

Abstract
Dozens of type A malyngamides, principally identified by a decorated six-membered cyclohexanone head group and methoxylated lyngbic acid tail, have been isolated over several decades. Their environmental sources include macro-and microbiotic organisms, including sea hares, red alga, and cyanobacterial assemblages but their true producing organism has remained enigmatic. Many type A analogs display potent bioactivity in human-health related assays, spurring an interest in this molecular class and its biosynthetic pathway. Here we present the discovery of the type A malyngamide biosynthetic pathway in the first sequenced genome of the cyanobacterial genus Okeania. Bioinformatic analysis of two cultured Okeania genome assemblies identified 62 and 68 kb polyketide synthase/non-ribosomal peptide synthetase (PKS/NRPS) pathways with unusual loading and termination genes. NMR data of malyngamide C acetate derived from 13C-substrate-fed cultures provided evidence that an intact octanoate moiety is transferred to the first KS module via a LipM homolog originally associated with lipoic acid metabolism and implicated an inactive ketoreductase (KR0) as critical for six-membered ring formation, a hallmark of the malyngamide family. Phylogenetic analysis and homology modeling of the penultimate KR0 domain inferred structural cofactor-binding and active site alterations as contributory to domain dysfunction, which was confirmed by recombinant protein expression and NADPH binding assay. The carbonyl retained from this KR0 ultimately enables an intramolecular Knoevenagel condensation to form the characteristic cyclohexanone ring. Understanding this critical step allows assignment of a biosynthetic model for all type A malyngamides, whereby well-characterized tailoring modifications explain the surprising proliferation and diversity of analogs.
INTRODUCTION
Marine microorganisms have been extraordinarily rich sources of bioactive natural products, and several have been approved as drug treatments.1 A modified synthetic derivative of dolastatin 10, originally isolated from the sea hare Dolabella auricularia but whose true source is likely cyanobacteria, has been approved to treat lymphoma.2 A family of frequently isolated marine bioactive molecules, the type A malyngamides, present an array of bioactive properties including anticancer and anti-inflammatory activity.3,4 The first type A malyngamides were isolated and elucidated in 1978, and in the intervening decades, 28 analogs have been characterized.5 Type A and B malyngamides differ in the head group structure; type A mostly features a modified six-membered ring with a ketone functionality, while type B analogs contain a pyrrolidone head group (Note S1).6 Related, hermitamides and serinol-derived analogs contain a lyngbic acid tail with a modified amino acid head group.7,8 Type A malyngamides differ from one another principally in the tailoring of the head group, with methylation and oxidations found disparately at nearly all carbon positions about the cyclohexanone ring, and include additional features often deriving from epoxidation or acetylation. Predicted amino acid linkage (Gly, Ser, Thr, or β-Ala), and lyngbic acid chain length (12–16 carbon) can vary as well.5,9–14 However, the majority of the malyngamides possess a 14-carbon lyngbic acid tail and glycine linkage as those in this study. Thus, the core structure of these malyngamide analogs is remarkably conserved across biogeographically diverse locales and sources.
The attributed environmental sources of the malyngamides include red algae,15 invertebrates,12 and most commonly, assemblages of filamentous cyanobacteria of the genus Moorea (previously Lyngbya), isolated in pantropical distribution.16,17 While a range of biological activities have been reported for the 28 analogs, no single target has been identified, though the epoxyketone and enone moieties present in most analogs are a well-studied pharmacophore for proteasome inhibition.18
Previous research suggested that non-axenic cultures of Okeania hirsuta PAB10Feb10–1 (“PAB”) and PAP21Jun06–1 (“PAP”) were producers of this natural product class, however, a detailed analytical characterization and genetic context for their production was not included in those studies (Note S2).19–21 Here we establish PAP as the producer of malyngamide I, and PAB as the producer of malyngamide C and C acetate, and identify and characterize their complete polyketide synthase/non-ribosomal peptide synthetase (PKS/NRPS) biosynthetic pathways from their genomic sequences (Figure 1).
Figure 1.
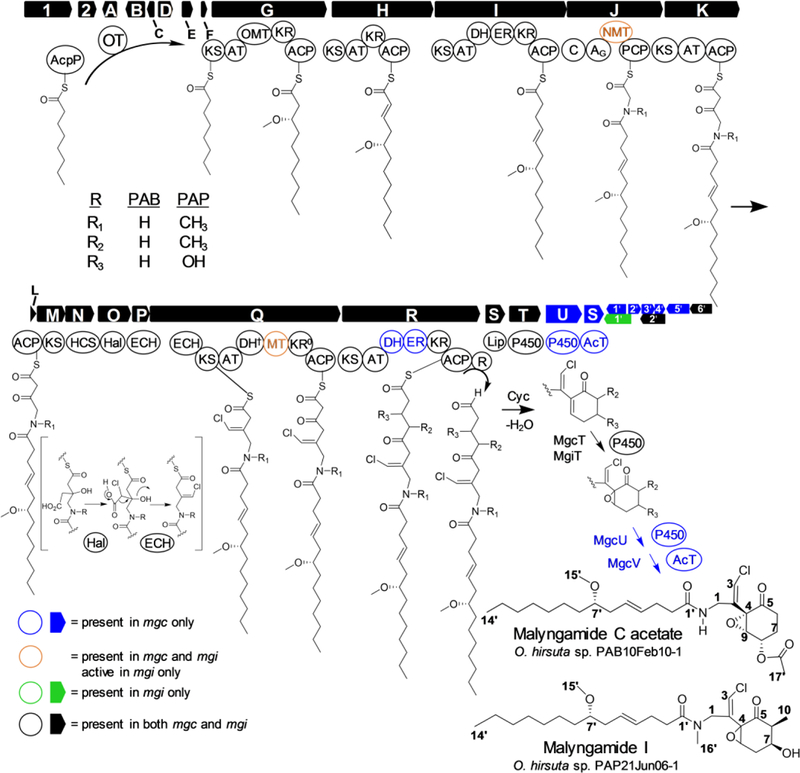
Combined malyngamide C acetate (mgc) and malyngamide I (mgi) biosynthesis pathways from Okeania hirsuta PAB and PAP. Abbreviations: acyl carrier protein (ACP), AcpP (fatty acid synthase acyl carrier protein), acetyltransferase (AcT), adenylation (A), acyltransferase (AT), condensation (C), C-methyltransferase (MT), dehydratase (DH), enoyl-CoA hydratase (ECH), enoyl reductase (ER), halogenase (Hal), hydroxymethylglutaryl-CoA synthase (HCS), ketosynthase (KS), ketoreductase (KR), lipocalin-like (Lip), N-methyltransferase (NMT), O-methyltransferase (OMT), octanoyltransferase (OT), cytochrome P450 (P450), peptidyl carrier protein (PCP), thioester reductase (R). Blue color indicates genes or domains found only in PAB. Orange indicates domains that are structurally present in both PAB and PAP, but active in PAP only. Green genes are present in PAP only. Black colored genes are present in both pathways. 0 – inactive domain. † – structurally present in PAP, catalytic residue absent in PAB, no activity predicted due to KR0.
