

Abstract
The extradiol dioxygenases are a large subclass of mononuclear non-heme Fe enzymes that catalyze the oxidative cleavage of catechols distal to their OH groups. These enzymes are important in bioremediation, and there has been significant interest in understanding how they activate O2. The extradiol dioxygenase homoprotocatechuate 2,3-dioxygenase (HPCD) provides an opportunity to study this process, as two O2 intermediates have been trapped and crystallographically defined using the slow substrate 4-nitrocatechol (4NC): a side-on Fe-O2-4NC species and a Fe-O2-4NC peroxy bridged species. Also with 4NC, two solution intermediates have been trapped in the H200N variant, where H200 provides a second-sphere hydrogen bond in the wild-type enzyme. While the electronic structure of these solution intermediates has been defined previously as FeIII-superoxo-catecholate and FeIII-peroxysemiquinone, their geometric structures are unknown. Nuclear resonance vibrational spectroscopy (NRVS) is an important tool for structural definition of non-heme Fe-O2 intermediates, as all normal modes with Fe displacement have intensity in the NRVS spectrum. In this study, NRVS is used to define the geometric structure of the H200N-4NC solution intermediates in HPCD as an end-on FeIII-superoxocatecholate and an end-on FeIII-hydroperoxo-semiquinone. Parallel calculations are performed to define the electronic structures and protonation states of the crystallographically defined wild-type HPCD-4NC intermediates, where the side-on intermediate is found to be a FeIII-hydroperoxo-semiquinone. The assignment of this crystallographic intermediate is validated by correlation to the NRVS data through computational removal of H200. While the side-on hydroperoxo semiquinone intermediate is computationally found to be nonreactive in peroxide bridge formation, it is isoenergetic with a superoxo catecholate species that is competent in performing this reaction. This study provides insight into the relative reactivities of FeIII-superoxo and FeIII-hydroperoxo intermediates in non-heme Fe enzymes and into the role H200 plays in facilitating extradiol catalysis.
Graphical Abstract
1. Introduction
Catechols are important intermediates in the microbial degradation of aromatic compounds.1 Two different subclasses of non-heme iron (NHFe) enzymes are involved in the dioxygenation and ring cleavage of these species: the intradiol dioxygenases and the extradiol dioxygenases (EDOs).2–4 The intradiol dioxygenases use an FeIII site to activate catechol for the spin forbidden reaction with triplet O2 and catalyze O2 incorporation between the two catechol OH groups in the cleavage of aromatic substrate C-C bond in forming cis-cis-muconic acids.5 In contrast, the EDOs, which are important in bioremediation6–8, use an FeII site to activate O2 for reaction with the catechol, catalyzing the oxidative cleavage of catechol distal to the OH groups in forming muconic semialdehyde products.4,9 There has been significant interest in the definition of their O2 activation mechanism on a molecular level.4,10,11 From crystallography, the resting FeII active site in these enzymes has the 2His/1 carboxylate facial triad ligation typical of NHFeII enzymes, and either two or three additional solvent-derived ligands.12–14 Crystallography of the EDO homoprotocatechuate 2,3-dioxygenase (HPCD), which is the subject of this study, reveals a 6-coordinate (6C) structure with one of the coordinated waters more weakly bound.15–17 Also from crystallography, bidentate binding of the native substrate, homoprotocatechuic acid (HPCA) (which binds as the monoanion with one hydroxyl group deprotonated18), leads to a 6C ferrous structure with a weakly coordinated water ligand (at 2.43 Å) that is primed for dissociation, enabling O2 binding to the Fe.19 It is important to note that catechols are redox active, capable of being reversibly oxidized by one electron to semiquinone and by two electrons to quinone.20 Further, unlike other subclasses of NHFeII enzymes, in the EDOs, all four electrons required for O2 activation are provided by the substrate.3,21,22
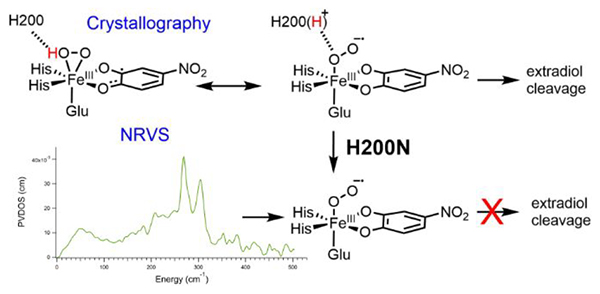
Crystallographic intermediates in the O2 reaction of HPCD with a slow substrate, 4-nitrocatechol (4NC), have been trapped and structurally characterized, offering mechanistic insight into EDO catalysis, including the nature of the initial activated O2 species.16 The first of these intermediates is a side-on bound Fe-O2 species in which the 4NC is distorted at one carbon, leading to its assignment as a semiquinone.16 From these observations, this initial O2 intermediate was assigned as a FeII-superoxosemiquinone species; note that the resolution of the crystal structure (1.95 Å) is not sufficient to distinguish superoxo from peroxo, or to determine whether the O2 is protonated. The second crystallographic intermediate is an Fe-O2-4NC bridged intermediate, which was assigned as a FeII-peroxy-quinone bridged species, thought to have formed from coupling of the superoxo and the semiquinone radicals in the initial O2 intermediate. A third species was also found in the crystal structure, consisting of the muconic semialdehyde product bound to the Fe, indicating that the O2 and peroxo bridged species are competent in extradiol dioxygenation.
In addition to these crystallographic intermediates, solution phase intermediates have also been trapped in active site variants where a histidine residue, H200, has been mutated.23–26 In particular, two O2 intermediates in the H200N variant using 4NC as the substrate are of interest in this study for correlation to the crystallographic intermediates.23 The first of these, Int-1, is an S = 2 species with a high-spin FeIII site as determined by Mössbauer and EPR spectroscopy. Also from EPR spectroscopy, 17Obroadening of the geff = 8.17 signal indicated radical character on the O2, leading to an electronic structure assignment of an S = 5/2 FeIII antiferromagnetically coupled to an S = ½ superoxo to give the S = 2 ground state. Geometric information, including the binding mode of the O2•-, remains unknown. The FeIII-superoxo-catecholate Int-1 decays slowly (k = 0.015 s−1 at 277 K) to Int-2. Mössbauer and EPR spectroscopy of Int-2 indicated that it also has an S = 2 ground state with a high-spin FeIII. In this case, only slight 17O broadening of the EPR signal was observed, indicating that the S = 2 ground state reflects antiferromagnetic coupling of the S = 5/2 FeIII with an S = ½ semiquinone radical (consistent with UV/Vis data indicating that the 4NC has been oxidized by one electron in Int-2). Int-2 was thus assigned as an FeIII-peroxo-semiquinone species, with the binding mode and protonation state of the peroxo unknown. Importantly, Int-2 does not perform extradiol cleavage, and instead, it persists until further equivalents of 4NC are added, which trigger the release of H2O2 and full oxidation of the 4NC to the quinone. H200 thus plays an important role in enabling extradiol cleavage, as the O2 reaction of wild-type HPCD with 4NC does produce an extradiol cleaved product.16,27 In wild-type, H200 is a second-sphere residue that is proposed to take a proton from the coordinated substrate and enable transfer of the proton for O-O cleavage.4 Correlation of the crystallographic intermediates in wild-type to the H200N Int-1 and Int-2 can provide insight into the role of H200; this requires further definition of the structures of Int-1 and Int-2.
Nuclear resonance vibrational spectroscopy (NRVS)28,29 is an ideal method for the definition of key structural features of NHFe oxygen intermediates.30 In NRVS, vibrational sidebands above the 14.4 keV 57Fe Mössbauer transition are probed using synchrotron radiation through counting fluorescence photons resulting from relaxation of the excited nuclear state. A partial vibrational density of states (PVDOS) spectrum is derived from the raw counts data.31 Importantly, the intensity of a feature in the PVDOS NRVS spectrum is directly proportional to the amount of Fe displacement in a normal mode at a given energy;32 thus, NRVS gives complete and selective information about the vibrations associated with the Fe active site. To interpret the NRVS data, density functional theory (DFT) calculations are used to simulate the NRVS spectrum for a given structure. Thus, to analyze the NRVS spectrum for an enzyme intermediate, it is important to, in parallel, collect NRVS data on similar model complexes that are structurally well-defined to both calibrate the DFT method and understand how known structural features influence the NRVS spectrum. This method of model-calibrated NRVS has been successfully applied to the low-spin FeIII-OOH intermediate in bleomycin,33 to the FeIV-oxo intermediate in SyrB2,34 to the FeIII2 bridged peroxide intermediate P’ in AurF,35 and to the high-spin FeIII-peroxy intermediate in the Rieske dioxygenase benzoate 1,2-dioxygenase.36
In the present study, NRVS is used to define the structures of Int-1 and Int-2 in H200N-4NC HPCD. To calibrate this analysis, NRVS data were also taken on the {FeNO}7 analogue (the 7 indicates the total valence electrons, 6 from FeII and 1 from NO)37 of Int-1, H200N-4NC-NO, and the computational method was calibrated using the {FeNO}7 model complex Fe-EDTA-NO, which has been structurally characterized by EXAFS.38,39 The wild-type HPCD-4NC crystallographic intermediates are also defined computationally in terms of protonation state and electronic structure and correlated to and calibrated by the experimental H200N Int-1 and Int-2 NRVS structures by removal of H200 in silico. Based on the crystallographic intermediate structures, the reaction coordinate for generating the peroxo bridged intermediate in HPCD is evaluated. Importantly, the crystallographic side-on O2 intermediate was found to be an FeIII-hydroperoxo-semiquinone that is not reactive; however, it is isoenergetic with a FeIII-superoxo species that is competent in electrophilic attack on its bound catecholate. Parallel calculations evaluating FeO2 attack on the coordinated substrate in H200N are also presented to offer insight into the observed lack of extradiol reactivity by Int-1 and Int-2. Note that several previous studies have computationally investigated this reaction, primarily considering the native substrate homoprotocatechuic acid (HPCA),10,11,40,41 though one has considered the 4NC reaction.42 These studies have obtained a variety of different results, necessitating that calculations be calibrated by additional experimental results. The present study focuses on the 4NC reaction and is experimentally calibrated by NRVS and other spectroscopic data for H200N and to the crystallographic structures for wild-type. The results of these experimentally calibrated calculations provide significant insight into the role of the catecholate in O2 activation, the relative reactivity of FeIII-superoxo and FeIII-hydroperoxo species, and the role H200 plays in extradiol catalysis.
2. Methods
2.1. Preparation of 57Fe H200N HPCD NRVS samples
57Fe (96.8 %) was purchased from Cambridge Isotopes. Sodium chlorite (80 %) was purchased from Sigma. Nitric oxide gas was purchased from Matheson Gas.
Recombinant H200N mutational variant of Brevibacterium fuscum HPCD was over-expressed and purified using previously described procedures.23,26,43 The 57Fe occupancy of HPCD was determined via ICP-MS (Earth Science, University of Minnesota).
Residual oxidized iron present following protein purification was reduced using sodium dithionite.23,44 Briefly, H200N HPCD was deoxygenated by flowing a stream of argon gas over gently stirring enzyme at 4 °C and transferred into a Coy anaerobic chamber. Fully reduced enzyme was prepared by addition of 0.3 – 0.5 equivalents of sodium dithionite followed by anaerobic gel-filtration (PD-10, GE Healthcare Lifesciences). Protein samples were concentrated using anaerobic centrifugal concentration. This procedure increased the specific activity of H200N HPCD, but the reaction cycle rate constants were unchanged.23
To generate the H200N-4NC enzyme-substrate complex, a concentrated solution of 4NC (0.1 M) was added to fully reduced H200N HPCD in 200 mM MOPS buffer, pH 7.5 to provide 1.2 equiv. of substrate over enzyme. Enzyme-substrate complexes were then either rapidly mixed with oxygen to generate reaction intermediates or exposed to nitric oxide gas to produce a nitrosyl complex.
Enzyme catalysis was initiated by generating millimolar concentrations of dissolved O2 using chlorite dismutase (Cld) (generous gift of Jennifer Dubois, Montana State University) and sodium chlorite.26,45,46 Cld (~ 15 μM) was added to the anaerobic HPCD solution and sodium chlorite was added to the O2-gas saturated buffer solution to provide a total of 1.2 equiv. of O2 over enzyme upon rapid mixing. Oxygen gas saturated buffer was produced by sparging buffer (4 °C) with oxygen gas (1.8 mM). Oxygen gas saturated buffer (1.8 mM, 4°C) was used for experiments at lower concentrations of HPCD.
The Int-1 and Int-2 reaction intermediate samples were produced using rapid freeze quench methods previously described.23 Briefly, anaerobic enzyme-4NC complexes (7.14 mM HPCD, 7.96 mM 4NC) with Cld (28 μM) were mixed with sodium chlorite (14.7 mM) oxygen-gas-saturated (1.8 mM) buffer using an Update Instruments model 1019 RFQ apparatus maintained at 4 °C. An additional set of samples was prepared using 5.0 mM anaerobic enzyme-4NC complex with Cld (20 μM) mixed with sodium chlorite (10 mM) oxygen gas-saturated buffer (1.8 mM). The Int-1 and Int-2, samples were collected directly into the NRVS cell and then frozen in liquid nitrogen at the appropriate time. Int-1 samples were frozen at 4 s, and Int-2 samples were frozen at 10 min.
To generate H200N-4NC-NO, nitric oxide gas was bubbled through 6 M NaOH and then water to remove NOx species. The H200N HPCD nitrosyl complex was produced by flowing nitric oxide gas over gently stirred anaerobic H200N-4NC (2.7 mM) solutions at 4 °C (3.32 mM dissolved NO47). The H200N HPCD nitrosyl complex was transferred to the NRVS cell within a Coy anaerobic chamber and then frozen in liquid nitrogen.
2.2. NRVS data collection and processing
57Fe NRVS data on H200N-4NC-NO, Int-1, and Int-2 were collected at Beamline 3-ID-D at APS and BL09XU at SPring-8 on multiple occasions. The NRVS energy scale was calibrated using [FeIII(Cl4)][Net4].48 Samples were maintained at ~60K on a copper sample mount in a liquid helium cryostat. Raw scans were added together using the PHOENIX software package31 until acceptable signal-to-noise in the inelastic peaks was achieved (at least 10:1). PHOENIX was also used to subtract the elastic peak from the raw data and convert the observed counts spectrum into the final vibrational density of states spectrum.
2.3. DFT calculations
For the DFT-calculated NRVS spectra, all DFT calculations used the BP86 functional49,50 with 10% Hartree-Fock exchange and the TZVP basis set.51 Geometry optimizations (using tight convergence criteria) and subsequent frequency calculations were performed using the Gaussian 09 software package.52 Implicit solvation effects were included using the polarizable continuum model as implemented in Gaussian 09,53 with water specified as the solvent for Fe-EDTA-NO and an epsilon of 4.0 used for the enzyme calculations. For the peroxy bridged intermediates, an epsilon of 20.0 was initially used to obtain optimized structures with the proton both on H200 and on the O2, and single-point energies were then calculated using an epsilon of 4.0. The crystal structure of FeII(EDTA)(OH2)54 was used as the starting point for Fe-EDTA-NO, with the water replaced by NO. Int-1 and Int-2 were modeled using the same cluster model that was used for the crystallographic calculations (vide infra) but with H200 removed as N200 is distant from the Fe-O2 moiety (5.2 Å) in the H200N structure.44 The amino acids truncated to methyl groups at the α-carbons and these carbons frozen during geometry optimization. H200N-4NC-NO was generated by changing the O2 in the optimized Int-1 structure to NO. For the final calculated NRVS spectra, frequencies were calculated with the masses of the hydrogens on the frozen carbons increased to 100 amu to mimic the effect of the protein backbone.55
For calculation of the crystallographic intermediates and evaluation of the peroxy bridge-forming reaction coordinate, the BP86 functional mixed with 10% Hartree-Fock exchanged was used along with a mixed basis set consisting of 6–311G*56–59 on the Fe and O2 and 6–31G*60–64 on the remaining atoms. The crystallographic intermediates were optimized starting from the crystal structure (PDB code 2IGA),16 and included the His155, His214, Glu267, His200, Tyr257, Asn157, His248, and Thr205 residues in addition to Fe, 4NC, and O2. Details on the choice of this cluster model are given in Section 3.2.1. The initial structures for the geometry optimization of the side-on O2 complex and the O2-bridged intermediate were taken from unit C and D of the crystallographic structure, respectively. Residues were truncated at β-carbon atoms or the closest carbon atoms to the functional groups—for example, glutamate was truncated to acetate—except for Asn157 and Thr205, which were truncated at α-carbon atoms for flexibility to allow for the proper hydrogen bonding interactions with the inner-coordination sphere. The carbon atoms at the truncation points were replaced with methyl groups and fixed as described above. The change in the positions of the fixed carbon atoms in unit C and D has only a small effect on the energetics: the calculated energy difference of the side-on O2 complex optimized with the fixed carbon positions in the unit D was–1.3 kcal/mol and the difference of the O2-bridge intermediate optimized with the fixed carbon positions in the unit C was–0.4 kcal/mol. Thermodynamic properties were calculated using Maxwell-Boltzmann statistics applied to harmonic frequencies, a rigid rotator and a translating particle, as implemented in Gaussian 09. To estimate hydrogen bonding energetics for H200, the total energy with H200 present was compared to the energy for 4-methylimidazolium or 4-methylimidazole (depending on the protonation of H200 in the initial structure) added to the energy of the rest of the system reoptimized with H200 deleted. These energetics were corrected for basis set superposition error (BSSE) using Boys counterpoise correction as implemented in Gaussian 09.
3. Results and Analysis
3.1. NRVS spectroscopy and calculations of H200N solution intermediates
To calibrate the NRVS analysis of Int-1 and Int-2, NRVS data were also collected on the {FeNO}7 complex of H200N-4NC. These NRVS data are shown in Figure 1A. The data have a weak feature at 419 cm−1 and two main peaks at 255 and 296 cm−1, with a low energy shoulder at 205 cm−1. To calibrate the DFT method used for simulating the NRVS spectra of H200N-4NCNO in terms of its accuracy with respect to structure and vibrational frequencies, geometry optimization and frequency calculations were performed on the {FeNO}7 model complex Fe-EDTA-NO. While no crystal structure exists for the model complex, geometric information is available from EXAFS,38 including the Fe-N-O angle from a multiple scattering analysis of the EXAFS data. Resonance Raman (rR) data have also been reported for Fe-EDTA-NO, giving the Fe-(NO) and N-O stretching energies.39,65 A previous study66 has found a combination of the BP86 functional mixed with 10% Hartree-Fock exchange (HFX) and a mixed basis set consisting of 6–311g* on the Fe-NO unit and 6–31g* on the remaining atoms performs well in describing the geometric and electronic structures of {FeNO}7 complexes. Thus, Fe-EDTA-NO was optimized using the BP86, BP86 with 10% Hartree Fock exchange (HFX), and B3LYP functionals, with this mixed basis set. The results of these calculations are given in Table 1. Of the three functionals, only BP86 + 10% HFX gave good agreement with the experimental structural and vibrational data. This BP86 + 10% HFX / mixed basis set method was then applied to H200N-4NCNO, using the DFT cluster model described in Section 3.2.1 with truncation at the α carbons. However, as shown in Figure S1, the experimentally calibrated DFT calculated NRVS spectrum of H200N-4NC-NO generated using this functional and basis set is in rather poor agreement with the experimental data, especially in the lower energy modes. The basis set used for the calculation was therefore expanded to TZVP on all atoms. From Table 1, this method applied to Fe-EDTA-NO using the 10% HFX functional still gives good agreement with the experimental data, though it gives a slightly longer Fe-N bond relative to the mixed basis set and thus underpredicts the νFe-N by 28 cm−1. Applying the BP86 + 10% HFX/TZVP method to H200N-4NC-NO, two isoenergetic orientations for the NO moiety were found: (1) Fe-NO bent towards and bisecting the two catecholic oxygen ligands, or (2) Fe-NO bent away from the catecholate substrate. The former agrees better with the NRVS data and is in much better agreement in the low energy modes than the corresponding calculated spectrum using the mixed basis set, as shown in Figure S1. The conformation with the Fe-NO unit bent towards the substrate is also consistent with a crystal structure of the NO-bound complex in 2,3-dihydroxybiphenyl 1,2-dioxygenase.67 This final calculated spectrum and its DFT structure are shown in Figure 1B and 1C, respectively (the calculated spectrum for the second conformation is given in Figure S2).
Figure 1.

