

Abstract
Modular type I polyketide synthases (PKSs) produce some of the most chemically complex metabolites in nature through a series of multi-enzyme modules. Each module contains a variety of catalytic domains to selectively tailor the growing molecule. PKS O-methyltransferases (O-MTs) are predicted to methylate β-hydroxyl or β-keto groups but their activity and structure have not been reported. We determined the domain boundaries and characterized the catalytic activity and structure of the StiD and StiE O-MTs, which methylate opposite β-hydroxyl stereocenters in the myxobacterial stigmatellin biosynthetic pathway. Substrate stereospecificity was demonstrated for the StiD O-MT. Key catalytic residues were identified in the crystal structures and investigated in StiE O-MT via site-directed mutagenesis and further validated with the cyanobacterial CurL O-MT from the curacin biosynthetic pathway. Initial structural and biochemical analysis of PKS O-MTs supplies a new chemoenzymatic tool, with the unique ability to selectively modify hydroxyl groups during polyketide biosynthesis.
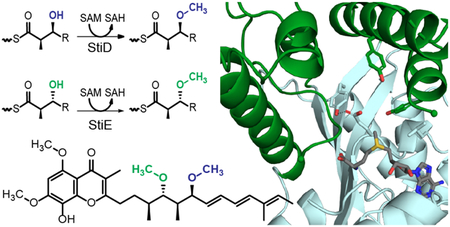
Graphical Abstract
Polyketides comprise a wide variety of bioactive natural products, including clinically effective antibiotic1, antiparasitic2, immunosuppressant3, and chemotherapeutic agents4. Modular type I polyketide synthases (PKSs) use a series of multienzyme modules to biosynthesize chemically complex polyketides with exquisite stereospecificity and regioselectivity from acyl coenzyme A (CoA) building blocks.5 First, the acyltransferase (AT) selects an acyl group (e.g. malonyl, methylmalonyl) from CoA and delivers it to the acyl carrier protein (ACP), which tethers polyketide intermediates via a phosphopantetheine (Ppant) cofactor throughout the enzymatic assembly line. The acyl group is condensed with the growing polyketide chain by the ketosynthase (KS), elongating the core scaffold of the polyketide by two carbons to produce a β-keto intermediate. The β-keto polyketide can undergo a series of chemical transformations based upon the nature of the modification domains within the module.
Modification domains give rise to the stereochemical complexity and rich chemical diversity of polyketides.6 C-methyltransferases (C-MTs) introduce an (R)-α-methyl to the β-keto intermediate.7 Ketoreductases (KRs) stereoselectively reduce the β-keto to a β-hydroxyl group.8 KR stereoselectivity can be correlated to sequence motifs that identify the KR as A-type or B-type, which produce β-hydroxyl groups in S- and R-configurations, respectively.9 The resulting β-hydroxyl can be dehydrated to a cis- or trans-α, β-double bond by a dehydratase (DH).10–13 Finally, an enoyl reductase (ER) can catalyze reduction of the α-enoyl polyketide and reintroduce a stereocenter in intermediates with an α-methyl substitutient.14, 15 The stereospecific and regioselective control provided by the modular nature of PKSs has sparked significant interest in the rational design of PKS pathways for the production of new pharmaceuticals and other high value commodity chemicals.16
O-methyltransferases (O-MTs) are not canonical PKS domains, but are embedded in PKS modules of several pathways generally of myxobacterial or cyanobacterial origin (Supporting Information Figures 1 and 2).17–28 O-MTs are predicted to methylate β-hydroxyl or β-keto groups yielding the respective β-methoxy or enol methylether products (Figure 1).17 Based upon the well established sequence motifs for binding the (S)-adenosylmethionine (SAM) methyl donor,29 the PKS O-MTs are members of the large and diverse class I MT superfamily. However, the sequences diverge significantly from MTs of known structure, including the PKS C-MTs.30, 31 The biosynthetic pathway for stigmatellin (Figure 2a), an electron transport inhibitor and antifungal agent from the myxobacterium Stigmatella aurantiaca, contains two O-MTs, which share 40% sequence identity.18 The structure of the terminal polyketide product and sequence analysis of the KR domains indicate that StiD O-MT methylates an (S)-ß-hydroxyl (Figure 2b) produced by the A-type KR encoded in stiD, whereas StiE O-MT methylates an (R)-ß-hydroxyl (Figure 2g) produced by the StiE B-type KR. As the two O-MTs act on opposite stereocenters, we chose the stigmatellin pathway as a model system to investigate PKS O-MT structure and function. Biochemical results were further validated with the CurL O-MT from the cyanobacterial curacin biosynthetic pathway20. Like StiE O-MT, CurL O-MT is predicted to methylate an (R)-β-hydroxyl group (Figure 2i).
