

Abstract
Trans-acting acyltransferases (trans-ATs) are standalone enzymes that select and deliver extender units to polyketide synthase assembly lines. Accordingly, there is interest in leveraging trans-ATs as tools to regioselectively diversify polyketide structures. Yet, little is known regarding the extender unit and acyl carrier protein (ACP) specificity of trans-ATs, particularly those that utilize unusual ACP-linked extender units. For example, the biosynthesis of the antibiotic zwittermicin involves the trans-AT ZmaF, which is responsible for installing a rare ACP-linked aminomalonyl extender unit. Here, we developed a method to access a panel of non-natural and non-native ACP-linked extender units and used it to probe the promiscuity of ZmaF, revealing one of the most promiscuous ATs characterized to date. Furthermore, we demonstrated that ZmaF is highly orthogonal with respect to its ACP specificity, and the ability of ZmaF to trans-complement non-cognate PKS modules was also explored. Together, these results set the stage for further engineering ZmaF as a tool for polyketide diversification.
Graphical Abstract
INTRODUCTION
Many polyketides are constructed via the action of modular assembly lines called type I polyketide synthases (PKSs) whereby each module is responsible for the installation and tailoring of an extender unit building block that forms the polyketide scaffold. Each extender unit is selected by an acyltransferase (AT) domain that in most cases is embedded within each module of the assembly line. Given that such cis-ATs define large portions of polyketide structure, there continues to be much interest in utilizing them to diversify the structures of natural products.1 In particular, ATs are often targeted for enzyme engineering in order to introduce non-native or non-natural extender units into polyketides.2, 3 Recently, the promiscuity of ATs has proven to be a useful platform for engineering ATs with new and orthogonal extender unit specificities,4–8 thus providing a strategy for potential regioselective modification of polyketide structure. An emerging class of ATs differ from their canonical cis-counterparts in that they are standalone “discrete” enzymes. These “trans-ATs” utilize malonyl-CoA (M-CoA) and transfer the malonyl unit to their cognate polyketide synthase module(s).9, 10 Yet, a small number of trans-ATs natively install more unusual extender units and can in principle be leveraged to diversify the structures of polyketide natural products. For example, the trans-AT KirCII in the biosynthesis of the antibiotic kirromycin utilizes ethylmalonyl-CoA (EM-CoA) and has been shown to display activity with several non-natural extender units.11, 12 This poly-specificity has been used to produce regioselectively modified kirromycin derivatives via precursor-directed biosynthesis, demonstrating the remarkable plasticity of the kirromycin assembly line.13 All other trans-ATs that use unusual (i.e. non-malonyl) extender units require ACP-linked extender units instead of CoA-linked substrates. However, nothing is known regarding the ability of trans-ATs to accept non-natural ACP-linked extender units. Indeed, the substrate scope of canonical cis-ATs that depend on ACP-linked extenders is also poorly described, while only a few examples of cis-ATs that use multiple extender units are known. For example, the cis-AT from the FK506 PKS accepts both ACP-linked and CoA-linked extender units in its natural pathway.14 The module 4 cis-AT of the FkbB PKS incorporates an allylmalonyl unit through an ACP-linked substrate while an ethylmalonyl unit is incorporated by the same AT through a CoA-linked substrate.15
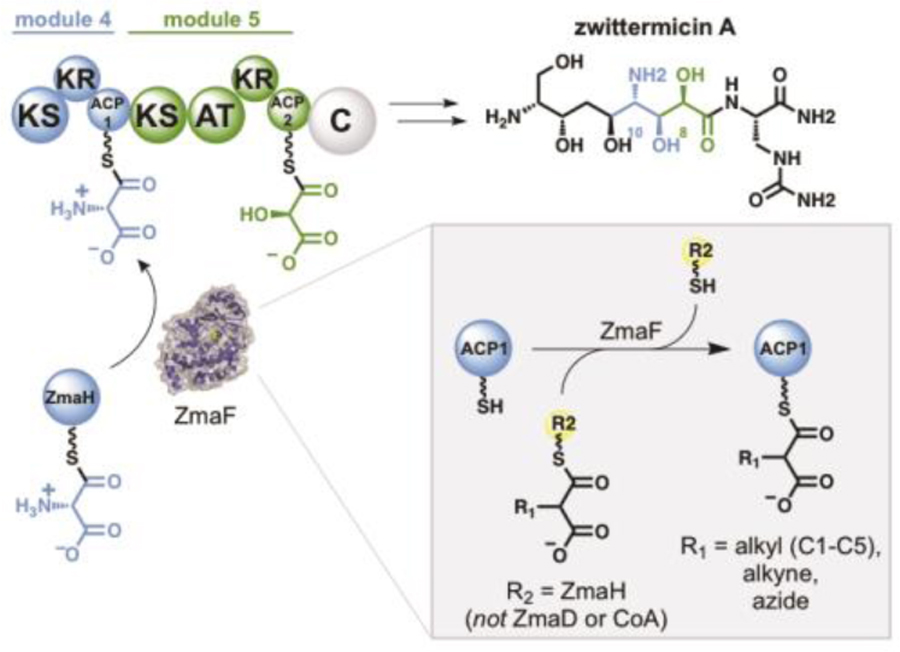
Interestingly, the biosynthesis of zwittermicin A (ZMA) includes two ATs that each use relatively rare and distinct ACP-linked extender units.16 Notably, the C10-amino group in ZMA is installed via the action of the trans-AT ZmaF which transfers an aminomalonyl unit from the ACP-linked substrate aminomalonyl-ZmaH (AmM-ZmaH) to a single carrier protein within the ZMA assembly line, ZmaA-ACP1 (ZACP1) (Figure 1).17 The next module in the ZMA assembly line incorporates a hydroxymalonyl unit, also through an ACP-linked extender unit (hydroxymalonyl-ZmaD) but through the cis-AT, ZmaA-AT (Figure 1).18 Aminomalonyl extender units are extremely rare and found in only two other biosynthetic pathways.19–21 The incorporation of an aminomalonyl into a non-cognate polyketide is attractive not only for the electronic impact of the amine, but also as a chemical handle for downstream diversification via semi-synthesis. Previous characterization of ZmaF suggested that it discriminates against hydroxymalonyl-ZmaD (the native substrate for the cis-AT in module 4) and is specific for ZmaH as the acyl-carrier.17 ZmaF was also inactive with reduced precursors of AmM-ZmaH, suggesting that ZmaF also discriminates against non-native acyl units. It was also shown that ZmaF was not able to undergo self-acylation or trans-acylation with M-CoA or methylmalonyl-CoA (MM-CoA), reinforcing the potential dependence of ZmaF specificity to recognition of the ZmaH-carrier.17 The presumed orthogonality of ZmaF and ZmaA-AT—with respect to their extender unit specificities and carrier protein specificities—provides an intriguing platform for polyketide diversification if the molecular basis for these specificities can be understood and harnessed.
Figure 1. ZMA biosynthesis by the PKS/NRPS hybrid pathway.

Each module is shown with the cognate building block tethered to the carrier protein. The pathway produces two metabolites (not shown), one of which is a prodrug that is cleaved to yield the active product, ZMA. The trans-AT ZmaF transfers an AmM extender unit to ZmaA-ACP1 through an ACP-linked extender unit, AmM-ZmaH. The subsequent PKS module also utilizes an ACP-linked extender unit (hydroxymalonyl-ZmaD) through the cis-AT, ZmaA-AT.
Herein, the substrate scope of ZmaF was probed by leveraging an engineered acyl-CoA ligase11, 22 and a promiscuous phosphopantetheinyl transferase (PPTase)23 to generate a panel of non-native and non-natural ACP-linked extender units. Characterization of ZmaF represents the first ACP-linked accepting trans-AT to be extensively probed for substrate promiscuity. The ability of ZmaF to trans-complement non-cognate PKS modules was also explored, which led to the first exploration of the molecular determinants responsible for recruitment of ZmaF to a cognate ACP. Collectively, this study provides a blueprint for further engineering ZmaF as a tool for polyketide diversification.
