

Abstract
A3 adenosine receptor (A3AR) agonists are effective at limiting injury caused by ischemia/reperfusion injury of the heart in experimental animal models. However, understanding of their mechanism of action, which is likely multifactorial, remains incomplete. In prior studies, it has been demonstrated that A3AR-mediated ischemic protection is blocked by glibenclamide and is absent in Kir6.2 gene ablated mice that lack the pore-forming subunit of the ATP-sensitive potassium (KATP) channel, suggesting one contributing mechanism may involve accelerated activation of KATP channels. However, presence of A3ARs in the myocardium has yet to be established. Utilizing a whole-cell recording technique, in this study we confirm functional expression of the A3AR in adult mouse ventricular cardiomyocytes, coupled to activation of ATP-dependent potassium (KATP) channels via Gi inhibitory proteins. We further show that ischemic protection provided by the selective A3AR agonist CP-532,903 in an isolated, buffer-perfused heart model is lost completely in Adora3LoxP/LoxP;Myh6-Cre mice, which is a newly developed model developed and comprehensively described herein whereby the A3AR gene (Adora3) is deleted exclusively in cardiomyocytes. Our findings, taken together with previously published work, are consistent with the hypothesis that A3AR agonists provide ischemic tolerance, at least in part, by facilitating opening of myocardial KATP channels.
Keywords: adenosine, adenosine receptors, ischemia/reperfusion, ATP-sensitive potassium channels
Graphical Abstract
1. Introduction
Adenosine is produced rapidly in large quantities in the myocardium during ischemia and during reperfusion where it acts in several ways to protect against injury and to promote repair [1–5]. It mediates its effects via a family of four G protein-coupled adenosine receptors designated A1, A2A, A2B, and A3 [4]. The A3 adenosine receptor (A3AR) is a Gi protein-coupled receptor that is widely distributed, although it is most abundantly expressed in granulocytic cells including mast cells, neutrophils, and eosinophils [6–10]. In some rodents, but not in other species, the A3AR stimulates degranulation of mast cells and subsequent release of a number of biological mediators including histamine [6, 11].
In prior studies, selective agonists for the A3AR have been shown to be effective at limiting injury of the heart caused by ischemia and reperfusion [5, 12–20]. These agents have high therapeutic potential because they produce less hemodynamic side effects compared to adenosine or agonists for the other AR subtypes. Several different A3AR agonists have proven effective in both in vitro (isolated-perfused hearts) and in vivo (infarction and ‘stunning’) models of ischemia/reperfusion injury in a number of different species (mice, rats, rabbits, and dogs), including the prototypical N6-benzyladenosine-5’-N-methylcarboxamide derivatives IB-MECA and Cl-IB-MECA [5, 12–17]. In a recent study, we reported cardioprotective efficacy by a multivalent dendrimeric conjugate (MRS5246) bearing a member of a new class of N6-benzyladenosine-5’-N-methylcarboxamide derivatives that contain a bicyclic methanocarba (bicyclo[3.1.0]-hexane) ring system in place of the ribose [19].
Although efficacy has been firmly established, the mechanisms by which A3AR signaling protect against myocardial ischemia/reperfusion injury remain unclear. While there is evidence for an anti-inflammatory effect [14, 16], A3AR agonist-mediated cardioprotection is blocked by the ATP-sensitive potassium (KATP) channel antagonist glibenclamide in reductionist model systems (i.e., isolated hearts and cardiomyocytes [5]). KATP channels are potassium-selective ion channels expressed in the myocardium that activate during ischemia as ATP levels drop [21–23]. Activation of KATP channels in metabolically compromised myocardium promotes cellular survival by reducing calcium overloading and depression of force development during muscle contraction secondary to action potential shortening [21–23]. It has previously been established that KATP channel activity in cardiomyocytes is enhanced by A1AR signaling through Gi inhibitory proteins and it is thought that A1AR agonists protect against ischemic injury by potentiating KATP channel opening [5, 22, 23]. Similar to other modifiers of the KATP channel (i.e. ADP, H+, certain potassium channel openers), Gi proteins activated by A1 receptors as well as certain other Gi protein-coupled receptors expressed in the myocardium such as muscarinic receptors reduce sensitivity of the channel to blockade by ATP [21–23]. Considering that A3ARs are also Gi protein-coupled receptors, it is plausible that A3AR agonists may act by a similar mechanism. However, this idea remains controversial because, unlike with the A1AR [24], there is little evidence for expression of A3ARs in adult cardiomyocytes.
CP-532,903 is an A3AR agonist with greater than 100-fold selectivity versus the A1AR and little if any activity versus A2A and A2BARs [18, 20, 25, 26]. Structurally, CP-532,903 is an N6-dichlorobenzylsubstituted adenosine derivative modified at the 5’ position of the ribose moiety as a methyluronamide and at the 3’ position with an amino group in place of the hydroxyl group [18, 25, 26]. In a previous study, we reported that CP-532,903 was highly cardioprotective in an isolated mouse heart model of global ischemia/reperfusion injury and that protection provided by CP-532,903 was absent in experiments using hearts from Adora3−/− mice or from mice lacking the pore-forming subunit (Kir6.2) of the KATP channel, implicating involvement of the A3AR and KATP channels [18]. Given remaining questions regarding the cellular expression of A3ARs in the heart, however, whether the cardiomyocyte is the primary site of action of CP-532,903 remains uncertain.
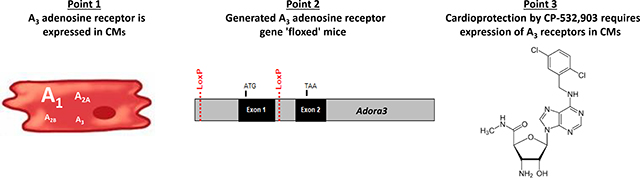
The goal of this study was to directly address these issues by creating a new mouse model described herein allowing for conditional deletion of the A3AR gene (Adora3) specifically in cardiomyocytes using the LoxP/Cre recombinase strategy. Utilizing this new tool and an isolated buffer-perfused mouse heart model, we describe complete loss of cardioprotective efficacy by CP-532,903. In parallel electrophysiological studies utilizing cardiomyocytes isolated from global Adora1 or Adora3-deleted mice, we demonstrate that CP-532,903 activates KATP channels in adult mouse ventricular cardiomyocytes via activation of A3ARs and pertussis toxin-sensitive inhibitory G proteins. Collectively, these results confirm the presence of functional A3ARs in ventricular cardiomyocytes of mice and confirm cardiomyocyte A3ARs as an important target for A3AR agonist-mediated cardioprotection.
2. Materials and Methods
2.1. Materials
Cell culture reagents and TRIzol reagent were from Invitrogen (Carlsbad, CA). CP-532,903 was provided by Pfizer Inc. (Groton, CT), Liberase Blendzyme I was from Roche Applied Biosciences (Indianapolis, IN), E-4031 was purchased from Wako (Osaka, Japan), nisoldipine was a gift from Miles-Pentex (West Haven, CT), SYBR Green Supermix was from Bio-Rad Laboratories (Hercules, CA), GoTaq G2 Green Mastermix was from Promega, ZM 241385 and PSB-603 were from Tocris Bioscience (Ellisville, MO), histamine radioimmunoassays were obtained from Immunotech (Marseille, France), and all other drugs and reagents were purchased from Sigma-Aldrich (St. Louis, MO).
2.2. Animal Care and Use
This investigation adhered to the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals (NIH Pub. Nos. 85–23, Revised 1996). All protocols utilizing mice were preapproved by the Medical College of Wisconsin’s Institutional Animal Care and Use Committee (IACUC; Animal Welfare Assurance #A3102–01).