Figure 1.

Reactions carried out by PKS O-MTs. O-MTs methylate (a) the (S)-β-hydroxyl produced by an A-type KR or (b) the (R)-β-hydroxyl produced by a B-type KR, resulting in a β methoxy group. Some PKS O-MTs methylate (c) a β-keto group, resulting in an enol methylether product. O-MTs in this study are indicated below their predicted reactions.
Figure 2.

Activity of PKS O-MTs on ACP linked substrates. (a) The metabolite stigmatellin contains two methoxy groups with opposite stereochemical configurations. The methoxy moieties introduced by O-MTs encoded in PKS modules are colored (StiD, blue; StiE, green). (b) Natural substrate of StiD O-MT. (c-f) Mass spectra of reaction mixtures of StiD O-MT with various triketide substrate mimics (1, 2, 3, 4) linked to StiD ACP monitored by the phosphopantetheine ejection assay.34,35 Reactions are shown in red, no enzyme controls are shown in black. Experimentally determined m/z values are in red, calculated m/z values are in black. A contaminant of m/z ~411 Da was present in all samples. The expected Ppant isotope patterns were observed for all substrates and products. (g) Natural substrate of StiE O-MT. (h) Mass spectra of reaction mixtures with StiE O-MT and StiE (3R)-3-hydroxy-5-methoxy-myristoyl-ACP substrate mimic (6). (i) Natural substrate of CurL O-MT. (j) Mass spectra of reaction mixtures of reactions with CurL O-MT and CurL (R)-3-hydroxydodecanoyl-ACP substrate mimic (8).
We first examined the O-MT domain boundaries to excise the domain from the module. O-MTs that methylate at a β-hydroxyl are flanked by AT and KR domains. Sequence motifs delineating the C-terminus of the AT (so-called post-AT linker) and N-terminal β-strand of the KR (Supporting Information Figure 1), guided the production of nine StiD O-MT constructs (Supporting Information Table 1). Three StiD fragments were soluble and two (956–1266, 976–1266) were purified to homogeneity. Soluble fragments of StiE (942–1257, 961–1257) and CurL (981–1315) were designed based on the experimentally determined domain boundaries for StiD. StiD was predominantly dimeric in solution as determined by size exclusion chromatography, whereas StiE O-MT was predominantly monomeric and CurL O-MT was monomeric (Supporting Information Figure 3a). Inclusion of an additional 30 amino acids between the CurL post-AT linker and O-MT (951–1315) (Supporting Information Figure 1) resulted in a dimeric CurL O-MT (Supporting Information Figure 3a). This region shares 30% sequence identity with the post-AT sequence of CurI (KS-AT-KR-ACP), which was essential for dimerization of the CurI KS-AT32, and occurs in many cyanobacterial PKS modules lacking DH domains (Supporting Information Figure 4). As this region promotes dimerization of both Curl and CurL fragments, we conclude that it is a cyanobacterial post-AT dimerization element (DE), which may be similar to the DE between AT and KR in the actinobacterial Spn module 333.
We next tested the ability of excised StiD, StiE, and CurL O-MT to methylate mimics of their predicted substrates. When StiD O-MT was presented with a truncated triketide version of the presumed (S)-ß-hydroxy substrate linked to N-acetylcysteamine (NAC), a common mimic for phosphopantetheine (Ppant), in the presence of S-adenosylmethionine (SAM) no methylation occurred. However, when presented with an excised StiD ACP-linked (S)-configured β-hydroxy triketide substrate (1), StiD O-MT produced the expected β-methoxy product (5) as detected by the mass spectrometry-based Ppant ejection assay (Figure 2c) in an overnight reaction (~0.01 μM ACP converted min−1).34,35 Additionally StiE O-MT and CurL O-MT, which are predicted to act on (R)-β-hydroxy substrates (Figures 2g, 2i), methylated the respective substrates (3R)-3-hydroxy-5-methoxy-myristoyl-ACP (6) (~1.2 μM ACP converted min−1) and (R)-3-hydroxydodecanoyl-ACP (8) (~0.03 μM ACP converted min−1) (Figures 2h, 2j).
Given the encoded stereospecificity of KR domains, we tested whether O-MTs select for stereocenters at the α- and β-positions. A series of ACP linked diastereomeric triketide substrates (2, 3, 4) with varying stereocenters at the α-methyl and β-hydroxyl groups were tested with StiD O-MT. StiD did not methylate any substrate with non-native stereocenters at the α- or β-positions (Figures 2d, 2e, 2f), indicating that PKS O-MTs display substrate stereospecificity.
