

Abstract
The methyl substituents in products of trans-acyltransferase assembly lines are usually incorporated by S-adenosyl-methionine (SAM)-dependent methyltransferase (MT) domains. The gem-dimethyl moieties within the polyketide disorazol are installed through the iterative action of an MT in the third module of its assembly line. The 1.75-Å-resolution crystal structure of this MT helps elucidate how it catalyzes the addition of two methyl groups. Activity assays of point mutants on β-ketoacyl chains linked to an acyl carrier protein and N-acetylcysteamine provide additional insights into the roles of active site residues. The replacement of an alanine with a phenylalanine at an apparent gatekeeping position resulted in more monomethylation than dimethylation. MTs may form an interface with ketoreductases (KRs) and even mediate the docking of trans-acyltransferase assembly line polypeptides through this association.
Graphical Abstract
Polyketides are bioactive secondary metabolites produced by diverse bacteria through the action of molecular factories called modular polyketide synthases (PKSs).1–4 The chemistry of a polyketide is determined by the types of modules within its assembly line. In order for a module to catalyze a two-carbon elongation of a polyketide intermediate, an acyltransferase (AT) that selects an α-carboxyacyl extender unit, a ketosynthase (KS) that performs the decarboxylative condensation of the extender unit to a growing chain, and an acyl carrier protein (ACP) that covalently shuttles polyketide intermediates between domains are required.1 Combinations of processing enzymes, such as a ketoreductase (KR), dehydratase (DH), and enoylreductase (ER), may also be present within the module to afford functionality such as a β-hydroxyl group, an α/β-double bond, or a β-methylene.
Assembly line installation of methyl groups on the α-carbon of polyketide intermediates is accomplished through two routes. In cis-AT assembly lines, which harbor embedded AT domains, it is primarily by condensation with a methylmalonyl extender unit.1 In trans-AT assembly lines, which rely on separately encoded, malonyl-specific ATs, it is primarily by the transfer of methyl groups by C-methyltransferases (MTs) from S-adenosylmethionine (SAM) to β-ketoacyl-ACP substrates.2 MT domains are present in ~2% of cis-AT assembly lines and ~80% of trans-AT assembly lines;2,3 they are also commonplace in iterative PKSs.
Four representatives of these MTs have been structurally elucidated—two catalytically inactive MTs from the porcine and human fatty acid synthases (FASs; PDBs 2VZ8, 4PIV),5,6 the MT from the citrinin iterative PKS (CitMT, PDB 5MPT),7 and the MT from the eighth module of the curacin cis-AT assembly line (CurMT8, PDB 5THZ; updated nomenclature is used throughout this manuscript).8–10 They are class I MTs that possess an N-terminal lid subdomain and a C-terminal SAM-binding, Rossmann-fold subdomain. The MTs from the mammalian FASs and the curacin assembly line are inserted into the structural subdomain of KR (KRs) after its first β-strand. Domain–domain contact with KRs is extensive within both the porcine and human FASs and thought to be similar within the curacin synthase.10 Within trans-AT assembly lines, MT domains are C-terminal to KR domains; however, KRs and MTs may associate here too as docking interactions seem to be mediated by KRs at the C-terminal ends of upstream polypeptides and MTs at the N-terminal ends of downstream polypeptides.11
The structures of CitMT and CurMT8 have provided clues for how methyl groups are transferred to β-ketoacyl substrates. An invariant glutamate/histidine pair is prominent within both active sites. This histidine has been proposed to serve as a general base that enables the α-carbon to nucleophilically attack the SAM methyl group.7,10 Bound S-adenosylhomocysteine (SAH) molecules reveal how SAM is positioned during catalysis. Previous work from both cis- and trans-AT assembly lines demonstrated that MTs generally operate on β-ketoacyl-ACPs instead of malonyl-ACPs and that the methyl group is installed with a d-orientation.12–15
The features that control whether zero, one, or two methyl groups are transferred to a substrate are also unknown. During citrinin biosynthesis, CitMT transfers a methyl group to the diketide, triketide, and tetraketide intermediates but not to the final pentaketide intermediate.7 Most characterized trans-AT PKS assembly lines contain monomethylating MTs. The calyculin, diaphorin, disorazol, elansolid, kirromycin, onnamide, oxazolomycin, pederin, phormidolide, psymberin, and rhizopodin each contain one gem-dimethylating MT, while the bryostatin assembly line contains two gem-dimethylating MTs (Figure 1).2,3,16 Except in the bryostatin and calyculin assembly lines, gem-dimethylating MTs are associated with a KR domain that operates only after two methyl groups have been transferred. Recent bioinformatic analysis on trans-AT KRs revealed that KRs associated with MTs clade separately from KRs not associated with MTs, suggesting that an MT-associated KR may be more active on a β-ketoacyl intermediate after one or two methyl groups have been transferred to its α-carbon.9
Figure 1.

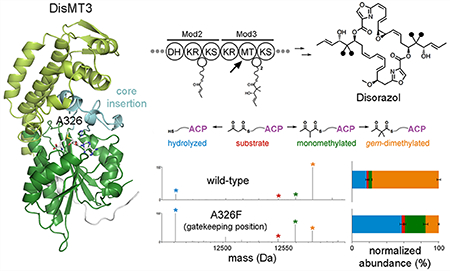
gem-Dimethylation by MTs in trans-AT assembly lines. (A) DisMT3 from the third module of the disorazol assembly line twice methylates the α-carbon of a triketide intermediate before the accompanying KR reduces its β-keto group. (B) Products of trans-AT assembly lines show the methyl groups, marked with red circles, incorporated by MTs. Some MTs operate iteratively to install gem-dimethyl moieties indicated with “2x.”