A) NRVS data for H200N-4NC-NO, with peak energies given and peaks assigned by correlation to B. Note that there is a discontinuity at 337 cm−1 in A due to splicing together two data sets, where the higher energy data set used a longer count rate from 300 to 630 cm−1 in order to resolve the peak at 419 cm−1 above the noise level. B) Best DFT calculated spectrum for H200N-4NC-NO, with modes labeled as in D. C) DFT-optimized structure of H200N-4NC-NO, with important structural parameters given. Fe is shown in orange, amino acid carbons in green, substrate carbons in purple, nitrogens in blue, oxygens in red, and hydrogens in white. D) Schematic depictions of the normal modes that appear in the NRVS data and calculated spectrum.
Table 1.
Experimental and calculated structural and vibrational parameters for Fe-EDTA-NO with several functionals and basis sets.
This calibrated, DFT calculated spectrum correlates with the data and provides a basis for assigning the experimental NRVS spectrum. The two intense peaks at 255 cm−1 and 296 cm−1 are, respectively, the in-plane transaxial bend (δ transaxialip, calculated at 259 cm−1, ν3a in Figure 1D), consisting of equatorial Fe motion in the Fe-NO plane, and the δFe-NO (calculated at 301 cm−1, ν2 in Figure 1D). The remaining transaxial bend, δ transaxialoop, consisting of equatorial Fe motion perpendicular to the Fe-NO plane, is calculated at 216 cm−1 (ν3b in Figure 1D) and would be under the broad low energy shoulder at 205 cm−1. While the pair of transaxial bends are typically degenerate, they are split in energy in this case due to mixing of the δ transaxialoop with an antisymmetric Fe-catecholate stretch calculated at 290 cm−1 (ν5 in Figure 1D), not discernible in the experimental data due to its small amount of Fe displacement. Previous studies have similarly found that the transaxial bends can be split due to mixing with nearby vibrational modes.33 Also under the shoulder at 205 cm−1 is a vibration that consists of z-translation of the entire Fe-NO unit, calculated at 191 cm−1 (ν4 in Figure 1D). Finally, the 419 cm−1 peak in the data can be correlated to the highest energy feature in the calculated spectrum, which is the νFe-N at 360 cm−1 (ν1 in Figure 1D). The experimental νFe-N of 419 cm−1 is 77 cm−1 lower in energy than the νFe-N in Fe-EDTA-NO, which can be attributed to strong donation from the catecholate to the Fe weakening its axial Fe-N bond. However, the calculated νFe-N is low by 59 cm−1 relative to experiment. This is consistent with the TZVP basis set overestimating the Fe-N bond length and thus underestimating the Fe-N bond strength, leading to an νFe-N that is too low in energy, as also observed above for Fe-EDTA-NO. Several structures with fixed Fe-N bond lengths were optimized (1.86, 1.84, and 1.82 Å), and fixing this bond at 1.82 Å was found to give a structure with νFe-N at 402 cm−1 that is only 1.1 kcal/mol higher in energy than the freely optimized structure. Importantly, this perturbation does not have a significant effect on the DFT calculated NRVS spectrum in the region below 350 cm−1 (see Figure S3 in the SI). The BP86 + 10% HFX/TZVP method is thus calibrated for application to the Int-1 and Int-2 NRVS data.
The electronic structures of {FeNO}7 EDTA and by correlation H200N-4NC-NO have previously been determined39 and are high-spin FeIII (S = 5/2) antiferromagnetically coupled to NO— (S = 1), leading to the S = 3/2 ground states observed experimentally for both. The H200N-4NC-NO NRVS data provide a well-defined reference for analysis of the NRVS data for the {FeO2}8 Int-1 and Int-2, presented in Figures 2 and 3. There are a number of possible structures for these Fe/O2 intermediates: side-on and end-on FeIII-superoxy-catecholate, side-on and end-on FeII-superoxy-semiquinone, side-on and end-on FeIII-peroxy-semiquinone, and side-on and end-on FeIII-hydroperoxy-semiquinone. From all of these potential starting structural guesses, only two geometry optimized structures could be obtained using the calibrated BP86 + 10%HFX / TZVP method: all unprotonated starting structures converged to an end-on FeIII-superoxy-catecholate, and all protonated starting structures converged to an end-on FeIII-hydroperoxy-semiquinone (electronic structure parameters for these optimized structures are given in Table S1). The end-on FeIII-superoxo-catecholate structure gives a calculated NRVS spectrum in good agreement with the NRVS data on Int-1 (Figure 2), and the end-on FeIII-hydroperoxo-semiquinone structure gives good agreement with the NRVS data of Int-2 (Figure 3). These structural assignments are consistent with EPR and Mössbauer data on these complexes,23 with the mode of binding and protonation state now defined by NRVS.
Figure 2.

A) NRVS data for Int-1 with modes assigned by correlation to B. B) DFT calculated spectrum for the Int-1 structure that best fits the NRVS data: an end-on FeIII-superoxo-catecholate with the Fe-OO unit bent towards and bisecting the catecholate. Modes are labeled as in D. C) Final DFT structure of Int-1, with important geometric parameters listed below the structure. Color scheme is as in Figure 1. D) Schematic depictions of the normal modes that appear in the NRVS data and calculated spectrum.
Figure 3.