To understand the structural basis of O-MT reactivity, we determined crystal structures of StiD and StiE O-MT with varying domain boundaries (Figure 3, Supporting Information Table 2). An initial structure of selenomethionine-labeled StiD O-MT (971–1266) was solved by single-wavelength anomalous diffraction (SAD) phasing, and this structure was used to solve subsequent StiD and StiE O-MT structures by molecular replacement. StiD and StiE O-MTs (Figure 3), which methylate opposing stereocenters, are highly similar in structure (RMSD 0.88 Å for 193 Cα atoms). PKS O-MTs are members of the class I methyltransferase superfamily, which share a seven β-stranded SAM binding core.29 The StiD dimer interface is mediated by hydrophobic and hydrogen bonding contacts (StiD Phe1134, Arg1173, Val1175, Glu1176, Gln1237, Asp1239, Val1242, Tyr1246, His1249, Ile1250, Trp1254). Although predominantly monomeric in solution, StiE forms a dimer in the crystal with the analogous interaction interface as StiD (StiE Ala1021, Asp1023, Ala1026, Gly1124, Leu1126, Arg1165, Val1167, Gln1229, Asp1231, Phe1233, Arg1235) (Supporting Information Figures 3b, 3c), suggesting that StiE is dimeric in the context of a module. The angle between monomers within the StiD and StiE dimers differs by ~20°. CurL O-MT (981–1315), which did not crystallize, is exclusively monomeric (Supporting Information Figure 3a). Thus, the CurL O-MT may rely on the post-AT DE for dimerization within the module (Supporting Information Figure 4).
Figure 3.

StiD and StiE O-MT structures. (a) StiD O-MT dimer. One protomer is colored as a rainbow from blue (N-terminus) to red (C-terminus). SAH is shown in sticks with atomic coloring (C, gray; O, red; N, blue; S, yellow). Termini are shown in spheres. (b) StiD O-MT active site colored by structural region (core, light blue; lid, dark blue). Key amino acids and SAH are shown in sticks with atomic coloring. The first ordered residue in the crystal structure (Glu980) is indicated with a sphere. (c) StiE O-MT dimer colored according to Cα B-factor from red (80 Å2) to white (20 Å2). SAM is shown in sticks. In both StiE and StiD, the lid and N-terminal helix have far greater mobility than the SAM binding core. (d) StiE O-MT active site colored by structural region (core, light blue; lid, green). Key amino acids and SAM are shown in sticks with atomic coloring. The last amino acid (Val958) of the N-terminal helix containing conserved aromatic amino acid Tyr954 is 17 Å away from the next ordered residue (His974).
Different branches of the class I MT superfamily can be distinguished by a lid domain, which lies over the common SAM binding site and is associated with substrate binding.36 StiD and StiE O-MT have a helical lid composed of two N-terminal helices and three helices inserted between β-strands six and seven of the MT core (Supporting Information Figure 1). The lid has substantially different positions (up to 8 Å displacement) in the six independent views of the StiD protomer within three crystal structures (Supporting Information Figure 5a), indicating that it is dynamic in the substrate-free state.
Despite a low sequence identity (<20%), the PKS O-MTs have greatest structural similarity to the phosphoethanolamine N-methyltransferase (PDB 3UJ8, RMSD 1.9 Å for 153 Cα atoms),37 mycolic acid cyclopropane synthase (PDB 1KPG, RMSD 1.6 Å for 152 Cα atoms),·38 RebM sugar O-methyltransferase (PDB 3BUS, RMSD 1.6 Å for 145 Cα atoms),39 and the geranyl diphosphate C-methyltransferase (GPPMT) (PDB 3VC2, RMSD 1.4 Å for 155 Cα atoms).40 Class I MTs on this branch of the superfamily have helical lids of similar structure, including helices at the N-terminus and inserted between β-strands 6 and 7 of the core (Supporting Information Figure 6). Interestingly, this MT superfamily branch includes the SpnF Diels-Alderase (PDB 4PNE, RMSD 3.5 Å for 193 Cα atoms), which was adapted to a new function and does not catalyze methyl transfer41. Unlike StiD and StiE O-MTs, the other members of this branch of the superfamily are monomeric and lack the C-terminal helix that forms part of the StiD and StiE dimer interface (Supporting Information Figure 6).
