

Abstract
Gloeostereum incarnatum is a precious edible mushroom that is widely grown in Asia and known for its useful medicinal properties. Here, we present a high-quality genome of G. incarnatum using the single-molecule real-time (SMRT) sequencing platform. The G. incarnatum genome, which is the first complete genome to be sequenced in the family Cyphellaceae, was 38.67 Mbp, with an N50 of 3.5 Mbp, encoding 15,251 proteins. Based on our phylogenetic analysis, the Cyphellaceae diverged ~174 million years ago. Several genes and gene clusters associated with lignocellulose degradation, secondary metabolites, and polysaccharide biosynthesis were identified in G. incarnatum, and compared with other medicinal mushrooms. In particular, we identified two terpenoid-associated gene clusters, each containing a gene encoding a sesterterpenoid synthase adjacent to a gene encoding a cytochrome P450 enzyme. These clusters might participate in the biosynthesis of incarnal, a known bioactive sesterterpenoid produced by G. incarnatum. Through a transcriptomic analysis comparing the G. incarnatum mycelium and fruiting body, we also demonstrated that the genes associated with terpenoid biosynthesis were generally upregulated in the mycelium, while those associated with polysaccharide biosynthesis were generally upregulated in the fruiting body. This study provides insights into the genetic basis of the medicinal properties of G. incarnatum, laying a framework for future characterization of bioactive proteins and pharmaceutical uses of this fungus.
Keywords: Gloeostereum incarnatum, whole genome sequencing, PacBio, secondary metabolite, cytochrome P450 enzyme (CYP), terpenoid
1. Introduction
Mushrooms are an important source of nutrition, and a growing body of evidence has indicated that mushrooms may have medicinal properties and human health benefits [1,2,3]. Gloeostereum incarnatum (family Cyphellaceae) is an edible mushroom, which grows as a saprophyte on broad-leaved trees [4]. G. incarnatum is native to China, but is popular in other regions in Asia too, such as Japan and Siberia [4]. Besides its savory taste, G. incarnatum is well-known for its medicinal properties. Antioxidant, immunomodulatory, anti-inflammatory, anti-proliferative, and antibacterial properties have been attributed to this mushroom [5,6,7]. Recent studies have shown that sesquiterpenes and polysaccharides are the main bioactive compounds underlying the beneficial effects of G. incarnatum [5,6,8].
With the rapid advancement of sequencing technologies, the number of available fungal genomes has increased [9,10]. However, genomes of medicinal mushrooms remain scarce. Recently, the genomes of a few medicinal mushrooms (e.g., Ganoderma lucidum, Antrodia cinnamomea, and Hericium erinaceus) were released, and proteins putatively associated with the pharmacological properties of these mushrooms were investigated [11,12,13]. Gene clusters associated with the synthesis of various bioactive secondary metabolites, such as terpenoids and polypeptides, have been identified in many medical mushroom genomes [11,13]. For instance, nine gene clusters associated with the cytochrome P450 (CYP) and triterpenoid pathways were identified in A. cinnamomea [11], while four gene clusters associated with terpene and polyketide biosynthesis were identified in H. erinaceus [13]. In G. lucidum, 24 physical CYP gene clusters, possibly involved in triterpenoid biosynthesis, were identified [12]. Although several bioactive compounds have been identified in G. incarnatum [5,6,8,14], the genetic basis of the medicinal benefits of this mushroom are largely unknown.
In this study, we used the Pacific Biosciences (PacBio) long-read sequencing platform [15] to perform the de novo assembly of the G. incarnatum genome. This is the first genome to be sequenced in the Cyphellaceae family. We also compared the transcriptome profiles of the mycelium and the fruiting body, the two major developmental stages of G. incarnatum. The sequenced genome of G. incarnatum presented herein is, to our knowledge, one of the most comprehensive assembled genomes of an edible mushroom. In this study, we aimed to (1) present a high-quality reference genome for G. incarnatum, which can be used for future analyses of genome function and genetic variation and (2) identify relevant functional genes, gene clusters, and signaling pathways associated with the saprophytic lifestyle and pharmaceutical properties of G. incarnatum. We specifically focused on terpene biosynthesis, cytochrome P450 enzyme biosynthesis, and polysaccharide production. Our study provides a valuable genomic and transcriptomic resource for future studies of the genetic basis of the medicinal properties of G. incarnatum. Such studies would represent a first step towards realizing the full potential of G. incarnatum as a source of pharmacologically active compounds on an industrial scale.