Typically, cyclic cyanobacterial lipopeptides are loaded by either fatty acid CoA-ligases,22 amino acid or acyl-AMP synthetases,23 GCN5-acetyltransferase (GNAT) domains,24 or pseudo-GNAT domains.25 However, both malyngamide pathways are predicted to initiate biosynthesis using MgcA/MgiA, a LipM-like octanoyltransferase (Note S3).26 As filamentous marine cyanobacteria have not yet proven to be genetically tractable, we sought to examine malyngamide biosynthesis via several alternative methods, including genomic analysis of the genus Okeania, stable-isotope labeled feeding of malyngamide subunits, pharmacological knockouts, bioinformatics and phylogeny of the MgcA octanoyltransferase, and homology modeling and biochemical analysis of the MgcQ inactive ketoreductase (KR0) domain.
Ultimately, JCC calculated between 13C-enriched cyclohexanone carbons clarified the biosynthesis of the head group in both pathways, which is not eminently predicted by the domain order within the PKS/NRPS modules.We deduced that a thioester reductase (R) domain in the final PKS module generates a terminal aldehyde, and its electrophilicity allows a Knoevenagel condensation by nucleophilic attack from the alpha carbon of the MgcK-installed acetate unit. Subsequent P450 activity and acetyltransfer is proposed to generate the final characteristic head groups in each molecule. We present a compelling model for the biosynthesis of type A malyngamides wherein the biosynthetic keystone is the KR0-enabled Knoevenagel condensation. With this new insight, well understood KS, NRPS, and tailoring enzymatic reactions easily explain all of the type A malyngamide analogs. Implications for PKS/NRPS biosynthesis strategies are discussed, as this pathway demonstrates a unique example of module dysfunction leading to a striking radiation of chemical diversity.
RESULTS AND DISCUSSION
Structure confirmation.
Chemical extracts of Okeania hirsuta strains PAB and PAP were analyzed by HPLC-ESI-MS/MS. Peaks with [M+H]+ 456.2 and 498.2 were identified in PAB, and one major peak with [M+H]+ 484.2 was found in PAP. MS/MS fragmentation patterns of [M+H]+ 456.2 and of [M+H]+ 484.2 matched the MS/MS fragmentation patterns of pure compound standards of malyngamide C and malyngamide I (Figure S1a, S1b), respectively, while the MS/MS pattern of [M+H]+ 498.2 suggested an acetylated analog of malyngamide C (Figure S1c, Note S4, Note S5). Putative malyngamide C acetate HR-ESI-MS was [M+H]+ = 498.2630, corresponding to molecular formula of C26H41ClNO6, and optical rotation (OR) –34.6 (c 0.1,CH3OH). Putative malyngamide I HR-ESI-MS was [M+H]+ = 484.2840, corresponding to formula of C26H43ClNO5, and OR + 40.0 (c 0.1, CH OH). HR-ESI-MS values and OR for each accord with previously obtained values.16,17 The structure of malyngamide C acetate was further confirmed by 1D 1H NMR and 13C NMR (Table S1), and malyngamide I structure was confirmed by 1H NMR and 13C NMR, as well as COSY, HSQC, and HMBC (Table S2).
Genomic information and pathway analysis.
PAB and PAP genome assembly information is summarized in Table S3. A phylogenomic tree generated from conserved genes in diverse cyanobacteria indicates Okeania is closely related to Trichodesmium and has considerable evolutionary distance from planktonic Synechococcus and Prochlorococcus (Figure S2). Unlike Moorea, the Okeania genome contains the nitrogen-fixing nif genes, and both Okeania were experimentally determined to fix nitrogen under N-starvation using the acetylene reduction assay (Figure S3). Two closely related PKS/NRPS pathways were detected in PAB and PAP via antiSMASH27 and DELTA-BLAST,28 and were predicted to encode for malyngamide C acetate (“mgc” cluster, 68 kb in length) and malyngamide I (“mgi” cluster, 62 kb in length), respectively. The predicted open reading frames and proteins for the biosynthesis of these pathways are discussed below (Figure 1, Table S4a and Table S4b). PCR was used to confirm correct pathway assembly and close short gaps (Table S5).
A LipM-like octanoyltransferase, MgcA, is predicted to initiate each pathway by transferring an octanoyl bound to fatty acid synthase (FAS) acyl carrier protein (AcpP, generally ACP) to the MgcG KS active site cysteine. MgcA is slightly promiscuous toward decanoyl-ACP, as trace amounts of compounds with +26 AMU, [M+H]+ 526 in PAB and [M+H] + 512 in PAP, respectively, were observed at a ratio of approximately 20:1 via MS (Figure S4). This substrate flexibility is not entirely surprising, as malyngamides D and E feature homologs of lyngbic acid that are extended by two saturated carbon atoms.5 Downstream of mgcA, the genes mgcB and mgcC appear to encode for transposases, possibly reflective of the combinatorial diversity of PKS/NRPS pathway loading genes. MgcD is an sfp-type phosphopantetheinylase that activates ACPs involved in PKS pathways with phosphopantetheine.29 MgcE is another transposase, and MgcF is a small membrane-bound protein with homology to a short noncatalytic portion of JamB in the jamaicamide pathway. MgcG is the first PKS module, whose KS active site is initially octanoyl-bound by action of MgcA. This octanoyl moiety undergoes acetate extension by the MgcG AT (acyltransferase)catalyzed condensation with malonyl-CoA. The resulting beta-carbonyl functionality is first reduced to a hydroxyl group by the MgcG KR, and then methylated by an O-methyltransferase (O-MT). MgcH adds an acetate unit and reduces the prior carbonyl to a trans double bond via a KR followed by dehydration; the latter transformation is likely performed by the downstream MgcI dehydratase (DH) domain, as observed in other PKS/NRPS pathways.24,30,31 MgcI lengthens the bound molecule by an acetate unit and fully reduces the beta-carbonyl by activity of a KR, DH, and enoylreductase (ER), thus completing the synthesis of ACP-bound lyngbic acid.
MgcJ is an NRPS module with an adenylation (A) domain that activates and then installs a glycine residue, and in the PAP pathway also contains an N-methyltransferase (N-MT) domain that methylates the newly introduced amino acid using S-adenosyl methionine (SAM). In the PAB pathway, the N-MT is structurally present but inactive. Subsequently, MgcK installs another intact acetate unit with no reduction of the preceding beta-carbonyl. MgcL through MgcP install a chlorinated exo-cyclic methylene group via an HMG-CoA synthase-like mechanism. 32,33 Another malonyl-CoA is condensed to an acetate by MgcQ, but unexpectedly, the MgcK-installed acetate carbonyl at C-5 is not reduced by the MgcQ KR. Therefore, the KR is an inactive KR0, discussed subsequently. The C-methyltransferase (C-MT) within MgcQ/MgiQ is active in the mgi pathway (in PAP), but not in the mgc pathway, (in PAB) which is missing a required active site His (Figure S5a). Lastly, the PAB MgcQ DH lacks a catalytic site His and thus is not predicted to be active; while the same domain in PAP does contain the active site His, this is a null point due to the pathway’s inactive KR0 (Figure 1, Figure S5b).
The final KS module, MgcR and MgiR, are structurally divergent in the two organisms; in the PAB pathway, the module reduces the MgcQ-installed carbonyl with a full reductive cassette (KR, ER, DH) whereas the PAP pathway contains solely a KR, resulting in a hydroxyl group at C-7. Crucially, in both strains, MgcR terminates with an R domain and is predicted to generate a terminal aldehyde via a 2e− transfer, thereby generating a suitable electrophile for cyclization, though there is some ambiguity as to which gene is specifically responsible for the mechanism, as discussed below in feeding studies and JCC analysis section (Figure 1, Figure 2a). MgcT, a P450 with significant homology between pathways, is then predicted to catalyze epoxidation of the newly generated double bond. Here, biosynthesis halts in PAP, producing malyngamide I. In PAB, MgcU, an additional P450 is predicted to oxidize C-8 to an alcohol, followed by acetylation with O-acetyltransferase MgcV, thereby producing malyngamide C acetate. Orf 1 and 2 are enzymes of unknown function found just upstream of MgcA, and are homologous to those in the mgi pathway. Several ORFs of unknown function are found downstream of each pathway, flanked downstream by a kinase regulator common to both pathways.