RESULTS AND DISCUSSION
Preparation of ZmaH-linked extender unit substrates
ZmaH, the ACP that carries the extender unit to ZmaF, was expressed and purified as both holo and apo protein for subsequent preparation of various acylated forms. Generation of ZmaH-linked substrates was enabled by the promiscuous acyl-CoA ligase, MatB,11, 22 and promiscuous PPTase, Sfp23 (Figure 2A). Neither wild-type MatB nor the best-performing available MatB mutants are able to generate AmM-CoA from CoA and aminomalonic acid (data not shown), thus AmM-ZmaH was generated in vitro using the enzymes responsible for biosynthesis of AmM-ZmaH in the native ZMA pathway (Figure 2B).21 The apo-ZmaH was converted to each respective acyl-ZmaH using Sfp and the corresponding acyl-CoA following confirmation via HPLC-UV/Vis that each malonic acid was converted to the acyl-CoA. Intact protein mass spectrometry (MS) was then employed to confirm the formation of each acyl-ZmaH (Figure 2C and Supplemental Table S1). In every case, formation of the desired acyl-ZmaH substrate was confirmed (Supplemental Table S1).
Figure 2. Generation and analysis of ZmaH-linked extender units.

(A) An acyl-CoA panel was generated using MatB or suitable MatB mutant. The formation of these intermediates was confirmed by HPLC-UV analysis and used in the subsequent Sfp reaction to convert apo-ZmaH to acyl-ZmaH. Following the confirmation of acyl-ZmaH formation by MS, each acyl-ZmaH substrate was assayed with ZmaF in the presence of holo-ZACP1. Formation of acyl-ZACP1 was monitored via intact protein MS. (B) In order to assay ZmaF with its natural substrate, AmM-ZmaH, the enzymes involved in ZMA biosynthesis were employed to generate AmM-ZmaH in vitro from holo-ZmaH. (C) As an example, mass spectra showing conversion of apo-ZmaH to ethylmalonyl-ZmaH (EM-ZmaH) are shown. Purified apo-ZmaH (bottom) is incubated with Sfp and EM-ZmaH to generate EM-ZmaH. Holo-ZmaH is also generated as a result of remaining CoA from the MatB reaction used in the subsequent Sfp reaction. Gluconylated protein is present due to either spontaneous or enzymatic in vivo modification in E. coli.
Extender unit promiscuity of ZmaF
In order to determine the extender unit specificity of ZmaF, in vitro reactions were performed with purified excised proteins. Using boundaries determined from secondary structure prediction and sequence alignments with previously characterized ACPs, the ACP partner for ZmaF, holo-ZACP1 (Figure 1), was provided as a standalone protein fused at the N-terminus to a hexa-histidine tag. Each substrate was tested for ZmaF trans-acylation activity by monitoring the formation of acyl-ACP1 (Figure 3A and Supplemental Table S1). In the case of the natural substrate AmM-ZmaH, which was enzymatically synthesized using the AmM-ZmaH biosynthetic pathway, high enzymatic conversion to the fully oxidized product was not achieved, and the reaction conditions did not allow for determination of the percent conversion. Each acyl-ZmaH was also incubated with holo-ZACP1 in the absence of ZmaF (Figure 3B) to monitor any background acylation of ZACP1. Propionyl-ZmaH was also prepared as a negative control, as it lacks the terminal carboxylate and was not expected to be recognized by ZmaF.
Figure 3. Activity of ZmaF with ZmaH-linked extender units.

(A) The ability of ZmaF to convert apo-ZACP1 to the corresponding acyl-ZACP1 with a panel of acyl-ZmaH was probed by intact protein MS. (B) Analysis of representative set of reactions to monitor ZmaF activity. EM-ZmaH was incubated with ZACP1 in the absence (top) and presence (bottom) of ZmaF to monitor ZmaF-dependent trans-acylation of ZACP1 to form EM-ZACP1. * gluconyl-holo-ZmaH; ** acetyl-holo-ZACP1 (C) Substrate specificity of ZmaF. Error bars represent standard deviation of duplicates performed.
Remarkably, ZmaF was active with all malonate-derived acyl-ZmaH substrates in this panel, as determined by the ZmaF-dependent formation of each acyl-ZACP1 (Figure 3B and Supplemental Table S1). ZmaF discriminated only against propionyl-ZmaH, which was not expected to be a substrate for the trans-AT. The activity of ZmaF with this panel of acyl-ZmaH’s makes it, to the best our knowledge, one of the most promiscuous ATs characterized to date. ZmaF was able to trans-acylate ZACP1 with R-malonyl substrates ranging from small (R = H) to large (R = isopentyl). The percent conversion from holo-ZACP1 to acyl-ZACP1 demonstrates that the activity of ZmaF increases as the malonyl side chain increases in carbon-chain length through propyl when the carbon chain is fully saturated and not branched (i.e. H, methyl, ethyl, and propyl). However, ZmaF demonstrates a substantial decrease in activity with the branched substrate, isopropyl, as compared to propyl. ZmaF also demonstrated a 4-fold lower activity with propargylmalonyl-ZmaH as compared to propylmalonyl-ZmaH, perhaps due to loss of flexibility of the alkyne-substrate. Indeed, ZmaF displayed a higher activity with allylmalonyl-ZmaH than with propargylmalonyl-ZmaH, suggesting that the active site can better accommodate the more flexible side chain. In the case of four-carbon side chains, ZmaF is more active with the linear butylmalonyl- than the branched isobutylmalonyl-ZmaH. Conversely, the trans-AT favored the branched isopentyl over the linear five-carbon side chain. This difference in substrate preference might be attributed to the length of each of the side chains; the isopentyl substrate is approximately the same length as the linear butyl. Based on this observation, the activity would be expected to continue to decrease as the extender unit C2 side chain length increases past four carbons. Notably, the natural substrate for ZmaF, AmM-ZmaH, was unable to be quantified under these conditions. Thus, the relative activity of the non-natural substrates compared to the cognate substrate could not be determined. Regardless, this data describes remarkable acyl-promiscuity for a previously uncharacterized trans-AT that natively uses ACP-linked extender units.
Thioester specificity requirements of ZmaF
Notably, the azidoethylmalonate unit provides an orthogonal handle to enable quick and efficient screening for further investigation into the activity of ZmaF. Accordingly, for an initial assessment of thioester specificity, ZmaF was tested with a small panel of thioester-activated azidoethylmalonates and analyzed by in-gel fluorescence monitoring after incubation with fluorescent dibenzocyclooctyne (DBCO) (Figure 4A).24 Azidoethylmalonate was chemoenzymatically linked to CoA (AzEM-CoA) or pantetheine (AzEM-pantetheine) via the MatBcatalyzed condensation of azidoethylmalonate and the corresponding thiol. The MatB-generated AzEM-CoA was also used to produce AzEM-ZmaH from apo-ZmaH using Sfp (Figure 2A). According to the proposed mechanism described by Thomas and colleagues,18 the presence of the ACP-carrier might enable acylation through opening of the active site, caused by association with the ACP. AzEM-pantetheine was therefore also tested in the presence of apo-ZmaH to determine if binding or association with ZmaH could support the self-acylation of ZmaF with AzEM-pantetheine (Figure 4A). The product mixtures were incubated with the fluorescent DBCO, and the presence of fluorescently labelled ZmaF (via self-acylation of AzEM-thioester) and ZAPC1 (the subsequent trans-acylation product) was determined by visualization of the SDS-PAGE gel under a trans-illuminator (Figure 4B).
Figure 4.

Azido-functionalized extender units as probes for ZmaF activity. (A) Thioester-linked azidoethylmalonates were tested with ZmaF and ZACP1. Following reaction with ZmaF, each product mixture was incubated with DBCO to fluorescently label any protein modified with the azide. (B) Each reaction was analyzed via SDS-PAGE gel followed by fluorescence-imaging. Signals on the fluorescence-imaged gel (top) indicate that the protein has been acylated with AzEM. The gel was then subjected to Coomassie staining (bottom) to visualize all proteins in the reaction.
Among the conditions tested, the only reaction that led to the identification of a fluorescently labelled ZACP1 was that with the Sfp-generated AzEM-ZmaH (Figure 4B, lane 1). Notably, the absence of labelled ZACP1 in lane 2 confirms that the trans-acylation of ZACP1 is ZmaF-dependent. This experiment and the data above suggests that the acyl unit must be carried by ZmaH, the natural carrier for ZmaF, as the CoA- and pantetheine-linked AzEM were not substrates. Consequently, further activity probing of various acyl substrates was performed with ZmaH-linked extender units.