A) NRVS data for Int-2 with normal modes assigned by correlation to B. B) DFT calculated spectrum for the Int-2 structure that best fits the NRVS data: an end-on FeIII-hydroperoxy-semiquinone with the hydroperoxy moiety bent towards and hydrogen bonding with the semiquinone oxygen with more O− character and a shorter Fe-O bond (O(C1)). Modes are labeled as in D. C) Final DFT structure of Int-2. Oxygens are shown in red, nitrogens in blue, iron in orange, hydrogens in white, protein carbons in green, and substrate carbons in purple. C1 and C2 are labeled, and important structural parameters are given below the structure. D) Schematic depictions of the normal modes (with labels) that appear in the NRVS data and calculated spectrum.
As shown in the overlay of the H200N-4NC-NO and Int-1 data in Figure S4, the Int-1 data (Figure 2A) are qualitatively similar to those for H200N-4NC-NO (Figure 1A): Int-1 has two intense peaks in the 250–330 cm−1 energy region with the same intensity ratio as H200N-4NC-NO and a broad low-energy shoulder from 200 to 250 cm−1. The primary difference between the spectra is a shift of the two main bands in Int-1 to higher energy. In addition, no peaks above the noise level appear in the Int-1 data at higher energies (above 330 cm−1). The DFT optimized structure for the FeIII-superoxo Int-1 is also similar to that for H200N-4NC-NO; its Fe-OO unit is also bent and oriented towards and bisecting the catecholate, though its Fe-O bond is longer than the Fe-N (1.96 vs 1.88 Å) and the Fe-O2 has a more acute Fe-OO angle (124° vs 132°). Note that a second end-on superoxo conformer could be geometry optimized, with the Fe-OO unit bent away from the catecholate; this is higher in energy (by 0.4 kcal/mol) and gives worse agreement with the NRVS spectrum (calculated spectrum is shown in Figure S5). The DFT calculated NRVS spectrum for the lower energy structure is given in Figure 2B (with the structure shown in Figure 2C) and is used to assign the Int-1 NRVS spectrum.
From the DFT calculated spectrum, the intense peaks at 270 cm−1 and 305 cm−1 are, respectively, assigned as the δ transaxialip (calculated at 267 cm−1, ν3a in Figure 3D) and the δFe-OO (calculated at 313 cm−1, ν in Figure 2D). The δ transaxialoop contributes intensity to the shoulder between 200 and 250 cm−1 (225 cm−1, ν3b in Figure 2D); as observed for H200N-4NC-NO, δ transaxialoop is also calculated at lower energy than δ transaxialip due to mixing with the antisymmetric νFe-catecholate, calculated at 291 cm−1 (ν5 in Figure 2D, again not discernible in the data due to its lack of Fe displacement). Also contributing intensity to the 200–250 cm−1 shoulder is the z-translation of the Fe-OO unit (calculated at 197 cm−1, ν4 in Figure 2D). Finally, the νFe-O is calculated at 351 cm−1 (ν1 in Figure 2D). While this could correlate to the peak at 353 cm−1 in the data, this feature is not significantly above the noise, and the νFe-O is calculated to have very little Fe displacement; thus, no assignment is made.
With the assignment of the NRVS spectrum of Int-1, the origins of the differences between its spectrum and that of H200N-4NC-NO can be defined. The largest change is the higher energy δ transaxialip in Int-1 relative to H200N-4NC-NO (270 cm−1 vs 255 cm−1). This difference is due to the change from NO− to O •- 2(i.e. {FeNO}7 vs {FeO2}8), where the extra antibonding electron eliminates some of the π bonding interaction between the N/O and Fe;65,66 this weakens the axial bond and leads to stronger equatorial bonding and increased vibrational energy (also reflected in the Fe-catecholate and Fe-N/O bond lengths given in Figures 1C and 2C). The δFe-(N/O)-O energy also increases from 296 to 305 cm−1, which can be attributed to the decrease in the Fe-(N/O)-O angle in going from NO− to O •−2. This angle change partially compensates for the loss of the π bonding interaction by increasing the σ overlap between the O •−2 π* orbital in the Fe-OO plane with dz2, as described in Ref 66.
With the structure of Int-1 assigned from its NRVS spectrum, we now turn to its decay product, Int-2. Its NRVS spectrum is shown in Figure 3A. In contrast with Int-1, the Int-2 data show one intense, broad peak at 267 cm−1 with a high-energy shoulder at 296 cm−1, but otherwise look similar to those for Int-1. (see an overlay of Int-2 and Int-1 NRVS data in Figure S6). Qualitatively, this suggests a similar assignment for Int-2 and Int-1, but with the δFe-OO shifted down in energy in Int-2 and merged with the intense δ transaxial peak (also clear from Figure S6). The DFT calculated NRVS spectrum for the end-on FeIII-hydroperoxide-semiquinone structure in Figure 3B supports this assignment. Note that this structure includes a hydrogen bonding interaction between the hydroperoxy proton and one O of the semiquinone that is more tightly bonded to the Fe (O(C1)), as shown in Figure 3C. (A structure without this hydrogen bond could also be optimized: this structure is 2.5 kcal/mol higher in energy than the hydrogen bonded conformation and gives worse agreement with the NRVS data, as shown by its DFT calculated spectrum in Figure S7.) The 296 cm−1 high-energy shoulder in the experimental data correlates with the calculated δFe-OO at 302 cm−1 (ν2 in Figure 3D). The intense 267 cm−1 experimental peak correlates with the calculated δ transaxialoop at 257 cm−1 (ν3b in Figure 3D). The peak at 217 cm−1 can be primarily assigned to the δ transaxialip, which mixes over several modes in this energy range and has an intensity-weighted average energy of 224 cm−1 (ν3a in Figure 3D). This mode is now mixed with both the δFe-OO and the antisymmetric νFe-semiquinone (calculated at 268 cm−1 with significant NRVS intensity [ν5 in Figure 3D], but overlapping the δ transaxialoop). This mixing of δ transaxialip with both the δFe-OO and the antisymmetric νFe-substrate (as was observed in H200N-4NC-NO) results from the structure (Figure 3C) where the Fe-OOH is hydrogen bonded to a substrate O rather than bisecting the semiquinone. Finally, the calculated νFe-O is at 434 cm−1 (ν1 in Figure 3D). This could correlate with the peak at 445 cm−1 in the data; however, this peak is not significantly above the noise level, and the calculated νFe-O intensity is low, making a definitive assignment unwarranted. It is important to note, however, that the calculated νFe-O energy is low relative to that previously observed experimentally for a 6C high-spin FeIII-alkylperoxo complex,68 which was at 637 cm−1. This is consistent with donation from the 4NC semiquinone substrate weakening the axial Fe-O bond.
From their NRVS assignments, the primary difference between Int-1 and Int-2 is the lower energy of the δFe-OO bend in the latter (296 [Figure 3A] vs 305 cm−1 [Figure 2A]). This effect is reproduced by the DFT calculations, which predict the Fe-OO bend energy decreasing by 11 cm−1, from 313 to 302 cm−1, in going from Int-1 to Int-2 (from Figures 2B and 3B). This energy decrease is due to an increased mixing between the δFe-OO and νO-O for Int-2 relative to Int-1 (3.3% vs 0.0%), which occurs because the calculated peroxy νO-O is lower in energy than that of the superoxo (892 vs 1129 cm−1).
The NRVS data, in combination with comparisons to NRVS spectra derived from DFT calculations, has thus provided a basis for geometric and electronic structure assignments for Int-1 and Int-2, including definition of the protonation state and orientation of the Fe-OO unit. The calculated electronic structures of Int-1 and Int-2 are also consistent with previous Mössbauer and EPR data23 and calculations.11,41 Importantly, the NRVS-derived structures for Int-1 and Int-2 are also fully consistent with the DFT structures obtained by removing H200 from the possible superoxo and hydroperoxo intermediates optimized for wild-type HPCD-4NC from the crystal structure in Section 3.2.2.
3.2. Computational correlation to crystallographic intermediates
3.2.1. Selection of the cluster model for DFT calculations
The crystallographic structure at the active site of the HPCD for the side-on O2 intermediate (PDB code 2IGA, subunit C) shows that the Fe center is coordinated by the 2His/1Glu facial triad, H155, H214 and E267, O2, and the 4NC dianion.16 The initial model for optimizing the side-on O2 intermediate consisted of just these residues; however, a side-on O2 complex could not be optimized using this simple facial triad model. The model was thus expanded to include second sphere residues. All second sphere residues hydrogen bonding to facial triad residues, the O2, or the 4NC oxygens coordinated to Fe with heavy atom distances 3.0 Å or less were included to generate an intermediate model I: these additional residues are H200, H248, Y257, and N157. While both side-on O2 and end-on O2 structures could be obtained using this model, the orientation of N157 was incorrect. To improve this, we generated a model where W192 was added (model II); this was not found to improve the N157 orientation. A third model was generated where W192 was removed and T205 was added, model III. Here, the N157 orientation was correct, as well as those of the other residues, and model III was used as the final cluster model. The remaining issue is the protonation state of H248; while most previous studies11,41 have included this as a doubly protonated imidazolium, we found that this lead to endergonic O2 binding in model III (+8.4 kcal/mol), while O2 binding was found to be exergonic with an imidazole H248 hydrogen bonding to E267 (−6.9 kcal/mol, where this tautomer was chosen due to the short N248-E267 heavy atom distance of 2.9 Å). The final cluster model used for this study thus consisted of H155, H214, E267, H200 (imidazole or imidazolium, vide infra), H248 (imidazole), Y257, N157, and T205, as shown in Scheme 1. Note that the relative energetics of the end-on and side-on structures (vide infra) are approximately the same for both protonation states of H248 in models I, II, and III.
Scheme 1.

Depiction of the final DFT cluster model, model III.
3.2.2. Side-on O2 intermediate
To correlate the H200N solution intermediates to the wild-type crystallographic intermediates in HPCD, the wild-type crystallographic side-on intermediate was evaluated computationally. This intermediate was previously assigned as an FeII-superoxo-semiquinone species based on its geometric structure. Model III with a neutral H248 hydrogen bonding to E267 was used for these calculations. H200 was included either doubly protonated or with one proton transferred to the O2 moiety (this proton likely derives from the 4NC substrate, which is a monoanion in solution and loses its proton upon binding to the Fe,18 or from a proton transfer pathway44). Geometry optimizing the first of these structures (proton on H200) on the S = 2 surface leads to the end-on O2 species 1a, in poor agreement with crystallography, while optimization of the second of these structures (proton on O2) gives the side-on O2H species 1b, which agrees well with the crystal structure. These DFT structures are shown in Figure 4, with important geometric and electronic structural parameters given in Table 2. From the bond lengths and spin densities in Table 2, 1a is best described as an end-on high-spin FeIII-superoxy catecholate species (O-O = 1.34 Å, spin 4NC = 0.05) with the protonated H200 hydrogen bonded to the proximal O (N-Oprox distance = 2.67 Å), with the superoxo oriented towards the substrate C2 by hydrogen bonds to H200 and N157. Structure 1b reproduces the crystallography well, as demonstrated by the overlay of 1b with the crystal structure given in Figure S8, and is a side-on high-spin FeIII-hydroperoxy semiquinone species, with the unpaired β spin density delocalized over the semiquinone (O-O = 1.44 Å, spin 4NC = −0.47; frontier molecular orbitals (FMOs) are given in the SI, Figure S10). One significant difference between 1b and the crystallography is their differing distortions from planarity for C2 (0.006 Å vs 0.23 Å). This distortion was used to argue for semiquinone character in the crystal structure. As described in the SI, a bound semiquinone should not have an out of plane distortion more than 0.0067 Å; thus, the structure of 1b is consistent with its semiquinone description. These two structures are nearly isoenergetic in free energy (0.4 kcal/mol favoring the side-on hydroperoxy structure, −1.4 kcal/mol in ΔH). The side-on intermediate observed in the crystal structure is thus assigned computationally as an FeIII-hydroperoxy semiquinone species, rather than having an FeII-superoxy semiquinone electronic structure. Protonation of the O2 moiety has induced electron transfer from the 4NC substrate to the O2, as was also found for the H200N intermediates (see Section 3.1). Transfer of this proton back to H200 strengthens the newly deprotonated Fe-O(1) bond, leading to loss of the Fe-O(2) bond proximal to the substrate and transfer of an electron from O2 back to the substrate, generating an end-on FeIII-superoxo with the O2 moiety oriented towards the catecholate, as shown in Scheme 2. We estimate a low barrier of 3.3 kcal/mol for this conversion on the basis of a calculated 2D potential energy surface (Figure S11 and Table S2) where the O(1)-H and Fe-O(2) distances were varied.
Figure 4.

DFT structures of the H200(H)+ end-on O2 species 1a, best described as a high-spin FeIII-superoxo-catecholate, antiferromagnetically coupled, and the H200 side-on O2H species 1b, best described as a high-spin FeIII-hydroperoxo-semiquinone, antiferromagnetically coupled. The iron center, fixed carbon atoms, and the phenyl ring of the substrate are colored with yellow, pale green, and gray, respectively.
Table 2.
Geometric and electronic structural parameters and energetics of 1a and 1b, as well as of the crystal structure of the side-on O2 intermediate1.
| 1a | 1b | crystallography | |
|---|---|---|---|
| energy (kcal/mol) | |||
| ΔE | −1.4 | 0.0 | |
| ΔH | −1.4 | 0.0 | |
| ΔG | 0.4 | 0.0 | |
| bond length (Å) | |||
| Fe—O(1) | 2.051 | 2.234 | 2.5 |
| Fe—O(2) | 2.971 | 2.005 | 2.4 |
| O—O | 1.344 | 1.442 | 1.3 |
| Fe—O(C1) | 2.017 | 2.026 | 2.2 |
| Fe—O(C2) | 1.969 | 2.220 | 2.2 |
| C1—O | 1.314 | 1.288 | 1.3 |
| C2—O | 1.332 | 1.285 | 1.4 |
| C1—C2 | 1.447 | 1.480 | 1.5 |
| C2(deviation from ring) | 0.007 | 0.006 | 0.23 |
| angle and dihedral angle (deg.) | |||
| ∠O(2)-O(1)-Fe | 120.7 | 61.7 | 70.5 |
| ∠O(1)-O(2)-Fe | 36.4 | 79.0 | 78.9 |
| ∠O(2)-O(1)-Fe-C(C2) | 6.3 | 31.9 | 3.9 |
| hydrogen bonds (Å) | |||
| O(1)-H | 1.615 | 1.046 | — |
| O(1)-N(H200) | 2.673 | 2.606 | 2.469 |
| spin density | |||
| Fe | 4.03 | 4.12 | |
| O(1) | −0.20 | 0.12 | |
| O(2) | −0.47 | 0.25 | |
| O2/O2H | −0.66 | 0.37 | |
| C6(4NC) | 0.05 | −0.47 |
The numbering of atoms are shown in Figure 4.
Scheme 2.