Figure 2.
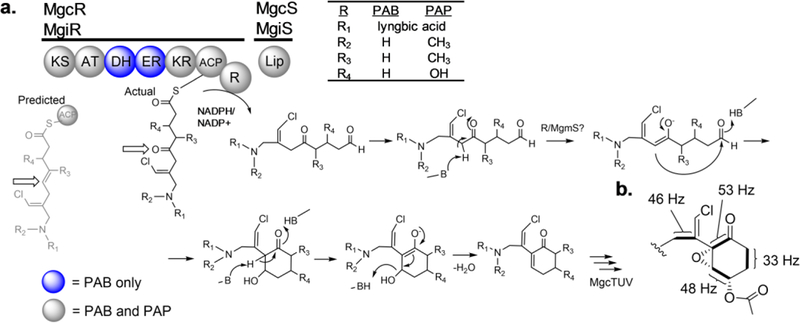
Cyclization scheme. a) Illustration of predicted cyclization mechanism of malyngamide C acetate, catalyzed by MgcR and MgcS. Blue colored domains are found only in PAB and not PAP. Active site acid-base chemistry deprotonates the carbon adjacent to the C-5 carbonyl, forming an oxyanion enoyl intermediate, which then collapses to nucleophilically attack the C-9 aldehyde, forming a transient alcohol at C-9. A second deprotonation at C-4 forms a second enolate intermediate, which collapses to lose hydroxide and forms the cyclohexenone core structure of malyngamide K. P450 and acetylation tailoring follows to form the final head group depicted in b) which indicates JCC values derived from malyngamide C acetate extracted and purified from [1,2-13C2]acetate-fed and [1,2-13C2]glycine-fed PAB cultures.
13C-octanoate feeding and MgcA phylogeny.
LipM-like octanoyltransferase functionality has not been observed before in the context of initiating a PKS or hybrid biosynthetic pathway. MgcA was examined by feeding of [1-13C]octanoate to O. hirusta PAB over a growth period of two weeks, followed by purification and 13C-NMR comparison of resonance integrals to unlabeled malyngamide C acetate. A threefold enrichment of the −8 (C-7’) resonance was observed compared to unlabeled malyngamide C acetate, indicating direct utilization of the intact octanoate moiety into the pathway (Figure 3a, Table S6). The modest enrichment of C-1’ (~50%) [1-13C]octanoate supplementation may results from beta-oxidation of C-1 – C-2 of the labeled octanoate into [1-13C]acetate building blocks.
Figure 3.
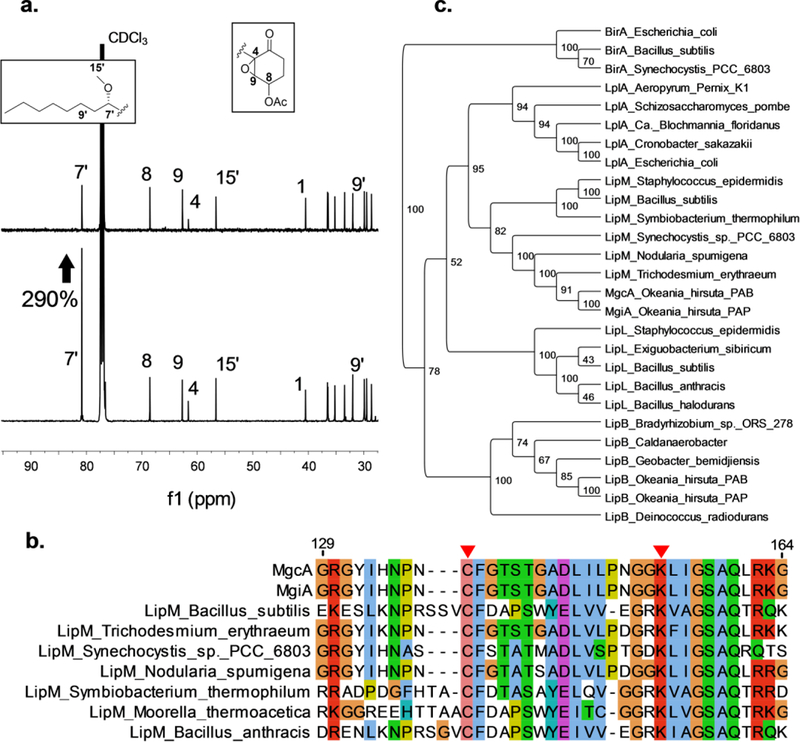
[1-13C]octanoate fed batch enrichment and LipM phylogeny. a) Comparison of 13C-NMR C-7', C-8, C-9, C-4, and C-15' resonances from malyngamide C acetate isolated from native abundance sample (top) and sample fed with [1-13C]octanoate (bottom). Percentage increase in intensity of C-7' signal depicted next to C-7' signal. b) alignment of LipM octanoyltransferases with MgcA/MgiA. Active site cysteine and lysine are indicated with red triangles. c) Phylogenetic tree of select PFAM03099 protein sequences. Topology threshold percentages depicted at node junctions.
MgcA and MgiA were analyzed by phylogenetic comparison within PFAM03099, the biotin/lipoate A/B protein ligase family. Key active site cysteine and lysine residues are conserved between MgcA, MgiA, B. subtilis LipM octanoyltransferase, and other putative cyanobacterial LipM analogs (Figure 3b). A phylogenetic tree was created based on the core structure of PFAM03099, and MgcA and MgiA clade with other putative LipM enzymes with a consensus support value of 95% vs. LipL, the nearest PFAM03099 clade (Figure 3c). LipM clearly falls outside the alternate family clades of BirA, LipB, Lpl, and LplA. In B. subtilis, LipM acts as an intermediate, transferring octanoate between octanoyl-FAS ACP and a lysine active site on the E2 lipoyl domain (LD) of 2-oxoacid dehydrogenase complexes.26 However, no such LD is found between mgc pathway Orf1 and MgcG. Therefore, it is unclear specifically how MgcA transfers the C8 moiety from octanoyl-ACP to the MgcG KS active site. However, based on results of the 13C-labeled acetate and octanoate feeding experiments, phylogenetic analysis, and close proximity to MgcG PKS machinery, we assign the activity of MgcA and MgiA as responsible for initiating the malyngamide pathways. Interestingly, both O. hirsuta strains also contain LipB; while this is a functionally homologous lipoic acid synthesis enzyme, it differs in structure and sequence homology to LipM.50 Indeed, many cyanobacteria contain both LipB and LipM homologs, including closely related Trichodesmium erythraeum, where a homologous LipM is not found adjacent to any biosynthesis pathway. In Okeania, we speculate that LipB satisfies the essential requirement for octanoyl transfer in lipoic acid synthesis, thus allowing the reprogramming of LipM to initiate a hybrid PKS/NRPS pathway; to our knowledge, this neofunctionalization of LipM is without precedent and represents a unique involvement of a member of this enzyme family to contribute to secondary metabolism.