The StiD 976–1266 and StiE 961–1257 O-MT structures have solvent-exposed active sites and also lack an N-terminal lid helix that lays over the SAM binding site in the structures of several homologs (Supporting Information Figure 6). The analogous site above SAM in the StiE 961–1257 structure is occupied by a cloning-artifact peptide from the TEV protease recognition site (Tyr-Phe-Gln-Ser-Asn-Ala, Supporting Information Figure 5b). The peptide sequence is similar to a sequence in StiE (953Phe-Tyr-Asp-Ser-Leu-Ala958) that precedes the crystallized fragment (961–1257). Moreover, the PKS O-MT sequences contain adjacent conserved hydrophobic (preferred aromatic) amino acids in the N-terminal region (Supporting Information Figure 1). Thus, we incorporated additional N-terminal amino acids in the StiD and StiE fragments and removed the TEV protease recognition sequence for additional crystallographic studies, resulting in a 1.7-Å structure of StiD 956–1266 and a 1.9-Å structure of StiE 942–1257. No density was observed for the additional N-terminal residues of StiD. However, the map for StiE 942–1257 had density for a partially ordered helix above the SAM binding site (Supporting Information Figure 5c). We modeled residues 948–958, including conserved Phe953-Tyr954, into the helical density. The helix is 17 Å away from the next ordered amino acid, His974 (Figure 3d).
We used site directed mutagenesis to test whether the conserved N-terminal aromatic amino acids play a role in catalytic activity of the robustly active StiE and CurL O-MTs, which methylate (R)-β-hydroxy substrates (Figures 4a, 4b, 4c). Relative activity was quantified based upon the abundance of Ppant ejection fragments of the substrates and products34, 35. A CurL Y1010F variant had barely detectable activity (50-fold less than wild-type) (Figure 4c). In contrast, a StiE Y954F variant (Figure 4a) had increased activity relative to the wild type, but deletion of the StiE N-terminal helix (StiE Δ942–960) abolished detectable activity (Figure 4b). Although the Tyr to Phe substitutions had differing effects on the catalytic activity of StiE and CurL O-MTs, the presence of the N-terminal helix was critical for activity, and the conserved aromatic residues are likely near or in the active site when substrate is bound. Additional evidence comes from the PKS O-MT homolog phosphoethanolamine N-MT, which contains an essential Tyr in the analogous N-terminal helix.37 The helical lid and the loop between β-strand 5 and α-helix 7 are the most poorly ordered parts of both StiD and StiE (Figure 3c), thus we hypothesize that these parts of the structure become ordered upon delivery of the ACP-bound substrate and that the N-terminal helix containing the conserved aromatic region serves as a “latch” to close the active site and facilitate catalysis.
Figure 4.

Relative methylation activities of CurL and StiE O-MT variants. (a) StiE O-MT active site colored as in Fig. 3d. Substituted amino acids are shown in sticks. (b) StiE O-MT activity on (3R)-3-hydroxy-5-methoxy-myristoyl-ACP substrate (6). (c) CurL O-MT activity on (R)-3 hydroxydodecanoyl-ACP substrate (8). Activity relative to wild-type was quantified using the LC-MS based Ppant ejection assay.34, 35 The mutagenesis sites in StiE and CurL O-MTs are analogous (StiE/CurL: Tyr954/Tyr1010, Glu1102/Glu1161, Leu1106/His1165, Tyr1209/Tyr1267, Tyr1223/Tyr1281) based on alignment of the 35% identical sequences. Error bars represent triplicate experiments. (d) StiE O-MT mechanism. The conserved Glu polarizes the β-hydroxyl for methylation. (e) Model of StiD module. Connections of the O-MT to neighboring domains are indicated with dashed lines. For the dimeric StiD and StiE O-MTs, the linker from AT is 30–35 amino acids and linker to KR is 14–18 amino acids.
SAM or the product (S)-adenosylhomocysteine (SAH) bound to both StiD and StiE in a similar orientation as in the structures of homologs (Figure 3, Supporting Information Figure 6, Supporting Information Table 2). In some of these structures, a His side chain adjacent to the SAM methyl is associated with activity (Figure 3b, Supporting Information Figure 1, Supporting Information Figure 7).37,39 The His is not conserved in PKS O-MTs, as the corresponding position in StiE is Leu1106 (Figures 3d, 4a). The CurL variants H1165A and H1165N had 10- to 20-fold decreased activity relative to wild type (Figure 4c), and StiE L1105H displayed a two-fold loss in activity (Figure 4b), indicating that the active site His may assist in substrate binding, but is not essential for catalysis.