Kinetic investigation of the ZmaF-catalyzed reaction with AzEM-ZmaH
The ability of ZmaF to utilize AzEM-ZmaH allows for a higher throughput analysis of ZmaF activity through the in-gel fluorescence assays as compared to MS analysis to monitor the acylation of the ACP. This novel activity was therefore harnessed to assess the kinetics of ZmaF to determine reaction conditions for additional acyl-ZmaH substrates. After creating a standard curve with Sfp-generated AzEM-ZmaH (Supplementary Figure S1A), a time-course of the ZmaF-catalyzed reaction was performed at presumed saturating conditions of AzEM-ZmaH to determine the appropriate time point representative of initial velocity (V0) for AzEM-ZmaH titration. AzEM-ZACP1 formation was linear with respect to time during the first ~30 minutes (Supplementary Figure S1B).
Next, a range of AzEM-ZmaH concentrations was tested to determine the kcat and KM of ZmaF with AzEM-ZmaH as the substrate. Fluorescence values for AzEM-ZACP1 formation were converted to concentration and then fitted to the Michaelis-Menten equation (Supplementary Figure S1C). The kcat was found to be 2.1 ± 0.3 × 10−3 s−1 and the KM of ZmaF for AzEM-ZmaH was 9 ± 4.6 µM. As a comparison, the trans-AT DszD from disorazole synthase was found to have a kcat of 28 s−1 and a KM of 7 µM with M-CoA, its natural substrate.25 The large difference between the kcat values of the two trans-ATs might be explained in part because AzEM-ZmaH is not the natural acyl substrate for ZmaF. The reaction with ZmaF and AmM-ZmaH could not be kinetically evaluated due to the complexity of the reaction and the low yield of AmM-ZmaH. Another potential contributing factor is the use of ACP- versus CoA-linked substrates of the respective trans-ATs. If the ACP-donor must bind to the AT to affect a conformational rearragement,18 the additional requirement for self-acylation could lead to reduced kinetic rates. Additionally,
Probing orthogonality within ZMA system
The cis-AT ZmaA-AT and the trans-AT ZmaF are presumed to be completely orthogonal.17 That is, the carrier protein and acyl specificities of each AT are completely non-overlapping. In the natural system, ZACP1 is trans-acylated by the trans-AT ZmaF with aminomalonyl carried by ZmaH (Figure 1). The module served by ZmaF also includes the cis-AT, ZmaA-AT, which transfers a hydroxylmalonyl unit from ZmaD to ZACP2. Here, this aspect of orthogonality was further probed by testing the ability of each AT to transfer methylmalonyl (MM) to ZACP1 or ZACP2 from MM-ZmaH or MM-ZmaD (Figure 5) via intact protein MS analysis of the corresponding acyl-ACP (Table 1).
Figure 5. Probing the orthogonality of the ZMA ATs.

The ability of each substrate (acyl-ZmaH or acyl-ZmaD) and enzyme (ZmaF or ZmaA-AT) pair to trans-acylate each ACP (ZACP1 or ZACP2) was determined using intact protein MS. To determine the ability of each enzyme (ZmaF or ZmaA-AT) to undergo self-acylation with AzEM-ZmaH or AzEM-ZmaD (R=AzEM), transfer of the azide to the AT active site was determined by in-gel fluorescence.
Table 1.
ZmaF and ZmaA-AT activity with acyl-ACP substrates.
| Entry | Enzyme | ACPa | ACP partner | Self-acylation | Trans-acylation |
|---|---|---|---|---|---|
| 1 | ZmaF | MM-ZmaH | ZACP1 | NDb | 23c |
| 2 | ZmaF | MM-ZmaH | ZACP2 | NDb | 3c |
| 3 | ZmaF | MM-ZmaD | ZACP1 | NDb | 0c |
| 4 | ZmaF | MM-ZmaD | ZACP2 | NDb | 0c |
| 5 | ZmaA-AT | MM-ZmaH | ZACP1 | NDb | 0c |
| 6 | ZmaA-AT | MM-ZmaH | ZACP2 | NDb | 0c |
| 7 | ZmaA-AT | MM-ZmaD | ZACP1 | NDb | 0c |
| 8 | ZmaA-AT | MM-ZmaD | ZACP2 | NDb | 0c |
| 9 | ZmaF | AzEM-ZmaH | ZACP1 | +d | NDe |
| 10 | ZmaF | AzEM-ZmaH | ZACP2 | +d | NDe |
| 11 | ZmaF | AzEM-ZmaD | ZACP1 | −d | NDe |
| 12 | ZmaF | AzEM-ZmaD | ZACP2 | −d | NDe |
| 13 | ZmaA-AT | AzEM-ZmaH | ZACP1 | −d | NDe |
| 14 | ZmaA-AT | AzEM-ZmaH | ZACP2 | −d | NDe |
| 15 | ZmaA-AT | AzEM-ZmaD | ZACP1 | −d | NDe |
| 16 | ZmaA-AT | AzEM-ZmaH | ZACP2 | −d | NDe |
MM, methylmalonyl; AzEM, azidoethylmalonyl
ND = not determined. The self-acylation with MM substrates was not determined due to limitations of the assay (see main text).
Percent conversion to the MM-ACP. The trans-acylation with MM substrates was determined by intact protein MS analysis of the ACP.
The self-acylation with AzEM substrates was determined by in-gel fluorescence identification of the labelled ZmaF or ZmaA-AT.
ND = not determined. The trans-acylation was not determined because only ZACP1 can be distinguished from AzEM-ZmaH. There is insufficient electrophoretic separation between every other combination of ACP extender unit (AzEM-ZmaD or AzEM-ZmaH) and target carrier protein (ZAPC1 or ZACP2) (data not shown) to allow detection by in-gel fluorescence.
As expected, ZmaF was able to trans-acylate ZACP1 in the presence of MM-ZmaH (Table 1, entry 1). ZmaF was also able to trans-acylate ZACP2 using MM-ZmaH, but at a significantly reduced level (Table 1, entry 2). As expected, ZmaF was not able to trans-acylate either ZACP1 or ZACP2 in the presence of MM-ZmaD (Table 1, entries 3 and 4). These results further support that ZmaF, while promiscuous towards non-natural acyl units, is highly specific for ZmaH and ZACP1 as its acyl donor and ACP partner, respectively.17 ZmaA-AT was not able to trans-acylate ZACP1 or ZACP2 in the presence of either MM-ZmaH or MM-ZmaD (Table 1, entries 5–8). The inability of ZmaA-AT to trans-acylate its cognate ACP partner (ZACP2) with MM-ZmaD suggests that ZmaA-AT is not active with the non-natural acyl unit and that the extender unit specificity of ZmaF and ZmaA-AT are likely orthogonal, even though ZmaA-AT might be self-acylated in the presence of MM-ZmaD.18
Next, to complement the trans-AT assays, the ability of ZmaF and ZmaA-AT to self-acylate with various cognate or non-cognate substrates was probed. It is difficult to use intact MS to determine whether self-acylation of ZmaF or ZmaA-AT is taking place due to the large size of the enzyme. However, self-acylation of each enzyme can be detected via in-gel fluorescence (Figure 4A) in the presence of each ACP carrier (ZmaD and ZmaH) and each ACP partner (ZACP1 and ZACP2) given the significant difference in electrophoretic mobility between each AT and ACP. The results of the self-acylation in-gel fluorescence assay with AzEM-substrates (Table 1, entries 9–16; Supplemental Figure S2) closely resemble those from the trans-acyltransferase assay with MM-substrates (Table 1, entries 1–8), i.e. ZmaF is active in the presence of AzEM-ZmaH and ZACP1 (Table 1, entry 9; Supplemental Figure S2, lane 1). Notably, and consistent with the ability of ZmaF to transfer MM to ZACP2 to some extent, self-acylation of ZmaF was also detected with AzEM-ZmaH in the presence of ZACP2 (Table 1, entry 10; Supplemental Figure S2, lane 3). As demonstrated with the MM-substrates, ZmaF was not self-acylated with AzEM-ZmaD in the presence of either ZACP1 or ZACP2 (Table 1, entries 11–12; Supplemental Figure S2, lanes 5 and 7). Additionally, ZmaA-AT was not self-acylated with AzEM-ZmaH or AzEM-ZmaD, regardless of the ACP partner (Table 1, entries 13–16; Supplemental Figure S2, lanes 9, 11, 13, and 15). These findings reinforce the orthogonality within the ZMA system and further suggest that ZmaA-AT is not promiscuous with non-natural acyl-partners.