Energetically similar structures 1b and 1a, obtained with the proton on the O2 moeity and on H200, respectively.
3.2.3. Effect of H200 on the relative energetics of a superoxo catecholate and hydroperoxo semiquinone
In wild-type HPCD-4NC, the computations described above show that the crystallographic intermediate 1b and the superoxo species 1a are approximately isoenergetic. To define the role H200 plays in tuning these relative energetics, and to correlate the crystal structure calculations to the H200N NRVS solution data in Section 3.1, H200 was removed from 1a and 1b, and both starting structures were reoptimized. Note that in the H200 asparagine variant, the side chain of N200 is far enough away from the Fe (5.2 Å) that it would not participate in hydrogen bonding with the O2 unit.44 The final structures resulting from this calculation are shown in Figure 5, and geometric and electronic structural parameters for both are given in Table S3 in the SI. Removing [H200(H)]+ from 1a gives an end-on FeIII-superoxo species (1a’) that is similar to the NRVS derived structure of Int-1 (Figure 2C), but with some quantitative geometric and electronic structural differences (see Table S4 and the overlay in Figure S9, left). The primary structural differences are a long Fe-O bond relative to Int-1 (2.08 vs. 1.96 Å) and a rotation of H248 in 1a’ relative to the crystallographic and Int-1 orientations. This rotation is precluded by Van der Waals interactions with residues present in the crystal structure but not included in the computational model. Recalculating 1a’ using the same basis set but with H248 constrained in the crystallographic position gave a geometry optimized 1a’ structure with the same electronic structure as Int-1 and a nearly equivalent Fe-O bond length (1.950 Å); structural parameters for this complex are also given in Table S4, and an overlay with Int-1 is given in Figure S9, center. This constrained 1a’ structure is only 1.2 kcal/mol higher in energy than the freely optimized structure and is shown in Figure 5, right. The constrained 1a’ structure is used for energetic comparisons in the rest of the text. Similarly, removing [H200]0 from 1b gives an end-on FeIII-hydroperoxo species (1b’) that equivalent to the NRVS-defined structure of Int-2 (Figure 3C; geometric and electronic structure details are given in Table S3). An overlay of Int-2 with 1b’ is shown in Figure S9, right, and Table S4 gives a comparison of structural parameters for both species. In contrast to 1a and 1b being at approximately the same energy in wild-type, with H200 removed the hydroperoxy FeIII-semiquinone 1b’ is 17.3 kcal/mol more stable in enthalpy (20.5 kcal/mol in free energy) than 1a’ (proton transferred to a histidine). This is depicted schematically in Figure 6. As the estimation of the H200 interaction energy involved optimizing H200 and the rest of the system in separate calculations (as shown on the left side of Figure 6), the energetics were corrected for basis set superposition error (BSSE) using Boys counterpoise method. From Figure 6, the FeIII-superoxo intermediate is destabilized by removing H200 by 17.3 kcal/mol, and the hydroperoxo is stabilized by 1.4 kcal/mol. The superoxo is destabilized because, upon removal of [H200(H)]+, the ionic interaction and hydrogen bond between [H200(H)]+ and the proximal superoxo O are lost, while the hydroperoxo is slightly destabilized because the loss of the H200 hydrogen bond to the hydroperoxo proton is compensated by gaining a stronger hydrogen bond to the oxygen atom on the substrate C1 (as shown in Figure 5, right). Thus, in wild-type HPCD H200 tunes the energies of the superoxo and hydroperoxo species, making them similar in energy. The side-on hydroperoxy intermediate is the one experimentally observed in the crystal, perhaps because it is further stabilized by crystal packing forces. In contrast to wild-type, in H200N the hydroperoxo species is significantly more stable than the superoxo (by 17.3 kcal/mol in enthalpy, 20.5 kcal/mol in free energy).
Figure 5.

Geometry optimized structures for 1a’ and 1b’ generated by removal of H200 from 1a and 1b, respectively. The representations are the same as Figure 4.
Figure 6.

Energy diagram showing the effect of H200 on the relative enthalpies of superoxy and hydroperoxy intermediates in HPCD. The energetics of wild-type 1a and 1b are shown on the right, and 1a’ + [H200(H)]+ and 1b’ + [H200]0 are shown on the left (which include a non-interacting H200 in a separate calculation as a proton donor/acceptor). Removing H200 destabilizes the FeIII-superoxo intermediate by 17.3 kcal/mol and stabilizes the FeIII-hydroperoxo intermediate by 1.4 kcal/mol. Here, enthalpy is shown rather than Gibbs free energy due to entropic differences introduced for the structures on the left through removal of the H200. Energies have been corrected for BSSE.
It should be noted that this assignment of the side-on O2 intermediate as a FeIII-hydroperoxysemiquinone is different from the previous assignment as a FeII-superoxy-semiquinone, and that FeIII has not been observed in solution spectroscopy of any stable or intermediate form of HPCD in which H200 is present.4,16It is essential to computationally consider whether it is possible that the intermediate in the crystal structure is indeed an FeII-superoxo-semiquinone. We thus attempted to geometry optimize the alternative FeII electromer. We found that it was necessary to increase the Hartree-Fock exchange included in our calculations to 50%, as the FeII state could not be optimized with either 10% or 20% HFX (all initial electronic states converged back to FeIII species). With 50% HFX, four structures for the initial Fe-O2 species could be optimized; the relative enthalpies of these species are given in Figure 7, left. Lowest in energy is the end-on FeIII-superoxo catecholate, with the side-on FeIII-hydroperoxo semiquinone at +8.1 kcal/mol, the end-on FeII-superoxo semiquinone at +10.7 kcal/mol, and the side-on FeII-superoxo-semiquinone at +16.3 kcal/mol. Note that with the overly large amount of HFX needed to obtain an FeII electromer, the side-on FeIII-hydroperoxy-semiquinone is significantly higher in energy than the end-on FeIII-superoxo-catecholate due to the decrease in covalency of both Fe-O2 bonds. To calibrate these results, we also optimized the end-on FeIII-superoxo-catecholate in H200N, Int-1, and its end-on FeII-superoxo-semiquinone electromer. As detailed above, Int-1 is well defined from spectroscopy to be an FeIII-superoxo-catecholate complex. As shown on the right side of Figure 7, these calculations found that the FeII-superoxo-semiquinone electromer is at +13.4 kcal/mol, higher in enthalpy relative to the FeIII-superoxo-catecholate, consistent with the experimental observation that Int-1 is FeIII. (Further supporting rejection of the FeII state, the FeII-superoxo-semiquinone electromer gives a DFT calculated NRVS spectrum that is not consistent with the experimental data for Int-1, as shown in Figure S12.) Given the similar difference in energy between the end-on FeIII-superoxo and FeII-superoxo species in wild-type (10.7 or 16.3 kcal/mol, depending on O2 binding mode, Figure 7, left), H200 is not found to play a significant role in stabilizing the FeII-superoxo-semiquinone state. Further, in the wild-type calculation, even with 50% HFX, the side-on FeIII-hydroperoxy-semiquinone species is 8.2 kcal/mol lower in energy than the side-on FeII-superoxy-semiquinone, consistent with the above assignment of the crystallographic intermediate as a side-on FeIII-hydroperoxy-semiquinone.
Figure 7.

Relative enthalpies of BP86+50%HFX geometry-optimized FeIII and FeII structures of the initial Fe-O2 intermediate in wild-type (left) and with H200 removed (right).
3.2.4. Peroxy bridge intermediate
The geometric and electronic structure of the peroxy bridged intermediate (subunit D in the wild-type crystal structure),16 which is the product of 1a/1b reacting with the substrate, was also evaluated. This intermediate was previously assigned as an FeII-peroxy-quinone bridged species.16 As with the side-on O2 structure evaluated in Section 3.2.2, this bridged intermediate can also be either unprotonated with [H200(H)]+ (2a) or protonated with [H200]0 (2b). The DFT structures of 2a and 2b are shown in Figure 8, and geometric and electronic structural parameters for both (as well as the geometric parameters for the crystal structure) are given in Table 3. Of these two structures, the unprotonated peroxy bridged species 2a is in better agreement with the crystal structure, as its calculated Fe-O bond length of 2.065 Å is consistent with the crystallographic value of 2.088 Å, whereas the 2.366 Å bond length of the hydroperoxy bridged species 2b (ε=20.0) is too long relative to experiment. These structures are also approximately isoenergetic (ΔG = 0.2 kcal/mol at ε=20.0, favoring the unprotonated bridged species). Thus, it is reasonable to assign the crystallographic structure as the unprotonated peroxy bridged intermediate 2a, where the peroxy O bound to the Fe is hydrogen bonded to the protonated H200. As with the side-on FeIII-hydroperoxy intermediate, it is likely that this intermediate (2a) is further stabilized by crystal packing forces, as it is experimentally observed. Figure 9 gives the important FMO for 2a and 2b. The total spin on 4NC (Table 3) provides a basis for assigning the electronic structures of these intermediates. From the FMO in Figure 9, the electronic structure of 2a is best described as a mixture of FeII-peroxy-quinone/FeIII-peroxy-semiquinone character. The hydroperoxy 2b has a similar electronic structure with slightly more FeII/quinone character due to the proton on the O proximal to the Fe that weakens the Fe-O bond and thus enhances charge donation from the substrate to the Fe. The effect of H200 on the relative energetics of 2a and 2b was also evaluated by removing [H200(H)]+ from 2a and reoptimizing (2a’) and removing [H200]0 from 2b and reoptimizing (2b’). The results of these calculations are summarized in Figure 10, with DFT optimized structures and geometric and electronic structural parameters for 2a’ and 2b’ given in the SI (Table S3). From these calculations, removing [H200(H)]+ from 2a leads to a peroxy bridged intermediate 2a’ that is less stable by 27.4 kcal/mol, and removing [H200]0 from 2b leads to a hydroperoxy bridged intermediate 2b’ that is less stable by 10.2 kcal/mol (structures are shown in Figure S13, with geometric and electronic structure parameters given in Table S2). Similar to the results for the initial O2 intermediates in Section 3.2.3, the presence of H200 in the wild-type structure makes the two protonation states of the peroxo bridged intermediate approximately the same in energy, while the protonated peroxy bridged species is more stable by 17.9 kcal/mol in the absence of H200 (as in H200N). As with Figure 6, these energies have been corrected for BSSE. The stabilization of the unprotonated 2a by H200 through the ionic interaction, including a hydrogen bond, between [H200(H)]+ and the negatively charged peroxy O in Figure 10 is even greater than the stabilization of the superoxy intermediate 1a; this is attributed to a stronger ionic interaction due to the greater negative Mülliken charge on the hydrogen-bonded O of the peroxo in 2a’ (–0.56) relative to the superoxo 1a’ (–0.37) (Mülliken charges given in the SI, Table S3).
Figure 8.

DFT structures of the [H200(H)]+ peroxy bridged species species 2a and the [H200]0 hydroperoxy bridged species 2b (ε = 4.0), with the Fe-OO in the plane of the page. The representations are the same as previous figures. Bond lengths are shown in Å. The bond lengths in parentheses are hydrogen bond heavy atom distances.
Table 3.
Geometric and electronic structural parameters and energetics of 2a and 2b, as well as of the crystal structure of the hydroperoxy bridged intermediate.1
| 2a2 | 2b | 2a3 | 2b | crystallography | |
|---|---|---|---|---|---|
| dielectric constant for PCM | 20.0 | 20.0 | 4.0 | 4.0 | |
| energy (kcal/mol) | |||||
| ΔE | 0.0 | 1.0 | 2.9 | 0.0 | |
| ΔH | 0.0 | 1.2 | 2.7 | 0.0 | |
| ΔG | 0.0 | 0.2 | 3.7 | 0.0 | |
| bond length (Å) | |||||
| Fe—O(1) | 2.065 | 2.366 | 2.058 | 2.376 | 2.1 |
| O—O | 1.463 | 1.470 | 1.469 | 1.470 | 1.5 |
| C2—O(2) | 1.518 | 1.504 | 1.501 | 1.505 | 1.4 |
| Fe—O(C1) | 2.137 | 2.046 | 2.127 | 2.050 | 2.2 |
| Fe—O(C2) | 1.990 | 1.997 | 2.005 | 2.000 | 2.3 |
| C1—O | 1.279 | 1.278 | 1.280 | 1.276 | 1.3 |
| C2—O | 1.370 | 1.363 | 1.369 | 1.361 | 1.5 |
| C1—C2 | 1.531 | 1.530 | 1.532 | 1.531 | 1.5 |
| C2(deviation from ring) | 0.380 | 0.385 | 0.380 | 0.385 | 0.46 |
| hydrogen bonds (Å) | |||||
| O(1)-H | 1.523 | 1.060 | 1.976 | 1.049 | — |
| O(1)-N(His200) | 2.621 | 2.623 | 3.024 | 2.642 | 3.0 |
| spin density | |||||
| Fe | 4.03 | 3.99 | 4.00 | 3.98 | |
| O(1) | 0.06 | 0.01 | 0.07 | 0.01 | |
| O(2) | −0.02 | −0.02 | −0.01 | −0.02 | |
| O2/O2H | 0.04 | −0.01 | 0.06 | −0.01 | |
| C6(4NC) | −0.43 | −0.38 | −0.38 | −0.36 |
The numbering of atoms is shown in Figure 4.
Note that geometry optimization with ε = 4.0 was found to converge to 2b from both possible starting structures. Single-point energy using ε = 4.0 with thermal corrections with ε = 20.0 were performed to compare energetics of 2a and 2b below.
The optimized structure using ε = 4.0 with O(1)-N(His200) fixed. The energies relative to 2b optimized under the same condition as 2a and the thermal corrections with ε = 20.0 were applied to both 2a and 2b.
Figure 9.