2.3. Source of Mice
Wild-type C57Bl/6J, Tg(Myh6-cre)2182Mds/J (#011038), and Tg(ACTFLPe)9205Dym (#003800) mice were purchased from The Jackson Laboratory. Global Adora1 null mice were a gift from Dr. Bertil Fredholm (Karolinska Institute) [27] and global Adora3 null mice were from Merck Research Laboratories [28].
2.4. Cardiomyocyte and Cardiac Fibroblast Isolation
Cardiomyocytes from the left ventricles of 10–14 week-old male or female adult mice were isolated as described previously (www.signalinggateway.org/data/ProtocolLinks.html; protocol no. PP000000125). In brief, hearts were excised from pentobarbital-anesthetized mice (75 mg/kg i.p.), cannulated via the aorta onto a blunted needle, and perfused for 10 min with warmed (37°C) perfusion buffer (in mM: 113 NaCl, 4.7 KCl, 0.6 KH2PO4, 0.6 Na2HPO4, 1.2 MgSO4-7H2O, 0.032 phenol red, 12 NaHCO3, 10 KHCO3, 10 HEPES [pH 7.4], 30 taurine, 10 2,3-butanedione monoxime, 5.5 glucose) containing 0.25 mg/ml Liberase Blendzyme I, 0.14 mg/ml trypsin, and 12.5 μM CaCl2. Following perfusion, the left ventricle was dissected free from the atria and right ventricle, and repeatedly passed through a plastic transfer pipette to disaggregate the cells into a single-cell suspension. Subsequently, myocytes were enriched by sedimentation in perfusion buffer containing 5% bovine calf serum while slowly exposing the cells to increasing concentrations of CaCl2 to achieve a final concentration of 1.2 mM. The final cell pellet containing calcium-tolerant ventricular cardiomyocytes was re-suspended in minimal essential medium containing Hanks’ salts, 2 mM L-glutamine, 5% bovine calf serum, 10 mM 2,3-butanedione monoxime, and 100 U/ml penicillin, which were used in electrophysiology studies. To further increase purity for the qPCR studies, the cardiomyocytes were allowed to attach to laminin-coated tissue culture plates for 1 h and then washed extensively with cell culture media to remove non-adherent cells. Cardiomyocyte purity after selective plating averaged 90–95%. Supernatants containing fibroblasts from the cardiomyocyte sedimentation procedure were plated onto plastic cell culture dishes and cultured in DMEM containing 10% fetal bovine serum, 100 units/ml penicillin, and 100 μg/ml streptomycin until confluent.
2.5. Quantitative RT-PCR (qPCR)
qPCR was performed, as described previously [8, 29], to assess mRNA levels of AR transcripts in isolated cardiomyocytes and cultured cardiac fibroblasts. Total RNA was obtained using TRIzol reagent. Subsequently, 1 μg of total RNA was reverse-transcribed using a mixture of random and poly-T primers, according to the manufacturer’s protocol (Invitrogen). Primers were designed for the mouse A1 (forward, 5′-TGGCTCTGCTTGCTATTG-3′; reverse, 5′-GGCTATCCAGGCTTGTTC-3′), A2A (forward, 5′ TCAGCCTCCGCCTCAATG-3′; reverse, 5′-CCTTCCTGGTGCTCCTGG-3′), A2B (forward, 5′-TTGGCATTGGATTGACTC-3′; reverse, 5′-TATGAGCAGTGGAGGAAG-3′), and A3AR (forward, 5′-CGACAACACCACGGAGAC-3′; reverse, 5′-GCTTGACCACCCAGATGAC-3′) using Beacon Design software (Bio-Rad Laboratories). PCR amplification (in SYBR Green Supermix) was performed using an iCycler iQ thermocycler (Bio-Rad Laboratories) for 40 cycles of 25 s at 95°C followed by 45 s at an optimized annealing temperature for each AR. The cycle threshold, determined as the initial increase in fluorescence above background, was ascertained for each sample. Melt curves were performed upon completion of the cycles to ensure that nonspecific products were absent. For quantification of AR transcripts, a standard curve plotting cycle threshold versus copy number was constructed for each receptor subtype by analyzing 10-fold serial dilutions of plasmids containing the full-length mouse AR cDNA clones. AR transcript levels were expressed as copies per 100 ng of total RNA.
2.6. Electrophysiology
Whole-cell KATP currents (IKATP) were recorded from isolated cardiomyocytes at room temperature in a sodium-free external solution (5 mM KCl, 132 mM N-methyl-D-glucamine, 1 mM CaCl2, 2 mM MgCl2, 10 mM HEPES [pH 7.4]) containing 4-aminopyridine (5 mM) and E-4031 (5 μM) to block voltage-activated potassium currents, nisoldipine (200 nM) to block voltage-activated calcium currents, ZM 241385 (100 nM) to block A2AARs, and PSB 603 (100 nM) to block A2BARs. For the pertussis toxin experiments, cardiomyocytes were incubated in external solution containing 2 μg/ml pertussis toxin for a minimum of 1 h and up to 6 h during the course of the day while experiments were being conducted. The pipette solution was also sodium-free (60 mM K-glutamate, 50 mM KCl, 1 mM MgCl2, 1 mM CaCl2, 11 mM EGTA, and 10 mM HEPES [pH 7.4]) and contained K2-ATP at a low concentration (0.2 mM) to reduce inhibition of KATP channels and 0.5 mM Na+-GTP to permit G protein activation. Recording pipettes were pulled from borosilicate glass capillary tubes (Garner Glass, Claremont, CA) using a horizontal two-stage puller (Sachs-Flaming P-97; Sutter Instruments, Novato, CA) and heat polished (microforge MF-830; Narishige, Tokyo, Japan). In standard solutions, the pipette resistance ranged from 3 to 5 MΩ. Current was measured using a patch clamp amplifier (Axopatch 200B; Axon Instruments, Foster City, CA) interfaced to a computer via a digitizer (Digidata 1322 A; Axon Instruments). Data acquisition and analysis were conducted using the pClamp software package version 9.0 (Axon Instruments). Additional analyses were performed on Origin version 7 (OriginLab, Northampton, MA).
2.7. Generation of Mice with LoxP-Flanked Adora3 Alleles
To generate mice with LoxP-flanked alleles, a targeting vector was prepared from a 13 kb bacterial artificial chromosome (BAC) clone containing Adora3 (Figure 1). The vector contained a LoxP site in the 5’ non-coding region, which was created by subcloning an insert extended with the LoxP sequence between XhoI and BlpI sites. In addition, the vector contained a neomycin phosphotransferase (neo) cassette (a gift from Stephen Duncan, PhD, Medical University of South Carolina) inserted into intron 1 by recombineering. The neo cassette was flanked on either side by Frt sites and on the 3’ end with a LoxP site. Correctly targeted embryonic stem cells (ESCs; line D3), confirmed by Southern blotting (Figure 1), were injected into C57Bl/6NCrl blastocysts, which implanted and transmitted the targeted allele via germline. The neo cassette in intron 1 was subsequently removed by mating with a mouse ubiquitously expressing a Flp recombinase leaving behind the LoxP site. Mice heterozygous for the targeted allele (Adora3LoxP/+) were back-crossed with wild-type C57Bl/6J mice for nine generations, and then bred to homozygosity resulting in the genotype Adora3LoxP/LoxP. Mice (C57Bl/6) containing the cardiac-specific Myh6-driven Cre-recombinase transgene, which is briefly activated during early development followed by strong and persistent expression in the ventricles following birth [30], were bred into the heterozygous Adora3LoxP/+ background. Adora3LoxP/LoxP;Myh6-Cre+ were bred with Adora3LoxP/LoxP mice to obtain the experimental (Adora3LoxP/LoxP;Myh6-Cre) and control (Adora3LoxP/LoxP) littermate genotypes that were compared in the study.