Ancymidol pharmacological knockout.
A P450 is theorized to epoxidize C-4 – C-9 in malyngamide I and malyngamide C acetate, and an additional P450 in PAB is predicted to hydroxylate C8. To evaluate this possibility, we provided cultures of Okeania hirsuta PAB with ancymidol, a P450 inhibitor, for a period of two weeks.34 LC/MS results indicated a relative decrease in malyngamide C acetate production and a concomitant relative increase in the production of shunt product [M+Na]+ peaks (Figure 4). To confirm that shunt products were indeed malyngamides, MS2 data was collected for each shunt product and compared to fragments derived from known masses malyngamide C and malyngamide C acetate. Differences in product ion masses by the same molecular weight change as the parent ion, an MS1 elevated [M+2] peak corresponding to chlorination, as well as MS2-based networking confirmed that the shunt products were malyngamides (Figure S6). A species corresponding to the mass of didehydro-deoxy-malyngamide C, m/z 474.2, showed a relative increase from near nonexistent to approximately 10% of total malyngamide pool, while didehydro-N-methyl-malyngamide C, m/z 490.2, increased from near baseline to approximately 5% of total malygamide pool. The relative increase of these species when treated with a P450 inhibitor is intriguing; we speculate that this may occur as a result of kinetic retardation of pathway throughput in which intermediate species are held longer in each KS/NRPS turnstile.35 This would enable domains with a structurally inefficient N-MT (the C-MT is catalytically inert) to have greater access to the MgcJ-bound intermediate, generating more of the N-methylated species.
Figure 4.

Ancymidol pharmacological knockout. Bar segments are labeled with the respective percentages of the total pool of each chemical species with standard deviation.
13C-labeled acetate, glycine feeding studies.
To explore KS loading, biosynthesis of the head group, and confirm construction of the skeletal core as predicted by informatics, [1,2-13C2]acetate and [1,2-13C2]glycine were provided to live cultures of O. hirsuta PAB. Incorporations of labeled acetate and glycine units were determined by the appearance of flanking shifts in the 13C NMR, indicating J-coupling between intact pairs of 13C-enriched atoms (Figure 2b, Table S7). [1,2-13C2]glycine supplementation resulted in the emergence of flanking peaks only between C-1 and C-2, as expected. By contrast, [1,2-13C2]acetate supplementation showed flanking peaks for all intact acetate-derived carbon atoms, with the exception of C-3 which is derived from C-2 of acetate via the MgcN HCS-mediated reaction. As expected, the appearance in malyngamide C acetate of flanking 13C NMR shifts at positions C-14’ – C-7’ from [1,2-13C2]acetate provision indicates that acetyl-CoA subunits are extended via FAS machinery until octanoate is preferentially transferred by MgcA to the first KS module of the pathway. Differing levels of [1,2-13C2]acetate incorporation support the conclusion that different biosynthetic processes are responsible for C-7’ to C-14’ versus the other acetate-deriving carbon atoms in the molecule. The coupled signals for C-4 – C-9, C-1’, C-3’ – C-5’, and C-16’ – C-17’ averaged 15.1% ± 3.0% of the unenriched signal intensity whereas those for C-7’ – C-14’ averaged 20.8% ± 2.2 % of the unenriched signal (P-value = 0.001 by one-way ANOVA) (Figure S7). The intensity of some flanking signals could not be accurately measured due to overlap with neighboring peaks and these were not included in the analysis. The results suggest higher enzyme processivity and turnover associated with the biosynthesis of the −1 to −8 section of the lyngbic acid tail versus the remainder of the malyngamide chain. This is possibly due to different processivity and turnover rates in standalone type II FAS systems versus type I PKS megasynthase modules, consistent with the hypothesis that MgcA directs a FAS-derived octanoyl-ACP to MgcG.
An analysis of JCC coupling values between adjacent carbons was pivotal to understanding the biosynthesis of the cyclohexanone head group. Intact acetate incorporations were identified for C-4 – C-5, C-6 – C-7, and C-8 – C-9, installed by MgcK, MgcQ, and MgcR by collinearity (Table S7). These results were not in accordance with initial predictions for formation of the cyclohexanone ring. Bioinformatic analysis of the gene cluster indicated that C-5 – C-6 should be reduced and converted to an alkene by a theoretically functional KR/DH domain in MgiQ (PAP strain), whereas C-5 should be reduced to a hydroxy group by a theoretically functional KR/DH0 (PAB strain). However, these predicted products did not provide for a reasonable mechanism for carbocyclization that involves the alpha carbon (C-4) of the MgcK-derived acetate unit. One possibility considered was that perhaps the MgcQ KR was a vestigial, inactive KR0, in which case the C-5 carbonyl would remain intact. In fact, this appears to be the case, as is detailed in the subsequent section (Figure 2).
The coupling of C-8 – C-9 infers that an intact acetate unit is loaded by MgcR, followed by chain release due to the R domain subsequent to the MgcR ACP. Some R domains such as CpaS are redox incompetent, and solely catalyze Dieckmann cyclization, a non-redox intramolecular Claisen condensation.36 In this latter case, these R domains substitute an L for Y in the YXXXK short-chain dehydrogenase (SDR) catalytic motif, and possess a conserved Asp residue which is required for cyclization. However, the MgcR/MgiR R domains contain the catalytic residues required for SDR reduction and NADPH binding (Figure S5c), as in LtxA and Lys2.37,38 Therefore, it is expected that an aldehyde is generated at C-9.37–39 As a result of the carbonyl derived from MgcK and MgcQ KR0, the pKa of the C-4 proton is significantly reduced, making it an attractive reactive carbon for cyclization with the C-9 aldehyde electrophile generated by MgcR R. Cyclization is proposed as follows: deprotonation of C-4 leads to a stabilized enolate anion species, which collapses back to reform the C-5 ketone such that the enolate double bond electrons attack the terminal C9 aldehyde, forming a C–C bond between C-4 and C-9 and a C-9 hydroxyl, which is then eliminated, representing a complete intramolecular Knoevenagel condensation (Figure 2).
The exact responsible proteins and timing of cyclization may be due to a combination of MgcR R domain and MgcS, a small lipocalin-like protein that has homologs implicated in PKS/NRPS cyclization in several microbial natural products, including the cyanobacterial metabolite anatoxin A.40–42 As lipocalins have been identified as mostly non-enzymatic fatty acid-binding and transport proteins of eukaryotes, we speculate their role in malyngamide biosynthesis is that of a stabilizing entity, positioning or conforming the molecular chain such that C-4 is proximal to C-9 so as to promote carbon-carbon bond formation. The intermediate product resulting from cyclization is malyngamide K, an analog independently isolated from environmental sources.14,21 Subsequent tailoring produces malyngamide C acetate and I, respectively, from the mgc and mgi pathways.
MgcQ KR expression, phylogeny, and modeling.
