells in acute injury-induced skeletal muscle regeneration, Development 138(17) (2011) 3639–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Murphy MM, Lawson JA, Mathew SJ, Hutcheson DA, Kardon G, Satellite cells, connective tissue fibroblasts and their interactions are crucial for muscle regeneration, Development 138(17) (2011) 3625–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Shadrach JL, Wagers AJ, Stem cells for skeletal muscle repair, Philos Trans R Soc Lond B Biol Sci 366(1575) (2011) 2297–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Cosgrove BD, Gilbert PM, Porpiglia E, Mourkioti F, Lee SP, Corbel SY, Llewellyn ME, Delp SL, Blau HM, Rejuvenation of the muscle stem cell population restores strength to injured aged muscles, Nat Med 20(3) (2014) 255–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Fry CS, Lee JD, Mula J, Kirby TJ, Jackson JR, Liu F, Yang L, Mendias CL, Dupont-Versteegden EE, McCarthy JJ, Peterson CA, Inducible depletion of satellite cells in adult, sedentary mice impairs muscle regenerative capacity without affecting sarcopenia, Nat Med 21(1) (2015) 76–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Sousa-Victor P, Gutarra S, Garcia-Prat L, Rodriguez-Ubreva J, Ortet L, Ruiz-Bonilla V, Jardi M, Ballestar E, Gonzalez S, Serrano AL, Perdiguero E, Munoz-Canoves P, Geriatric muscle stem cells switch reversible quiescence into senescence, Nature 506(7488) (2014) 316–21. [DOI] [PubMed] [Google Scholar]
- [20].Jang YC, Sinha M, Cerletti M, Dall’Osso C, Wagers AJ, Skeletal muscle stem cells: effects of aging and metabolism on muscle regenerative function, Cold Spring Harb Symp Quant Biol 76 (2011) 101–11. [DOI] [PubMed] [Google Scholar]
- [21].Keefe AC, Lawson JA, Flygare SD, Fox ZD, Colasanto MP, Mathew SJ, Yandell M, Kardon G, Muscle stem cells contribute to myofibres in sedentary adult mice, Nat Commun 6 (2015) 7087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Goody MF, Henry CA, A need for NAD+ in muscle development, homeostasis, and aging, Skelet Muscle 8(1) (2018) 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Canto C, Menzies KJ, Auwerx J, NAD(+) metabolism and the control of energy homeostasis: A balancing act between mitochondria and the nucleus, Cell Metab 22(1) (2015) 31–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Braidy N, Guillemin GJ, Mansour H, Chan-Ling T, Poljak A, Grant R, Age related changes in NAD+ metabolism oxidative stress and Sirt1 activity in wistar rats, PLoS One 6(4) (2011) e19194. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [25].Gomes AP, Price NL, Ling AJ, Moslehi JJ, Montgomery MK, Rajman L, White JP, Teodoro JS, Wrann CD, Hubbard BP, Mercken EM, Palmeira CM, de Cabo R, Rolo AP, Turner N, Bell EL, Sinclair DA, Declining NAD(+) induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging, Cell 155(7) (2013) 1624–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Imai S, Guarente L, NAD+ and sirtuins in aging and disease, Trends Cell Biol 24(8) (2014) 464–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Prolla TA, Denu JM, NAD+ deficiency in age-related mitochondrial dysfunction, Cell Metab 19(2) (2014) 178–80. [DOI] [PubMed] [Google Scholar]
- [28].Frederick DW, Loro E, Liu L, Davila A Jr., Chellappa K, Silverman IM, Quinn WJ, Gosai SJ, Tichy ED, Davis JG, e. al., Loss of NAD homeostasis leads to progressive and reversible degeneration of skeletal muscle, Cell Metab 24 (2016) 269–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Zhang H, Ryu D, Wu Y, Gariani K, Wang X, Luan P, D’Amico D, Ropelle ER, Lutolf MP, Aebersold R, Schoonjans K, Menzies KJ, Auwerx J, NAD(+) repletion improves mitochondrial and stem cell function and enhances life span in mice, Science 352(6292) (2016) 1436–43. [DOI] [PubMed] [Google Scholar]
- [30].Fang EF, Lautrup S, Hou Y, Demarest TG, Croteau DL, Mattson MP, Bohr VA, NAD(+) in aging: Molecular mechanisms and translational implications, Trends Mol Med 23(10) (2017) 899–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Mills KF, Yoshida S, Stein LR, Grozio A, Kubota S, Sasaki Y, Redpath P, Migaud ME, Apte RS, Uchida K, Yoshino J, Imai SI, Long-term administration of nicotinamide mononucleotide mitigates age-associated physiological decline in mice, Cell Metab 24(6) (2016) 795–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].van de Weijer T, Phielix E, Bilet L, Williams EG, Ropelle ER, Bierwagen A, Livingstone R, Nowotny P, Sparks LM, Paglialunga S, Szendroedi J, Havekes B, Moullan N, Pirinen E, Hwang JH, Schrauwen-Hinderling VB, Hesselink MK, Auwerx J, Roden M, Schrauwen P, Evidence for a direct effect of the NAD+ precursor acipimox on muscle mitochondrial function in humans, Diabetes 64(4) (2015) 1193–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Pissios P, Nicotinamide N-methyltransferase: More than a vitamin B3 clearance enzyme, Trends Endocrinol Metab 28(5) (2017) 340–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Kim HC, Mofarrahi M, Vassilakopoulos T, Maltais F, Sigala I, Debigare R, Bellenis I, Hussain SN, Expression and functional significance of nicotinamide N-methyl transferase in skeletal muscles of patients with chronic obstructive pulmonary disease, Am J Respir Crit Care Med 181(8) (2010) 797–805. [DOI] [PubMed] [Google Scholar]