Figure 1. Gene targeting strategy.

The targeting vector, containing a 13.0 kb fragment of Adora3 isolated from a mouse BAC clone library, was prepared to introduce two LoxP sites into Adora3, one into the 5’ non-coding region proximal to exon 1 and the second into intron 1. This permits Cre recombinase-mediated excision of exon 1 including the ATG start site along with ~2 kb of upstream sequence containing promoter elements. This targeting strategy avoids depletion of the transmembrane and immunoglobulin domain containing 3 splice variant (TMIGD3 or Adora3i3) that shares exon 2 of Adora3 with the A3AR [42]. Correctly targeted ESCs (line D3 derived from blastocysts of a 129S2/SvPas mouse) were injected into C57Bl/6 blastocysts, generating 60–90% chimeric male mice estimated from coat color that transmitted the floxed allele via germline, providing the genotype Adora3LoxP(FrtneoFrt)LoxP/+). The neo gene in intron 1 was removed by mating with a mouse expressing the Flp recombinase transgene while leaving the LoxP site behind. A. Restriction map of the LoxP-targeted Adora3 allele. Bars denote Southern blot probes named 5’ probe and 3’ probe. B. Southern blotting of a correctly targeted ESC line used for blastocyst injections. Left panel. Digestion with NheI followed by hybridization with the 5’ probe reveals a 10.8 kb band diagnostic of the floxed allele due to presence of a NheI site within the neo cassette and a 16.6 kb band diagnostic of the wild-type allele. Right panel. Digestion with BglI and hybridization with the 3’ probe yields a 19.5 kb band from the wild-type allele and a 16.6 kb band from the floxed allele created by a BglI site within the neo cassette. An additional Southern blot was performed by hybridizing a BglI digest with a probe corresponding to the neo cassette, which revealed a single 16.6 kb band ensuring that the targeting vector did not randomly insert into non-endogenous targets (not shown). Appropriate targeting was further confirmed by PCR amplification to detect presence of the LoxP sites, the neo cassette, and site-specific insertion (not shown).
2.8. Genotyping
Genotyping of Adora3LoxP mice was performed by PCR in 20 μl reactions that included 1x GoTaq G2 Green Mastermix (Promega), primers (0.5 μM), MgCl2 at the concentrations listed in Table 1, and 4 μl template. Templates consisted of DNA extracted from ear samples that had been boiled for 10 minutes in 0.3 ml 10 mM NaOH/1 mM EDTA. PCR products were amplified as indicated in Table 1, followed by separation and imaging of amplicons on ethidium bromide-stained 1.5% agarose gels. An example of genotyping results is shown in Figure 2.
Table 1.
Primer pairs for PCR genotyping.
| Primer Set | Primer Sequences (5’-3’) | Amplicon (bp) | Purpose | ||
|---|---|---|---|---|---|
| 5’ LoxP | FWD REV |
GGTTTGAATGTCAGTTTGTCAC GCATACATTATACGAAGTTATCCC |
wt allele | none | detect 5’ LoxP |
| floxed allele | 820 | ||||
| 3’ LoxP | FWD REV |
GGAAGGTTTGTTTAAGCAGAAAT GCCTCCACTCAACCTTAGC |
wt allele | 581 | distinguish het. vs homozygous |
| floxed allele | 615 | ||||
| Recombined LoxP | FWD REV |
CCACACCTTCTAATAGTGCCACT CTATCTGTCCCTGTCCCCTTTC |
intact floxed allele | 4,317 | detect Cre-mediated recombination |
| recomb floxed allele | 190 | ||||
| Myh6-Cre | FWD REV |
ATGACAGACAGATCCCTCCTATCTCC CTCATCACTCGTTGCATCATCGAC |
wt allele | none | detect Myh6-Cre transgene |
| Myh6-Cre | 300 | ||||
Reactions (20 μl) contained 1x GoTaq G2 Mastermix (Promega), primers (0.5 μM), 1.5 mM MgCl2 (2.0 mM for Myh6-Cre amplification), and 4 μl DNA template prepared as described in METHODS. Cycling conditions were as follows: 95 °C for 15 min followed by 35 cycles of 94 °C for 30 s/60 °C for 45 s/72 °C for 30 s, and then concluding with 72 °C for 10 min.
Figure 2. Genotyping protocol.

Mice are genotyped using two sets of primers to identify the 5’ LoxP site upstream of exon 1 and the 3’ LoxP site within intron 1. A third primer set is used to detect presence of the Myh6-Cre transgene. Lane 1: The reverse primer of the 5’ LoxP primer set anneals to the LoxP sequence thereby generating an 820 bp amplicon from the floxed allele or no product with the wild-type allele. Lane 2: The 3’ LoxP primer set flanks the 3’ LoxP site thereby generating a 581 bp amplicon diagnostic of the wild-type allele and a 615 bp amplicon diagnostic of the floxed allele. The presence of both amplicons with the 3’ LoxP primer set indicates heterozygosity. Lane 3: The Myh6-Cre primer set amplifies a 300 bp product from the Myh6-Cre transgene.
2.9. Assessment of Cre-Mediated Adora3 Recombination
Cre-mediated recombination of the Adora3 allele was assessed by PCR on DNA samples of the spleen, lung, brain, liver, kidney, small intestine, large intestine, skeletal muscle (quadriceps), and left ventricle using the primer pairs and reaction conditions listed in Table 1. These primers, which anneal to the 5’ non-coding region and intron 1 amplify a 190 bp segment following recombination.
2.10. Measurement of Plasma Histamine Levels
To ensure that placement of LoxP sequences did not interfere with normal expression and function of the A3AR in AdoraLoxP mice, Cl-IB-MECA-induced increases in plasma histamine levels was assessed. In prior studies we have shown that both Cl-IB-MECA and CP-532,903 increase plasma histamine levels in mice due to A3AR-mediated mast cell degranulation [13, 14, 18]. We used Cl-IB-MECA in this study to conserve our limited supply of CP-532,903. Mice were anesthetized with sodium pentobarbital (75 mg/kg i.p.), intubated, respirated with room air supplemented with 100% oxygen (tidal volume 225 μl; rate ~100 strokes/minute; model 845 Hugo Sachs Elektronic), and subjected to a thoracotomy to expose the heart. A saline-filled cannula (stretched PE-10 tubing) was inserted into the left carotid artery for drug administration. Blood samples (~150 μl) were collected by cardiac puncture into EDTA-coated syringes containing the histamine N-methyltransferase inhibitor SKF-91488 (final concentration = 20 μM). The blood samples were centrifuged (1000 x g for 10 min at 4° C) and the plasma was analyzed for histamine content by enzyme-linked immunosorbant assay (Immuno-Biological Laboratories).