The hallmark C-5 ketone which is present in all type A malyngamides is installed by MgcK; however, as discussed, an initial bioinformatic analysis of MgcQ/MgiQ inferred C-4 – C-5 reduction to an alkene in PAB and generation of a C-5 hydroxyl group in PAP. A series of active site and tertiary structure alterations in the MgcQ/MgiQ KR0 appears to have rendered this domain nonfunctional. We explored the viability of the mgc and mgi KR catalytic domain (KRc) via phylogeny and homology modeling. Key KRc regions and active site residues have been studied in detail previously and indicate regions important for activity and stereochemistry: the NADPH binding site, stereocontrol by the presence of either the “LDD” motif or the “W” motif, catalytic site, and “lid” helix region which encloses the active site.43–45 Alignment of MgcQ domains from PAB and PAP to known active microbial KR domains as well as MgcG, H, and I show an unusual E variation in the first glycine of the conserved GGXGXXG diphosphate-binding “P-loop” in the NADPH binding site (Figure 5), in addition to other, unshared variations in residues 7–13. The inactive domain with the most similar NADPH binding site variant that has been characterized is EpoE,46 which contains a G7D mutation at the same location, though EpoE also contains an Arg at the position of the Tyr proton donor. An analysis of the PAB MgcQ KR catalytic site compared to active and inactive KR domains reveals some heterogeneity in sequence: the PAB MgcQ KRc contains a catalytic Tyr155, while the PAP MgcQ KRc contains a His at the same position. However, active KRc domains from CurJ, JamJ, AprI also contain the His alteration, implying that the His may also serve as a proton donor in KRc reduction (Figure 5). KRc domains from AmphI, NysI, and PimS2 are inactive examples as well; they contain a catalytic site Glu among other noncanonical residue changes. StiH contains an active site His, yet is inactive, possibly due to a Glu substitution at the Lys118 position (MgcQ KRc numbering) required for catalysis.40,47 Overall, catalytic activity is known to be dependent on several steric and electrostatic interactions between enzyme and cofactor reaction participants. As such, alignment of the active site lid sequence in MgcQ indicates a deletion of several residues relative to active KRc domains, a modification that is also observed in the inactive KRc Raps2–6 involved in the biosynthesis of rapamycin.48 However, the MgiQ KR in PAP shows no such truncation. To summarize, while MgcQ and MgiQ contain catalytic site Tyr155/His155 and Lys118, they lack either the conserved LDD or W stereocontrol motifs, a bulky Glu7 in the NADPH binding P-loop, and MgcQ contains a truncated lid.
Figure 5.

Sequence alignment of key regions of the KRc domain. Numbering accords with the sequence of the KRc0 of MgcQ. G7E highlighted in red box. Catalytic site residues Lys118 and Tyr155 indicated with red triangles. Stereocontrol Trp site indicated with blue triangle. * indicates inactive domains. MgcQ lid residue alignment gap in light blue dashed box.
We initially used homology modeling to examine the NADPH binding site and other tertiary structure elements as potential sources of MgcQ KRc inactivity. A homology model of the MgcQ and MgiQ KRc were generated based on the X-ray crystal structure of the active fungal amphotericin AmphB KRc, with bound NADPH (Figure 6). In both MgcQ and MgiQ, models revealed that the G7E substitution of a polar glutamate residue within the NADPH binding site introduces considerable steric and electrostatic bulk, potentially hindering binding; Ser10 in MgcQ also appears to contribute minor steric bulk. Modeling also suggested tertiary structure changes to the positioning of the active site lid; however, the amino acid heterogeneity in lid sequence does not allow for reliable prediction of specific residues or motifs responsible for inactivity in this region.
Figure 6.
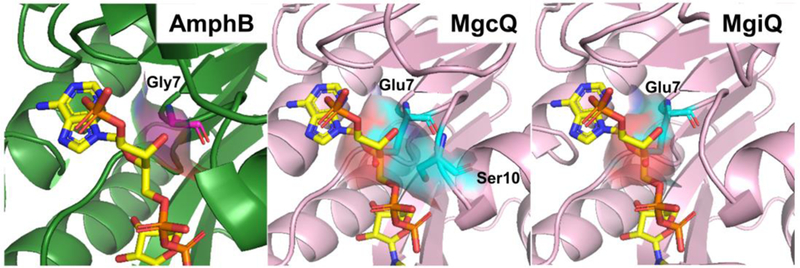
Homology modeling of MgcQ and MgiQ KRc. From left to right: AmphB KRc crystal structure (green cartoon, Gly7 backbone and transparent surface in magenta), MgcQ homology model (pink cartoon, Glu7/Ser10 rendered as sticks and transparent surface in cyan/red), and MgiQ homology model (pink cartoon, Glu7 rendered as sticks and transparent surface in cyan/red). NADPH rendered in stick form.
To explore the hypothesized inactivity of MgcQ KR0, we expressed and purified residues 1751–2679 of native MgcQ as a His-tagged MT-KR0 didomain and assessed NADPH binding with fluorescence polarization.49 Residues encompassing the catalytic KR domain (KRc) are numbered 1–245 in this study, corresponding to residues 2391–2635 of MgcQ and 662–906 of the recombinant MT-KR0 didomain. To understand the role of the NADPH binding site in KR0 inactivity, we additionally prepared two didomain constructs with mutations in the KR0 NADPH binding sites: E7G, imparting a reversion to a canonical binding site in MgiQ and MgcQ KRc, and double mutant E7G S10G, with the Ser10 found only in MgcQ as comparison. Based on these unusual modifications to the P-loop, we tested our hypothesis that cofactor binding to MgcQ KR is impaired by directly measuring NADPH binding to intact didomain via fluorescence polarization. Results showed that compared to the CurJ MT-KR control didomain, NADPH showed no significant increase in polarization in the presence of increasing concentration of the native or altered PAB MgcQ MT-KR0 didomains, indicating total lack of measurable NADPH binding to the KRc (Figure 7). Thus, we theorize that Glu7 and Ser10 are likely not the only modifications to MgcQ and MgiQ which confer inactivity: this likely results from uncharacterized changes in tertiary domain structure as well. Overall, evidence strongly suggests NADPH cannot bind to MgcQ, rendering the KR0 redox incompetent.
Figure 7.
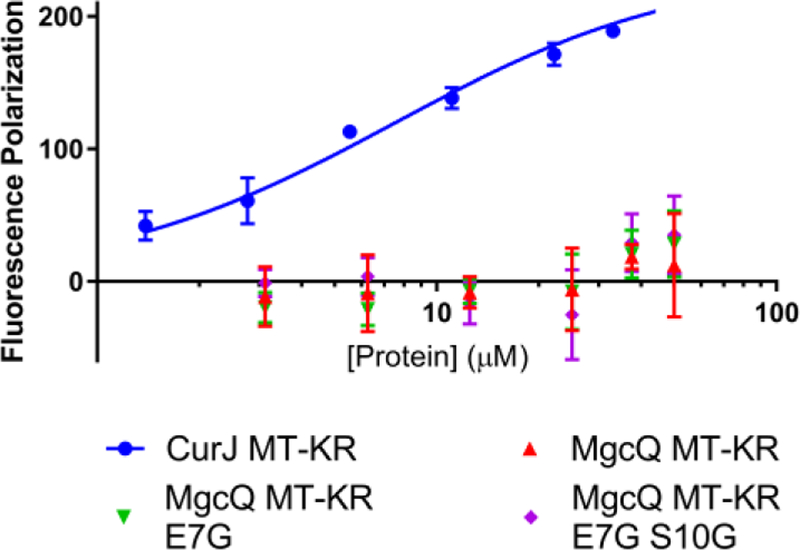
Fluorescence polarization of light when NADPH solution incubated with increasing concentrations of native MgcQ MT-KR0, single mutant McgQ MT-KR0 E7G, and double mutant MgcQ MT-KR0 E7G S10G compared to CurJ MT-KR positive control.