β LUMOs for 2a and 2b calculated at the level of BP86 mixed with 10% Hartree-Fock exchange, using the 6–311G* basis set on Fe-O2 and 6–31G* on the remaining atoms. The isosurfaces are plotted with values of ±0.06 a.u.–3/2. These MOs that show their electronic structures as having mixed FeIII-hydroperoxy-semiquinone / FeII-hydroperoxy-quinone character, with 2b having more FeII-quinone character due to the protonated proximal O. The numbers in parentheses are weights of orbitals of the Fe 3d, the O2, and 4NC, in that order.
Figure 10.

Energy diagram showing the effect of H200 on the relative enthalpies of peroxy and hydroperoxy bridged intermediates in HPCD. The energetics of wild-type 2a and 2b are shown on the right, and 2a’ + [H200(H)]+ and 2b’ + [H200]0 are shown on the left. Removing H200 destabilizes the Feperoxo bridged intermediate by 27.4 kcal/mol and the Fe-hydroperoxo bridged intermediate by 10.2 kcal/mol. In Figure 10, all the electronic energy calculations were carried out using ε=4.0. 2b was geometry optimized and its thermal correction calculated using ε=4.0. For 2a, the optimized geometry and thermal correction of 2a were calculated with ε=20.0, with a single-point ε=4.0 calculation used on this optimized structure to obtain the electronic energy.
3.3. Reaction coordinate for forming the peroxo bridge in wild-type HPCD
The reaction coordinate for generating the peroxy bridged intermediate in wild-type was investigated, with both 1a and 1b considered as possible reactants, as these would be at very similar energy and in an equilibrium, and both 2a and 2b are considered as possible products for the same reason. Reaction coordinates for the FeIII-superoxo 1a forming both the non-protonated 2a and the protonated 2b peroxo bridged species were obtained. These reaction coordinates are shown in Figure 11, and geometric and electronic structural parameters for the transition states are given in Table 4. Both reaction coordinates for 1a pass through a low barrier and have product-like transition states. With respect to Gibbs free energy, the reaction coordinate for generating the unprotonated bridge (Reaction 1) has a slightly lower barrier (7.5 kcal/mol) than the reaction coordinate for generating the protonated bridge (Reaction 2, 8.2 kcal/mol) (though as shown in Figure 11 and from Table 4, Reaction 2 has the lower barrier in electronic energy (7.8 kcal/mol, vs 8.8 kcal/mol for Reaction 1)). The geometric and electronic structures for both transition states are similar; the primary difference is that the proton is already transferred from H200 to the proximal O (O-H distance = 1.059 Å) in the transition state for reaction 2 (TS2, structure shown in the SI, Figure S14) and remains on H200 (O-H distance = 1.414 Å) in the TS for Reaction 1 (TS1, structure shown in the SI, Figure S14). TS1 has a shorter Fe-O(proximal) bond length than TS2 (2.102 vs. 2.247 Å, respectively). Both of these barriers are lower than the ~17.5 kcal/mol ΔG‡ associated with the reaction rate of 0.05 s−1,27 indicating that the superoxo species 1a is competent to form the peroxo bridge. Further, the barrier for both reactions is primarily due to the thermodynamics of peroxo bridge formation, given the late transition states (from Figure 11) and similar energetics of the transition states and products (from Table 4).
Figure 11.

Reaction coordinate for the superoxo intermediate 1a forming the peroxo bridged intermediate 2a (reaction 1, black) and the hydroperoxo bridged intermediate 2b (reaction 2, red) (with electronic energies shown) plotted against the distance between the distal oxygen O(2) and the bridge head C2(3) shown in Figure 4. Electronic energies are quoted relative to the side-on hydroperoxo complex, 1b (i.e. the energy of 1b is 0 kcal/mol). IRCs to the reactant 1a for both of the 2a and 2b led to lower energy structures close to, but not identical to, 1a because the constraint on the positions of the carbon atoms fixed at the crystallographic structure are slightly relaxed along the IRC. The proton transfer in Reaction 2 occurred automatically during the IRC scan from TS2 back to the reactants.
Table 4.
Geometric and electronic structural parameters for the transition state between 1a and 2a (TS1) and for the transition state between 1a and 2b (TS2), as well as for the products 2a and 2b.1
| TS1 | 2a2 | TS2 | 2b | |
|---|---|---|---|---|
| energy (kcal/mol) | ||||
| ΔE‡/ΔE0 | 8.8 | 8.3 | 7.8 | 6.1 |
| ΔH‡/ΔH0 | 6.1 | 5.3 | 5.4 | 4.6 |
| ΔG‡/ΔG0 | 7.5 | 10.53 | 8.2 | 7.0 |
| bond length (A)1 | ||||
| Fe—O(1) | 2.102 | 2.065 | 2.247 | 2.376 |
| O—O | 1.429 | 1.463 | 1.432 | 1.470 |
| C2—O(2) | 1.753 | 1.518 | 1.812 | 1.505 |
| Fe—O(C1) | 2.112 | 2.137 | 2.070 | 2.050 |
| Fe—O(C2) | 2.011 | 1.990 | 2.016 | 2.000 |
| C1—O | 1.281 | 1.279 | 1.279 | 1.276 |
| C2—O | 1.331 | 1.370 | 1.321 | 1.361 |
| C1—C2 | 1.504 | 1.531 | 1.499 | 1.531 |
| C2(deviation from ring) | 0.261 | 0.380 | 0.239 | 0.385 |
| hydrogen bonds (Å)1 | ||||
| O(1)-H | 1.414 | 1.523 | 1.059 | 1.049 |
| O(1)-N(His200) | 2.556 | 2.621 | 2.606 | 2.642 |
| spin density1 | ||||
| Fe | 4.02 | 4.03 | 4.01 | 3.98 |
| O(1) | 0.03 | 0.06 | 0.00 | 0.01 |
| O(2) | −0.11 | −0.02 | −0.13 | −0.02 |
| C6(4NC) | −0.31 | −0.43 | −0.30 | −0.36 |
The numbering of atoms (the numbers in parentheses in the left column) is as shown in Figure 8.
Single-point energy using ε = 4.0 with thermal corrections with ε = 20.0 were performed.
2a in ε = 20.0 has a small entropy (390.0 cal/mol K) relative to TS1 at ε = 4.0 (402.5 cal/mol K), because the number of imaginary modes irrelevant to the reaction barrier, resulting from the steric constraint imposed on the geometry optimization, in TS1 is less than in 2a. As a result, this entropy effect lowers TS1 more than 2a with respect of Gibbs free energy. This entropy effect arises from a tight electrostatic interaction between protonated H200 and the bare anionic O2 moiety, and because of this we consider enthalpy in energetic comparisons involving 2a below.
The FMOs (i.e. the α and β LUMOs) involved in the slightly lower-energy Reaction 1 provide insight into the electronic structural changes that occur over the course of attack on the substrate and are given in Figure 12; the similar Reaction 2 FMOs are given in the SI (Figure S15). The FMO used by the superoxo intermediate to attack the substrate is its π*σ FMO (i.e. the O2•- π* orbital in the Fe-OO plane, the α LUMO in Figure 12, upper-left), which has its lobes in the Fe-OO plane and thus good overlap with the catecholate π cloud. From the FMOs in Figure 12, at the TS, a total of 0.61 α electrons have been transferred from the substrate to the bound superoxo and 0.20 β electrons from the substrate to the Fe, and at the product a total of 0.70 α electrons have been transferred from the substrate to the O2 and 0.17 β electrons have been transferred from the substrate to the Fe(dπ). This is consistent with the description given in Section 3.2.4 of 2a; the FeIII-superoxy-catecholate reactant changes into the peroxo bridged FeII-quinone/FeIII-semiquinone mixed first product. The superoxo species 1a is thus a competent reactant to form the peroxo bridge in that it can perform an electrophilic attack on the catecholate through its π*σ FMO, passing through a low barrier of 7.5 kcal/mol (Figure 11) and taking an electron from the substrate π cloud to generate the peroxo bridge. This result is most similar to those previously reported in Refs 11 and 42.
Figure 12.

FMOs for reaction 1 calculated at the level of BP86 mixed with 10% Hartree-Fock exchange, using the 6–311G* basis set on Fe-O2 and 6–31G* on the remaining atoms. The isosurfaces are plotted with values of ±0.06 a.u.–3/2 The numbers in parentheses are the weights of the Fe 3d, O2, and 4NC orbitals, in that order. The weights of the α- and βLUMO show the initial FeIII-superoxo catecholate reactant attacking the π cloud of the catecholate through its π*σ FMO.
The alternative possible reaction pathway to generate the peroxo bridge involves direct attack by the non-protonated O of the side-on hydroperoxo intermediate 1b to generate the hydroperoxo bridged intermediate 2b. To search for a TS along this concerted side-on hydroperoxo attack pathway, a 2D potential energy surface (PES) was generated, with the Fe-O(nonprotonated, np) distance serving as one coordinate and the O(np)-C2(substrate) distance serving as the other. In addition, the Fe-O(protonated) distance was fixed at 2.3 Å, as this bond length does not change significantly between the reactant (2.234 Å) and the product (2.376 Å). This PES is shown in Figure 13. As can be seen in Figure 13, performing a concerted attack by decreasing the O(np)-C2(substrate) distance without breaking the Fe-O(np) bond is highly unfavorable energetically; at Fe-O(np) lengths less than 2.6 Å, decreasing the O(np)-C2(substrate) bond results in a barrier of at least 20 kcal/mol (upper-middle section of the PES), far higher than the 8.8 kcal/mol barrier for superoxo attack (TS1, Figure 11). To understand why the Fe-O(np) bond must break to allow attack on C2, it is important to consider the FMO that 1b would use to attack the substrate (Figure 14) and how this develops along the reaction coordinate. As shown in Figure 14, the concerted reaction is a nucleophilic attack by the occupied peroxo π*v orbital (i.e. the π* orbital perpendicular to the Fe-OO plane) on the unoccupied 4NC π* orbital. This is reflected by the peroxo π*v character in the α LUMO (Figure 14, right) of the structure with an Fe-O(np) distance of 2.380 Å and O(np)-C2(substrate) distance of 2.110 Å (green star on PES in Figure 13, left; structure shown on Figure 13, right). To react with this FMO, the substrate would need to distort and approach perpendicular to the O-O vector to allow for overlap between the unoccupied semiquinone π orbital and the peroxo π*v; this distortion is unfavorable, as the substrate is bound bidentate to the Fe in the equatorial plane. To visualize this distortion, the strained transition state-like structure denoted by green star on Figure 13 obtained from DFT is shown in the SI (in Figure S17), in addition to the FMO shown in Figure 14 and the scheme in Figure 13, right.
Figure 13.

A 3D plot of the 2D PES for generating a hydroperoxy bridged intermediate (2b) from the side-on FeIII-hydroperoxy-semiquinone intermediate 1b. The Fe-O(np) bond length is increased along the y-axis, while the O(np)-C2(substrate) distance is increased along the x-axis. All energies shown on the z-axis are electronic energies in kcal/mol with the energy of 1b set to 0 kcal/mol. The reactant 1b and product 2b are not shown on this PES as these would appear at the bottom-left and upper-right portions of the PES, respectively. The dashed red line shows the direct path connecting 1b and 2b, and a 2D contour map showing the positions of 1b and 2b is given in the SI (Figure S16). The black and gray circles indicate species having β-spin density on the distal oxygen atom: the gray circles have β-spin density less than 0.1 on O(np) and thus primarily peroxy character, and the black circles have β-spin density more than 0.1 and thus significant superoxy character. The green and blue stars denote specific structures on the 2D PES that are described in the text; these structures are shown on the right.
Figure 14.