2.11. Langendorff-Perfused Isolated Heart Model
Male or female mice (10–12 weeks of age) were anesthetized with sodium pentobarbital (75 mg/kg i.p.). As soon as deep anesthesia was achieved, the hearts were removed, arrested in ice-cold perfusion solution, and then rapidly cannulated via the aorta and perfused by the Langendorff method at continuous pressure (80 mm Hg) with Krebs-Henseleit (KH) buffer containing 118 mM NaCl, 4.7 mM KCl, 1.2 mM MgCl2, 2.5 mMCaCl2, 1.2 mM KH2PO4, 0.5 mM EDTA, 25 mM NaHCO3, and 11 mM glucose. The KH buffer was equilibrated with 95% O2/5% CO2 at 37°C to achieve a pH of 7.4 and filtered through an in-line Sterivex 0.22 μm filter unit (Millipore, Bedford, MA) to remove particulate matter. With each heart, polyethylene tubing was inserted into the apex to drain the left ventricle, after which a custom-prepared fluid-filled plastic film balloon connected to a pressure transducer (ADInstruments, Colorado Springs, CO) was inserted into the left ventricle via the left atria for continuous measurement of left ventricular pressure. The hearts were immersed in perfusion buffer maintained at 37°C, and the balloons were inflated to achieve end-diastolic pressures of 5–10 mm Hg. The left ventricular pressure signals were acquired continuously using a PowerLab data acquisition system (ADInstruments) and processed (Chart software) to yield heart rates and left ventricular dP/dts. Coronary flows were monitored by an ultrasonic in-line flow-probe connected to a T206 flowmeter (Transonic Systems, Inc., Ithaca, NY). To allow for pacing, the left ventricles were pierced with 0.125 mm silver electrodes connected to a Grass SD9 stimulator (Astro-Med, Inc., West Warwick, RI).
The experimental protocol was as follows. Once prepared, the hearts were perfused for 20 min while being paced at 420 bpm (2 ms square waves 20% above threshold) to allow for stabilization and acquisition of baseline parameters. Subsequently, they were subjected to 20 minutes of no-flow global ischemia followed by 45 minutes of reperfusion, achieved by closing and opening an in-line stopcock. Hearts were randomly assigned to be perfused with buffer containing either CP-532,903 (100 nM) or equivalent vehicle (less than 0.1 %) for 10 min before the induction of ischemia. Left ventricular function was continually assessed prior to ischemia and for 45 minutes after flow was resumed. All experiments and data analyses were conducted in a blinded manner without knowledge of drug treatment or mouse heart genotype. Headrick and colleagues have previously documented that adenosine efflux increases ~150-fold following global, normothermic ischemia (20 min) using a similar isolated mouse heart model [31].
2.12. Data Analysis
All data are reported as means ± SEM. Left ventricular functional parameters and IKATP densities were compared by an unpaired Student’s t-test or a one-way ANOVA, as appropriate. P<0.05 was considered statistically significant.
3. Results
3.1. Identification of A3ARs in Cardiomyocytes: mRNA Expression and Electrophysiology
Due to lack of useful commercially available antibodies, it is currently not possible to assess expression of ARs in cells/tissues by conventional immunological methods. By qPCR, A3AR transcripts were detected in total RNA prepared from isolated cardiomyocyte preparations, although the level of expression was very low compared to that of A1 (150-fold less) and A2AAR (28-fold less) mRNAs (Table 2). Low-level expression of A2BAR transcripts was also detected in cardiomyocytes.
Table 2.
Adenosine Receptor mRNA content in isolated cardiomyocytes and cardiac fibroblasts determined by qPCR.
| Cardiomyocytes (n=4) | Cardiac Fibroblasts (n=3) | |
|---|---|---|
| A1 | 12,830 ± 2,534 | 8 ± 4 |
| A2A | 2,388 ± 304 | 4,083 ± 141 |
| A2B | 330 ± 45 | 40,940 ± 2,123 |
| A3 | 85 ± 5 | 19 ± 3 |
copy number (mean ± SEM) per 100 ng total RNA
Because we have previously observed that A3AR agonist-mediated cardioprotection is blocked in some model systems by glibenclamide [12, 18], we proceeded to assess coupling of the A3AR to KATP channel opening. IKATP was recorded in freshly isolated mouse left ventricular cardiomyocytes using the whole-cell configuration of the patch clamp technique. Basal whole-cell current was monitored for 20 minutes to allow for diffusional exchange of ATP between the pipette solution, present at a low concentration (0.2 mM), and the intracellular milieu. This protocol reduced inhibition of KATP channels by ATP and mimicked reductions in ATP that occur during metabolic compromise. At this time, the effect of applying CP-532,903 (1 μM) or the A1AR agonist CPA (1 μM) on IKATP was measured by 100-ms test pulses from −120 to +60 mV from a holding potential of −40 mV. To confirm specificity of the agonists, recordings were obtained from myocytes isolated from global Adora1−/− or global Adora3−/− mice. Myocytes that exhibited spontaneous activation of outward current immediately upon establishing whole-cell configuration prior to drug application were discarded.
As shown in Figure 3, application of CPA to wild-type myocytes increased IKATP, defined by glibenclamide (1 μM)-sensitivity. This effect of CPA was inhibited by exposure to pertussis toxin (Figure 5) and was also apparent in myocytes obtained from Adora3−/− mice, but not from Adora1−/− mice (Figure 3). Currents evoked in the presence of CPA were subtracted from those in the absence of the agonist to yield the drug-induced contribution to the current. Current densities, normalized to cell capacitance, were determined to be 2.70 ± 0.65 and 2.51 ± 0.76 pA/pF at test potentials of 0 and +40 mV, respectively (Figure 3). As demonstrated in Figures 4 and 5, CP-532,903 also increased IKATP via a pertussis toxin-sensitive mechanism in wild-type cardiomyocytes, although it was slightly less in magnitude (1.86 ± 0.31 and 1.95 ± 0.55 pA/pF at test potentials of 0 and +40 mV) compared to that elicited by CPA. Based on findings with gene-deleted mice, this effect of CP-532,903 was dependent on expression of the A3AR (but not the A1AR). These findings indicate that both A1 and A3ARs, coupled to activation of KATP channels, are expressed in mouse ventricular cardiomyocytes. As hypothesized previously [22, 23], these findings are consistent with the idea that stimulation of these receptors activate Gi proteins in cardiomyocytes, which reduces sensitivity of KATP channels to ATP thereby causing them to open.
Figure 3. Evidence for coupling of A1ARs to KATP channel activation in adult mouse cardiomyocytes.

A and B. Representative whole-cell current traces recorded from an isolated cardiomyocyte from a wild-type (WT) C57B/6 mouse after application of CPA (1 μM) in the presence or absence of glibenclamide (1 μM). The glibenclamide-sensitive current shown in B was obtained by digitally acquiring the CPA-elicited current that was sensitive to inhibition by glibenclamide. C and D. Representative whole-cell currents recorded from an isolated cardiomyocyte from either an Adora1−/− mouse (C) or and Adora3−/− mouse (D) after application of CPA with corresponding current-voltage relationships of the CPA-elicited current obtained by digital subtraction. E) Summary (n=5–7; means ± SEM) of the effects of CPA on IKATP showing dependence on expression of the A1 but not the A3AR. Current density was determined at the 0 and +40 mV test pulses from a −40 mV holding potential. Current density, determined by normalizing current amplitude to cell capacitance, is presented to account for differences in cell size. All recordings were obtained as described in METHODS in the presence of ZM241385 (100 nM) and PSB603 (100 nM) to block A2A and A2BARs. *P<0.05 vs WT.
Figure 5. CPA- and CP-532,903-elicted IKATP are blocked by treatment with pertussis toxin.

Representative whole-cell currents recorded from pertussis toxin-treated cardiomyocytes (2 μg/ml for a minimum of 1 h) isolated from wild-type C57Bl/6 mice after application of CPA (1 μM; A) or CP-532,903 (1 μM; B). Both agonists failed to significantly elicit IKATP after pertussis toxin treatment. Similar results were obtained in recordings from 4–6 additional cells per treatment group. All recordings were obtained as described in METHODS in the presence of ZM241385 (100 nM) and PSB603 (100 nM) to block A2A and A2BARs.