- [35].Rudolphi B, Zapp B, Kraus NA, Ehebauer F, Kraus BJ, Kraus D, Body weight predicts nicotinamide N-methyltransferase activity in mouse fat, Endocr Res (2017) 1–9. [DOI] [PubMed]
- [36].Giresi PG, Stevenson EJ, Theilhaber J, Koncarevic A, Parkington J, Fielding RA, Kandarian SC, Identification of a molecular signature of sarcopenia, Physiol Genomics 21(2) (2005) 253–63. [DOI] [PubMed] [Google Scholar]
- [37].Kraus D, Yang Q, Kong D, Banks AS, Zhang L, Rodgers JT, Pirinen E, Pulinilkunnil TC, Gong F, Wang YC, Cen Y, Sauve AA, Asara JM, Peroni OD, Monia BP, Bhanot S, Alhonen L, Puigserver P, Kahn BB, Nicotinamide N-methyltransferase knockdown protects against diet-induced obesity, Nature 508(7495) (2014) 258–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Neelakantan H, Vance V, Wetzel M, Wang H-YL, McHardy SF, Finnerty C, Hommel JD, Watowich SJ, Selective and membrane permeable small molecule inhibitor of nicotinamide N-methyltransferase reverses diet-induced obesity in mice, Biochemical Pharmacology 147 (2018) 141–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Neelakantan H, Wang HY, Vance V, Hommel JD, McHardy SF, Watowich SJ, Structure-activity relationship for small molecule inhibitors of nicotinamide N-methyltransferase, J Med Chem 60(12) (2017) 5015–5028. [DOI] [PubMed] [Google Scholar]
- [40].Iyer SR, Valencia AP, Hernandez-Ochoa EO, Lovering RM, In vivo assessment of muscle contractility in animal studies, Methods Mol Biol 1460 (2016) 293–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Call JA, Eckhoff MD, Baltgalvis KA, Warren GL, Lowe DA, Adaptive strength gains in dystrophic muscle exposed to repeated bouts of eccentric contraction, J Appl Physiol (1985) 111(6) (2011) 1768–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Erratum to “A Guideline of Selecting and Reporting Intraclass Correlation Coefficients for Reliability Research” [J Chiropr Med 2016;15(2):155–163], J Chiropr Med 16(4) (2017) 346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Koo TK, Li MY, A Guideline of Selecting and Reporting Intraclass Correlation Coefficients for Reliability Research, J Chiropr Med 15(2) (2016) 155–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Trammell SA, Brenner C, Targeted, LCMS-based metabolomics for quantitative measurement of NAD(+) metabolites, Comput Struct Biotechnol J 4 (2013) e201301012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Choi JY, Hwang CY, Lee B, Lee SM, Bahn YJ, Lee KP, Kang M, Kim YS, Woo SH, Lim JY, Kim E, Kwon KS, Age-associated repression of type 1 inositol 1, 4, 5-triphosphate receptor impairs muscle regeneration, Aging (Albany NY) 8(9) (2016) 2062–2080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Cousin W, Ho ML, Desai R, Tham A, Chen RY, Kung S, Elabd C, Conboy IM, Regenerative capacity of old muscle stem cells declines without significant accumulation of DNA damage, PLoS One 8(5) (2013) e63528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Price NL, Gomes AP, Ling AJ, Duarte FV, Martin-Montalvo A, North BJ, Agarwal B, Ye L, Ramadori G, Teodoro JS, Hubbard BP, Varela AT, Davis JG, Varamini B, Hafner A, Moaddel R, Rolo AP, Coppari R, Palmeira CM, de Cabo R, Baur JA, Sinclair DA, SIRT1 is required for AMPK activation and the beneficial effects of resveratrol on mitochondrial function, Cell Metab 15(5) (2012) 675–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Ryall JG, Dell’Orso S, Derfoul A, Juan A, Zare H, Feng X, Clermont D, Koulnis M, Gutierrez-Cruz G, Fulco M, Sartorelli V, The NAD(+)-dependent SIRT1 deacetylase translates a metabolic switch into regulatory epigenetics in skeletal muscle stem cells, Cell Stem Cell 16(2) (2015) 171–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Ryall JG, Metabolic reprogramming as a novel regulator of skeletal muscle development and regeneration, FEBS J 280(17) (2013) 4004–13. [DOI] [PubMed] [Google Scholar]
- [50].Repele A, Lupi R, Eaton S, Urbani L, Coppi PD, Campanella M, Cell metabolism sets the differences between subpopulations of satellite cells (SCs), BMC Cell Biology 14 (2013) 24–31. [DOI] [PMC free article] [PubMed] [Google Scholar]