Relevant FMOs for 1b and the transition state-like strained structure (TS’) along the PES (denoted by the green star on Figure 13) calculated at the level of BP86 mixed with 10% Hartree-Fock exchange, using the 6–311G* basis set on Fe-O2 and 6–31G* on the remaining atoms. 1b has an α unoccupied FMO with primarily semiquinone character (upper-left). As the substrate approaches the peroxo, this unoccupied FMO gains some peroxo π*v character, as shown in the α LUMO on the right. The peroxo π*v orbital is occupied in 1b, as shown in the bottom left; this reaction is thus a nucleophilic attack.
Given that the concerted attack to form 2b is high in energy, 1b would need to use a nonconcerted mechanism wherein the Fe-O(np) bond breaks prior to decreasing the O(np)-C2(substrate) distance. Breaking this bond without concerted decrease of the O(np)-C2(substrate) bond (Fe-O(np) = 2.980 Å, O(np)-C2(substrate) = 2.110 Å, blue star at the bottom-right corner of the 2D PES in Figure 13, left, with scheme shown in Figure 13, right) generates an FeIII-hydrosuperoxo-catecholate species (an end-on superoxo with the proton on the O bound to the Fe) at 6.5 kcal/mol, higher in energy than the end-on superoxo 1a (H+ on H200). This promotes transfer of the proton back to H200 and reformation of 1a, and the reaction would then occur through the superoxo attack mechanisms described in Figure 11. Thus, no low energy reaction pathway exists for formation of a peroxy bridged intermediate by the side-on FeIII-hydroperoxy-semiquinone 1b; instead, the isoenergetic end-on FeIII-superoxy-catecholate 1a is calculated to be the reactive species.
3.4. Lack of peroxy bridge formation by Int-1: reaction coordinate for H200N-4NC
As presented in the Introduction, Int-1 in H200N does not perform extradiol cleavage, and instead decays slowly to Int-2 (k = 0.015 s−1 at 277 K),23 presumably through delivery of a solvent proton.44 Since it was proposed in Section 3.3 that a FeIII-superoxo-catecholate species similar to Int-1 is the reactive intermediate in the wild-type enzyme reaction with 4NC, it is necessary to consider why Int-1 in H200N is not reactive in direct attack on the substrate. A reaction coordinate for formation of a peroxy bridged structure by Int-1 was computationally generated, using the NRVS structure of Int-1 as a starting point (shown in Figure 2C); the results of this calculation are summarized in Table 5, with structures of the species along the reaction coordinate given in the SI (Figure S18). As shown in Table 5, peroxy bridge formation by Int-1 proceeds through a barrier of 18.5 kcal/mol (TSInt−1) and generates a FeIII-peroxy-semiquinone product at 17.4 kcal/mol in free energy (PInt−1, calculated geometric and electronic structure parameters for these species are also given in Table 5). This barrier is much higher than the 7.5 kcal/mol barrier calculated for superoxo attack in wild-type (Table 4) and comparable to the ~18 kcal/mol barrier for the Int-1 to Int-2 conversion estimated from the experimental rate, which can be considered as the barrier associated with delivery of the proton to Int-1. The high barrier for this superoxide attack on the coordinated 4NC is due primarily to the unfavorable thermodynamics for formation of the peroxy bridged product, and from Sections 3.2.3 and 3.2.4, this unfavorable reaction energy is due to the lack of H200, which stabilizes the unprotonated peroxy bridged intermediate by 10.1 kcal/mol more than it stabilizes the superoxo reactant (17.3 vs 27.4 kcal/mol, from Figures 6 and 10) due to its stronger ionic interaction that includes a hydrogen bond between H200(H)+ and the negatively charged peroxy bridge O.
Table 5.
Energies and structural parameters for species along the reaction coordinate for peroxy bridge formation by Int-1.
| Int-1 | TSInt−1 | PInt−1 | |
|---|---|---|---|
| energy (kcal/mol) | |||
| ΔE | 0.0 | 17.2 | 16.3 |
| ΔH | 0.0 | 13.7 | 13.3 |
| ΔG | 0.0 | 18.5 | 17.4 |
| bond length(Å)1 | |||
| Fe—O(1) | 1.926 | 1.946 | 1.808 |
| O—O | 1.314 | 1.394 | 1.474 |
| C(3)—O(2) | 2.878 | 1.750 | 1.501 |
| spin density1 | |||
| Fe | 3.50 | 4.05 | 2.78 |
| O(1) | −0.07 | 0.19 | 0.10 |
| O(2) | −0.32 | −0.05 | 0.05 |
| C6(4NC) | 0.00 | −0.46 | 0.66 |
The numbering of atoms are as shown in Figure 4.
Finally, the reactivity of the end FeIII-hydroperoxo Int-2 with semiquinone substrate was also considered. This attack would involve the distal, protonated O attacking carbon 2 of the substrate to generate a distally protonated hydroperoxy bridged intermediate. While a transition state for this attack could not be located, a 1D linear transit (Table S5) involving systematic decrease of the O(distal)-C2(substrate) bond length shows that this attack would proceed through a barrier of ~32 kcal/mol. Further, geometry optimization of the distally protonated hydroperoxy bridged species was found to converge back to the Int-2 reactant. The lack of reactivity of the end-on hydroperoxy Int-2 can be understood by considering the small coefficient of the protonated O in the occupied π*σ FMO (11.9%, Figure S19) that would be involved in nucleophilic attack it would use for nucleophilic attack on 4NC. Thus, Int-1 is unreactive with substrate and decays via protonation to Int-2, which is similarly unreactive and persists until further equivalents of substrate are added, which triggers the release of H2O2 and 4NC-quinone.23
4. Discussion
The above results offer new insights into the structures of intermediates in the extradiol dioxygenases and how these relate to reactivity. We used NRVS to define the structure of Int-2 in H200N as an end-on FeIII-hydroperoxo-semiquinone (consistent with a previous computational study based on calculated Mössbauer parameters11), and define the first wild-type crystallographic intermediate as a side-on FeIII-hydroperoxo-semiquinone. The presence of H200 in wild-type thus stabilizes side-on hydroperoxo binding. Importantly, Int-2 is a dead-end species, and the side-on hydroperoxo was also found to be unreactive despite having different FMOs available for nucleophilic attack than the end-on hydroperoxo Int-2 (vide infra). The lack of reactivity for the side-on hydroperoxo is due to the unfavorable distortion of the substrate that would be required for nucleophilic attack. Despite this, the side-on hydroperoxo is not a dead-end species due to the presence of H200, which can deprotonate the hydroperoxo O to generate an isoenergetic end-on FeIII-superoxo-catecholate species. This is in contrast with previous work42 that also optimized a side-on FeIII-hydroperoxo species, but found it to be 12.3 kcal/mol higher in energy than the FeIII-superoxo. This difference is likely due to the use of a protonated H248 in the former study, as we similarly find the hydroperoxo to be 11.3 kcal/mol higher in energy than the superoxo using our computational method and model with a protonated H248. Our result using a neutral H248 presented in this study is consistent with the experimental result that the side-on intermediate is observed crystallographically. In addition, previous computational studies11,42 have found that an FeIII-superoxo-catecholate is competent in performing electrophilic attack on the catecholate, which we confirmed from our experimentally calibrated calculations. This is in contrast to other computational studies that argued for FeII-superoxo-semiquinone10 or mixed FeII-superoxosemiquinone/FeIII-superoxo-catecholate41 reactive species. Through correlation to the crystal structure, we assign peroxo bridged species as an unprotonated delocalized FeIII-peroxo-semiquinone/FeII-peroxoquinone intermediate. The full mechanism for peroxo bridge formation evaluated in this study is shown in Scheme 3. Note that our assignment of the peroxo bridge as unprotonated is in contrast with previous computational studies that assigned the bridge as protonated,11,41 though one other study did also find this to be unprotonated.10 The importance of protonation of the bridging peroxo for O-O cleavage in the EDOs is an important issue and is presently being evaluated through spectroscopically calibrated calculations on a relevant intermediate in wild-type HPCD with the native substrate.
Scheme 3.

Mechanism of peroxy bridge formation in wild-type HPCD.
There has been general interest in understanding the factors that drive the formation of the initial activated O2 intermediates in several classes of NHFe enzymes. In isopenicillin N-synthase (IPNS), a combined MCD/DFT study on the {FeNO}7-substrate complex predicted that binding of the thiolate substrate tunes down the FeII/FeIII reduction potential, making the one electron reduction of O2 in the formation of an initial FeIII-superoxo intermediate thermodynamically favorable.69 Consistent with the MCD/DFT prediction and earlier speculation,70 an FeIII-superoxo intermediate has recently been observed in IPNS.71 In contrast, recent studies on the Rieske dioxygenase benzoate 1,2-dioxygenase, where neither substrate nor cofactor coordinate to the NHFe site, indicated that the FeII / O2 reaction proceeds through an initial FeIII-superoxo intermediate that is thermodynamically uphill by ~10 kcal/mol. In this case, reactivity is driven by the favorable thermodynamics of a subsequent proton-coupled electron transfer step.36,72 The extradiol dioxygenases present a potential third situation, as here the substrate directly coordinates to the Fe as in IPNS, but unlike IPNS, the substrate is redox active, and it has previously been thought4 that the substrate provides an electron to O2 in its binding process. The results reported in Section 3.2.2 for the initial crystallographic O2 intermediate of the wild-type EDO with 4NC as the catecholate substrate are consistent with this prediction, as this substrate and the Fe have each been oxidized by one electron to generate an FeIII-hydroperoxo-semiquinone species. However, this is calculated in the wild-type enzyme to be very similar in energy to the end-on FeIII-superoxo-catecholate species, where no electron has been transferred from the substrate to drive its formation. Further, from the NRVS results in Section 3.1 and from earlier EPR and Mössbauer studies,23 a long-lived FeIII-superoxo-4NC catecholate intermediate (Int-1) is clearly formed in the absence of H200. Thus, in the EDO/substrate species, the catecholate binding plays a role in tuning the FeII/FeIII reduction potential through donation to the Fe, similar to the role the substrate plays in IPNS. This donor ability of the catecholate is reflected experimentally in the NRVS data for H200N-4NC-NO relative to past vibrational data for Fe-EDTA-NO (Section 3.1), where catecholate binding greatly weakens the axial Fe-NO bond, as reflected by the decrease in the νFe-N energy. Computationally, 4NC binding to the FeII makes O binding as FeIII-superoxide more favorable by 9.3 kcal/mol in free energy relative to O2 binding to a 5C FeII facial triad (see Table S6). The dianionic 4NC is thus a strong donor, enabling formation of a FeIII-superoxo-catecholate species, and the nitro group disfavors electron transfer from the 4NC to the FeIII to form a FeII-superoxo-semiquinone electromer (see Section 3.2.3).
It is important to note that, unlike 4NC, the native substrate in HPCD, HPCA, binds as a monoanion.18 However, if HPCA becomes deprotonated by H200 upon O2 binding, it would become stronger donor to the Fe than the 4NC, potentially even resulting in an FeII-semiquinone-superoxo structure. To date, transient kinetic experiments have failed to provide any insight into the oxidation state of the active site iron in an intermediate that can react with HPCA in the wild-type enzyme. The first trapped intermediate in the wild-type/HPCA reaction has been shown by Mössbauer spectroscopy to have a high-spin FeII site,24 but this species occurs after reaction with HPCA. It has been assigned as either the peroxy bridged intermediate or a subsequent intermediate prior to product formation. The observed intermediate does show that if an FeIII reactive species forms in the reaction, a full β electron must be transferred from the substrate to the high-spin FeIII (α spins) during the reaction cycle. The timing of this electron transfer would depend on the relative reduction potentials of the substrate and the metal or metal-O2 complex, providing insight into the comparable reactivities of Mn and Co-substituted HPCD with HPCA relative to the native Fe enzyme.73 Future efforts will focus on evaluating this ferrous intermediate in wild-type with HPCA.
The results described above also offer insight into the differing reactivities of FeIII-superoxy and FeIII-hydroperoxy species. From Section 3.3, a side-on FeIII-hydroperoxy species, while observed crystallographically in wild-type, is unreactive with its bound substrate, and from Section 3.4 an end-on FeIII-hydroperoxy intermediate (Int-2) is similarly unreactive in H200N. In contrast, the end-on FeIII-superoxy intermediate in wild-type, which is not observed in the crystal, is able to react with bound 4NC substrate through a reasonable barrier. Insight into this reactivity difference can be gained through consideration of the reactive FMOs. As shown schematically in Figure 15, both the side-on hydroperoxy (left) and end-on hydroperoxy (middle) would perform nucleophilic attack on the semiquinone substrate through an occupied π*v (perpendicular to the O-O plane) or occupied π*σ (in-plane) orbital, respectively. In the former case, this FMO is poorly oriented to attack the π cloud of the substrate, which is bound to the Fe in an equatorial position (Section 3.3, Figure 14). In the latter case, the occupied π*σ has a poor orbital coefficient on the attacking O. In contrast, for the end-on superoxy species the attack is electrophilic, involving (Figure 15, right) an unoccupied π*σ orbital with lobes in the Fe-OO plane oriented toward the occupied π cloud of the substrate. It should be noted that, while in wild-type and H200N, a hydroperoxo is not active in attack on 4NC, in Y257F a hydroperoxy FeII species is observed to slowly react with a bound quinone.25 Factors leading to FeII stabilization in this variant and its relative reactivity still need to be explored.25
Figure 15.