Figure 4. Evidence for coupling of A3ARs to KATP channel activation in adult mouse cardiomyocytes.

Representative whole-cell currents recorded from an isolated cardiomyocyte from either a wild-type (WT) C57B/6 (A), Adora1−/− (B), or Adora3−/− (C) mouse after application of CP-532,903 (1 μM) with corresponding current-voltage relationships of the CP-532,903-elicited current obtained by digital subtraction. D. Summary (n=5–7; means ± SEM) of the effects of CP-532,903 on IKATP showing dependence on expression of the A3 but not the A1AR. Current density was determined at the 0 and +40 mV test pulses from a −40 mV holding potential. All recordings were obtained as described in METHODS in the presence of ZM241385 (100 nM) and PSB603 (100 nM) to block A2A and A2BARs. *P<0.05 vs WT.
3.2. Production of Adora3LoxP/LoxP;Myh6-Cre Mice
Mice with the experimental genotype Adora3LoxP/LoxP;Myh6-Cre and the control genotype Adora3LoxP/LoxP were prepared (Figure 1). The LoxP sites located in the 5’ non-coding region and in the first intron were designed to undergo recombination by Cre recombinase, the Myh6-Cre driver for which becomes strongly expressed in ventricular cardiomyocytes near the time of birth [30]. This resulted in ablation of the first exon, along with ~2 kb of non-coding DNA upstream of exon 1 that contained the Adora3 promoter [32]. As shown in Figure 6, PCR amplification of the Adora3 allele in Adora3LoxP/LoxP;Myh6-Cre mice revealed that the LoxP-targeted allele was disrupted in a tissue-specific manner, as revealed by the 190 bp product indicative of recombination in the heart, which was not detected in samples from seven other organs or from skeletal muscle. Placement of LoxP sites into the Adora3 allele did not interfere with expression or function of the A3AR, demonstrated by retained ability of the A3AR agonist Cl-IB-MECA to increase plasma histamine levels in Adora3LoxP/LoxP mice resultant from A3AR-induced mast cell degranulation (Figure 6). Cl-IB-MECA also increased plasma histamine levels in Adora3LoxP/LoxP;Myh6-Cre mice to an equivalent extend as compared to both Adora3LoxP/LoxP and C57BL/6 wild-type mice, indicating specificity of the Myh6-Cre transgene (Figure 6).
Figure 6. Myh6-Cre disrupts the LoxP-flanked A3AR gene (Adora3) specifically in myocardium.

Panel A: Depiction of Adora3LoxP allele before and after Myh6-Cre-mediated recombination. Recombination was assessed by PCR amplification of genomic DNA obtained from a panel of organs of a 12 week-old adult male Adora3LoxP/LoxP;Myh6-Cre mouse. Each lane contains a PCR reaction of genomic DNA obtained from the indicated tissues. The positive control lane used DNA isolated from cultured ear fibroblasts from an Adora3LoxP/LoxP mouse infected with an adenovirus that expresses Cre recombinase. The negative control lane contained a PCR reaction using genomic DNA obtained from heart tissue of a 10 week-old male Adora3LoxP/LoxP mouse. An amplicon was only generated with heart DNA from the Adora3LoxP/LoxP;Myh6-Cre mouse, indicating cardiomyocyte-selective recombination. Panel B: Administration of Cl-IB-MECA (100 μg/kg i.p.) to Adora3LoxP/LoxP or Adora3LoxP/LoxP;Myh6-Cre mice (n=3–7) increased plasma histamine levels resultant from A3AR-mediated mast cell degranulation to an equivalent extent as compared to wild-type C56Bl/6 mice.
3.3. Isolated Heart Studies
Hearts were perfused by the Langendorff procedure, as described in METHODS. The experimental designed involved comparing the effects of treatment with CP-532,903 on hearts from mice with the experimental genotype Adora3LoxP/LoxP;Myh6-Cre or control mice having the Adora3LoxP/LoxP genotype while pacing at a rate of 420 beats/min. CP-532,903 was infused into the coronary circulation of the hearts for 10 min immediately before 20 min of global ischemia. Recovery of left ventricular contractile function (developed pressure, maximal rate of contraction, and maximal rate of relaxation) was used as an index of the extent of ischemia/reperfusion injury.
As shown in Table 3, baseline functional parameters and coronary flow rates were not significantly different between Adora3LoxP/LoxP;Myh6-Cre and Adora3LoxP/LoxP hearts. Neither coronary flow rate nor contractile properties were changed during infusion of CP-532,902 into hearts of either genotype (Table 3). These observations concur with our previous study, which demonstrated that infusion of CP-532,903 into spontaneously beating isolated mouse hearts up to a concentration of 100 nM did not alter left ventricular function, coronary flow rate, or heart rate, whereas infusion of an A1 (CCPA) or an A2AAR (CGS 21680) agonist produced prominent changes in all of these parameters [18].
Table 3.
Functional parameters of isolated hearts at baseline and after infusion of CP-532,903 or vehicle while pacing at 420 beats/min.
| Genotype | N | Treatment | Devel. Pressure (mmHg) | +dP/dt (mmHg/s) | −dP/dt (mmHg/s) | Coronary Flow (ml/min/g) |
|---|---|---|---|---|---|---|
| Adora3LoxP/LoxP | Male = 9 | baseline | 93.3 ± 2.0 | 3,704 ± 263 | −2,006 ± 406 | 22.4 ± 1.8 |
| Female = 4 | vehicle | 94.9 ± 1.8 | 3,765 ± 253 | −1,985 ± 401 | 22.1 ± 2.0 | |
| Total = 13 | ||||||
| Male = 7 | baseline | 86.5 ± 3.5 | 3,098 ± 467 | 2,254 ± 118 | 23.1 ± 1.1 | |
| Female = 3 | CP-532,903 | 86.2 ± 3.2 | 3,238 ± 476 | 2,273 ± 116 | 22.3 ± 1.0 | |
| Total = 10 | ||||||
| Adora3LoxP/LoxP;Myh6-Cre | Male = 7 | baseline | 79.2 ± 7.0 | 3,248 ± 408 | −2,108 ± 226 | 21.9 ± 1.1 |
| Female = 6 | vehicle | 80.5 ± 6.7 | 3,299 ± 390 | −2,127 ± 229 | 21.0 ± 0.9 | |
| Total = 13 | ||||||
| Male = 7 | baseline | 88.6 ± 1.8 | 3,553 ± 197 | −2,249 ± 111 | 25.2 ± 1.8 | |
| Female = 6 | CP-532,903 | 88.8 ± 1.9 | 3,571 ± 192 | −2,255 ± 112 | 24.6 ± 2.1 | |
| Total = 13 | ||||||
With all hearts, contractile function diminished upon the onset of ischemia with complete arrest within 5 min. With the onset of reperfusion, the hearts resumed spontaneous contraction within ~2 min, and then after a short period of decline contractile function progressively improved for the remainder of the reperfusion period. Left ventricular developed pressure data during reperfusion of control Adora3LoxP/LoxP hearts treated with either vehicle or CP-532,903 is shown in Figure 7 (panel a). As is common practice with the isolated mouse heart model [15, 18, 19, 33], the data in Figure 7 are expressed as a percentage of pre-ischemic values to control for interindividual variability within the experimental groups. As reported previously [18], control Adora3LoxP/LoxP hearts treated with CP-532,903 exhibited better recovery of left ventricular developed pressure compared to those treated with vehicle at nearly all time-points assessed during reperfusion. Final recovery of developed pressure was 70.1 ± 2.7% of baseline in CP-532,903-treated hearts versus 58.1 ± 3.3% in vehicle-treated hearts (P<0.05). Other contractile parameters including maximal +dP/dt and −dPdt were also significantly improved by CP-532,903 by ~15–20% (P<0.05), whereas the rate of coronary flow was unaffected (Figure 7a). Although group sizes were too small to show significant differences, when analyzed individually there was a clear trend for CP-532,903 to provide improvement in hearts from both male and female Adora3LoxP/LoxP mice (not shown).