Here, we report the 1.75 Å-resolution crystal structure of the gem-dimethylating MT from the third module of the trans-AT assembly line that Sorangium cellulosum So ce12 utilizes to biosynthesize the antimitotic polyketide disorazol (DisMT3; Figure 1).2,17 DisMT3 is the first MT from a trans-AT assembly line as well as the first gem-dimethylating MT to be structurally determined. Assays of active site point mutants toward β-ketoacyl chains bound to both ACP and N-acetylcysteamine (NAC) provide insight into how the iterative transfer of methyl groups is accomplished. Modeling of the substrate and monomethylated intermediate in the active site show the positioning required for each methylation. How MT and KR domains associate to dock adjacent assembly line polypeptides is also discussed.
RESULTS AND DISCUSSION
Domain Boundaries and Structure Solution.
In contrast to the MT domains of cis-AT assembly lines or mammalian FASs that are embedded within the KR domain, the MT domains of the trans-AT assembly line are completely C-terminal to KR domains.5,12,13 The boundaries of DisKR3, DisMT3, and DisACP3 were hypothesized through sequence alignments and known structures of KRs and ACPs (Figure S1).11,18–20 The DNA encoding the putative MT domain (His3097 to Ala3498 of DisA) was then amplified and cloned into the expression vector pET28b. DisMT3 was expressed in E. coli BL21(DE3) cells, purified by nickel affinity and size-exclusion chromatography, and crystallized. The 1.75-Å-resolution structure of DisMT3 (numbering beginning with the first visible residue, equivalent to Ala3099 in DisA) was solved by molecular replacement using CurMT8 (PDB 5THZ) as a search model (Table S1).10,21
Overall Structure.
The model of DisMT3 contains 392 contiguous residues that form an N-terminal lid subdomain and a SAM-binding core subdomain like CurMT8 and CitMT (1.76 and 1.74 Å Cα RMSD, respectively; Figures 2 and S2). The lid (residues 23–173) is comprised of eight helices as well as a three-stranded, antiparallel β-sheet that has so far only been observed in MTs from assembly lines and iterative PKSs (the lid from the catalytically inactive MT of the mammalian FAS is half the size, and the lid of CitMT has one fewer helix than CurMT8 and DisMT3). The core subdomain (residues 174–392) possesses a Rossmann fold that contains motifs I–VI of class I MTs.22,23 Following its fifth β-strand, a large insertion termed the “core insertion” plays a significant role in forming the active site.10 The N-terminal 22 residues of DisMT3 form loops and a helix that interact with the core subdomain; the interactions made by these first residues in DisMT3, CurMT8, and CitMT are distinct from one another.
Figure 2.

Structure of an MT from a trans-AT assembly line. (A) A stereodiagram shows the architecture of DisMT3. The 22 N-terminal residues, the lid subdomain, the core subdomain, and the core insertion are indicated by color. SAM was modeled into the active site based on the location of SAH in CitMT (PDB 5MPT). Residues thought to position the substrate for catalysis are shown as sticks. (B) A sequence alignment shows the similarity of MTs from trans-AT assembly lines (DisMT3, BaeMT10, and DifMT2), cis-AT assembly lines (CurMT8 and GphMT4), and iterative PKSs (CitMT). The invariant glutamate/histidine dyad as well as a highly conserved tyrosine are indicated with circles, while residues that help form the substrate tunnel are indicated with squares. DisMT3, disorazol, Sorangium cellulosum So ce12, CAI43932; BaeMT10, bacillaene, Bacillus amyloliquefaciens FZB42, CAG23959; DifMT2, difficidin, Bacillus amyloliquefaciens FZB42, CAG23977; CurMT8, curacin, Moorea producens 3L, AEE88280; GphMT4, gephronic acid, Archangium violaceum Cb vi76, AHA38200; CitMT, citrinin, Monascus purpureus, BAD44749.
The Active Site.
Neither SAM nor SAH were observed in the DisMT3 active site even though crystals were grown in 1 mM SAM (crystals soaked in 3 mM sinefungin were also unbound). Thus, the uncomplexed structure of DisMT3 was compared with the binary complex of CurMT8 (bound with SAH) and the ternary complex of CitMT (bound with SAH and an unknown substrate mimic).7,10 Through a superposition with the CitMT structure, SAM was modeled into the DisMT3 active site (Figures 2a and S2a). In the CitMT structure, a highly conserved threonine in Motif I forms a hydrogen bond with the SAH carboxylate, while in CurMT8 structures the residue does not consistently form the same interaction. The equivalent threonine of DisMT3 (Thr213) is not positioned to form a hydrogen bond with the SAM carboxylate. A highly conserved active site tyrosine (Tyr170 in DisMT3) possesses high B-factors in both unbound DisMT3 and SAH-bound CurMT8 yet is well-ordered in CitMT. Thus, the active site of an MT may not be ordered for catalysis until both SAM and a substrate are bound.