FMOs used by side-on (left) and end-on (center) FeIII-hydroperoxy species, as well as for an end-on FeIII-superoxy species (right), in forming a peroxy bridged intermediate. Only the end-on FeIII-superoxy π*σ FMO has a good orbital coefficient on the attacking O and overlap with the π cloud of the substrate for electrophilic attack.
The results above also shed light on the role of H200 in extradiol catalysis. Currently, the role of H200 is thought to be to facilitate uptake of a proton from the substrate and its delivery to the peroxo bridged intermediate to facilitate O-O cleavage.4,10,11 From Sections 3.2.4 and 3.4, H200 also plays an essential role in stabilizing the peroxo bridged intermediate (by 10.2 kcal/mol relative to the superoxo reactant, see Section 3.4). This stabilization lowers the barrier for peroxy bridge formation from 18.5 to 7.5 kcal/mol due to the strong ionic interaction, including a hydrogen bond, between protonated H200 and the proximal O of the peroxo. The lack of this stabilization in H200N raises the barrier for bridge formation, resulting in the observed lack of extradiol cleavage by Int-1.
As a final comment, O2 intermediates have been trapped in the H200N and H200C variants of HPCD using the native substrate HPCA, and these do perform extradiol cleavage.24,26 Currently available spectroscopic data, including Mössbauer and UV/Vis absorption, suggest that these intermediates are both FeIII-(hydro)peroxy-semiquinone species. These putative FeIII-(hydro)peroxy intermediates can react with HPCA, in contrast to the behavior of the H200N FeIII-hydroperoxy with 4NC semiquinone Int-2 (Section 3.4), and in these intermediates there is also no H200 present to stabilize the peroxy bridged species. NRVS studies of these intermediates are underway to further define their structures and elucidate their activities in extradiol cleavage of the native substrate in the absence of H200.
5. Conclusion
We have used NRVS to define the structures of two O2 intermediates in the H200N variant of HPCD using the slow substrate 4NC and computationally correlated these to the initial crystallographic O2 intermediate in wild-type HPCD with 4NC. From calculations correlated to the structure, this initial crystallographic intermediate is assigned as a side-on FeIII-hydroperoxy-semiquinone species. Our computational studies further defined the second crystallographic intermediate as an unprotonated delocalized FeIII-peroxy-semiquinone/FeII-peroxy-quinone bridged species. The FeIII-hydroperoxysemiquinone intermediate was calculated not to form the peroxo bridged intermediate; however, it is at a similar energy to an end-on FeIII-superoxy-catecholate species that was calculated to directly react to form the peroxo bridge. Correlation of these results to the H200N solution NRVS data also revealed that, in wild-type HPCD, H200 plays a key role in stabilizing the peroxy bridged intermediate, enabling extradiol reactivity.
Supplementary Material
Acknowledgments
Funding for this work was provided by the National Institutes of Health (GM-40392 to E.I.S. and GM 118030 to J.D.L.), and JSPS KAKENHI (Grant No. 24221005 to M. Seto) and JSPS “Strategic Young Researcher Overseas Visits Program for Accelerating Brain Circulation” (Project No. R2304 and R2606 supporting to Y. Wasada-Tsutsui). Synchrotron experiments at SPring-8 were performed with the approval of the Japan Synchrotron Radiation Research Institute (JASRI; proposal no. 2013B0105), and use of the Advanced Photon Source was supported by the Department of Energy, Office of Science, contract DE-AC-02–06CH11357. Electronic structure calculations of the crystallographic intermediates and peroxy bridge forming reaction coordinate were performed on the Fujitsu CX400 system at the Nagoya University Information Technology Center.
Footnotes
Publisher's Disclaimer: This document is confidential and is proprietary to the American Chemical Society and its authors. Do not copy or disclose without written permission. If you have received this item in error, notify the sender and delete all copies.
Supporting information. Supporting results and analysis. NRVS DFT calculated spectra for H200N-4NCNO using the 6–311g*/6–31g* and TZVP basis sets. DFT calculated NRVS spectrum and structure for alternate H200N-4NC-NO conformation. DFT calculated NRVS spectrum for H200N-4NC-NO with shortened Fe-N bond. Overlay of H200N-4NC-NO and Int-1 NRVS data. DFT calculated NRVS spectrum and structure for alternative Int-1 conformation. Overlay of Int-1 and Int-2 NRVS data. DFT calculated NRVS spectrum and structure for alternative Int-2 conformation. Overlays of the crystallographic structure with relevant computational structures. Overlays of the NRVS structures with 1a’ and 1b’. FMOs for 1b. 2D potential energy surface for the 1a to 1b interconversion. DFT calculated NRVS spectrum for FeII-superoxo-semiquinone electromer. DFT structures for 2a’ and 2b’. DFT structures for TS1 and TS2. FMOs for reaction 2. 2D contour map of hydroperoxy bridge formation reaction from the side-on hydroperoxy species. DFT structure for a species on the 2D surface. DFT structures for species along the Int-1 bridge formation reaction. FMO of Int-2. Electronic structure parameters for Int-1 and Int-2. Geometric and electronic structure parameters for 1a’, 1b’, 2a’ and 2b’. Table of energies for species on the 2D potential energy surface for the 1a to 1b interconversion. Comparison of geometric parameters for Int-1, 1a’, and 1a’ using the Int-1 truncation. 1D linear transit energies for Int-2 attack on the bound semiquinone. Crystallographic and DFT-optimized geometric parameters for Mg(II) semiquinone model complex. Crystallographic and DFT-optimized geometric parameters for Ga(III) semiquinone model complex. Supporting references.
References
- (1).Dagley S Catabolism of Aromatic Compounds by Micro-Organisms In Advances in microbial physiology; Elsevier, 1971; Vol. 6, pp 1–46. [DOI] [PubMed] [Google Scholar]
- (2).Vaillancourt FH; Bolin JT; Eltis LD The Ins and Outs of Ring-Cleaving Dioxygenases. Crit. Rev. Biochem. Mol. Biol 2006, 41 (4), 241–267. [DOI] [PubMed] [Google Scholar]
- (3).Solomon EI; Brunold TC; Davis MI; Kemsley JN; Lee SK; Lehnert N; Neese F; Skulan AJ; Yang YS; Zhou J Geometric and Electronic Structure/function Correlations in Non-Heme Iron Enzymes. Chem. Rev 2000, 100 (1), 235–349. [DOI] [PubMed] [Google Scholar]
- (4).Lipscomb JD Mechanism of Extradiol Aromatic Ring-Cleaving Dioxygenases. Curr. Opin. Struct. Biol 2008, 18 (6), 644–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Lipscomb JD; Orville AM Mechanistic Aspects of Dihydroxybenzoate Dioxygenases. Met. Ions Biol. Syst 1992, 28, 243–298. [Google Scholar]
- (6).Hrywna Y; Tsoi TV; Maltseva OV; Quensen JF; Tiedje JM Construction and Characterization of Two Recombinant Bacteria That Grow on Ortho- and Para-Substituted Chlorobiphenyls. Appl. Environ. Microbiol 1999, 65 (5), 2163–2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Gibson DT; Parales RE Aromatic Hydrocarbon Dioxygenases in Environmental Biotechnology. Curr. Opin. Biotechnol 2000, 11 (3), 236–243. [DOI] [PubMed] [Google Scholar]
- (8).Furukawa K Engineering Dioxygenases for Efficient Degradation of Environmental Pollutants. Curr. Opin. Biotechnol 2000, 11 (3), 244–249. [DOI] [PubMed] [Google Scholar]
- (9).Kovaleva EG; Lipscomb JD Versatility of Biological Non-Heme Fe(II) Centers in Oxygen Activation Reactions. Nat. Chem. Biol 2008, 4 (3), 186–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Siegbahn PEM; Haeffner F Mechanism for Catechol Ring-Cleavage by Non-Heme Iron Extradiol Dioxygenases. J. Am. Chem. Soc 2004, 126 (29), 8919–8932. [DOI] [PubMed] [Google Scholar]
- (11).Christian GJ; Ye S; Neese F Oxygen Activation in Extradiol Catecholate Dioxygenases - a Density Functional Study. Chem. Sci 2012, 3 (5), 1600–1611. [Google Scholar]
- (12).Han S; Eltis LD; Timmis KN; Muchmore SW; Bolin JT Crystal Structure of the Biphenyl-Cleaving Extradiol Dioxygenase from a PCB-Degrading Pseudomonad. Science 1995, 270 (5238), 976–980. [DOI] [PubMed] [Google Scholar]
- (13).Cho HJ; Kim K; Sohn SY; Cho HY; Kim KJ; Kim MH; Kim D; Kim E; Kang BS Substrate Binding Mechanism of a Type I Extradiol Dioxygenase. J. Biol. Chem 2010, 285 (45), 34643–34652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Uragami Y; Senda T; Sugimoto K; Sato N; Nagarajan V; Masai E; Fukuda M; Mitsui Y Crystal Structures of Substrate Free and Complex Forms of Reactivated BphC, an Extradiol Type Ring-Cleavage Dioxygenase. J. Inorg. Biochem 2001, 83 (4), 269–279. [DOI] [PubMed] [Google Scholar]
- (15).Vetting MW; Wackett LP; Que L; Lipscomb JD; Ohlendorf DH Crystallographic Comparison of Manganese- and Iron-Dependent Homoprotocatechuate 2,3-Dioxygenases. J. Bacteriol 2004, 186 (7), 1945–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Kovaleva EG; Lipscomb JD Crystal Structures of Fe2+ Dioxygenase Superoxo, Alkylperoxo, and Bound Product Intermediates. Science 2007, 316 (5823), 453–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Fielding AJ; Kovaleva EG; Farquhar ER; Lipscomb JD; Que L A Hyperactive Cobalt-Substituted Extradiol-Cleaving Catechol Dioxygenase. J. Biol. Inorg. Chem 2011, 16 (2), 341–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Vaillancourt FH; Barbosa CJ; Spiro TG; Bolin JT; Blades MW; Turner RFB; Eltis LD Definitive Evidence for Monoanionic Binding of 2,3-Dihydroxybiphenyl to 2,3-Dihydroxybiphenyl 1,2-Dioxygenase from UV Resonance Raman Spectroscopy, UV/Vis Absorption Spectroscopy, and Crystallography. J. Am. Chem. Soc 2002, 124 (11), 2485–2496. [DOI] [PubMed] [Google Scholar]
- (19).Kovaleva EG; Lipscomb JD Structural Basis for the Role of Tyrosine 257 of Homoprotocatechuate 2,3-Dioxygenase in Substrate and Oxygen Activation. Biochemistry 2012, 51 (44), 8755–8763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Horner L; Geyer E Zur Kenntnis Der O-Chinone, XXVII: Redoxpotentiale von Brenzcatechin-Derivaten. Eur. J. Inorg. Chem 1965, 98 (6), 2016–2045. [Google Scholar]
- (21).Solomon EI; Light KM; Liu LV; Srnec M; Wong SD Geometric and Electronic Structure Contributions to Function in Non-Heme Iron Enzymes. Acc. Chem. Res 2013, 46 (11), 2725–2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Solomon EI; Goudarzi S; Sutherlin KD O2 Activation by Non-Heme Iron Enzymes. Biochemistry 2016, 55 (46). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Mbughuni MM; Chakrabarti M; Hayden JA; Bominaar EL; Hendrich MP; Muenck E; Lipscomb JD Trapping and Spectroscopic Characterization of an Fe-III-Superoxo Intermediate from a Nonheme Mononuclear Iron-Containing Enzyme. Proc. Natl. Acad. Sci. U. S. A 2010, 107 (39), 16788–16793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Mbughuni MM; Chakrabarti M; Hayden JA; Meier KK; Dalluge JJ; Hendrich MP; Muenck E; Lipscomb JD Oxy Intermediates of Homoprotocatechuate 2,3-Dioxygenase: Facile Electron Transfer between Substrates. Biochemistry 2011, 50 (47), 10262–10274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Mbughuni MM; Meier KK; Münck E; Lipscomb JD Substrate-Mediated Oxygen Activation by Homoprotocatechuate 2,3-Dioxygenase: Intermediates Formed by a Tyrosine 257 Variant. Biochemistry 2012, 51 (44), 8743–8754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Meier KK; Rogers MS; Kovaleva EG; Mbughuni MM; Bominaar EL; Lipscomb JD; Münck E A Long-Lived FeIII-(Hydroperoxo) Intermediate in the Active H200C Variant of Homoprotocatechuate 2, 3-Dioxygenase: Characterization by Mössbauer, Electron Paramagnetic Resonance, and Density Functional Theory Methods. Inorg. Chem 2015, 54 (21), 10269–10280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Groce SL; Lipscomb JD Aromatic Ring Cleavage by Homoprotocatechuate 2,3-Dioxygenase: Role of His200 in the Kinetics of Interconversion of Reaction Cycle Intermediates. Biochemistry 2005, 44 (19), 7175–7188. [DOI] [PubMed] [Google Scholar]