Computational prediction of the ZmaF/ZmaH/ZACP1 interaction epitopes
Structures have not yet been reported for ZmaF, ZACP1, or ZmaH. To provide a working model to understand these proteins, homology models of ZmaF, ZACP1, and ZmaH were built using the I-TASSER server26–28 and refined using the Fragment-Guided Molecular Dynamics (FG-MD) simulation server (Supplemental Figure S3).29 It should be noted that no structure has been reported for a trans-AT that has a standalone ACP as its polyketide acyl donor which likely increases the difficultly of predicting a correct structure for ZmaF. Nevertheless, most of the ZmaF sequence aligned well with the threading templates and the resulting structural model is suitable for our preliminary studies. Homology models for both ZACP1 and ZmaH resulted in the expected three-helix bundle with the conserved phosphopantetheine (Ppant) attachment site (Ser43 and Ser36, respectively) at the N-terminus of α-helix 2 (αHII).
The homology models were used to perform docking studies between ZmaH and ZmaF and between ZACP1 and ZmaF using ClusPro.30–32 ClusPro generated 29 and 28 models for each pair of proteins, respectively. Each docking model was initially assessed by examining the distance between the active site serine of ZmaF and the Ppant attachment site of the ACP. The length of Ppant is approximately 17 Å and this is the maximum approximate distance during transfer of an extender unit from the AT to ACP. ZmaF was also viewed as a surface model (Figure 6A) to identify the active site chamber accessible to the Ppant arm to further assess the docking models. According to these criteria, the best docking models revealed that ZmaH (Figure 6B) and ZACP1 (Figure 6C) appear to dock in the same general surface region of ZmaF. This suggests that a binary complex between ZmaH and ZmaF facilitates self-acylation of the enzyme which is followed by the departure of ZmaH and then binding of ZACP1 to ZmaF to enable trans-acylation.
Figure 6. Docking studies with homology models using ClusPro.

(A) Surface view of ZmaF to visualize the active site chamber. The active site serine is yellow and circled. (B) The top three docking locations of ZmaH. (C) The top three selected docking locations of ZACP1. The Ppant attachment sites of each ACP structure are shown as yellow spheres. In all cases, for clarity, the poorly resolved C-terminus of ZmaF was removed from the structure.
Trans-complementation of modules from the 6-deoxyerythronolide B synthase with ZmaF and DszD
Given the interest in accessing erythromycin derivatives with improved pharmacological properties, and the wealth of data available that describes the ability to apply combinatorial biosynthesis efforts to the erythromycin biosynthetic pathway,1 modules of the 6-deoxyerythronolide B synthase (DEBS) are often tested for their ability to be trans-complemented by orthogonal ATs.8, 25, 33–35 To benchmark the ability of ZmaF to trans-complement DEBS, AT-knockout mutants of DEBS module 6-TE (DM6) and DEBS module 3-TE fusion (DM3) were constructed by mutagenesis of the active site serine to an alanine to generate DEBS mod6TE-ATº (DM6º) and DEBS mod3TE-ATº (DM3º), respectively. The ability of each purified holo-DEBS protein to be trans-acylated by ZmaF with MM-ZmaH as the extender unit substrate (Figure 7A, R = ZmaH; R’ = Me) and by DszD with M-CoA (Figure 7A, R = CoA; R’ = H) was tested by high performance liquid chromatography (HPLC) and liquid chromatography-mass spectrometry (LC-MS) analysis of the corresponding product mixtures. In this single module reaction, diketide-N-acetylcysteamine (DKS) acts as a mimic of an upstream polyketide intermediate and is loaded onto the KS of the module. Upon successful complementation in the presence of DKS, ZmaF is expected to produce methylpyrone (Figure 7A, R’ = Me), whereas DszD is expected to produce the corresponding pyrone (Table 2, R’ = H). As expected, DszD was able to complement DM3 (Table 2, entry 3; Supplementary Figure S4A), DM3º (Table 2, entry 8; Supplementary Figure S4B), DM6 (Table 2, entry 13; Supplementary Figure S4C), and DM6º (Table 2, entry 18; Supplementary Figure S4C) in the presence of M-CoA.
Figure 7.

Trans-complementation assays of DEBS modules. (A) General reaction scheme showing the use of DKS and a CoA- or ACP-linked extender unit, to probe the ability of trans-ATs to complement a DEBS module. (B) Summary of the key results obtained from the trans-complementation assays.
Table 2.
Trans-complementation reactions of DEBS modules. Reactions were monitored via HPLC/UV-Vis and presence of pyrone was confirmed by MS.
| Entry | Module construct | Substratea | Trans-AT | Pyroneb | Methylpyroneb |
|---|---|---|---|---|---|
| 1 | DM3 | MM-CoA | − | ++ | |
| 2 | DM3 | M-CoA | + | − | |
| 3 | DM3 | M-CoA | DszD | ++ | − |
| 4 | DM3 | MM-ZmaH | − | − | |
| 5 | DM3 | MM-ZmaH | ZmaF | − | − |
| 6 | DM3º | MM-CoA | − | + | |
| 7 | DM3º | M-CoA | − | − | |
| 8 | DM3º | M-CoA | DszD | ++ | − |
| 9 | DM3º | MM-ZmaH | − | − | |
| 10 | DM3º | MM-ZmaH | ZmaF | − | − |
| 11 | DM6 | MM-CoA | − | ++ | |
| 12 | DM6 | M-CoA | − | − | |
| 13 | DM6 | M-CoA | DszD | ++ | − |
| 14 | DM6 | MM-ZmaH | − | − | |
| 15 | DM6 | MM-ZmaH | ZmaF | − | − |
| 16 | DM6º | MM-CoA | − | − | |
| 17 | DM6º | M-CoA | − | − | |
| 18 | DM6º | M-CoA | DszD | ++ | − |
| 19 | DM6º | MM-ZmaH | − | − | |
| 20 | DM6º | MM-ZmaH | ZmaF | − | − |
M, malonyl; MM, methylmalonyl
Significant product formation is indicated with “++”. Detection of minimal product is indicated with “+” while “-ˮ indicates that product was not detected.
Also, and as expected, methylpyrone was detected in reactions containing MM-CoA and active DEBS AT (Table 2, entries 1, 11; Supplemental Figure S4A/C) but was either minimally produced (Table 2, entry 6; Supplemental Figure S4B) or not detected (Table 2, entry 16; Supplemental Figure S4D) in reactions containing an AT-null DEBS construct. Interestingly, the level of pyrone production by DM3º in the presence of M-CoA and DszD (Supplemental Figure S4B) was higher than that of the methylpyrone produced by DM3 in the presence of MM-CoA (Supplemental Figure S4A). Under the conditions tested, DszD was provided in 5-fold molar excess of the DEBS construct, likely accounting for the corresponding increased product formation. This suggests that under these conditions, trans-acylation of the ACP might be the rate-limiting step. This result also holds true in the case of DM6, which also produced more pyrone than methylpyrone in the presence of M-CoA/DszD and MM-CoA, respectively (Supplemental Figure S4C).
In contrast to the reactions with DszD, ZmaF was unable to complement any of the four DEBS constructs, as judged by the inability to detect methylpyrone in those reactions (Table 2, entries 5, 10, 15, and 20; Figure 7B). It is possible that ZmaF can complement the module at low levels, but the provided extender unit concentration prevents detection of methylpyrone (the minimum detection limit is ~1% of the methylpyrone produced when DM3 is incubated with MM-CoA). The attempted ZmaF-catalyzed complementation of the DEBS modules with MM-ZmaH is limited by the concentration of MM-ZmaH. The method of preparation of the substrate via Sfp prevents supplying the extender unit at concentrations comparable to that of reactions with CoA-linked extender units. Furthermore, it is not possible to regenerate the extender unit in situ because the holo-ACP product cannot be phosphopantetheinylated. In addition to the attempted complementation of the DEBS modules, ZmaF was not able to trans-acylate any standalone DEBS ACP in vitro (data not shown). This suggests that the DEBS ACP’s lack sufficient molecular information to be recognized by ZmaF on their own, and/or that other recognition determinants from the cognate module, ZmaA, are required for efficient utilization by ZmaF, as has been proposed for other trans-ATs.36–38
Chimeragenesis of DEBS Module 3
The ACP partner housed within a given module (in the case of ZmaF, ZACP1) must be involved in protein interactions between the module and the trans-AT, as the trans-AT must dock at the ACP for the trans-acylation of the acyl unit to the Ppant arm of the ACP. Accordingly, to begin to assess whether other recognition determinants in ZmaA are required to enable complementation of DEBS modules by ZmaF, the ACP of DM3 was exchanged with ZACP1 of ZmaA (Figure 7B). The DM3 system was chosen in preference to DM6 given that in the pyrone assays described above, the DM3 constructs performed several-fold better than the DM6 constructs, according to both the trans-complementation experiments with DszD and the direct incorporation of MM-CoA by the DEBS module, as judged by relative HPLC product peak areas (Supplemental Figure S4A-D). This can be explained in part because the DKS is a better mimic of the precursor natively accepted by DM3 than that of DM6. Additionally, in the case of trans-complementation with DszD, the trans-AT is known to have higher affinity with DEBS ACP3 than with DEBS ACP6.39 MM-ZmaH was chosen as the substrate for ZmaF in the trans-complementation assays with the DM3 chimeras, given that ZmaF is active with MM-ZmaH and ZACP1. Moreover, MM-CoA is the natural extender unit substrate for module 3 of DEBS. Thus, once loaded onto the ACP via trans-complementation, the resulting polyketide intermediate produces a product that is identical to that produced by the native activity of DM3 in this assay system. Accordingly, any inability to trans-complement by a trans-AT must be a result of faulty protein-protein interactions (or protein folding), as it is known that the other domains within the construct (the KS and TE) can process the introduced methylmalonyl unit. Notably though, the use of the ACP-linked extender unit, as required by ZmaF, limits the concentration of the substrate, as the solubility limit of ZmaH does not approach that of the acyl-CoA’s. Therefore, acyl-CoA concentrations were reduced compared to those used above to match the concentration of MM-ZmaH.