Figure 7. CP-532,903 does not protect hearts from Adora3LoxP/LoxP;Myh6-Cre mice in which Adora3 is deleted specifically in cardiomyocytes form ischemia/reperfusion injury.

Effect of CP-532,903 on recovery of left ventricular developed pressure (expressed as a percentage of baseline) of isolated hearts from Adora3LoxP/LoxP (Panel A) and Adora3LoxP/LoxP;Myh6-Cre (Panel B) mice subjected to 20 min of global ischemia and 45 min of reperfusion. Hearts were treated with either CP-532,903 (100 nM) or equivalent vehicle for 10 min prior to induction of ischemia. Insets show final recovery of left ventricular maximal ±dP/dt and coronary flow rate data at the end of the 45-min reperfusion period. Values shown are means ± SEM. *P<0.05 vs. vehicle-treated group. n = 10–13.
The effect of CP-532,903 on hearts from Adora3LoxP/LoxP;Myh6-Cre mice in which Adora3 is deleted specifically in cardiomyocytes is shown in Figure 7 (panel b). In these studies, CP-532,903 did not improve functional recovery. If anything, the rate of recovery of CP-532,903-treated hearts appeared to be delayed compared to vehicle-treated hearts.
4. Discussion
A3AR agonists hold promise as therapeutic agents for the treatment of several diseases including myocardial ischemia/reperfusion injury and have the advantage of being effective while producing no adverse hemodynamic effects. In the current study, we provide definitive evidence for the presence of A3ARs coupled to activation of KATP channels in adult mouse ventricular cardiomyocytes. We subsequently found that the selective A3AR agonist CP-532,903 lost effectiveness to reduce ischemia/reperfusion injury in an isolated, buffer-perfused heart model when Adora3 was deleted in cardiomyocytes. These findings identify cardiomyocyte A3ARs as an important target of A3AR agonist-mediated cardioprotection. Taking into consideration results of prior published work [12, 18], our findings are consistent with the hypothesis that A3AR agonists provide ischemic tolerance, at least in part, by facilitating opening of myocardial KATP channels.
The current study utilized an isolated, buffer-perfused heart model to exclude contribution of blood components and a complete inflammatory response to the injury process. It is well established that the A3AR is expressed in several different leukocyte populations, in particular granulocytic cells including neutrophils, where A3AR signaling modulates chemotaxis and oxidant production by the NADPH oxidase complex [6–10]. In a previous study, we found that the A3AR agonist Cl-IB-MECA reduced infarct size in an in vivo mouse model of ischemia/reperfusion when given at the time of reperfusion, and that this was dependent on expression of A3ARs in bone marrow-derived cells [14]. In a separate study, IB-MECA reduced contractile dysfunction caused by infusion of neutrophils into the coronary circulation of post-ischemic, buffer-perfused isolated rat hearts [16]. Thus, the cardioprotective mechanisms of A3AR agonists are likely multifactorial. Current evidence suggests some contribution of inflammation suppression and reduced immune cell-mediated reperfusion injury. Because A3AR agonists stimulate mast cell degranulation in some rodents including mice [6, 11], another contributing mechanism in these species may relate to potential benefit from depleting mast cells of their contents prior to the onset of ischemia [6, 17]. With the isolated heart model, it is important to recognize that post-ischemic contractile dysfunction results from a combination of both reversible injury processes (i.e., myocardial ‘stunning’) and cell death (i.e., infarction). Whether CP-532,903 protected against one or both of these forms of injury was not addressed in the present investigation, although we speculate a broad effect considering A3AR agonists have been shown in a number of different model systems to reduce infarct size, to promote cardiomyocyte survival, and to attenuate myocardial stunning [5, 12–20].
Traditionally, the A1AR has been viewed as the only AR subtype expressed in cardiac muscle. In ventricular muscle cells, A1AR signaling reduces the positive inotropic effects of β adrenergic receptor stimulation, by coupling through Gi proteins to multiple effectors including adenylyl cyclase, potassium channels, and calcium channels [24]. As reported previously using neonatal (rat)/juvenile (guinea pig) cells [22, 23] and as demonstrated in this study using adult mouse cells, the A1AR also signals to facilitate opening of KATP channels in ventricular cardiomyocytes. With the development of more selective pharmacological tools and genetically modified mice, however, it has become apparent that multiple ARs are expressed in cardiomyocytes [2]. For instance, it is now well accepted that A2AARs are expressed in the myocardium that modulate contractile responses and potentially the release of pro-growth/pro-inflammatory factors [2]. There is evidence for cardiomyocyte expression of A2BARs that may modulate metabolism and responses to hypoxic stress [34]. Our finding in the current study for presence of A3ARs in mouse cardiomyocytes, despite low transcript levels, needs to be confirmed in other species. It will be important to determine whether the A3AR may couple to additional signaling pathways in cardiomyocytes.
It is necessary to point out in our studies that post-ischemic recovery of contractile function of vehicle-treated Adora3LoxP/LoxP;Myh6-Cre hearts with Adora3 deleted in cardiomyocytes tended to be better compared to vehicle-treated Adora3LoxP/LoxP hearts. Final recovery of developed pressure was 58.1 ± 3.3% of baseline in vehicle-treated Adora3LoxP/LoxP hearts vs. 70.0 ± 2.2% in vehicle-treated Adora3LoxP/LoxP;Myh6-Cre hearts. Recovery of other contractile parameters was also better (Figure 7). It is tempting to conclude from this comparison that cardiomyocyte A3ARs play a damaging role during the pathogenesis of ischemia/reperfusion injury even though these same receptors are the molecular site of action of A3AR agonist-mediated cardioprotection. Perhaps both A3AR agonism and antagonism, depending on timing, may attenuate ischemia/reperfusion injury, potentially explained by the unique biology of the A3AR such as exceptionally rapid receptor desensitization [35, 36] or susceptibility to biased agonism [37, 38]. However, we believe that it is not prudent to make direct comparisons between these genotypes in our studies due to the presence of the Myh6-Cre transgene in Adora3LoxP/LoxP;Myh6-Cre hearts. It is also possible that unanticipated compensatory changes may arise from cardiomyocyte-specific deletion of Adora3 [15, 39]. For example, it is theoretically possible for expression or cellular localization of other members of the AR family to be impacted by loss of A3AR signaling [40]. This observation requires further investigation and must be aided by experimental data with A3AR antagonists as they become available. Interestingly, mice with global deletion of Adora3 have also been reported to be more resistant to ischemia/reperfusion injury, but whether this is attributed to loss of A3AR signaling, non-specific compensatory changes, or genetic background effects remains uncertain [15, 33, 39, 41]. Our targeting strategy for depletion of the A3AR in the present investigation was designed to avoid disruption of the transmembrane and immunoglobulin domain containing 3 splice variant (TMIGD3i3 or Adora3i3) that shares exon 2 of Adora3 with the A3AR [42].