Biosynthetic model and conclusion
From two strains of the tropical marine cyanobacterium Okeania hirsuta (PAB and PAP), a highly similar pair of hybrid PKS/NRPS biosynthetic pathways were discovered that are predicted to encode for malyngamide C acetate and malyngamide I. Biosynthesis is initiated through a LipM octanoyltransferase from lipoic acid synthesis, then extended with PKS/NRPS modules, including a chlorinated β-branching methylation. In the penultimate PKS module, an unexpected KR0 generates a carbonyl which later contributes to carbocyclization via attack on a terminal aldehyde generated by an R-domain in the final PKS module. P450 and acetyltransfer reactions complete the biosynthesis.
Examining the 28 type A malyngamides characterized to date, we have assigned each molecule to one of four pathway groups, A1–A4 (Figure 8), with numbering of the 6-membered ring using equivalent positions to figure 1. Groupings are assigned from the predicted methylation, reductive domain activity, and chain release mechanism of the predicted final two modules, which define the PKS-derived architecture of the head group. To summarize, group A1 molecules have no methylations on the head group and a saturated C-7 position. Group A2 molecules are methylated at C-6 and feature either a hydroxyl, acetyl, or unsaturated C-7 position. Group A3 malyngamides are methylated at C9, which offers the possibility of an intriguing potential alternate cyclization strategy (Figure S8), while group A4 molecules are methylated at C8 only, and not at C9, implying retention of Knoevenagel cyclization, but with C-MT activity on the head group. Each pathway group produces a base malyngamide species: A1; K, A2; L, A3; G, A4; E, upon which head group tailoring proceeds. Some analogs, such as malyngamide C and malyngamide N, may in fact be shunt products from malyngamide C acetate or malyngamide I, or whose presence is induced by environmental or stability factors, or a combination of both.
Figure 8.
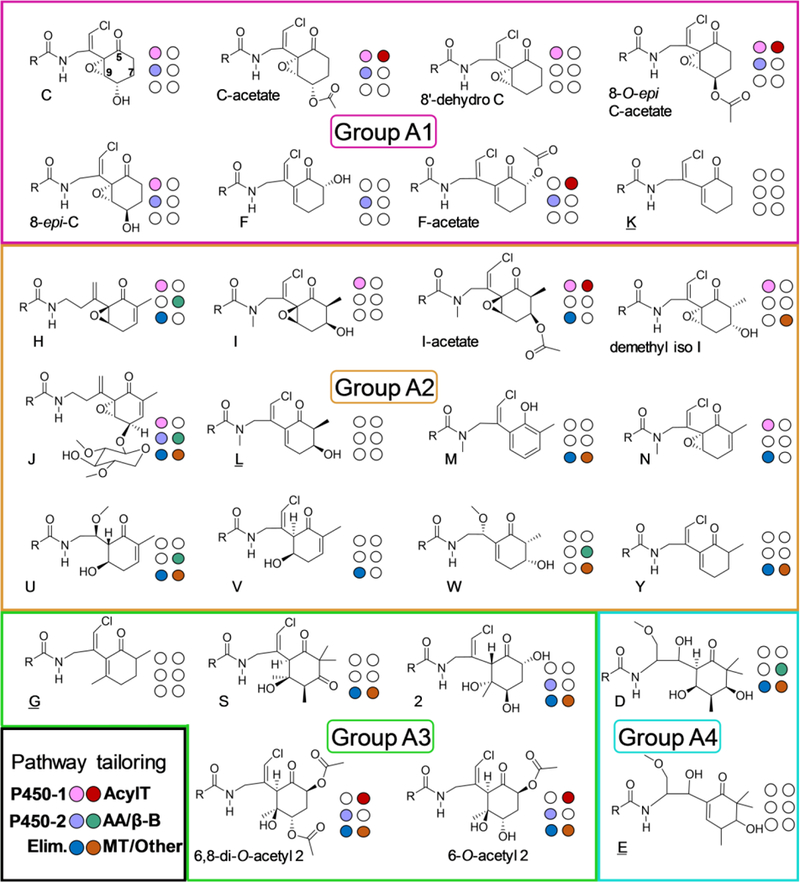
Biosynthetic model for type A malyngamides: Group A1 in pink, Group A2 in orange, Group A3 in light green, and group A4 in aqua. Untailored “base” chemical species are noted in each category by six uncolored circles and underline. Predicted tailoring differences from base species due to pathway diversity are indicated by coloration of the six circles next to each head group, with key to tailoring predictions in lower left. Differences to “base’ group molecule are organized into six main categories: P450–1; P450 equivalent to MgcT (epoxidation), P450–2; P450 equivalent to MgcU (hydroxylation), Elim; pattern of post-cyclization hydroxyl elimination, AcylT; acetyltransferase activity, AA/β-B; amino acid incorporation or β-branching/halogenation cassette, and MT/Other; methyltransferase, cyclization, or other transformations.
Most of the components of malyngamide biosynthesis, namely short chain fatty acid and amino acid activation and incorporation, polyketide assembly, chlorination, P450 activity, β-branching, O-methylation and acetylation, and glycosylation, are well-understood tailoring reactions in PKS and NRPS pathways. Thus, it is surprising that the “keystone” of type A malyngamide biosynthesis and radiative chemogenomics in Okeania is not a biochemical transformation at all, but rather the lack thereof. The altered MgcQ KR0 and R domains are required to enable the critical Knoevenagel cyclization. In this regard, the MgcQ module and domains appear to be in a state of catalytic disrepair, yet with retention of genetic information and structural integrity. Though MgcQ by sequence contains KS-AT-DH-MT-KR-ACP, the DH, MT, and KR are catalytically inactive in PAB, while only the KR is catalytically inactive in the PAP strain. Because inactive domains are normally excised over evolutionary time as they do not contribution to fitness, it is curious that Okeania has retained these partially non-functional MgcQ and modules. As all type A malyngamides possess cyclohexyl ring structures, there appears to be considerable selective pressure to maintain this moiety, as its presence requires both a partially non-functional MgcQ KR0 and a functional MgcR R. Thus, we speculate that excision of individual domains from MgcQ may render the module structurally and functionally defunct and unable to interact with the downstream MgcR. Further, since biosynthesis groups A1-A4 varyingly contain such diversity in tailored motifs, it would appear that the KR0 has persisted through evolutionary time. Indeed, the global distribution of Okeania, widespread isolation of malyngamides, and shared bioactivity among numerous of analogs speaks to the KR0 representing an ancient and important dysfunctionality that gave rise to this intriguing family of molecules.
METHODS
Methods and materials for collection, culturing, genomic DNA extraction, genome assembly, bioinformatic pathway analysis, LipM and domain phylogeny, homology modeling, biomass chemical extraction, malyngamide isolation and purification, stable isotope feeding scheme, ancymidol feeding and analysis, mass spectrometry, NMR and enrichment analysis, recombinant protein expression, NADPH binding assay, cyanobacterial culturing for nitrogen fixation, and nitrogen fixation measurements are detailed in supplementary information. Accession numbers for alignments and phylogeny can be found in Table S8.
Supplementary Material
Acknowledgments
We would like to acknowledge the help of M. Skiba and Q. Dan for construct design and recombinant protein biochemistry, F. Fu for advisement on the acetylene reduction assay, C. Larsen for genomic DNA extraction, Y. Su and L. Gross for high resolution mass spectrometry, and M. Kissick, A.-M. Hoskins, P. Kanjanakantorn, and B. Ni for cyanobacterial culturing and extraction.