Two amino acids in the active site are conserved among PKS O-MTs. In StiE, conserved Glu1102 (StiD Glu1110, CurL Glu1161) is positioned with one carboxylate oxygen 4 Å from the SAM methyl group and the other carboxylate oxygen hydrogen bonded with the hydroxyl of invariant Tyr1223 (StiD Tyr1231; CurL Tyr 1281) (Figures 3b, 3d, and 4a, Supporting Information Figure 1). Several homologs of the PKS O-MT have a Glu or Asp within hydrogen bonding distance of a Tyr or Trp at the analogous positions (Supporting Information Figure 7). Substitutions of the Glu or Tyr were highly deleterious to catalysis in both StiE and CurL O-MTs (StiE E1102A, E1102Q, Y1223F; CurL E1161A, E1161Q, Y1281F) (Figures 4b, 4c), suggesting that the conserved Glu-Tyr pair is essential for catalysis in all PKS O-MTs that methylate β-hydroxyl groups. The proximity to the SAM methyl makes Glu the primary candidate for deprotonation of the substrate β-hydroxyl during the methyl transfer reaction (Figure 4a).
Finally, a Tyr just outside the active site of StiE O-MT (Tyr1209, Figure 3d, 4a) occurs in many O-MTs that are predicted to methylate R-hydroxyl substituents, but not those acting on S-hydroxyl or β-keto groups (Supporting Information Figure 1). Substitution of the Tyr (CurL Y1267F; StiE Y1209F) resulted in a six-fold decrease in CurL activity (Figure 4c), but only had a modest effect on StiE activity (Figure 4b). Although conserved in most R-hydroxyl methylating O-MTs, this residue is not essential for StiE activity.
Based on the mutagenesis results and sequence comparison, we propose that binding of an ACP-linked substrate triggers ordering of the N-terminal helix and lid to form the Michaelis complex by positioning the appropriate substrate oxygen atom adjacent to the SAM methyl group and active site Glu (Figure 4d). The conserved Glu-Tyr pair (Figures 3b, 3d, and 4a) is essential for activity and likely deprotonates the β-hydroxyl. We modeled the Ppant-linked substrate in the active site between the SAM methyl donor and active site Glu, with the Ppant near β-strands 4 and 5 and α-helix 10 (Supporting Information Figure 8).
Interestingly, O-MTs with β-keto substrates (Figure 1c), which do not require β-hydroxy deprotonation, have a Gln in the position analogous to the essential Glu (Supporting Information Figure 1). We tested whether a Gln substitution for the active site Glu would convert the β-hydroxyl methylating StiE O-MT or CurL O-MT to a β-keto methylase. However, neither StiE E1102Q nor CurL E1161Q methylated the simple β-keto substrate acetoacetyl-ACP. Additional active site amino acids with non-essential roles in StiE or CurL O-MT activity may play a role in substrate recognition and stereospecificity. Methylation of a β-keto would require deprotonation of the α-carbon. An active site His is conserved in β-keto methylating O-MTs and may play a role in α-proton abstraction, similar to the active site His in PKS C-MTs30.
The structural differences between PKS O-MTs and C-MTs are striking. Although both contain the seven β-stranded SAM-binding core, their lid domains are topologically different and are inserted into different regions of the seven β-stranded MT core. Additionally, insertion of O-MTs and C-MTs into PKS modules differs. Like the “pseudo” methyltransferase in the PKS homolog metazoan fatty acid synthase (mFAS),42 PKS C-MTs are embedded after the first N-terminal β-strand of the KR domain30, whereas the O-MT precedes all elements of the KR. The O-MT seems to be a later addition in the evolution of PKS modules, whereas the last PKS-mFAS common ancestor likely contained a functional C-MT. As PKS modules are obligate dimers, the O-MT may assist dimer formation in PKS modules lacking DH domains. This is evident in the existence of small dimerization element (951–980) that precedes the monomeric CurL O-MT (981–1315) (Supporting Information Figure 3a, Supporting Information Figure 4). The presence of a dimerization element prior to the O-MT in cyanobacterial CurL O-MT, but not the myxobacterial O-MTs from the stigmatellin pathway, suggests that the overall architecture of PKS modules with KS-AT-OMT-KR-ACP could differ between pathways. In StiD and StiE, the conserved O-MT N and C-termini occur ~30–35 amino acids after the AT and ~16 amino acids prior to the start of the KR (Figure 4e). The AT-OMT linker is twice as long as typical AT-DH linkers, whereas the OMT-KR linker is ~5 amino acids longer than typical DH-KR linkers. The relatively long AT-OMT linker, suggests that AT and O-MT may not be closely associated, whereas the OMT-KR linker length is of similar length to typical PKS interdomain linkers.