2. Materials and Methods
2.1. Fungal Material, Sequencing, and Genome Assembly
We isolated protoplast-derived monokaryons from the dikaryotic strain of the G. incarnatum commercial strain CCMJ2665. The monokaryons were obtained as described previously [16], except that the dikaryotic mycelia were incubated for 240 min at 30 °C in lywallzyme lysing enzyme. The single-nucleated genomic DNA of the G. incarnatum monokaryon strain was then used for genome sequencing and annotation. Genomic DNA was extracted using NuClean Plant Genomic DNA Kits (CWBIO, Beijing, China). The genome of G. incarnatum was sequenced on a PacBio Sequel long-read sequencing platform with a library insert size of 20 kb, at the Engineering Research Center of the Chinese Ministry of Education for Edible and Medicinal Fungi, Jilin Agricultural University (Changchun, China). Raw data were assembled with SMARTdenovo (https://github.com/ruanjue/smartdenovo). The completeness of the genome assembly was evaluated using Core Eukaryotic Genes Mapping Approach (CEGMA) [17] and Benchmarking Universal Single-Copy Orthologs (BUSCO; [18]). The whole-genome sequence of G. incarnatum has been deposited in GenBank (in submission). The genome reported in this study has been deposited in GenBank under the accession RZIO00000000.
2.2. Genome Annotation
Three different strategies were used to predict genes in the G. incarnatum genome: Sequence homologies with four representative mushrooms; ab initio with Augustus [19], Genescan [20], GlimmerHMM [21], and SNAP [22]; and combining extrinsic and ab initio approaches with GLEAN (http://sourceforge.net/projects/glean-gene). GLEAN gene prediction results were used for subsequent analyses. Protein-coding genes were annotated by GLEAN using both ab initio and evidence-based methods [23]. Predicted genes were functionally annotated against several databases—National Center for Biotechnology Information (NCBI) non-redundant (nr), Swiss-Prot, and InterPro—using BLASTP searches (e-value ≤ 1 × 10−5). Gene annotations were refined using the following databases: Gene Ontology (GO) [24], Clusters of Orthologous Groups (KOG) [25], and Kyoto Encyclopedia of Genes and Genomes (KEGG) [26]. Transposon sequences were identified by aligning the assembled genome to the Repbase database [27] with RepeatMasker (version 3.3.0; http://www.repeatmasker.org/; [28]) and RepeatProteinMasker [22]. Tandem repeat sequences (TRF) were predicted with Tandem Repeat Finder [29]. Ribosomal RNA (rRNA) sequences were identified, based on sequence homology and also through use of de novo prediction strategies with rRNAmmer [30]. Transfer RNA (tRNA) genes were identified using tRNAscan-SE [31]. Non-coding RNAs, such as small nuclear RNA (snRNAs) and microRNAs (miRNAs), were predicted with Rfam [32].
2.3. Evolutionary Analysis and Phylogeny
The phylogenetic analysis was performed using single-copy genes shared across G. incarnatum and another nine fungal species (Omphalotus olearius, Gymnopus luxurians, Laccaria bicolor, Coprinopsis cinerea, Armillaria ostoyae, Lentinula edodes, Schizophyllum commune, Serpula lacrymans and Coniophora puteana). The “all against all” BLASTP searches were performed with a cutoff e-value of 1 × 10−7 for proteins from all species. The alignments of gene pairs were conjoined by solar. Only gene pairs with an alignment ratio (aligned region by total length) of more than 30% in both homologous genes were kept for the following gene family construction. Gene families were clustered using a sparse graph of gene relationships using the hierarchical clustering algorithm hcluster_sg 0.5.1 package. Finally, we identified single-copy genes which had only one homolog per taxon, and those genes were used to construct the phylogenetic tree. The protein sequences of these single-copy genes were aligned using MUSCLE [33] and the protein alignments were transformed into codon alignments with PAL2NAL. Gblocks was used to refine each codon alignment, and all refined alignments were concatenated to a super codon alignment. RAxML software (version 7.2.3) [34] was used to construct the phylogenetic tree using the maximum likelihood (ML) algorithm. The best-scoring ML tree was inferred using the rapid bootstrap analysis after 1000 runs. The divergence times among species were estimated using the mcmctree module in PAML [35] with the calibration time of Serpula lacrymans and Coniophora puteana according to Floudas et al. (2012).