The histidine/glutamate dyad of DisMT3 (His288/Glu314) is structurally equivalent to the dyads of CurMT8 and CitMT, with the glutamate likely perturbing the pKa of the histidine (Figure 2). A similar dyad is thought to help prepare the enolate of acetyl-coenzyme A (CoA) for an attack on α-ketoglutarate in the Saccharomyces cerevisiae homocitrate synthase.24 In serine proteases, the pKa of the active site histidine is 7.5 due to a neighboring aspartate and is elevated to 10–12 as an oxyanion develops during catalysis.25 Thus, a positively charged histidine may bind the enolate of the β-ketoacyl substrate and help position it for an attack on the SAM methyl group. As seen in the ternary complex of CitMT, Tyr170 is likely to be positioned next to one of the CH bonds of the SAM methyl group to stabilize the transition state that forms as the methyl group migrates to the α-carbon of the substrate.26
A large tunnel in DisMT3 can accommodate the tail of the polyketide intermediate. Hydrophobic side chains lining it include Tyr132, Met166, Leu169, and Tyr170 from the lid; Met315 from the core; as well as Phe321, Leu322, Ala326, and Trp322 from the core insertion (Figure 2a). While the DisMT3 substrate can fit into this cavity, many substrates for assembly line MTs are significantly longer and would likely need to double-back toward the phosphopantetheinyl arm.
DisMT3 Activity Assays.
The activity of the isolated DisMT3 domain was evaluated through in vitro assays. β-Ketobutyryl-DisACP3, generated using β-ketobutyryl-CoA and Sfp (the surfactin phosphopantetheinyl transferase27), was incubated with DisMT3 and SAM. Combined liquid chromatography and ultraviolet photodissociation tandem mass spectrometry (UVPD) was then used to quantify and locate transferred methyl groups (Figure 3).28 The reaction with wild-type DisMT3 yielded 77% of the anticipated gem-dimethylated product (as well as 4% monomethylated and 2% unmethylated products and 17% hydrolyzed byproduct). Reactions with malonyl-DisACP3, generated using malonyl-CoA and Sfp, did not yield any gem-dimethylated product (although 4% monomethylated product was apparently formed), reaffirming that the preferred substrates of MTs from trans-AT assembly lines are β-ketoacyl-ACPs (Figure S3).12–15 UVPD analysis revealed that all modifications are attached to the phosphopantetheinylated serine (Figure S4).1,9
Figure 3.

Functional assays of DisMT3 and point mutants. Deconvoluted MS1 spectra of DisACP3 constructs were obtained by averaging over each LCMS elution profile. Roles of active site residues were analyzed with point mutants Y170F, N285A, H288A, H288N, E314A, E314Q, and A326F. The mutation of His288 to either alanine or asparagine resulted in a large decrease in gem-dimethylation, indicating that this residue plays a key role. The A326F mutant yielded a greater percentage of monomethylation compared to gem-dimethylation. The bar graph shows the normalized abundance of each species (error bars from duplicate experiments).
The contributions of active site residues toward catalysis were evaluated with the point mutants Y170F, N285A, H288A, H288N, E314A, and E314Q (Figure 3). While the Y170F, N285A, E314A, and E314Q mutants yielded amounts of gem-dimethylated product similar to wild-type DisMT3, the His288 mutants were significantly impaired. The H288A mutant yielded 12% gem-dimethylated and 5% monomethylated product, and the H288N mutant gave just 2% gem-dimethylated and 4% monomethylated product. The CurMT8 and CitMT point mutants that showed the greatest abrogation of methylation activity were those with substitutions to the active site histidine.
Additional enzymatic assays were performed on the small molecule mimic β-ketopentanoyl-S-N-acetylcysteamine (SNAC). Wild-type DisMT3 did not yield any gem-dimethylated product but did produce some monomethylated product, α-methyl-β-ketopentanoyl-SNAC (Figure S5). Only the Y170F point mutant generated detectable levels of the monomethylated product. The absence of gem-dimethylation in these assays suggests that the interaction between these small molecule mimics and DisMT3 provides insufficient binding energy to catalyze the second methylation reaction. Thus, protein–protein interactions between the MT and ACP domains may greatly aid in completing the transfer of both methyl groups. Perhaps because small molecules rely more on interactions with active site residues, alterations to the active site through the N285A, E314A, and E314Q substitutions resulted in a greater loss of activity toward the SNAC-bound substrate than the ACP-bound substrate.
Association of MTs and KRs.
Assembly lines are split into several polypeptides that noncovalently associate. To accomplish this, the polypeptides of trans-AT assembly lines commonly utilize split domains located partially on the upstream polypeptide and partially on the downstream polypeptide. Such docking interactions include a four-helix bundle formed by two helices on each polypeptide and a DH domain formed by ~10 residues on the upstream polypeptide with the remainder on the downstream polypeptide.11,19,29,30 Since many trans-AT assembly lines possess polypeptide disconnections between KR and MT domains, a similar docking interaction is likely to exist between the KRs of upstream polypeptides and the MTs of downstream polypeptides.
In both the porcine and human FASs, the KR domain and the catalytically inactive MT domain form a large interface (Figure 4). The first helix of the core subdomain of MT is inserted into a large groove on the KR surface, and its side chains make ionic and polar contacts with complementary KR side chains (Figure 4a). The same interface has been proposed for the MT/KR interactions within cis-AT assembly lines because of the similarity of CurMT8 with the catalytically inactive MTs of mammalian FASs and its equivalent insertion into the KR domain immediately following its first β-strand (Figure 4a and b).4,10 Because the MT domains of trans-AT assembly lines are C-terminal to KR domains, one might expect a docking interface that is distinct from the mammalian FAS and cis-AT assembly lines. However, modeling reveals that the end of the KR domain is close to the beginning of the MT domain.5,6 In trans-AT assembly lines, the number of residues between KR and MT is the same whether the domains are embedded in the same or separate polypeptides (Figure 4c). The C-terminus of an upstream polypeptide may often provide the N-terminal residues of the downstream MT domain. However, the seventh module of the difficidin assembly line and the 11th module of the elansolid assembly line reveal this is not always the case. Thus, domain–domain interactions may make the major contribution to KR/MT docking and facilitate the noncovalent assembly of split modules (Figure 4d).5,6,10
Figure 4.