O-MTs are essential biosynthetic tools for the production of O-methoxy groups in polyketides. Identification of domain boundaries and amino acids necessary for catalysis provides fundamental information for the inclusion of O-MTs in PKS engineering efforts. O-MTs could be particularly valuable biocatalysts, as they are capable of selectively modifying hydroxyl groups prior to downstream synthetic transformations. Additionally, this information allows for the ablation of pathway O-MT activity by mutagenesis, providing a potential route to O-desmethyl polyketide products while maintaining the structural integrity of the PKS module.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health (NIH) grants DK042303 to J.L.S., CA100887 to P.W., and CA108874 to D.H.S., W.H.G., and J.L.S. M.A.S. was supported by a predoctoral fellowships from an NIH Cellular Biotechnology Training Program (GM008353) and the University of Michigan Rackham Graduate School. M.M.B was supported by a Perrigo Fellowship from the University of Michigan Life Sciences Institute and a grant from the University of Michigan College of Literature, Science, and the Arts honors program. GM/CA@APS is supported by the NIH National Institute of General Medical Sciences (AGM-12006) and National Cancer Institute (ACB-12002).
Footnotes
SUPPORTING INFORMATION
The Supporting information is available free of charge on the ACS Publications website at http://pubs.acs.org
Detailed Experimental Procedures, Supporting Information Figures 1–8, Supporting Information Tables 1–3.
ACCESSION CODES
The atomic coordinates and structure factors have been deposited in the Protein Data Bank (PDB ID codes 6ECU, SeMet StiD 976–1266; 6ECV, StiD 976–1266; 6ECW, StiD 956–1266; 6ECT, StiE 961–1257; 6ECX, StiE 942–1257).
REFERENCES
- [1].Cortes J, Haydock SF, Roberts GA, Bevitt DJ, and Leadlay PF (1990) An unusually large multifunctional polypeptide in the erythromycin-producing polyketide synthase of Saccharopolyspora erythraea, Nature 348, 176–178. [DOI] [PubMed] [Google Scholar]
- [2].Ikeda H, Nonomiya T, Usami M, Ohta T, and Omura S (1999) Organization of the biosynthetic gene cluster for the polyketide anthelmintic macrolide avermectin in Streptomyces avermitilis, Proc Natl Acad Sci U S A 96, 9509–9514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Schwecke T, Aparicio JF, Molnar I, Konig A, Khaw LE, Haydock SF, Oliynyk M, Caffrey P, Cortes J, Lester JB, and et al. (1995) The biosynthetic gene cluster for the polyketide immunosuppressant rapamycin, Proc Natl Acad Sci U S A 92, 7839–7843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Grimm A, Madduri K, Ali A, and Hutchinson CR (1994) Characterization of the Streptomycespeucetius ATCC 29050 genes encoding doxorubicin polyketide synthase, Gene 151, 1–10. [DOI] [PubMed] [Google Scholar]
- [5].Donadio S, Staver MJ, McAlpine JB, Swanson SJ, and Katz L (1991) Modular organization of genes required for complex polyketide biosynthesis, Science 252, 675–679. [DOI] [PubMed] [Google Scholar]
- [6].Keatinge-Clay AT (2016) Stereocontrol within polyketide assembly lines, Nat Prod Rep 33, 141–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Xie X, Khosla C, and Cane DE (2017) Elucidation of the stereospecificity of C-methyltransferases from trans-AT polyketide synthases, J Am Chem Soc 139, 6102–6105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Zheng J, and Keatinge-Clay AT (2013) The status of type I polyketide synthase ketoreductases, Med. Chem. Commum 4, 34–40. [Google Scholar]
- [9].Caffrey P (2003) Conserved amino acid residues correlating with ketoreductase stereospecificity in modular polyketide synthases, Chembiochem 4, 654–657. [DOI] [PubMed] [Google Scholar]