In conclusion, we achieved three important goals in this study. First, we provide evidence for functional expression of A3ARs in the mouse myocardium, which are coupled to KATP channel activation. Second, and perhaps most importantly, we successfully prepared a genetically modified line of mice that allows for conditional depletion of the A3AR in mice by means of the LoxP-Cre recombinase strategy. This new tool will be a valuable resource for future investigation of the complex biology of the A3AR in mice. Finally, utilizing this tool we provide new information regarding the underlying causes of A3AR agonist-mediated cardioprotection. Additional studies are needed to assess additional A3AR agonists with different signaling profiles [37, 38], to examine the effect of deletion of Adora3 in other cell types, and to expand the analyses to in vivo models. Our findings further support the idea that A3AR agonism may be a useful approach for limiting ischemia/reperfusion injury of the heart.
Acknowledgements
We thank Dr. Jeffrey Conroy, PhD (Roswell Cancer Institute, SUNY-Buffalo, NY) for providing mouse BAC clones, Stephen Duncan, PhD (Medical University of South Carolina, Charlestown, SC) for assistance with designing and preparing the Adora3 targeting construct, and the Transgenic Core Facility at the Medical College of Wisconsin and the Blood Research Institute of Southeastern Wisconsin for embryonic stem cell targeting and blastocyst injections.
Funding
This work was supported by the National Institutes of Health (R01 HL077707, HL133589, and HL111392) and by a research award from the Cardiovascular Center at the Medical College of Wisconsin.
Abbreviations:
- AR
(adenosine receptor)
- IB-MECA
[N6-(3-iodobenzyl)adenosine-5’-N-methyluronamide]
- Cl-IB-MECA
[2-chloro-N6-(3-iodobenzyl)adenosine-5’-N-methyluronamide]
- KATP
(ATP-dependent potassium channel)
- CP-532,903
(N6-(2,5-dichlorobenzyl)-3’-aminoadenosine-5’-N-methylcarboxamide]
- CCPA
(2-chloro-N6-cyclopentyladenosine)
- CPA
(N6-cyclopentyladenosine)
- Adora3
(A3 adenosine receptor gene)
- ZM241385
[4-[2-[7-(2-furyl)[1,2,4]triazolo-[2,3-a][1,3,5]triazine-5-ylamino]ethyl]phenol]
- PSB603
([8–4-[4-[4-[chlorophenzyl)piperazide-1-sulfonyl)phenyl]]-1-propylxanthine]
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Auchampach JA, Adenosine receptors and angiogenesis, Circ Res 101(11) (2007) 1075–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Auchampach JA, Bolli R, Adenosine receptor subtypes in the heart: therapeutic opportunities and challenges, Am J Physiol 276(3 Pt 2) (1999) H1113–6. [DOI] [PubMed] [Google Scholar]
- [3].Ely SW, Berne RM, Protective effects of adenosine in myocardial ischemia, Circulation 85(3) (1992) 893–904. [DOI] [PubMed] [Google Scholar]
- [4].Fredholm BB, Adenosine, an endogenous distress signal, modulates tissue damage and repair, Cell Death Differ 14(7) (2007) 1315–23. [DOI] [PubMed] [Google Scholar]
- [5].McIntosh VJ, Lasley RD, Adenosine receptor-mediated cardioprotection: are all 4 subtypes required or redundant?, J Cardiovasc Pharmacol Ther 17(1) (2012) 21–33. [DOI] [PubMed] [Google Scholar]
- [6].Linden J, Cloned adenosine A3 receptors: pharmacological properties, species differences and receptor functions, Trends Pharmacol Sci 15(8) (1994) 298–306. [DOI] [PubMed] [Google Scholar]
- [7].Chen Y, Corriden R, Inoue Y, Yip L, Hashiguchi N, Zinkernagel A, Nizet V, Insel PA, Junger WG, ATP release guides neutrophil chemotaxis via P2Y2 and A3 receptors, Science 314(5806) (2006) 1792–5. [DOI] [PubMed] [Google Scholar]
- [8].van der Hoeven D, Wan TC, Auchampach JA, Activation of the A3 adenosine receptor suppresses superoxide production and chemotaxis of mouse bone marrow neutrophils, Mol Pharmacol 74(3) (2008) 685–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Morschl E, Molina JG, Volmer JB, Mohsenin A, Pero RS, Hong JS, Kheradmand F, Lee JJ, Blackburn MR, A3 adenosine receptor signaling influences pulmonary inflammation and fibrosis, Am J Respir Cell Mol Biol 39(6) (2008) 697–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Walker BA, Jacobson MA, Knight DA, Salvatore CA, Weir T, Zhou D, Bai TR, Adenosine A3 receptor expression and function in eosinophils, Am J Respir Cell Mol Biol 16(5) (1997) 531–7. [DOI] [PubMed] [Google Scholar]
- [11].Van Schaick EA, Jacobson KA, Kim HO, AP IJ, Danhof M, Hemodynamic effects and histamine release elicited by the selective adenosine A3 receptor agonist 2-Cl-IB-MECA in conscious rats, Eur J Pharmacol 308(3) (1996) 311–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Auchampach JA, Rizvi A, Qiu Y, Tang XL, Maldonado C, Teschner S, Bolli R, Selective activation of A3 adenosine receptors with N6-(3-iodobenzyl)adenosine-5’-N-methyluronamide protects against myocardial stunning and infarction without hemodynamic changes in conscious rabbits, Circ Res 80(6) (1997) 800–9. [DOI] [PubMed] [Google Scholar]
- [13].Ge ZD, Peart JN, Kreckler LM, Wan TC, Jacobson MA, Gross GJ, Auchampach JA, Cl-IB-MECA [2-chloro-N6-(3-iodobenzyl)adenosine-5’-N-methylcarboxamide] reduces ischemia/reperfusion injury in mice by activating the A3 adenosine receptor, J Pharmacol Exp Ther 319(3) (2006) 1200–10. [DOI] [PubMed] [Google Scholar]
- [14].Ge ZD, van der Hoeven D, Maas JE, Wan TC, Auchampach JA, A3 adenosine receptor activation during reperfusion reduces infarct size through actions on bone marrow-derived cells, J Mol Cell Cardiol 49(2) (2010) 280–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Harrison GJ, Cerniway RJ, Peart J, Berr SS, Ashton K, Regan S, Matherne G. Paul, Headrick JP, Effects of A3 adenosine receptor activation and gene knock-out in ischemic-reperfused mouse heart, Cardiovasc Res 53(1) (2002) 147–55. [DOI] [PubMed] [Google Scholar]
- [16].Jordan JE, Thourani VH, Auchampach JA, Robinson JA, Wang NP, Vinten-Johansen J, A3 adenosine receptor activation attenuates neutrophil function and neutrophil-mediated reperfusion injury, Am J Physiol 277(5 Pt 2) (1999) H1895–905. [DOI] [PubMed] [Google Scholar]
- [17].Tian Y, Marshall M, French BA, Linden J, Yang Z, The infarct-sparing effect of IB-MECA against myocardial ischemia/reperfusion injury in mice is mediated by sequential activation of adenosine A3 and A 2A receptors, Basic Res Cardiol 110(2) (2015) 16. [DOI] [PubMed] [Google Scholar]