Each purified module was assayed as summarized in Figure 7A/B, Supplementary Figure S5A-D, and Supplementary Table S2. As a positive control, intact DM3 was used with its natural substrate, MM-CoA. As a positive control for trans-complementation, the trans-AT DszD was used with its natural substrate (M-CoA) and the AT-null mutant DM3°, whereby the active site serine of the AT within DM3 is mutated to alanine. To investigate the effect of the full ACP swap, the entire ACP domain of DM3 was replaced with ZACP1 in DM3 and DM3° to yield DM3-ZACP1 and DM3°-ZACP1, respectively (Figure 7B).
As expected, even at reduced concentrations of extender unit, DM3 produced methylpyrone (Figure 7B, R’ = CH3) in the presence of MM-CoA (Figure 7B; Supplementary Figure S5A, and Supplementary Table S2, entry 1). Interestingly, this construct also produced a trace amount of pyrone in reactions containing MM-ZmaH and MM-ZmaH with ZmaF (Supplementary Figure S5A and Supplementary Table S2, entries 2 and 3). In the chemoenzymatic synthesis of MM-ZmaH, apo-ZmaH is incubated with Sfp and excess MM-CoA. Therefore, when MM-ZmaH is added to the reaction, trace MM-CoA is likely present. The lack of methylpyrone production in reactions containing MM-ZmaH and ZmaF and DM3° (Supplementary Figure S5B and Supplementary Table S2, entries 5 and 6), also support that methylpyrone formation in the reactions with DM3 is due to the DEBS AT acylation using MM-CoA and is not driven by trans-complementation of ZmaF.
In addition, and as expected, DM3° produced pyrone (Figure 7B, R’ = H) in the presence of DszD and M-CoA (Figure 7B; Supplementary Table S2, entry 8; Supplementary Figure S5B), confirming the ability of the M-CoA specific trans-AT to complement the non-cognate module. However, the ACP-swapped DM3 chimeras were unable to support trans-complementation with ZmaF, as judged by the inability to detect the corresponding methylpyrone product (Figure 7B; Supplementary Table S2, entries 11, 14, 19, 24; Supplementary Figure S5C/D). Interestingly though, in the presence of DM3°-ZACP1 and M-CoA, DszD was able to drive pyrone formation, albeit at low yields (Figure 7B; Supplementary Figure S5D and Supplementary Table S2, entry 16). Conversely, DM3-ZACP1 containing an active DEBS AT was not able to produce methylpyrone in the presence of MM-CoA (Supplementary Figure S5C and Supplementary Table S2, entry 9). Based on the activity of DszD and DM3°-ZACP1, it might be expected that DM3-ZACP1 should be capable of producing methylpyrone in the presence of MM-CoA, but this was not observed. However, the relatively high concentration of trans-AT in the complementation reactions (DszD is supplied at a two-fold molar excess over the module) likely drives the reaction forward. This, coupled with the successful trans-complementation of DM3°-ZACP1 with DszD, indicates that (1) the DM3°-ZACP1 module is properly folded and capable of catalytic condensation, and (2) the failure of trans-complementation with ZmaF is likely caused by the inability of ZmaF to access the ACP within the module or to be recruited by it.
DISCUSSION
Previous characterization of the trans-AT ZmaF was limited to a small panel of non-cognate substrates.17 Here, we sought to gain a better understanding of the activity and specificity of this trans-AT to begin to assess its candidacy for trans-complementation. We were able to mimic the previously characterized orthogonality within the ZMA pathway to determine that ZmaF is not self-acylated with ZmaD-linked extender units and does not trans-acylate ZACP2 at a high level. Enabled by our previously engineered MatB mutants and the promiscuous Sfp, the activity of ZmaF was probed with a large panel of acyl extender units, revealing that the trans-AT demonstrated remarkable promiscuity towards ZmaH-linked units. Notably, ZmaF was active with AzEM-ZmaH, which provided a handle to increase throughput of the enzyme assays and provided a snapshot of the kinetics of ZmaF. In comparison to other characterized trans-ATs,25 it appears that ZmaF has a similar KM with its extender unit substrate but is slower with respect to its trans-acylation activity.
The acyl-promiscuity demonstrated by ZmaF makes it, to the best of our knowledge, the most promiscuous trans-AT characterized to date. The previously first and only known promiscuous trans-AT KirCII, whose natural substrate is EM-CoA, is active with just propargylmalonyl-CoA, AM-CoA, phenylmalonyl-CoA, and AzEM-CoA.11, 13 Notably, while the KirCII assays were performed under similar conditions to the ones with ZmaF described here, KirCII converted approximately 15% of holo-ACP to the corresponding ethyl-ACP, which is lower than that of many of the non-natural acyl units assayed with ZmaF here (Figure 3C).
The promiscuity of ZmaF is likely due to recognition and subsequent binding of ZmaH. For example, of the substrates tested with ZmaF, the trans-AT did not utilize a potential extender unit unless the acyl group was carried by ZmaH. In addition, of those ZmaH-linked substrates tested, ZmaF was shown to trans-acylate all malonyl derivatives and was inactive only with propionyl-ZmaH, which lacks the terminal carboxylate. In the natural biosynthetic system, ZmaF needs only select against the reduced versions of AmM-ZmaH, both of which lack the terminal carboxylate. Accordingly, ZmaF presumably experienced no evolutionary pressure to discriminate against substrates carried by ZmaH that include a terminal carboxylate. In this manner, perhaps other ACP-linked accepting ATs possess similar promiscuity to ZmaF. However, our initial assays with ZmaA-AT, a cis-AT that uses ACP-linked extender units, suggested that ZmaA-AT was inactive with non-natural acyl substrates carried by ZmaD, the natural acyl-donor of ZmaA-AT. Therefore, ZmaH recognition may be the dominant feature that confers ZmaF promiscuity, but there is also something else unique about the trans-AT that supports such broad substrate scope. These results also suggest that ZmaA-AT does not have the same level of acyl-promiscuity as ZmaF. Previous studies by Thomas and co-workers indicate that ZmaA-AT is at least self-acylated in the presence of MM-ZmaD18 or aminomalonyl-ZmaH,17 but any role of the trans-acylation step in determining specificity has not been explored.
While ZmaF demonstrated broad substrate scope with ZmaH-linked substrates, activity was not observed with CoA- or pantetheine-linked extender units. This feature renders the remarkable promiscuity of ZmaF challenging to leverage for polyketide diversification in vivo because of the difficulty of providing the suitably ACP-linked extenders units. To address this, a two-component approach was tested with AzEM-pantetheine in the presence of apo-ZmaH. It was rationalized that the binding of ZmaH causes a shift in secondary structure that allows for the opening of the ZmaF active site for acylation. Thus, the provided apo-ZmaH might allow for the subsequent acylation of ZmaF with the acyl-pantetheine substrate. This approach, however, was unsuccessful, revealing that ZmaF requires the covalent attachment of the acyl unit to ZmaH. The failure of this reaction might be due to ZmaH blocking the active site chamber upon association, never allowing for the acyl-pantetheine to access the active site, and this would require further investigation to address. Another approach to overcome this feature includes surface mutagenesis of ZmaF to engineer utilization of CoA-linked extender units in preference to ACP-linked ones, something that has been accomplished with other ACP-utilizing enzymes.40 Regardless, in vitro approaches such as cell-free transcription translation41, 42 will enable a simplified method to probe the promiscuity of biosynthetic steps downstream of ZmaF and to rapidly access novel non-natural analogues of ZMA and other polyketide-based natural products through an approach analogous to precursor directed biosynthesis.