Funding sources
This work was supported by: NIH GM107550-03, NIH GM118815-01A1, NIH CA108874, NIH GM067550, NIH R01 DK042303, and the Margaret J. Hunter Collegiate Professorship. AK was supported by St. Petersburg State University grant 15.61.951.2015.
Footnotes
Additional files
Supporting Information Available: This material is available free of charge via the Internet.
Accession codes
Okeania hirsuta PAB10Feb10-1 genome assembly: RCBY00000000. mgc pathway: MK142792. Okeania hirsuta PAP21Jun06-1 genome assembly: RCBZ00000000. mgi pathway: MK142793
References
- (1).Gerwick WH, and Moore BS (2012) Lessons from the past and charting the future of marine natural products drug discovery and chemical biology. Chem. Biol 19, 85–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Francisco JA, Cerveny CG, Meyer DL, Mixan BJ, Klussman K, Chace DF, Rejniak SX, Gordon KA, DeBlanc R, Toki BE, Law CL, Doronina SO, Siegall CB, Senter PD, and Wahl AF (2003) cAC10-vcMMAE, an anti-CD30-monomethyl auristatin E conjugate with potent and selective antitumor activity. Blood 102, 1458–1465. [DOI] [PubMed] [Google Scholar]
- (3).Gross H, McPhail KL, Goeger DE, Valeriote FA, and Gerwick WH (2010) Two cytotoxic stereoisomers of malyngamide C, 8-epi-malyngamide C and 8-O-acetyl-8-epi-malyngamide C, from the marine cyanobacterium Lyngbya majuscula. Phytochemistry 71, 1729–1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Villa FA, Lieske K, and Gerwick L (2010) Selective MyD88-dependent pathway inhibition by the cyanobacterial natural product malyngamide F acetate. Eur. J. Pharmacol 629, 140–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Mynderse JS, and Moore RE (1978) Malyngamides D and E, Two trans-7-Methoxy-9-methylhexadec-4-enamides from a Deep Water Variety of the Marine Cyanophyte Lyngbya majuscula. J. Org. Chem 43, 4359–4363. [Google Scholar]
- (6).Tidgwell K, Clark BR, and Gerwick WH (2010) Comprehensive Natural Products II, in Chemistry and Biology (Lewis M, Liu H-W, Townsend C, and Ebizuka Y, Eds.), pp 142–183. Elsevier Ltd. [Google Scholar]
- (7).Tan LT, Okino T, and Gerwick WH (2000) Hermitamides A and B, toxic malyngamide-type natural products from the marine cyanobacterium Lyngbya majuscula. J. Nat. Prod 63, 952–955. [DOI] [PubMed] [Google Scholar]
- (8).Wan F, and Erickson KL (1999) Serinol-derived malyngamides from an Australian cyanobacterium. J. Nat. Prod 62, 1696–1699. [DOI] [PubMed] [Google Scholar]
- (9).Praud A, Valls R, Piovetti L, and Banaigs B (1993) Malyngamide G : Proposition de structure pour un nouvel amide chloré d’une algue bleu-verte epiphyte de Cystoseira crinita. Tetrahedron Lett 34, 5437–5440. [Google Scholar]
- (10).McPhail KL, and Gerwick WH (2003) Three new malyngamides from a Papua New Guinea collection of the marine cyanobacterium Lyngbya majuscula. J. Nat. Prod 66, 132–135. [DOI] [PubMed] [Google Scholar]
- (11).Malloy KL, Villa FA, Engene N, Matainaho T, Gerwick L, and Gerwick WH (2011) Malyngamide 2, an oxidized lipopeptide with nitric oxide inhibiting activity from a Papua New Guinea marine cyanobacterium. J. Nat. Prod 74, 95–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Appleton DR, Sewell MA, Berridge MV, and Copp BR (2002) A new biologically active malyngamide from a New Zealand collection of the sea hare Bursatella leachii. J. Nat. Prod 65, 630–631. [DOI] [PubMed] [Google Scholar]
- (13).Orjala J, Nagle D, and Gerwick WH (1995) Malyngamide H, an ichthyotoxic amide possessing a new carbon skeleton from the caribbean cyanobacterium lyngbya majuscula. J. Nat. Prod 58, 764–768. [DOI] [PubMed] [Google Scholar]
- (14).Wu M, Milligan KE, and Gerwick WH (1997) Three new malyngamides from the marine cyanobacterium Lyngbya majuscula. Tetrahedron 53, 15983–15990. [Google Scholar]
- (15).Kan Y, Fujita T, Nagai H, Sakamoto B, and Hokama Y (1998) Malyngamides M and N from the Hawaiian red alga Gracilaria coronopifolia. J. Nat. Prod 61, 152–155. [DOI] [PubMed] [Google Scholar]
- (16).Ainslie RD, Barchi JJ, Kuniyoshi M, Moore RE, and Mynderse JS (1985) Structure of Malyngamide C. J. Org. Chem 50, 2859–2862. [Google Scholar]
- (17).Todd JS, and Gerwick WH (1995) Malyngamide I from the tropical marine cyanobacterium Lyngbya majuscula and the probable structure revision of stylocheilamide. Tetrahedron Lett 36, 7837–7840. [Google Scholar]
- (18).Meng L, Mohan R, Kwok BHB, Elofsson M, Sin N, and Crews CM (1999) Epoxomicin, a potent and selective proteasome inhibitor, exhibits in vivo antiinflammatory activity. Proc. Natl. Acad. Sci 96, 10403–10408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Engene N, Paul VJ, Byrum T, Gerwick WH, Thor A, and Ellisman MH (2013) Five chemically rich species of tropical marine cyanobacteria of the genus Okeania gen. nov. (Oscillatoriales, Cyanoprokaryota). J. Phycol 49, 1095–1106. [DOI] [PubMed] [Google Scholar]
- (20).Petitbois JG, Casalme LO, Lopez JAV, Alarif WM, Abdel-Lateff A, Al-Lihaibi SS, Yoshimura E, Nogata Y, Umezawa T, Matsuda F, and Okino T (2017) Serinolamides and Lyngbyabellins from an Okeania sp. Cyanobacterium Collected from the Red Sea. J. Nat. Prod 80, 2708–2715. [DOI] [PubMed] [Google Scholar]
- (21).Winnikoff JR, Glukhov E, Watrous J, Dorrestein PC, and Gerwick WH (2014) Quantitative molecular networking to profile marine cyanobacterial metabolomes. J. Antibiot. (Tokyo) 67, 105–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Zhu X, Liu J, and Zhang W (2015) De novo biosynthesis of terminal alkyne-labeled natural products. Nat. Chem. Biol 11, 115–120. [DOI] [PubMed] [Google Scholar]
- (23).Kleigrewe K, Almaliti J, Tian IY, Kinnel RB, Korobeynikov A, Monroe EA, Duggan BM, Di Marzo V, Sherman DH, Dorrestein PC, Gerwick L, and Gerwick WH (2015) Combining Mass Spectrometric Metabolic Profiling with Genomic Analysis: A Powerful Approach for Discovering Natural Products from Cyanobacteria. J. Nat. Prod 78, 1671–1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Chang Z (2004) Biosynthetic pathway and gene cluster analysis of curacin A, an antitubulin natural product from the tropical marine cyanobacterium Lyngbya majuscula. J. Nat. Prod 67, 1356–1367. [DOI] [PubMed] [Google Scholar]