- [18].Wan TC, Ge ZD, Tampo A, Mio Y, Bienengraeber MW, Tracey WR, Gross GJ, Kwok WM, Auchampach JA, The A3 adenosine receptor agonist CP-532,903 [N6-(2,5-dichlorobenzyl)-3’-aminoadenosine-5’-N-methylcarboxamide] protects against myocardial ischemia/reperfusion injury via the sarcolemmal ATP-sensitive potassium channel, J Pharmacol Exp Ther 324(1) (2008) 234–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Wan TC, Tosh DK, Du L, Gizewski ET, Jacobson KA, Auchampach JA, Polyamidoamine (PAMAM) dendrimer conjugate specifically activates the A3 adenosine receptor to improve post-ischemic/reperfusion function in isolated mouse hearts, BMC Pharmacol 11 (2011) 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Tracey WR, Magee WP, Oleynek JJ, Hill RJ, Smith AH, Flynn DM, Knight DR, Novel N6-substituted adenosine 5’-N-methyluronamides with high selectivity for human adenosine A3 receptors reduce ischemic myocardial injury, Am J Physiol Heart Circ Physiol 285(6) (2003) H2780–7. [DOI] [PubMed] [Google Scholar]
- [21].Burke MA, Mutharasan RK, Ardehali H, The sulfonylurea receptor, an atypical ATP-binding cassette protein, and its regulation of the KATP channel, Circ Res 102(2) (2008) 164–76. [DOI] [PubMed] [Google Scholar]
- [22].Kirsch GE, Codina J, Birnbaumer L, Brown AM, Coupling of ATP-sensitive K+ channels to A1 receptors by G proteins in rat ventricular myocytes, Am J Physiol 259(3 Pt 2) (1990) H820–6. [DOI] [PubMed] [Google Scholar]
- [23].Terzic A, Tung RT, Inanobe A, Katada T, Kurachi Y, G proteins activate ATP-sensitive K+ channels by antagonizing ATP-dependent gating, Neuron 12(4) (1994) 885–93. [DOI] [PubMed] [Google Scholar]
- [24].Belardinelli L, Shryock JC, Song Y, Wang D, Srinivas M, Ionic basis of the electrophysiological actions of adenosine on cardiomyocytes, Faseb J 9(5) (1995) 359–65. [DOI] [PubMed] [Google Scholar]
- [25].DeNinno MP, Masamune H, Chenard LK, DiRico KJ, Eller C, Etienne JB, Tickner JE, Kennedy SP, Knight DR, Kong J, Oleynek JJ, Tracey WR, Hill RJ, 3’-Aminoadenosine-5’-uronamides: discovery of the first highly selective agonist at the human adenosine A3 receptor, J Med Chem 46(3) (2003) 353–5. [DOI] [PubMed] [Google Scholar]
- [26].DeNinno MP, Masamune H, Chenard LK, DiRico KJ, Eller C, Etienne JB, Tickner JE, Kennedy SP, Knight DR, Kong J, Oleynek JJ, Tracey WR, Hill RJ, The synthesis of highly potent, selective, and water-soluble agonists at the human adenosine A3 receptor, Bioorg Med Chem Lett 16(9) (2006) 2525–7. [DOI] [PubMed] [Google Scholar]
- [27].Johansson B, Halldner L, Dunwiddie TV, Masino SA, Poelchen W, Gimenez-Llort L, Escorihuela RM, Fernandez-Teruel A, Wiesenfeld-Hallin Z, Xu XJ, Hardemark A, Betsholtz C, Herlenius E, Fredholm BB, Hyperalgesia, anxiety, and decreased hypoxic neuroprotection in mice lacking the adenosine A1 receptor, Proc Natl Acad Sci U S A 98(16) (2001) 9407–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Salvatore CA, Tilley SL, Latour AM, Fletcher DS, Koller BH, Jacobson MA, Disruption of the A3 adenosine receptor gene in mice and its effect on stimulated inflammatory cells, J Biol Chem 275(6) (2000) 4429–34. [DOI] [PubMed] [Google Scholar]
- [29].Kreckler LM, Wan TC, Ge ZD, Auchampach JA, Adenosine inhibits tumor necrosis factor-α release from mouse peritoneal macrophages via A2A and A2B but not the A3 adenosine receptor, J Pharmacol Exp Ther 317(1) (2006) 172–80. [DOI] [PubMed] [Google Scholar]
- [30].Ng WA, Grupp IL, Subramaniam A, Robbins J, Cardiac myosin heavy chain mRNA expression and myocardial function in the mouse heart, Circ Res 68(6) (1991) 1742–50. [DOI] [PubMed] [Google Scholar]
- [31].Headrick JP, Peart J, Hack B, Garnham B, Matherne GP, 5’-Adenosine monophosphate and adenosine metabolism, and adenosine responses in mouse, rat and guinea pig heart, Comp Biochem Physiol A Mol Integr Physiol 130(4) (2001) 615–31. [DOI] [PubMed] [Google Scholar]
- [32].Zhao Z, Francis C, Ravid K, Characterization of the mouse A3 adenosine receptor gene: exon/intron organization and promoter activity, Genomics 57(1) (1999) 152–5. [DOI] [PubMed] [Google Scholar]
- [33].Cerniway RJ, Yang Z, Jacobson MA, Linden J, Matherne GP, Targeted deletion of A3 adenosine receptors improves tolerance to ischemia-reperfusion injury in mouse myocardium, Am J Physiol Heart Circ Physiol 281(4) (2001) H1751–8. [DOI] [PubMed] [Google Scholar]
- [34].Eckle T, Hartmann K, Bonney S, Reithel S, Mittelbronn M, Walker LA, Lowes BD, Han J, Borchers CH, Buttrick PM, Kominsky DJ, Colgan SP, Eltzschig HK, Adora2b-elicited Per2 stabilization promotes a HIF-dependent metabolic switch crucial for myocardial adaptation to ischemia, Nat Med 18(5) (2012) 774–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Palmer TM, Benovic JL, Stiles GL, Agonist-dependent phosphorylation and desensitization of the rat A3 adenosine receptor. Evidence for a G-protein-coupled receptor kinase-mediated mechanism, J Biol Chem 270(49) (1995) 29607–13. [DOI] [PubMed] [Google Scholar]
- [36].Trincavelli ML, Tuscano D, Marroni M, Falleni A, Gremigni V, Ceruti S, Abbracchio MP, Jacobson KA, Cattabeni F, Martini C, A3 adenosine receptors in human astrocytoma cells: agonist-mediated desensitization, internalization, and down-regulation, Mol Pharmacol 62(6) (2002) 1373–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Baltos JA, Paoletta S, Nguyen AT, Gregory KJ, Tosh DK, Christopoulos A, Jacobson KA, May LT, Structure-Activity Analysis of Biased Agonism at the Human Adenosine A3 Receptor, Mol Pharmacol 90(1) (2016) 12–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Vecchio EA, Baltos JA, Nguyen ATN, Christopoulos A, White PJ, May LT, New paradigms in adenosine receptor pharmacology: allostery, oligomerization and biased agonism, Br J Pharmacol (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Yang T, Zollbrecht C, Winerdal ME, Zhuge Z, Zhang XM, Terrando N, Checa A, Sallstrom J, Wheelock CE, Winqvist O, Harris RA, Larsson E, Persson AE, Fredholm BB, Carlstrom M, Genetic Abrogation of Adenosine A3 Receptor Prevents Uninephrectomy and High Salt-Induced Hypertension, J Am Heart Assoc 5(7) (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Moriyama K, Sitkovsky MV, Adenosine A2A receptor is involved in cell surface expression of A2B receptor, J Biol Chem 285(50) (2010) 39271–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Guo Y, Bolli R, Bao W, Wu WJ, Black RG Jr., Murphree SS, Salvatore CA, Jacobson MA, Auchampach JA, Targeted deletion of the A3 adenosine receptor confers resistance to myocardial ischemic injury and does not prevent early preconditioning, J Mol Cell Cardiol 33(4) (2001) 825–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Burnett LA, Blais EM, Unadkat JD, Hille B, Tilley SL, Babcock DF, Testicular expression of Adora3i2 in Adora3 knockout mice reveals a role of mouse A3Ri2 and human A3Ri3 adenosine receptors in sperm, J Biol Chem 285(44) (2010) 33662–70. [DOI] [PMC free article] [PubMed] [Google Scholar]