Association of MT and KR domains. (A) Domain/domain interactions may be similar for each of the discussed synthases. Stereodiagrams show the structure of the MT+KR fragment of the human FAS (PDB 4PIV) and models for the association of MT and KR within cis-AT and trans-AT assembly lines based on the mammalian FAS. Dotted lines indicate the expected connectivity of linkers between the two domains. (B) While the MT domain is completely C-terminal to the KR domain in trans-AT assembly lines, it is embedded within the KR domain (after its β1) in cis-AT assembly lines and mammalian FASs. The location that the ER domain inserts into the FAS KR domain is shown. (C) The KR and MT domains of trans-AT assembly lines occur either in the same polypeptide (“together”) or in adjacent polypeptides (“split”). Initiating methionines are highlighted green to mark polypeptide disconnections. Split modules often also contain DH and ER domains. (D) A stereodiagram suggests how KR and MT make contact in the context of a module, either on the same polypeptide or split between polypeptides. Spheres indicate cofactors of KR and MT, the phosphopantetheinyl arm of ACP, and the reactive cysteine of KS. BaeMT15, bacillaene, Bacillus amyloliquefaciens FZB42, CAG23961; BasMT2/BasMT3, basiliskamide, Brevibacillus laterosporus PE36, ERM18798/ERM18797; BonMT3/BonMT10, bongkrekic acid, Burkholderia gladioli, AFN27480/AFN27483; CalMT24/CalMT25, calyculin, Candidatus Entotheonella, BAP05595; DifMT7/DifMT14, difficidin, Bacillus amylolique-faciens FZB42, CAJ57409/CAG23983; DisMT3, disorazol, Sorangium cellulosum So ce12, CAI43932; ElaMT11, elansolid, Chitinophaga sancti, AEC04363; EtnMT8, etnangien, Sorangium cellulosum So ce56, CAN93349; LumMT7, luminaolide, Planktothrix paucivesiculata PCC 9631, AKQ22682; OocMT13, oocydin, Serratia marcescens, AFX60318; PedMT8, pederin, symbiont bacterium of Paederus fuscipes, AAS47564; PhmMT4, phormidolide, Leptolyngbya sp. ISBN3-Nov-94–8, AMH40421; RhiMT7, rhizoxin, Paraburkholderia rhizoxinica, CAL69890; SorMT15, sorangicin, Sorangium cellulosum So ce12, ADN68480.
KRs from trans-AT modules containing an MT form a clade separate from KRs from modules not containing an MT9 [fingerprint differences include YAAG(C/I) vs YAAAN and WG(F/Y)Wx5 vs WPxWxxGGM].18,19 These KRs may contain structural elements that interface with the MT domain. They may also react faster on α-substituted β-ketoacyl intermediates compared with α-unsubstituted β-ketoacyl intermediates, thus promoting methylation prior to reduction. Within cis-AT assembly lines MT-associated KRs often contain histidine in place of the catalytic tyrosine.4
Model for Mono- and Dimethylation.
The following mechanism is proposed for how a trans-AT assembly line MT adds a methyl group to the re face of an α-unsubstituted, β-ketoacyl substrate to generate a d-oriented α-methyl substituent (Figure 5).14 Within the active site the substrate tautomerizes between its keto and enol forms (similar dicarbonyl compounds enolize to a greater extent in less polar environments).31,32 The active site histidine deprotonates the enol form of the substrate to generate the enolate. With the enolate and the imidazolium of the histidine forming a salt bridge (the decreased activity of the H288A and H288N mutants highlight the importance of this interaction), the re face of the α-carbon is positioned for an attack on the methyl group of SAM to generate a monomethylated d-α-methyl-β-ketoacyl product (Figure 5a). Transfer of the methyl group is facilitated by a CH–O bond between a conserved active site tyrosine (Tyr170 in DisMT3) and the SAM methyl group; the backbone carbonyl of an active site asparagine (Asn285 in DisMT3) may also form CH–O bonds with the methylenes neighboring the SAM sulfonium.26 The methylated acyl chain and SAH then exit the active site.
Figure 5.