- (25).Skiba MA, Sikkema AP, Moss NA, Lowell AN, Su M, Sturgis RM, Gerwick L, Gerwick WH, Sherman DH, and Smith JL (2018) Biosynthesis of t-Butyl in Apratoxin A: Functional Analysis and Architecture of a PKS Loading Module. ACS Chem. Biol 13, 1640–1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Christensen QH, and Cronan JE (2010) Lipoic acid synthesis: A new family of octanoyltransferases generally annotated as lipoate protein ligases. Biochemistry 49, 10024–10036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Blin K, Wolf T, Chevrette MG, Lu X, Schwalen CJ, Kautsar SA, Suarez Duran HG, De Los Santos ELC, Kim HU, Nave M, Dickschat JS, Mitchell DA, Shelest E, Breitling R, Takano E, Lee SY, Weber T, and Medema MH (2017) AntiSMASH 4.0 - improvements in chemistry prediction and gene cluster boundary identification. Nucleic Acids Res 45, W36–W41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Boratyn GM, Schäffer AA, Agarwala R, Altschul SF, Lipman DJ, and Madden TL (2012) Domain enhanced lookup time accelerated BLAST. Biol. Direct 7, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Beld J, Sonnenschein EC, Vickery CR, Noel JP, and Burkart MD (2014) The phosphopantetheinyl transferases: Catalysis of a post-translational modification crucial for life. Nat. Prod. Rep 31, 61–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Edwards DJ, Marquez BL, Nogle LM, Mcphail K, Goeger DE, Roberts MA, Gerwick WH, (2004) Structure and Biosynthesis of the Jamaicamides, New Mixed Polyketide-Peptide Neurotoxins from the Marine Cyanobacterium Lyngbya majuscula. Chem. Biol 11, 817–813. [DOI] [PubMed] [Google Scholar]
- (31).Fiers WD, Dodge GJ, Sherman DH, Smith JL, and Aldrich CC (2016) Vinylogous Dehydration by a Polyketide Dehydratase Domain in Curacin Biosynthesis. J. Am. Chem. Soc 138, 16024–16036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Gu L, Wang B, Kulkarni A, Geders TW, Grindberg RV, Gerwick L, Hkansson K, Wipf P, Smith JL, Gerwick WH, and Sherman DH (2009) Metamorphic enzyme assembly in polyketide diversification. Nature 459, 731–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Geders TW, Gu L, Mowers JC, Liu H, Gerwick WH, Håkansson K, Sherman DH, and Smith JL (2007) Crystal structure of the ECH2 catalytic domain of CurF from Lyngbya majuscula: Insights into a decarboxylase involved in polyketide chain β-branching. J. Biol. Chem 282, 35954–35963. [DOI] [PubMed] [Google Scholar]
- (34).Park NS, Myeong JS, Park HJ, Han K, Kim SN, and Kim ES (2005) Characterization and culture optimization of regiospecific cyclosporin hydroxylation in rare actinomycetes species. J. Microbiol. Biotechnol 15, 188–191. [Google Scholar]
- (35).Lowry B, Li X, Robbins T, Cane DE, and Khosla C (2016) A turnstile mechanism for the controlled growth of biosynthetic intermediates on assembly line polyketide synthases. ACS Cent. Sci 2, 14–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Liu X, and Walsh CT (2009) Cyclopiazonic acid biosynthesis in Aspergillus sp.: Characterization of a reductase-like R* domain in cyclopiazonate synthetase that forms and releases cyclo-acetoacetyl-L-tryptophan. Biochemistry 48, 8746–8757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Ehmann DE, Gehring AM, and Walsh CT (1999) Lysine biosynthesis in Saccharomyces cerevisiae: Mechanism of α-aminoadipate reductase (Lys2) involves posttranslational phosphopantetheinylation by Lys5. Biochemistry 38, 6171–6177. [DOI] [PubMed] [Google Scholar]
- (38).Edwards DJ, and Gerwick WH (2004) Lyngbyatoxin biosynthesis: Sequence of biosynthetic gene cluster and identification of a novel aromatic prenyltransferase. J. Am. Chem. Soc 126, 11432–11433. [DOI] [PubMed] [Google Scholar]
- (39).Li L, Deng W, Song J, Ding W, Zhao QF, Peng C, Song WW, Tang GL, and Liu W (2008) Characterization of the saframycin a gene cluster from Streptomyces lavendulae NRRL 11002 revealing a nonribosomal peptide synthetase system for assembling the unusual tetrapeptidyl skeleton in an iterative manner. J. Bacteriol 190, 251–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Gaitatzis N, Silakowski B, Kunze B, Nordsiek G, Blöcker H, Höfle G, and Müller R (2002) The biosynthesis of the aromatic myxobacterial electron transport inhibitor stigmatellin is directed by a novel type of modular polyketide synthase. J. Biol. Chem 277, 13082–13090. [DOI] [PubMed] [Google Scholar]
- (41).Liu X, Biswas S, Berg MG, Antapli CM, Xie F, Wang Q, Tang MC, Tang GL, Zhang L, Dreyfuss G, and Cheng YQ (2013) Genomics-guided discovery of thailanstatins A, B, and C as pre-mRNA splicing inhibitors and antiproliferative agents from Burkholderia thailandensis MSMB43. J. Nat. Prod 76, 685–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Jiang Y, Song G, Pan Q, Yang Y, and Li R (2015) Identification of genes for anatoxin-a biosynthesis in Cuspidothrix issatschenkoi. Harmful Algae 46, 43–48. [Google Scholar]
- (43).Keatinge-Clay AT (2007) A Tylosin Ketoreductase Reveals How Chirality Is Determined in Polyketides. Chem. Biol 14, 898–908. [DOI] [PubMed] [Google Scholar]
- (44).Keatinge-Clay AT (2016) Stereocontrol within polyketide assembly lines. Nat. Prod. Rep 33, 141–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Caffrey P (2003) Conserved amino acid residues correlating with ketoreductase stereospecificity in modular polyketide synthases. ChemBioChem 4, 654–657. [DOI] [PubMed] [Google Scholar]
- (46).Julien B, Shah S, Ziermann R, Goldman R, Katz L, and Khosla C (2000) Isolation and characterization of the epothilone biosynthetic gene cluster from Sorangium cellulosum. Gene 249, 153–160. [DOI] [PubMed] [Google Scholar]
- (47).Aparicio JF, Caffrey P, Gil JA, and Zotchev SB (2003) Polyene antibiotic biosynthesis gene clusters. Appl. Microbiol. Biotechnol 61, 179–188. [DOI] [PubMed] [Google Scholar]
- (48).Aparicio JF, Molnár I, Schwecke T, König A, Haydock SF, Khaw LE, Staunton J, and Leadlay PF (1996) Organization of the biosynthetic gene cluster for rapamycin in Streptomyces hygroscopicus: Analysis of the enzymatic domains in the modular polyketide synthase. Gene 169, 9–16. [DOI] [PubMed] [Google Scholar]
- (49).Jarabak J, and Sack GH (1969) A Soluble 17β-Hydroxysteroid Dehydrogenase From Human Placenta. The Binding of Pyridine Nucleotides and Steroids. Biochemistry 8, 2203–2212. [DOI] [PubMed] [Google Scholar]
- (50).Cronan JE (2016) Assembly of Lipoic Acid on Its Cognate Enzymes: an Extraordinary and Essential Biosynthetic Pathway. Microbiol. Mol. Biol. Rev 80, 429–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.