- (28).Seto M; Yoda Y; Kikuta S; Zhang XW; Ando M Observation of Nuclear Resonant Scattering Accompanied by Phonon Excitation Using Synchrotron Radiation. Phys. Rev. Lett 1995, 74 (19), 3828–3831. [DOI] [PubMed] [Google Scholar]
- (29).Sturhahn W; Toellner TS; Alp EE; Zhang X; Ando M; Yoda Y; Kikuta S; Seto M; Kimball CW; Dabrowski B Phonon Density-of-States Measured by Inelastic Nuclear Resonant Scattering. Phys. Rev. Lett 1995, 74 (19), 3832–3835. [DOI] [PubMed] [Google Scholar]
- (30).Scheidt WR; Li J; Sage JT What Can Be Learned from Nuclear Resonance Vibrational Spectroscopy: Vibrational Dynamics and Hemes. Chem. Rev 2017, 117 (19), 12532–12563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Sturhahn W CONUSS and PHOENIX: Evaluation of Nuclear Resonant Scattering Data. Hyperfine Interact. 2000, 125 (1–4), 149–172. [Google Scholar]
- (32).Sage JT; Paxson C; Wyllie GRA; Sturhahn W; Durbin SM; Champion PM; Alp EE; Scheidt WR Nuclear Resonance Vibrational Spectroscopy of a Protein Active- Site Mimic. J. Physics-Condensed Matter 2001, 13 (34), 7707–7722. [Google Scholar]
- (33).Liu LV; Bell CB I. I. I.; Wong SD; Wilson SA; Kwak Y; Chow MS; Zhao J; Hodgson KO; Hedman B; Solomon EI Definition of the Intermediates and Mechanism of the Anticancer Drug Bleomycin Using Nuclear Resonance Vibrational Spectroscopy and Related Methods. Proc. Natl. Acad. Sci. U. S. A 2010, 107 (52), 22419–22424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Wong SD; Srnec M; Matthews ML; Liu LV; Kwak Y; Park K; Bell CB III.; Alp EE; Zhao J; Yoda Y; Kitao S; Seto M; Krebs C; Bollinger JM Jr; Solomon EI Elucidation of the Fe(IV)=O Intermediate in the Catalytic Cycle of the Halogenase SyrB2. Nature 2013, 499 (7458), 320–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Park K; Li N; Kwak Y; Srnec M; Bell CB; Liu LV; Wong SD; Yoda Y; Kitao S; Seto M; Hu M; Zhao J; Krebs C; Bollinger JM; Solomon EI Peroxide Activation for Electrophilic Reactivity by the Binuclear Non-Heme Iron Enzyme AurF. J. Am. Chem. Soc 2017, 139 (20), 7062–7070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Sutherlin KD; Rivard BS; Boettger LH; Liu LV; Rogers MS; Srnec M; Yoda Y; Kitao S; Kobayashi Y; Saito M; Seto M; Hu M; Zhao J; Lipscomb JD; Solomon EI NRVS Studies of the Peroxide Shunt Intermediate in a Rieske Dioxygenase and Its Relation to the Native FeII O2 Reaction. J Am Chem Soc [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Enemark JH; Feltham RD Principles of Structure, Bonding, and Reactivity for Metal Nitrosyl Complexes. Coord. Chem. Rev 1974, 13 (4), 339–406. [Google Scholar]
- (38).Westre TE; Dicicco A; Filipponi A; Natoli CR; Hedman B; Solomon EI; Hodgson KO Determination of the Fe-N-O Angle in {FeNO}7 Complexes Using Multiple-Scattering EXAFS Analysis by GNXAS. J. Am. Chem. Soc 1994, 116 (15), 6757–6768. [Google Scholar]
- (39).Brown CA; Pavlosky MA; Westre TE; Zhang Y; Hedman B; Hodgson KO; Solomon EI Spectroscopic and Theoretical Description of the Electronic Structure of S=3/2 Iron-Nitrosyl Complexes and Their Relation to O2 Activation by Non-Heme Iron Enzyme Active Sites. J. Am. Chem. Soc 1995, 117 (2), 715–732. [Google Scholar]
- (40).Georgiev V; Borowski T; Blomberg MRA; Siegbahn PEM A Comparison of the Reaction Mechanisms of Iron- and Manganese-Containing 2,3-HPCD: An Important Spin Transition for Manganese. J. Biol. Inorg. Chem 2008, 13 (6), 929–940. [DOI] [PubMed] [Google Scholar]
- (41).Dong G; Shaik S; Lai W Oxygen Activation by Homoprotocatechuate 2,3-Dioxygenase: A QM/MM Study Reveals the Key Intermediates in the Activation Cycle. Chem. Sci 2013, 4 (9), 3624–3635. [Google Scholar]
- (42).Dong G; Lai W Reaction Mechanism of Homoprotocatechuate 2,3-Dioxygenase with 4-Nitrocatechol: Implications for the Role of Substrate. J. Phys. Chem. B 2014, 118 (7), 1791–1798. [DOI] [PubMed] [Google Scholar]
- (43).Wang YZ; Lipscomb JD Cloning, Overexpression, and Mutagenesis of the Gene for Homoprotocatechuate 2, 3-Dioxygenase Frombrevibacterium Fuscum. Protein Expr. Purif 1997, 10 (1), 1–9. [DOI] [PubMed] [Google Scholar]
- (44).Kovaleva EG; Rogers MS; Lipscomb JD Structural Basis for Substrate and Oxygen Activation in Homoprotocatechuate 2,3-Dioxygenase: Roles of Conserved Active Site Histidine 200. Biochemistry 2015, 54 (34), 5329–5339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Streit BR; DuBois JL Chemical and Steady-State Kinetic Analyses of a Heterologously Expressed Heme Dependent Chlorite Dismutase. Biochemistry 2008, 47 (19), 5271–5280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Krebs C; Dassama LMK; Matthews ML; Jiang W; Price JC; Korboukh V; Li N; Bollinger JM Jr Novel Approaches for the Accumulation of Oxygenated Intermediates to Multi-Millimolar Concentrations. Coord. Chem. Rev 2013, 257 (1), 234–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Hagen WR Biomolecular EPR Spectroscopy; CRC Press, 2008. [Google Scholar]
- (48).Smith MC; Xiao Y; Wang H; George SJ; Coucouvanis D; Koutmos M; Sturhahn W; Alp EE; Zhao J; Cramer SP Normal-Mode Analysis of FeCl4-and Fe2S2Cl42-via Vibrational Mössbauer, Resonance Raman, and FT-IR Spectroscopies. Inorg. Chem 2005, 44 (16), 5562–5570. [DOI] [PubMed] [Google Scholar]
- (49).Becke AD Density-Functional Exchange-Energy Approximation with Correct Asymptotic Behavior. Phys Rev A 1988, 38 (6), 3098–3100. [DOI] [PubMed] [Google Scholar]
- (50).Perdew JP Density-Functional Approximation For The Correlation-Energy Of The Inhomogeneous Electron-Gas. Phys. Rev. B 1986, 33 (12), 8822–8824. [DOI] [PubMed] [Google Scholar]
- (51).Schafer A; Huber C; Ahlrichs R Fully Optimized Contracted Gaussian-Basis Sets Of Triple Zeta Valence Quality For Atoms Li To Kr. J. Chem. Phys 1994, 100 (8), 5829–5835. [Google Scholar]
- (52).Frisch MJ; Trucks GW; Schlegel HB; Scuseria GE; Robb MA; Cheeseman JR; Scalmani G; Barone V; Mennucci B; Petersson GA; Nakatsuji H; Caricato M; Li X; Hratchian HP; Izmaylov AF; Bloino J; Zheng G; Sonnenberg JL; Hada M; Ehara M; Toyota K; Fukuda R; Hasegawa J; Ishida M; Nakajima T; Honda Y; Kitao O; Nakai H; Vreven T; Montgomery JA Jr.; Peralta JE; Ogliaro F; Bearpark M; Heyd JJ; Brothers E; Kudin KN; Staroverov VN; Kobayashi R; Normand J; Raghavachari K; Rendell A; Burant JC; Iyengar SS; Tomasi J; Cossi M; Rega N; Millam NJ; Klene M; Knox JE; Cross JB; Bakken V; Adamo C; Jaramillo J; Gomperts R; Stratmann RE; Yazyev O; Austin AJ; Cammi R; Pomelli C; Ochterski JW; Martin RL; Morokuma K; Zakrzewski VG; Voth GA; Salvador P; Dannenberg JJ; Dapprich S; Daniels AD; Farkas Ö; Foresman JB; Ortiz JV; Cioslowski J; Fox DJ 09, Revision D. 01, Gaussian. Inc., Wallingford, CT: 2009. [Google Scholar]
- (53).Cramer CJ; Truhlar DG Implicit Solvation Models: Equilibria, Structure, Spectra, and Dynamics. Chem. Rev 1999, 99 (8), 2161–2200. [DOI] [PubMed] [Google Scholar]
- (54).Lind MD; Hoard JL; Hamor MJ; Hamor TA Stereochemistry of Ethylenediaminetetraacetato Complexes. II. The Structure of Crystalline Rb[Fe(OH2)Y]·H2O. III. The Structure of Crystalline Li[Fe(OH2)Y·2H2O. Inorg. Chem 1964, 3 (1), 34–43. [Google Scholar]
- (55).Park K; Solomon EI Modeling Nuclear Resonance Vibrational Spectroscopic Data of Binuclear Nonheme Iron Enzymes Using Density Functional Theory. Can. J. Chem 2014, 92 (February), 975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).McLean AD; Chandler GS Contracted Gaussian Basis Sets for Molecular Calculations. I. Second Row Atoms, Z =11–18. J. Chem. Phys 1980, 72 (10), 5639–5648. [Google Scholar]
- (57).Krishnan R; Binkley JS; Seeger R; Pople JA Self-consistent Molecular Orbital Methods. XX. A Basis Set for Correlated Wave Functions. J. Chem. Phys 1980, 72 (1), 650–654. [Google Scholar]
- (58).Hay PJ Gaussian Basis Sets for Molecular Calculations. The Representation of 3 D Orbitals in Transition-metal Atoms. J. Chem. Phys 1977, 66 (10), 4377–4384. [Google Scholar]
- (59).Wachters AJH Gaussian Basis Sets for Molecular Wavefunctions Containing Third-Row Atoms. Theor. Chim. Acta 1971, 20 (1), 1–11. [Google Scholar]
- (60).Hehre WJ; Ditchfie R; Pople J. A. c: Further Extensions Of Gaussian-Type Basis Sets For Use In Molecular-Orbital Studies Of Organic-Molecules. J. Chem. Phys 1972, 56 (5), 2257-. [Google Scholar]
- (61).Ditchfie R; Hehre WJ; Pople JA Self-Consistent Molecular-Orbital Methods 9: Extended Gaussian-Type Basis For Molecular-Orbital Studies Of Organic Molecules. J. Chem. Phys 1971, 54 (2), 724-. [Google Scholar]
- (62).Hariharan PC; Pople JA The Influence of Polarization Functions on Molecular Orbital Hydrogenation Energies. Theor. Chim. Acta 1973, 28 (3), 213–222. [Google Scholar]
- (63).Hariharan PC; Pople JA Accuracy of AH N Equilibrium Geometries by Single Determinant Molecular Orbital Theory. Mol. Phys 1974, 27 (1), 209–214. [Google Scholar]
- (64).Francl MM; Pietro WJ; Hehre WJ; Binkley JS; Gordon MS; DeFrees DJ; Pople JA Self-Consistent Molecular Orbital Methods. XXIII. A Polarization-Type Basis Set for Second-Row Elements. J. Chem. Phys 1982, 77 (7), 3654–3665. [Google Scholar]
- (65).Solomon EI; Sutherlin KD; Srnec M High-Spin and Low-Spin States in (FeNO)7, FeIV=O, and FeIII-OOH Complexes and Their Correlations to Reactivity In Spin States in Biochemistry and Inorganic Chemistry: Influence on Structure and Reactivity; Swart M, Costas M, Eds.; John Wiley & Sons: Chichester, UK, 2015. [Google Scholar]
- (66).Sato N; Uragami Y; Nishizaki T; Takahashi Y; Sazaki G; Sugimoto K; Nonaka T; Masai E; Fukuda M; Senda T Crystal Structures of the Reaction Intermediate and Its Homologue of an Extradiol-Cleaving Catecholic Dioxygenase. J. Mol. Biol 2002, 321 (4), 621–636. [DOI] [PubMed] [Google Scholar]
- (67).Schenk G; Pau MYM; Solomon EI Comparison between the Geometric and Electronic Structures and Reactivities of {FeNO}(7) and {FeO2}(8) Complexes: A Density Functional Theory Study. J. Am. Chem. Soc 2004, 126 (2), 505–515. [DOI] [PubMed] [Google Scholar]
- (68).Lehnert N; Ho RYN; Que L; Solomon EI Electronic Structure of High-Spin Iron(III)-Alkylperoxo Complexes and Its Relation to Low-Spin Analogues: Reaction Coordinate of O-O Bond Homolysis. J. Am. Chem. Soc 2001, 123 (51), 12802–12816. [DOI] [PubMed] [Google Scholar]
- (69).Brown CD; Neidig ML; Neibergall MB; Lipscomb JD; Solomon EI VTVH-MCD and DFT Studies of Thiolate Bonding to {FeNO}(7)/{FeO2}(8) Complexes of Isopenicillin N Synthase: Substrate Determination of Oxidase versus Oxygenase Activity in Nonheme Fe Enzymes. J. Am. Chem. Soc 2007, 129 (23), 7427–7438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Roach PL; Clifton IJ; Hensgens CMH; Shibata N; Schofield CJ; Hajdu J; Baldwin JE Structure of Isopenicillin N Synthase Complexed with Substrate and the Mechanism of Penicillin Formation. Nature 1997, 387 (6635), 827–830. [DOI] [PubMed] [Google Scholar]
- (71).Tamanaha EY; Zhang B; Guo Y; Chang W; Barr EW; Xing G; St. Clair J; Ye S; Neese F; Bollinger JM Jr.; Krebs CJ Spectroscopic Evidence for the Two CH-Cleaving Intermediates of Aspergillus Nidulans Isopenicillin N Synthase. J. Am. Chem. Soc 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Rivard BS; Rogers MS; Marell DJ; Neibergall MB; Chakrabarty S; Cramer CJ; Lipscomb JD Rate-Determining Attack on Substrate Precedes Rieske Cluster Oxidation during Cis-Dihydroxylation by Benzoate Dioxygenase. Biochemistry 2015, 54 (30), 4652–4664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Fielding AJ; Lipscomb JD; Que L A Two-Electron-Shell Game: Intermediates of the Extradiol-Cleaving Catechol Dioxygenases Topical Issue in Honor of Ivano Bertini. Guest Editors: Lucia Banci, Claudio Luchinat. J. Biol. Inorg. Chem 2014, 19 (4–5), 491–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.