Models for mono- and dimethylation. (A) Stereodiagrams and a reaction scheme suggest how DisMT3 catalyzes the first and second methylations of its triketide substrate. His288 may be positively charged and form a salt bridge with the enolate of the substrate. The re face of the α-carbon can acquire the methyl group of SAM to generate a d-oriented methyl group in the product. The side chain of Tyr170 and the backbone carbonyl of Asn285 may form CH–O hydrogen bonds with SAM to help catalyze methyltransfer. Residues from the CitMT ternary complex structure are shown in transparent sticks—the Thr213 equivalent shifts to contact the SAM carboxylate, the Tyr170 equivalent shifts to contact the SAM methyl group, and the Ala326 equivalent is a phenylalanine that may sterically prevent a second methyltransfer. If MTs catalyze enolization, it may be through the cooperation of water molecules and His288. (B) Residues in the core insertion, especially those in the positions of Ser325, Ala326, and Trp333, are often different in dimethylating MTs compared to monomethylating MTs. SmdMT6, 9-methylstreptimidone, Steptomyces himastatinicus ATCC 53653, CCC21123; AtcMT8, anthracimycin, Streptomyces sp. T676, CTQ34882; BaeMT15, bacillaene, Bacillus amyloliquefaciens FZB42, CAG23961; BasMT2/BasMT3, basiliskamide, Brevibacillus laterosporus PE36, ERM18797/ERM18798; BatMT2, batumin, Pseudomonas fluorescens, ADD82939; BryMT6/BryMT11, bryostatin, Candidatus Endobugula sertula, ABM63527/ABM63528; CalMT18/CalMT25, calyculin, Candidatus Entotheonella, BAP05593/BAP05595; ChiMT3/ChiMT4/ChiMT9/ChiMT14/ChiMT15/ChiMT18, chivosazol, Sorangium cellulosum So ce56, AAY89049/AAY89050/AAY89052/AAY89053; CorMT13, corallopyronin, Corallococcus coralloides, ADI59534; ChxMT6, cycloheximide, Streptomyces sp. YIM 56141, AFO59866; DipMT2/DipMT8, diaphorin, Candidatus Profftella armatura, AGS06823/AGS06887; DifMT2/DifMT7, difficidin, Bacillus amyloliquefaciens FZB42, CAG23977/CAJ57409; DisMT3, disorazol, Sorangicin cellulosum So ce12, CAI43932; ElaMT3/ElaMT4/ElaMT11, elansolid, Chitinophaga sancti, AEC04357/AEC04363; EtnMT8/EtnMT19, etnangien, Sorangium cellulosum So ce56, CAN93349/CAN93352; KirMT4/KirMT9, kirromycin, Streptomyces collinus Tu 365, CAN89632/CAN89634; LtmMT6, lactimidomycin, Streptomyces amphibiosporus, ACY01401; MgsMT6, migrastatin, Streptomyces platensis subsp. rosaceus, ACY01391; MisMT18, misakinolide, Candidatus Entotheonella serta, AKQ22696; MmpMT2/MmpMT4, mupirocin, Pseudomonas fluorescens NCIMB 10586, AAM12913; NspMT2/NspMT7, nosperin, Nostoc sp. ‘Peltigera membranacea cyanobiont,’ ADA69237/ADA69239; OnnMT2/OnnMT8, onnamide, symbiont bacterium of Theonella swinhoei, AAV97870/AAV97877; OzmMT6/OzmMT7, oxazolomycin, Streptomyces albus, ABS90475/ABS90470; PtzMT12/PtzMT14/PtzMT18, patellazole, Candidatus Endolissoclinum faulkneri L5, AFK83819/AHC73995; PedMT8, pederin, symbiont bacterium of Paederus fuscipes, AAS47564; PhmMT4/PhmMT6, phormidolide, Leptolyngbya sp. ISBN3-Nov-94–8, AMH40421; RizMT3/RizMT9, rhizopodin, Stigmatella aurantiaca Sg a15, CCA89326; RhiMT5/RhiMT7, rhizoxin, Paraburkholderia rhizoxinica, CAL69889/CAL69890; SorMT4/Sor15/SorMT18, sorangicin, Sorangium cellulosum So ce12, ADN68476/ADN68480/ADN68482; Fr9MT9, spliceostatin, Burkholderia sp. FERM BP-3421, AIC32693; VirMT2, virginiamycin, Streptomyces virginiae, BAF50727.
gem-Dimethylating MTs like DisMT3 bind another molecule of SAM and the monomethylated product of the first reaction to transfer a second methyl group (Figure 5a). The enol form of the α-methyl-β-ketoacyl substrate is deprotonated by the active site histidine (the methyl group of the keto form would clash with the SAM methyl group). The increased pKa of the monomethylated substrate compared to the unmethylated substrate (10.6 vs 9.0) may have an impact on kinetics.33 Within monomethylating MTs, sterics may be the most significant factor inhibiting a second methylation, as a clash would form between the methyl group of an α-methyl, β-ketoacyl enolate, and an invariant phenylalanine in the core insertion of these MTs (CurMT8 and CitMT also possess this residue). This predicted clash would not occur in DisMT3 as the equivalent residue is an alanine (Ala326 in DisMT3). gem-Dimethylating MTs contain significant differences from core insertion residues that are invariant in monomethylating MTs (Figure 5b). The most obvious fingerprints are substitutions within the core insertion of the aforementioned phenylalanine, the threonine immediately upstream, and the tryptophan seven residues downstream. To test the hypothesis that a phenylalanine in place of Ala326 can gatekeep to prevent a second methylation, the A326F point mutant was generated and assayed (Figure 3). Reactions with β-ketobutyryl-DisACP3 yielded 60% greater monomethylation than dimethylation, indicating that in monomethylating MTs this phenylalanine residue helps prevent iterative methylation.
CONCLUSION
The 1.75-Å-resolution structure of DisMT3 reveals the architecture of MTs from trans-AT assembly lines as well as gem-dimethylating MTs for the first time. Accompanying assays of point mutants demonstrate the importance of active site residues toward catalysis and specificity. Binding modes for β-ketoacyl substrates and roles of residues during catalysis are proposed. Fingerprint residues of gem-dimethylating MTs are appropriately positioned on the core insertion to enable binding of the singly methylated intermediate. KRs that accompany MTs belong to a unique clade of KRs that may be catalytically delayed relative to methyltransfer. A significant interface may be formed between these KRs and the downstream MTs, even across polypeptide disconnections to mediate docking.
METHODS
Cloning, Expression, and Purification.