To help guide our engineering and characterization efforts, homology models of ZmaF, ZACP1, and ZmaH were constructed and used to perform computational docking studies. According to the models generated, ZmaH and ZACP1 dock in approximately the same region of ZmaF. This would be expected if both carrier proteins use the same entrance to the active site chamber, as defined by the active site serine of ZmaF. This suggested that rather than a single ternary complex, two consecutive binary complexes are required to accommodate binding of the ZmaH-linked extender unit followed by the target carrier protein, ZACP1. This prediction could help future efforts to leverage ZmaF as a tool for polyketide diversification. For example, according to this model. the ZmaF:ZmaH and ZmaF:ZACP1 interaction epitopes are partially overlapping, and this should facilitate identification of the recognition motifs that drive recruitment of acyl-ZmaF to the target (ZACP1 in ZmaA) because they likely share similar recognition features.
Efforts to leverage the promiscuity of ZmaF to diversify other polyketides focused on defining the ability of ZmaF to trans-complement non-cognate PKS modules. This included the design of DEBS Mod3TE chimeras in which the native ACP was replaced with ZACP1 from the ZMA PKS. However, ZmaF was unable to trans-acylate the target module even though the chimera was active in other assays. Future work will focus on identification of the molecular determinants in ZmaA that drive recruitment of ZmaF to the target module. In addition, chimeragenesis with other PKS modules and further optimization of the chimera boundaries2 will be carried out in an effort to identify the molecular interaction epitope. Cumulatively, this study represents the first comprehensive analysis of the substrate specificity and utility of a unique trans-AT and is a critical first step in elucidating the molecular basis for its carrier protein orthogonality and extender unit promiscuity, thus providing a blueprint for establishing ZmaF as a tool for polyketide diversification.
MATERIALS AND METHODS
Strains, Media, and Chemicals
All other chemicals were purchased from Sigma-Aldrich unless stated otherwise. PCR products were extracted with a Bio Basic Gel Extraction Kit. Restriction enzymes were purchased from New England Biolabs. Plasmids were isolated using a plasmid miniprep kit from Bio Basic. Other DNA preparation kits (genomic, and gel extraction) were purchased from (NEB). Oligos were synthesized by Integrated DNA Technologies (IDT) and purified by IDT using standard desalting. Polymerases were purchased from Fisher Scientific; all restriction enzymes were purchased from New England Biolabs (NEB). LB media was purchased from Fisher Scientific. Polyacrylamide gels were homemade and prepared using reagents purchased from Fisher Scientific; gels were prepared using 4% acrylamide stacking gel and 20% acrylamide running gel. Dibenzocyclooctyne-fluor (DBCO) 488 reagent was purchased from Sigma Aldrich. Reagents used for buffer preparation were purchased from VWR. Isopropyl-β-D-thiogalactopyranoside (IPTG) was purchased from CalBioChem. Bacillus thuringiensis 4BD1 strain was ordered from the Bacillus Genetic Stock Center (BGSC). For protein expression, E. coli strain BL21(DE3) was used unless stated otherwise. For DNA storage and manipulation, E. coli TOP10 (Invitrogen) was used.
Preparation of Substrates for MatB Reactions
Malonic acid substrates that were purchased as diesters were saponified before using in MatB reactions. Diethyl propylmalonate, dimethyl propargylmalonate, dimethyl isobutylmalonate, diethyl pentylmalonate, and diethyl isopentylmalonate were each saponified using the same protocol, adapted from previously published methods.11 Malonate diester (6.2 mmol) and NaOH (60 mmol) were added to round bottom flask containing 10 mL water and the mixture was stirred overnight at 65 °C. The solution was cooled on ice and slowly acidified on ice with concentrated HCl to pH ~3. The solution was extracted six times with 10 mL diethyl ether and the ether layers were combined, dried over MgSO4, and concentrated to yield a white solid in all cases. Pantetheine for MatB reaction was prepared fresh through reduction of commercially available pantethine. Reduction was performed according to previously published methods.43 Briefly, pantethine (0.21 mmol) was dissolved in 1 mL 20% w/v glycerol along with 1,4-dithiothreitol (DTT, 0.22 mmol). The solution was incubated at 60 °C for 15 min, and the resulting pantetheine solution was stored at −20 °C until use.
Chemo-Enzymatic Preparation of Aminomalonyl-ZmaH
Preparation of aminomalonyl-ZmaH was adapted from previous published methods.44 The cloning, expression, and purification of the required aminomalonyl-ZmaH biosynthetic genes are described in the Supplemental Methods. A 500 µL reaction contained the following: 75 mM Tris pH 7.5, 10 mM MgCl2, 1 mM tris(2-carboxyethyl)phosphine (TCEP), 12.5 µM holo-ZmaH, 1 µM ZmaJ, 200 µM NAD+, 100 µM FAD, 1 µM ZmaG, 1 µM ZmaI, 250 µM L-serine, and 5 mM ATP. The reaction was incubated at room temperature for 2 h before use. MS analysis confirmed the presence of aminomalonyl-ZmaH, but conversion was incomplete.
Preparation of Malonyl-Thioester Substrates
Malonyl-CoAs and other malonyl-thioesters were chemoenzymatically synthesized using MatB from Rhizobium trifolii. Wild-type MatB was used for malonyl- and methylmalonyl-CoA. Previously described double mutant MatB-T207G-M306I was used to make ethylmalonyl-, propylmalonyl-, isopropylmalonyl-, propargylmalonyl-, allylmalonyl-, butylmalonyl-, isobutylmalonyl-, pentylmalonyl-, isopentylmalonyl-, and azidoethylmalonyl-CoA.11 Each 800 µL reaction contained 16 mM of malonate or malonate analogue, 16 mM ATP, 8 mM CoA or pantetheine, 100 mM phosphate (pH 7.4), 10 mM MgCl2, and 10 µg of wild-type or mutant MatB. Reactions were incubated at room temperature overnight. Propionyl-CoA was generated using the propionyl-CoA ligase AcsA45 using the same reaction conditions as for MatB; conversion was confirmed via LC-MS (Supplementary Table S1). The conversion from CoA to each malonyl-CoA was confirmed by HPLC-UV-Vis analysis. The appropriate malonyl-CoA analogue was then incubated with apo-ZmaH and Sfp to convert ZmaH to malonyl-ZmaH. Each reaction contained 200 µM ZmaH; each malonyl-CoA was supplied in excess at 500 µM in an attempt to achieve complete conversion. Reactions were performed in 50 mM Tris buffer, pH 8.8, containing 10 mM MgCl2, 10% v/v glycerol, and 20 µg Sfp per 200 µL. Reactions were incubated for 4 h at room temperature.
Determination of ZmaF Substrate Scope
Conditions for ZmaF assays were adapted from previously published methods.24 Reactions prepared for MS analysis with the panel of acyl-ZmaH substrates contained 3 µM ZmaF, 30 µM ZACP1, and 60 µM acyl-ZmaH (except in the case of aminomalonyl-ZmaH, which was provided at ~4 µM). Reactions were incubated at room temperature for 25 min and then immediately diluted 10-fold into 100 mM NH4OAc. Samples were analyzed the same day by mass spectrometry on a Thermo Fisher Exactive Plus using heated electron spray ionization (HESI) according to the following parameters: spray voltage, 3.5 kV; capillary temperature, 350 °C; heater temperature, 200 °C; S lens RF level, 70 V; sheath gas flow rate, 45 au; resolution, 70,000 FWHM; scan range, 400–3,000 m/z. The flow rate was 500 µL/minute. Gradient was performed using 10% methanol in water with 0.1% formic acid (A) and 90:10 methanol:water with 0.1% formic acid as follows: 0–5 minutes, 25–100% B; 5–9 minutes, 100% B; 9–15 minutes, 25% B. The LC column was an Agilent Poroshell 300SB-C3, 2.1 × 75 mm, 5 µm. Isotopic distributions for all species (R-ZmaH and R-ZACP1) were calculated using a web-based isotope pattern calculator (IPC), provided by Pacific Northwest National Laboratory (omics.pnl.gov). These isotopic distributions were used to find the mass range for extracted ion counts for each acyl-ZmaH and acyl-ZACP1 and to obtain the calculated monoisotopic mass for ppm calculation. The resolution was set to 70,000. When applicable, Ppant arm and malonyl units were added as modifications. Percent conversion from holo-ZACP1 to acyl-ZACP1 was calculated by dividing the extracted ion count for malonyl-ZACP1 by the sum of the extracted ion counts of malonyl-ZACP1 and holo-ZACP1. The same mass ranges were used in the corresponding negative controls lacking ZmaF, and this percentage was subtracted from the percentage obtained from the reaction containing ZmaF. Most substrates were performed in duplicate, but as standard deviations were very low (≤2% in most cases), poorly performing substrates were analyzed only once.