- [10].Valenzano CR, You YO, Garg A, Keatinge-Clay A, Khosla C, and Cane DE (2010) Stereospecificity of the dehydratase domain of the erythromycin polyketide synthase, J Am Chem Soc 132, 14697–14699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Gay D, You YO, Keatinge-Clay A, and Cane DE (2013) Structure and stereospecificity of the dehydratase domain from the terminal module of the rifamycin polyketide synthase, Biochemistry 52, 8916–8928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Fiers WD, Dodge GJ, Sherman DH, Smith JL, and Aldrich CC (2016) Vinylogous dehydration by a polyketide dehydratase domain in curacin biosynthesis, J Am Chem Soc 138, 16024–16036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Xie X, and Cane DE (2018) Stereospecific formation of Z-trisubstituted double bonds by the successive action of ketoreductase and dehydratase domains from trans-AT polyketide synthases, Biochemistry 57, 3126–3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Zhang L, Ji J, Yuan M, Feng Y, Wang L, Deng Z, Bai L, and Zheng J (2018) Stereospecificity of enoylreductase domains from modular polyketide synthases, ACS Chem Biol 13, 871–875. [DOI] [PubMed] [Google Scholar]
- [15].Kwan DH, Sun Y, Schulz F, Hong H, Popovic B, Sim-Stark JC, Haydock SF, and Leadlay PF (2008) Prediction and manipulation of the stereochemistry of enoylreduction in modular polyketide synthases, Chem Biol 15, 1231–1240. [DOI] [PubMed] [Google Scholar]
- [16].Klaus M, and Grininger M (2018) Engineering strategies for rational polyketide synthase design, Nat Prod Rep. [DOI] [PubMed] [Google Scholar]
- [17].Silakowski B, Schairer HU, Ehret H, Kunze B, Weinig S, Nordsiek G, Brandt P, Blocker H, Hofle G, Beyer S, and Muller R (1999) New lessons for combinatorial biosynthesis from myxobacteria. The myxothiazol biosynthetic gene cluster of Stigmatella aurantiaca DW4/3–1, J Biol Chem 274, 37391–37399. [DOI] [PubMed] [Google Scholar]
- [18].Gaitatzis N, Silakowski B, Kunze B, Nordsiek G, Blocker H, Hofle G, and Muller R (2002) The biosynthesis of the aromatic myxobacterial electron transport inhibitor stigmatellin is directed by a novel type of modular polyketide synthase, J Biol Chem 277, 13082–13090. [DOI] [PubMed] [Google Scholar]
- [19].Weinig S, Hecht HJ, Mahmud T, and Muller R (2003) Melithiazol biosynthesis: further insights into myxobacterial PKS/NRPS systems and evidence for a new subclass of methyl transferases, Chem Biol 10, 939–952. [DOI] [PubMed] [Google Scholar]
- [20].Chang Z, Sitachitta N, Rossi JV, Roberts MA, Flatt PM, Jia J, Sherman DH, and Gerwick WH (2004) Biosynthetic pathway and gene cluster analysis of curacin A, an antitubulin natural product from the tropical marine cyanobacterium Lyngbya majuscula, J Nat Prod 67, 1356–1367. [DOI] [PubMed] [Google Scholar]
- [21].Edwards DJ, Marquez BL, Nogle LM, McPhail K, Goeger DE, Roberts MA, and Gerwick WH (2004) Structure and biosynthesis of the jamaicamides, new mixed polyketide-peptide neurotoxins from the marine cyanobacterium Lyngbya majuscula, Chem Biol 11, 817–833. [DOI] [PubMed] [Google Scholar]
- [22].Feng Z, Qi J, Tsuge T, Oba Y, Kobayashi T, Suzuki Y, Sakagami Y, and Ojika M (2005) Construction of a bacterial artificial chromosome library for a myxobacterium of the genus Cystobacter and characterization of an antibiotic biosynthetic gene cluster, Biosci BiotechnolBiochem 69, 1372–1380. [DOI] [PubMed] [Google Scholar]
- [23].Buntin K, Weissman KJ, and Muller R (2010) An unusual thioesterase promotes isochromanone ring formation in ajudazol biosynthesis, Chembiochem 11, 1137–1146. [DOI] [PubMed] [Google Scholar]
- [24].Kwan JC, Donia MS, Han AW, Hirose E, Haygood MG, and Schmidt EW (2012) Genome streamlining and chemical defense in a coral reef symbiosis, Proc Natl AcadSci U S A 109, 20655–20660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Wakimoto T, Egami Y, Nakashima Y, Wakimoto Y, Mori T, Awakawa T, Ito T, Kenmoku H, Asakawa Y, Piel J, and Abe I (2014) Calyculin biogenesis from a pyrophosphate protoxin produced by a sponge symbiont, Nat Chem Biol 10, 648–655. [DOI] [PubMed] [Google Scholar]
- [26].Muller S, Rachid S, Hoffmann T, Surup F, Volz C, Zaburannyi N, and Muller R (2014) Biosynthesis of crocacin involves an unusual hydrolytic release domain showing similarity to condensation domains, Chem Biol 21, 855–865. [DOI] [PubMed] [Google Scholar]