DNA encoding the DisMT3 domain was amplified from S. cellulosum So ce12 genomic DNA with primers DW490 and DW495, gel extracted, and Gibson-assembled (New England Biolabs) into the pET28b expression vector (all primer sequences are in Table S2). N-terminally His6-tagged DisMT3 was expressed in E. coli BL21(DE3) (6 L of LB media), grown to an OD600 of 0.6 at 37 °C, induced with 0.5 mM isopropyl β-d-1-thiogalactopyranoside, and shaken overnight at 15 °C. Protein was purified from cell lysate using affinity chromatography (HisPur nickel-NTA resin, Thermo Scientific) and gel filtration chromatography (GE Superdex 200 Increase 10/300 column equilibrated in 150 mM NaCl and 15 mM HEPES at pH 7.5). DisMT3 was concentrated to 5 mg mL−1 and flash-frozen in 100 μL aliquots. Point mutations of DisMT3 were generated following standard site-directed mutagenesis techniques. DNA encoding DisACP3 was amplified as described for DisMT3 using primers DisACP-FWD and DisACP-REV. DisACP3 and DisMT3 mutants were expressed and purified in the same fashion as wild-type DisMT3.
Crystallization.
After adding 1 mM SAM to 5 mg mL−1 DisMT3 for 15 min, 2 μL of this mixture was added to 1 μL of 20% (w/v) PEG 6000 and 0.1 M Bicine (pH 8.5). Crystals grew between 2 days and 4 months by sitting drop diffusion. They were cryoprotected in 20% (w/v) PEG 6000, 20% (v/v) glycerol, and 0.1 M Bicine (pH 8.5) prior to flash freezing in liquid nitrogen. Diffraction was obtained at ALS Beamline 5.0.3. The structure was solved using molecular replacement in Phaser with CurMT8 (PDB 5THZ) as a search model and refined using Refmac, ARP/wARP, and Phenix Refine.21,34–36 DisMT3 crystallized in P21 with one molecule per asymmetric unit. A resolution cutoff of 1.75 Å was selected to keep CC1/2 above 0.70 in the highest resolution bin.
Activity Assays.
Acyl-S-phosphopantetheinylation of DisACP3 was conducted with 50 mM HEPES (pH 7.5), 5 mM MgCl2, 2 nM Sfp, 300 μM DisACP3, and either 500 μM acetoacetyl-CoA or malonyl-CoA at 22 °C for 2 h. Following the acylation reaction, 50 μM acylated DisACP3 was incubated with 50 mM HEPES (pH 7.5), 5 mM MgCl2, 10 mM SAM, and 50 μM DisMT3 for 18 h at 22 °C. Protein solutions were desalted and concentrated to 10 μM in 50% (v/v) acetonitrile with 0.1% formic acid using a 3 kDa molecular weight cutoff filter device (MilliporeSigma, Burlington, MA). Solutions were loaded into Aucoated borosilicate emitters and infused with a nano-ESI source or introduced by chromatographic separation using a Dionex UltiMate 3000 RPLC nanoLC system integrated with a nano-ESI source. A solvent gradient of 15% to 55% B over 45 min at a flow rate of 300 nL min−1 using water (mobile phase A) and acetonitrile (mobile phase B), each containing 0.1% formic acid, was used for chromatographic separations. Agilent PLRP-S resin (5 μm, 1000 Å pore size) was used to pack in-house a trap column (3 cm × 100 μm) and analytical column with an integrated emitter (20 cm × 75 μm). A Thermo Fisher Scientific Orbitrap Elite mass spectrometer (Bremen, Germany) modified to perform UVPD in the higher-energy collisional dissociation cell with a Coherent Excistar 193 nm ArF excimer laser (Santa Cruz, CA) was used for all mass spectrometry experiments. UV photoactivation was achieved using one pulse at 2 mJ per pulse. MS1 and MS/MS spectra (60–500 scans) were acquired at 240 K resolving power (at m/z 400). Xtract (Thermo Fisher Scientific) and ProSight Lite v1.4 were used to process MS/MS spectra and identify fragment ions.37 Extracted ion chromatogram peak areas were divided by the sum of the observed areas for the species of interest and reported as the normalized abundance.
Small molecule activity assays with 50 mM HEPES (pH 7.5), 150 mM NaCl, 10 mM β-ketopentanoyl-SNAC, 30 mM SAM, 5% (v/v) DMSO, and 50 μM DisMT3 were incubated for 18 h at 22 °C. Post-incubation, reactions were thrice extracted with 100 μL of ethyl acetate, evaporated, and resuspended in 50 μL of methanol. From this solution, 20 μL was diluted into 1.5 mL of 5% methanol for LCMS analysis (Agilent 1260 HPLC with an Agilent 6120 Quadrupole APCI/ ESI instrument in positive mode; ZORBAX Eclipse Plus 95 Å C18 column, 2.1 mm × 50 mm, 5 μm; column temperature of 40 °C; flow rate of 0.7 mL min−1; gradient from 0 to 12 min of 5 to 100% methanol and from 12 to 15 min of 100% methanol). β-Ketopentanoyl-SNAC and the α-methyl-β-ketopentanoyl-SNAC standard were synthesized as previously reported.38
Supplementary Material
ACKNOWLEDGMENTS
The authors thank R. Müller for providing the genomic DNA for Sorangium cellulosum So ce12. Instrumentation and technical assistance for crystallographic work were provided by A. Monzingo and the Macromolecular Crystallography Facility, with financial support from the College of Natural Sciences, the Office of the Executive Vice President and Provost, and the Institute for Cellular and Molecular Biology at the University of Texas at Austin. The Berkeley Center for Structural Biology is supported in part by the National Institutes of Health, National Institute of General Medical Sciences, and the Howard Hughes Medical Institute. The Advanced Light Source is a Department of Energy Office of Science User Facility under Contract No. DE-AC02–05CH11231. We also would like to thank the NIH (GM121714 to J.S.B., GM106112 to A.T.K.) and the Welch Foundation (F-1155 to J.S.B., F-1712 to A.T.K.) for supporting this research.
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschembio.8b00733.