Thioester specificity assays
In most cases, 10 µL reactions contained 30 µM holo-ACP, 3 µM AT, and 300 µM CoA- or pantetheine-linked extender or 60 µM ACP-linked extender unit. Final buffer concentration for all ZmaF reactions was 75 mM Tris, pH 7.5 and 10 mM MgCl2. Reactions were run for 2 h, at which point a 5 µL aliquot was removed and added to 0.5 µL of 2 mM dibenzocyclooctyne-fluor 488 (DBCO). The labelling reaction was incubated at room temperature for 30 min before preparing samples for SDS-PAGE analysis. 7 µL of water was added to dilute reactions followed by 3 µL of 5 × SDS dye. Samples were boiled for 5 min at 95 °C. 2 µL of each reaction was loaded onto a 4%/15% discontinuous SDS-PAGE gel and run at 220 V for approximately 1 h. The gel image was captured using Typhoon FLA 7000 using the 473 nm laser and Y520 filter. Pixel size was set to 200 µm, and the photomultiplier tube (PMT) was set to 500, the lowest available setting. Analysis of bands was performed using ImageQuant TL software, version 7.0. Rubberband background subtraction method was used.
ZmaA-AT and ZmaF orthogonality
Reactions (10 µL) contained 30 µM holo-ACP (ZACP1 or ZACP2), 3 µM AT (ZmaA-AT or ZmaF), and 60 µM ACP-linked extender unit (MM-ZmaH, MM-ZmaD, AzEM-ZmaH, or AzEM-ZmaD). Final buffer concentration was 75 mM Tris, pH 7.5 and 10 mM MgCl2. Reactions were run for 2 h, at which point a 5 µL aliquot was removed and added to 0.5 µL of 2 mM dibenzocyclooctyne-fluor 488 (DBCO), worked-up and analyzed by LC-MS and in-gel fluorescence as described above.
DEBS Module Reactions
For the trans-complementation experiments of DEBS modules and DszD or ZmaF, the following conditions were used: 8 µM module, 5 mM diketide-SNAC (DKS), 50 µM extender unit (M-CoA, MM-CoA, or MM-ZmaH), 100 mM phosphate pH 7, 5 mM TCEP and 10 µM trans-AT (DszD or ZmaF) as indicated. Reactions were incubated benchtop for ~20 h, at which point a 40 µL aliquot was removed and quenched with 40 µL cold methanol. Quenched reactions were centrifuged at 13,000 rpm for 20 min. Supernatant was removed and analyzed via HPLC on a Varian ProStar. Gradient was performed at 1 mL/min using the following gradient: 0.1% TFA in H2O (A) and 0.1% TFA in acetonitrile (B) as follows: 0–2 minutes 0% B; 2–25 minutes 0–50% B; 25–30 minutes 100% B; 30–35 minutes 0% B.
Supplementary Material
Acknowledgments
This study was supported in part by National Institutes of Health grant GM104258 (G.J.W.) and an NSF grant CHE-1151299 (G.J.W.). The authors would like to thank K. Castelli for cloning the AcsA gene and S. Elliot for construction of DEBS Mod3TE and designing the sequencing primers for DEBS Mod3TE-ATº.
Footnotes
Competing Financial Interests
None to declare.
Supporting Information
Supplementary Tables S1-S6
Supplementary Figures S1-S5
Supplemental Methods including: cloning procedures for ZMA biosynthetic genes, protein expression and purification of ZMA proteins, expression and purification of AcsA, kinetic studies, homology modeling and docking studies, DEBS chimera design and construction, and protein expression and purification of DEBS modules and chimeras.
References
- 1.Kalkreuter E, and Williams GJ (2018) Engineering enzymatic assembly lines for the production of new antimicrobials, Curr Opin Microbiol 45, 140–148. [DOI] [PubMed] [Google Scholar]
- 2.Yuzawa S, Deng K, Wang G, Baidoo EE, Northen TR, Adams PD, Katz L, and Keasling JD (2017) Comprehensive in vitro analysis of acyltransferase domain exchanges in modular polyketide synthases and its application for short-chain ketone production, ACS Synth Biol 6, 139–147. [DOI] [PubMed] [Google Scholar]
- 3.Musiol-Kroll EM, and Wohlleben W (2018) Acyltransferases as tools for polyketide synthase engineering, Antibiotics (Basel, Switzerland) 7, 5137–5141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Koryakina I, Kasey C, McArthur JB, Lowell AN, Chemler JA, Li S, Hansen DA, Sherman DH, and Williams GJ (2017) Inversion of extender unit selectivity in the erythromycin polyketide synthase by acyltransferase domain engineering, ACS Chem Biol 12, 114–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bravo-Rodriguez K, Klopries S, Koopmans KR, Sundermann U, Yahiaoui S, Arens J, Kushnir S, Schulz F, and Sanchez-Garcia E (2015) Substrate flexibility of a mutated acyltransferase domain and implications for polyketide biosynthesis, Chem Biol 22, 1425–1430. [DOI] [PubMed] [Google Scholar]
- 6.Bravo-Rodriguez K, Ismail-Ali AF, Klopries S, Kushnir S, Ismail S, Fansa EK, Wittinghofer A, Schulz F, and Sanchez-Garcia E (2014) Predicted incorporation of non-native substrates by a polyketide synthase yields bioactive natural product derivatives, ChemBioChem 15, 1991–1997. [DOI] [PubMed] [Google Scholar]
- 7.Sundermann U, Bravo-Rodriguez K, Klopries S, Kushnir S, Gomez H, Sanchez-Garcia E, and Schulz F (2013) Enzyme-directed mutasynthesis: a combined experimental and theoretical approach to substrate recognition of a polyketide synthase, ACS Chem Biol 8, 443–450. [DOI] [PubMed] [Google Scholar]
- 8.Vogeli B, Geyer K, Gerlinger PD, Benkstein S, Cortina NS, and Erb TJ (2018) Combining promiscuous acyl-CoA oxidase and enoyl-CoA carboxylase/reductases for atypical polyketide extender unit biosynthesis, Cell Chem Biol 25, 833–839 [DOI] [PubMed] [Google Scholar]
- 9.Ray L, and Moore BS (2016) Recent advances in the biosynthesis of unusual polyketide synthase substrates, Nat Prod Rep 33, 150–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Helfrich EJ, and Piel J (2016) Biosynthesis of polyketides by trans-AT polyketide synthases, Nat Prod Rep 33, 231–316. [DOI] [PubMed] [Google Scholar]
- 11.Koryakina I, McArthur J, Randall S, Draelos MM, Musiol EM, Muddiman DC, Weber T, and Williams GJ (2013) Poly specific trans-acyltransferase machinery revealed via engineered acyl-CoA synthetases, ACS Chem Biol 8, 200–208. [DOI] [PubMed] [Google Scholar]
- 12.Musiol EM, Hartner T, Kulik A, Moldenhauer J, Piel J, Wohlleben W, and Weber T (2011) Supramolecular templating in kirromycin biosynthesis: the acyltransferase KirCII loads ethylmalonyl-CoA extender onto a specific ACP of the trans-AT PKS, Chem Biol 18, 438–444. [DOI] [PubMed] [Google Scholar]
- 13.Musiol-Kroll EM, Zubeil F, Schafhauser T, Hartner T, Kulik A, McArthur J, Koryakina I, Wohlleben W, Grond S, Williams GJ, Lee SY, and Weber T (2017) Polyketide bioderivatization using the promiscuous acyltransferase KirCII, ACS Synth Biol 6, 421–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jiang H, Wang YY, Guo YY, Shen JJ, Zhang XS, Luo HD, Ren NN, Jiang XH, and Li YQ (2015) An acyltransferase domain of FK506 polyketide synthase recognizing both an acyl carrier protein and coenzyme A as acyl donors to transfer allylmalonyl and ethylmalonyl units, FEBS J 282, 2527–2539. [DOI] [PubMed] [Google Scholar]