- [27].Krastel P, Roggo S, Schirle M, Ross NT, Perruccio F, Aspesi P Jr., Aust T, Buntin K, Estoppey D, Liechty B, Mapa F, Memmert K, Miller H, Pan X, Riedl R, Thibaut C, Thomas J, Wagner T, Weber E, Xie X, Schmitt EK, and Hoepfner D (2015) Nannocystin A: an elongation factor 1 inhibitor from myxobacteria with differential anti-cancer properties, Angew Chem Int Ed Engl 54, 10149–10154. [DOI] [PubMed] [Google Scholar]
- [28].Steinmetz H, Li J, Fu C, Zaburannyi N, Kunze B, Harmrolfs K, Schmitt V, Herrmann J, Reichenbach H, Hofle G, Kalesse M, and Muller R (2016) Isolation, structure elucidation, and (bio)synthesis of haprolid, a cell-type-specific myxobacterial cytotoxin, Angew Chem Int Ed Engl 55, 10113–10117. [DOI] [PubMed] [Google Scholar]
- [29].Kozbial PZ, and Mushegian AR (2005) Natural history of S-adenosylmethionine-binding proteins, BMC Struct Biol 5, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Skiba MA, Sikkema AP, Fiers WD, Gerwick WH, Sherman DH, Aldrich CC, and Smith JL (2016) Domain organization and active site architecture of a polyketide synthase C-methyltransferase, ACS Chem Biol 11, 3319–3327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Storm PA, Herbst DA, Maier T, and Townsend CA (2017) Functional and structural analysis of programmed C-methylation in the biosynthesis of the fungal polyketide citrinin, Cell Chem Biol 24, 316–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Whicher JR, Smaga SS, Hansen DA, Brown WC, Gerwick WH, Sherman DH, and Smith JL (2013) Cyanobacterial polyketide synthase docking domains: a tool for engineering natural product biosynthesis, Chem Biol 20, 1340–1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Zheng J, Fage CD, Demeler B, Hoffman DW, and Keatinge-Clay AT (2013) The missing linker: a dimerization motif located within polyketide synthase modules, ACS Chem Biol 8, 1263–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Dorrestein PC, Bumpus SB, Calderone CT, Garneau-Tsodikova S, Aron ZD, Straight PD, Kolter R, Walsh CT, and Kelleher NL (2006) Facile detection of acyl and peptidyl intermediates on thiotemplate carrier domains via phosphopantetheinyl elimination reactions during tandem mass spectrometry, Biochemistry 45, 12756–12766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Meluzzi D, Zheng WH, Hensler M, Nizet V, and Dorrestein PC (2008) Top-down mass spectrometry on low-resolution instruments: characterization of phosphopantetheinylated carrier domains in polyketide and non-ribosomal biosynthetic pathways, BioorgMed Chem Lett 18, 3107–3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Liscombe DK, Louie GV, and Noel JP (2012) Architectures, mechanisms and molecular evolution of natural product methyltransferases, Nat Prod Rep 29, 1238–1250. [DOI] [PubMed] [Google Scholar]
- [37].Lee SG, Kim Y, Alpert TD, Nagata A, and Jez JM (2012) Structure and reaction mechanism of phosphoethanolamine methyltransferase from the malaria parasite Plasmodium falciparum: an antiparasitic drug target, J Biol Chem 287, 1426–1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Huang CC, Smith CV, Glickman MS, Jacobs WR Jr., and Sacchettini JC (2002) Crystal structures of mycolic acid cyclopropane synthases from Mycobacterium tuberculosis, J Biol Chem 277, 11559–11569. [DOI] [PubMed] [Google Scholar]
- [39].Singh S, McCoy JG, Zhang C, Bingman CA, Phillips GN Jr., and Thorson JS (2008) Structure and mechanism of the rebeccamycin sugar 4’-O-methyltransferase RebM, J Biol Chem 283, 22628–22636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Koksal M, Chou WK, Cane DE, and Christianson DW (2012) Structure of geranyl diphosphate C-methyltransferase from Streptomyces coelicolor and implications for the mechanism of isoprenoid modification, Biochemistry 51, 3003–3010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Fage CD, Isiorho EA, Liu Y, Wagner DT, Liu HW, and Keatinge-Clay AT (2015) The structure of SpnF, a standalone enzyme that catalyzes [4 + 2] cycloaddition, Nat Chem Biol 11, 256–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Maier T, Leibundgut M, and Ban N (2008) The crystal structure of a mammalian fatty acid synthase, Science 321, 1315–1322. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.