Figures S1–S5 and Tables S1 and S2 (PDF)
Accession Codes
The DisMT3 coordinates have been deposited in the Protein Data Bank under accession code 6CCA.
REFERENCES
- (1).Keatinge-Clay AT (2012) The Structures of Type I Polyketide Synthases. Nat. Prod. Rep 29, 1050–1073. [DOI] [PubMed] [Google Scholar]
- (2).Piel J (2010) Biosynthesis of Polyketides by Trans-at Polyketide Synthases. Nat. Prod. Rep 27, 996–1047. [DOI] [PubMed] [Google Scholar]
- (3).Helfrich EJ, and Piel J (2016) Biosynthesis of Polyketides by Trans-at Polyketide Synthases. Nat. Prod. Rep 33, 231–316. [DOI] [PubMed] [Google Scholar]
- (4).Keatinge-Clay AT (2017) The Uncommon Enzymology of Cis-Acyltransferase Assembly Lines. Chem. Rev 117, 5334–5366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Maier T, Leibundgut M, and Ban N (2008) The Crystal Structure of a Mammalian Fatty Acid Synthase. Science 321, 1315–1322. [DOI] [PubMed] [Google Scholar]
- (6).Hardwicke MA, Rendina AR, Williams SP, Moore ML, Wang L, Krueger JA, Plant RN, Totoritis RD, Zhang G, Briand J, Burkhart WA, Brown KK, and Parrish CA (2014) A Human Fatty Acid Synthase Inhibitor Binds Beta-Ketoacyl Reductase in the Keto-Substrate Site. Nat. Chem. Biol 10, 774–779. [DOI] [PubMed] [Google Scholar]
- (7).Storm PA, Herbst DA, Maier T, and Townsend CA (2017) Functional and Structural Analysis of Programmed C-Methylation in the Biosynthesis of the Fungal Polyketide Citrinin. Cell Chem. Biol 24, 316–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Zhang L, Hashimoto T, Qin B, Hashimoto J, Kozone I, Kawahara T, Okada M, Awakawa T, Ito T, Asakawa Y, Ueki M, Takahashi S, Osada H, Wakimoto T, Ikeda H, Shin-Ya K, and Abe I (2017) Characterization of Giant Modular Pkss Provides Insight into Genetic Mechanism for Structural Diversification of Aminopolyol Polyketides. Angew. Chem., Int. Ed 56, 1740–1745. [DOI] [PubMed] [Google Scholar]
- (9).Vander Wood DA, and Keatinge-Clay AT (2018) The Modules of Trans-Acyltransferase Assembly Lines Redefined with a Central Acyl Carrier Protein. Proteins: Struct., Funct., Genet 86, 664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Skiba MA, Sikkema AP, Fiers WD, Gerwick WH, Sherman DH, Aldrich CC, and Smith JL (2016) Domain Organization and Active Site Architecture of a Polyketide Synthase C-Methyltransferase. ACS Chem. Biol 11, 3319–3327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Zeng J, Wagner DT, Zhang Z, Moretto L, Addison JD, and Keatinge-Clay AT (2016) Portability and Structure of the Four-Helix Bundle Docking Domains of Trans-Acyltransferase Modular Polyketide Synthases. ACS Chem. Biol 11, 2466–2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Wagner DT, Stevens DC, Mehaffey MR, Manion HR, Taylor RE, Brodbelt JS, and Keatinge-Clay AT (2016) Alpha-Methylation Follows Condensation in the Gephyronic Acid Modular Polyketide Synthase. Chem. Commun. (Cambridge, U. K.) 52, 8822–8825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Stevens DC, Wagner DT, Manion HR, Alexander BK, and Keatinge-Clay AT (2016) Methyltransferases Excised from Trans-at Polyketide Synthases Operate on N-Acetylcysteamine-Bound Substrates. J. Antibiot 69, 567–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Xie X, Khosla C, and Cane DE (2017) Elucidation of the Stereospecificity of C-Methyltransferases from Trans-at Polyketide Synthases. J. Am. Chem. Soc 139, 6102–6105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Poust S, Phelan RM, Deng K, Katz L, Petzold CJ, and Keasling JD (2015) Divergent Mechanistic Routes for the Formation of Gem-Dimethyl Groups in the Biosynthesis of Complex Polyketides. Angew. Chem., Int. Ed 54, 2370–2373. [DOI] [PubMed] [Google Scholar]
- (16).Bertin MJ, Vulpanovici A, Monroe EA, Korobeynikov A, Sherman DH, Gerwick L, and Gerwick WH (2016) The Phormidolide Biosynthetic Gene Cluster: A Trans-at Pks Pathway Encoding a Toxic Macrocyclic Polyketide. ChemBioChem 17, 164–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Wong FT, Chen AY, Cane DE, and Khosla C (2010) Protein-Protein Recognition between Acyltransferases and Acyl Carrier Proteins in Multimodular Polyketide Synthases. Biochemistry 49, 95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Piasecki SK, Zheng J, Axelrod AJ, Detelich ME, and Keatinge-Clay AT (2014) Structural and Functional Studies of a Trans-Acyltransferase Polyketide Assembly Line Enzyme That Catalyzes Stereoselective Alpha- and Beta-Ketoreduction. Proteins: Struct., Funct., Genet 82, 2067–2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Wagner DT, Zeng J, Bailey CB, Gay DC, Yuan F, Manion HR, and Keatinge-Clay AT (2017) Structural and Functional Trends in Dehydrating Bimodules from Trans-Acyltransferase Polyketide Synthases. Structure 25, 1045–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Haines AS, Dong