- 15.Mo S, Kim DH, Lee JH, Park JW, Basnet DB, Ban YH, Yoo YJ, Chen SW, Park SR, Choi EA, Kim E, Jin YY, Lee SK, Park JY, Liu Y, Lee MO, Lee KS, Kim SJ, Kim D, Park BC, Lee SG, Kwon HJ, Suh JW, Moore BS, Lim SK, and Yoon YJ (2011) Biosynthesis of the allylmalonyl-CoA extender unit for the FK506 polyketide synthase proceeds through a dedicated polyketide synthase and facilitates the mutasynthesis of analogues, J Am Chem Soc 133, 976–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chan YA, Boyne MT, 2nd, Podevels AM, Klimowicz AK, Handelsman J, Kelleher NL, and Thomas MG (2006) Hydroxymalonyl-acyl carrier protein (ACP) and aminomalonyl-ACP are two additional type I polyketide synthase extender units, Proc Natl Acad Sci U S A 103, 14349–14354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chan YA, and Thomas MG (2010) Recognition of (2S)-aminomalonyl-acyl carrier protein (ACP) and (2R)-hydroxymalonyl-ACP by acyltransferases in zwittermicin A biosynthesis, Biochemistry 49, 3667–3677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Park H, Kevany BM, Dyer DH, Thomas MG, and Forest KT (2014) A polyketide synthase acyltransferase domain structure suggests a recognition mechanism for its hydroxymalonyl-acyl carrier protein substrate, PLoS One 9, e110965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Holmes TC, May AE, Zaleta-Rivera K, Ruby JG, Skewes-Cox P, Fischbach MA, DeRisi JL, Iwatsuki M, Omura S, and Khosla C (2012) Molecular insights into the biosynthesis of guadinomine: a type III secretion system inhibitor, J Am Chem Soc 134, 17797–17806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brachmann AO, Garcie C, Wu V, Martin P, Ueoka R, Oswald E, and Piel J (2015) Colibactin biosynthesis and biological activity depend on the rare aminomalonyl polyketide precursor, Chem Commun (Camb) 51, 13138–13141. [DOI] [PubMed] [Google Scholar]
- 21.Zha L, Wilson MR, Brotherton CA, and Balskus EP (2016) Characterization of polyketide synthase machinery from the pks island facilitates isolation of a candidate precolibactin, ACS Chem Biol 11, 1287–1295. [DOI] [PubMed] [Google Scholar]
- 22.Koryakina I, and Williams GJ (2011) Mutant malonyl-CoA synthetases with altered specificity for polyketide synthase extender unit generation, ChemBioChem 12, 2289–2293. [DOI] [PubMed] [Google Scholar]
- 23.Quadri LE, Weinreb PH, Lei M, Nakano MM, Zuber P, and Walsh CT (1998) Characterization of Sfp, a Bacillus subtilis phosphopantetheinyl transferase for peptidyl carrier protein domains in peptide synthetases, Biochemistry 37, 1585–1595. [DOI] [PubMed] [Google Scholar]
- 24.Ye Z, Musiol EM, Weber T, and Williams GJ (2014) Reprogramming acyl carrier protein interactions of an zcyl-CoA promiscuous trans-acyltransferase, Chem Biol 21, 636–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dunn BJ, Watts KR, Robbins T, Cane DE, and Khosla C (2014) Comparative analysis of the substrate specificity of trans- versus cis-acyltransferases of assembly line polyketide synthases, Biochemistry 53, 3796–3806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang Y (2008) I-TASSER server for protein 3D structure prediction, BMC Bioinformatics 9, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Roy A, Kucukural A, and Zhang Y (2010) I-TASSER: a unified platform for automated protein structure and function prediction, Nat Protoc 5, 725–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang J, Yan R, Roy A, Xu D, Poisson J, and Zhang Y (2015) The I-TASSER Suite: protein structure and function prediction, Nat Methods 12, 7–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang J, Liang Y, and Zhang Y (2011) Atomic-level protein structure refinement using fragment-guided molecular dynamics conformation sampling, Structure 19, 1784–1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kozakov D, Beglov D, Bohnuud T, Mottarella SE, Xia B, Hall DR, and Vajda S (2013) How good is automated protein docking?, Proteins 82, 2159–2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Comeau SR, Gatchell DW, Vajda S, and Camacho CJ (2004) ClusPro: a fully automated algorithm for protein-protein docking, Nucleic Acids Res 32, W96–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Comeau SR, Gatchell DW, Vajda S, and Camacho CJ (2004) ClusPro: an automated docking and discrimination method for the prediction of protein complexes, Bioinformatics 20, 45–50. [DOI] [PubMed] [Google Scholar]
- 33.Thuronyi BW, and Chang MC (2015) Synthetic biology approaches to fluorinated polyketides, Acc Chem Res 48, 584–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Walker MC, Thuronyi BW, Charkoudian LK, Lowry B, Khosla C, and Chang MC (2013) Expanding the fluorine chemistry of living systems using engineered polyketide synthase pathways, Science 341, 1089–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kumar P, Koppisch AT, Cane DE, and Khosla C (2003) Enhancing the modularity of the modular polyketide synthases: transacylation in modular polyketide synthases catalyzed by malonyl-CoA:ACP transacylase, J Am Chem Soc 125, 14307–14312. [DOI] [PubMed] [Google Scholar]
- 36.Miyanaga A, Ouchi R, Ishikawa F, Goto E, Tanabe G, Kudo F, and Eguchi T (2018) Structural basis of protein-protein interactions between a trans-acting acyltransferase and acyl carrier protein in polyketide disorazole biosynthesis, J Am Chem Soc 140, 7970–7978. [DOI] [PubMed] [Google Scholar]
- 37.Gay DC, Gay G, Axelrod AJ, Jenner M, Kohlhaas C, Kampa A, Oldham NJ, Piel J, and Keatinge-Clay AT (2014) A close look at a ketosynthase from a trans-acyltransferase modular polyketide synthase, Structure 22, 444–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lohman JR, Ma M, Osipiuk J, Nocek B, Kim Y, Chang C, Cuff M, Mack J, Bigelow L, Li H, Endres M, Babnigg G, Joachimiak A, Phillips GN Jr., and Shen B (2015) Structural and evolutionary relationships of “AT-less” type I polyketide synthase ketosynthases, Proc Natl Acad Sci U S A 112, 12693–12698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wong FT, Jin X, Mathews II, Cane DE, and Khosla C (2011) Structure and mechanism of the trans-acting acyltransferase from the disorazole synthase, Biochemistry 50, 6539–6548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dong SH, Frane ND, Christensen QH, Greenberg EP, Nagarajan R, and Nair SK (2017) Molecular basis for the substrate specificity of quorum signal synthases, Proc Natl Acad Sci U S A 114, 9092–9097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Perez JG, Stark JC, and Jewett MC (2016) Cell-free synthetic biology: Engineering beyond the cell, Cold Spring Harbor perspectives in biology 8, pii: a023853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Moore SJ, Lai HE, Needham H, Polizzi KM, and Freemont PS (2017) Streptomyces venezuelae TX-TL - a next generation cell-free synthetic biology tool, Biotechnol J 12, 1600678–1600678. [DOI] [PubMed] [Google Scholar]
- 43.Hughes AJ, and Keatinge-Clay A (2011) Enzymatic extender unit generation for in vitro polyketide synthase reactions: structural and functional showcasing of Streptomyces coelicolor MatB, Chem Biol 18, 165–176. [DOI] [PubMed] [Google Scholar]
- 44.Chan YA, and Thomas MG (2009) Formation and characterization of acyl carrier protein-linked polyketide synthase extender units, Methods Enzymol 459, 143–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hashimoto Y, Hosaka H, Oinuma K, Goda M, Higashibata H, and Kobayashi M (2005) Nitrile pathway involving acyl-CoA synthetase: overall metabolic gene organization and purification and characterization of the enzyme, J Biol Chem 280, 8660–8667. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.