X, Song Z, Farmer R, Williams C, Hothersall J, Ploskon E, Wattana-Amorn P, Stephens ER, Yamada E, Gurney R, Takebayashi Y, Masschelein J, Cox RJ, Lavigne R, Willis CL, Simpson TJ, Crosby J, Winn PJ, Thomas CM, and Crump MP (2013) A Conserved Motif Flags Acyl Carrier Proteins for Beta-Branching in Polyketide Synthesis. Nat. Chem. Biol 9, 685–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, and Wilson KS (2011) Overview of the Ccp4 Suite and Current Developments. Acta Crystallogr., Sect. D: Biol. Crystallogr 67, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Liscombe DK, Louie GV, and Noel JP (2012) Architectures, Mechanisms and Molecular Evolution of Natural Product Methyltransferases. Nat. Prod. Rep 29, 1238–1250. [DOI] [PubMed] [Google Scholar]
- (23).Ansari MZ, Sharma J, Gokhale RS, and Mohanty D (2008) Silico Analysis of Methyltransferase Domains Involved in Biosynthesis of Secondary Metabolites. BMC Bioinf. 9, 454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Qian J, Khandogin J, West AH, and Cook PF (2008) Evidence for a Catalytic Dyad in the Active Site of Homocitrate Synthase from Saccharomyces Cerevisiae. Biochemistry 47, 6851–6858. [DOI] [PubMed] [Google Scholar]
- (25).Harris TK, and Turner GJ (2002) Structural Basis of Perturbed Pka Values of Catalytic Groups in Enzyme Active Sites. IUBMB Life 53, 85–98. [DOI] [PubMed] [Google Scholar]
- (26).Horowitz S, Dirk LM, Yesselman JD, Nimtz JS, Adhikari U, Mehl RA, Scheiner S, Houtz RL, Al-Hashimi HM, and Trievel RC (2013) Conservation and Functional Importance of Carbon-Oxygen Hydrogen Bonding in Adomet-Dependent Methyl-transferases. J. Am. Chem. Soc 135, 15536–15548. [DOI] [PubMed] [Google Scholar]
- (27).Quadri LE, Weinreb PH, Lei M, Nakano MM, Zuber P, and Walsh CT (1998) Characterization of Sfp, a Bacillus Subtilis Phosphopantetheinyl Transferase for Peptidyl Carrier Protein Domains in Peptide Synthetases. Biochemistry 37, 1585–1595. [DOI] [PubMed] [Google Scholar]
- (28).Shaw JB, Li W, Holden DD, Zhang Y, Griep-Raming J, Fellers RT, Early BP, Thomas PM, Kelleher NL, and Brodbelt JS (2013) Complete Protein Characterization Using Top-Down Mass Spectrometry and Ultraviolet Photodissociation. J. Am. Chem. Soc 135, 12646–12651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Dorival J, Annaval T, Risser F, Collin S, Roblin P, Jacob C, Gruez A, Chagot B, and Weissman KJ (2016) Characterization of Intersubunit Communication in the Virginiamycin Trans-Acyl Transferase Polyketide Synthase. J. Am. Chem. Soc 138, 4155–4167. [DOI] [PubMed] [Google Scholar]
- (30).Jenner M, Kosol S, Griffiths D, Prasongpholchai P, Manzi L, Barrow AS, Moses JE, Oldham NJ, Lewandowski JR, and Challis GL (2018) Mechanism of Intersubunit Ketosynthase-Dehydratase Interaction in Polyketide Synthases. Nat. Chem. Biol 14, 270–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Emsley J, and Freeman NJ (1987) Beta-Diketone Interactions: Part 5. Solvent Effects on the Keto Enol Equilibrium. J. Mol. Struct 161, 193–204. [Google Scholar]
- (32).Bertolasi V, Ferretti V, Gilli P, Yao X, and Li C (2008) Substituent Effects on Keto-Enol Tautomerization of Beta-Diketones from X-Ray Structural Data and Dft Calculations. New J. Chem 32, 694–704. [Google Scholar]
- (33).Middleton B, and Bartlett K (1983) The Synthesis and Characterisation of 2-Methylacetoacetyl Coenzyme a and Its Use in the Identification of the Site of the Defect in 2-Methylacetoacetic and 2-Methyl-3-Hydroxybutyric Aciduria. Clin. Chim. Acta 128, 291–305. [DOI] [PubMed] [Google Scholar]
- (34).Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, and Zwart PH (2010) Phenix: A Comprehensive Python-Based System for Macromolecular Structure Solution. Acta Crystallogr., Sect. D: Biol. Crystallogr 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Langer G, Cohen SX, Lamzin VS, and Perrakis A (2008) Automated Macromolecular Model Building for X-Ray Crystallography Using Arp/Warp Version 7. Nat. Protoc 3, 1171–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Murshudov GN, Vagin AA, and Dodson EJ (1997) Refinement of Macromolecular Structures by the Maximum-Likelihood Method. Acta Crystallogr., Sect. D: Biol. Crystallogr 53, 240–255. [DOI] [PubMed] [Google Scholar]
- (37).Fellers RT, Greer JB, Early BP, Yu X, LeDuc RD, Kelleher NL, and Thomas PM (2015) Prosight Lite: Graphical Software to Analyze Top-Down Mass Spectrometry Data. Proteomics 15, 1235–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Piasecki SK, Taylor CA, Detelich JF, Liu J, Zheng J, Komsoukaniants A, Siegel DR, and Keatinge-Clay AT (2011) Employing Modular Polyketide Synthase Ketoreductases as Biocatalysts in the Preparative Chemoenzymatic Syntheses of Diketide Chiral Building Blocks. Chem. Biol 18, 1331–1340. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.