2.4. Carbohydrate-Active Enzyme (CAZyme) Family Classification
The CAZymes in the G. incarnatum genome were identified by mapping the annotated protein sequences to the CAZy database (http://www.cazy.org/) [36] using BLASTP (cut-off e-value ≤ 1 × 10−5, identity ≥ 40% and coverage ≥ 40%). The recovered CAZymes were classified as glycoside hydrolases (GHs), auxiliary activities (AAs), carbohydrate-binding modules (CBMs), glycosyl transferases (GTs), polysaccharide lyases (PLs), and carbohydrate esterases (CEs).
2.5. Cytochrome P450 (CYP) Predictions
CYP proteins were predicted by aligning the gene models to the fungal P450 database (http://p450.riceblast.snu.ac.kr/index.php?a=view;) with BLASTP (e-value ≤ 1 × 10−5, matrix = BLOSUM62). CYP proteins were assigned to protein families based on Nelson’s nomenclature [37]. For protein sequences that aligned with multiple families, the top hit was chosen.
2.6. Secondary Metabolite Annotations
Secondary metabolite gene clusters were predicted with fungal AntiSMASH 3.0 (https://fungismash.secondarymetabolites.org/) [38], with the default parameter values.
2.7. RNA Sequencing of the Two Major Developmental Stages
Samples of the two major fungal developmental stages (mycelium and fruiting body) from the G. incarnatum strain CCMJ2665 were provided by the mushroom section of the Engineering Research Center of the Chinese Ministry of Education for Edible and Medicinal Fungi, Jilin Agricultural University (Changchun, China). RNA extraction and quality control were performed following the processes of Fu et al. [16]. cDNA libraries were constructed, and 150 paired-end sequencing was performed on an Illumina HiSeq 4000 platform at Novogene Co., LTD (Beijing, China). Sequencing data have been deposited in the NCBI SRA (accession no. PRJNA510218).
Raw data were filtered to remove adapter sequences and low-quality reads for downstream analyses. The trimmed reads were mapped to the G. incarnatum genome using TopHat v2.0.12 [39]. The number of reads mapped to each gene was counted using HTSeq v0.6.1 [40]. Fragments per kilobase of transcript per million mapped reads (FPKM) values were used to calculate gene expression. Genes differentially expressed between developmental stages were identified using the DESeq package (1.18.0) [41] in R with adjusted p-value set to <0.05.
3. Results and Discussion
3.1. Genome Sequencing and Assembly
A high-quality reference genome for G. incarnatum was generated from a protoplast monokaryon isolated from the dikaryotic strain of a commercial G. incarnatum cultivar (CCMJ2665; see Table 1). The genomic DNA of G. incarnatum was sequenced on PacBio SMRT Sequel platform generating ~94× coverage of 3,642 Mbp of clean data, as shown in Table S1. Compared to other edible and medicinal mushrooms, the assembled genome of G. incarnatum (38.7 Mbp), as shown in Figure 1, was of an intermediate size; the Wolfiporia cocos genome was the largest (50.5 Mbp); and the Agaricus bisporus var. bisporus genome was the smallest (30.2 Mbp), as shown in Table 1 [11,12,13,42,43,44,45,46,47,48]. G. incarnatum had a guanine-cytosine (GC) content of 49%; GC content in the other mushroom genomes examined was 45.3–55.9% (Table 1). The genome of G. incarnatum was one of the most complete assembled genomes across all representative edible and medicinal mushrooms examined, consisting of 20 scaffolds with an N50 of 3.5 Mbp (Table 1; Figure 1). The completeness of the G. incarnatum genome assembly was analyzed with the CEGMA [17] and the single-copy orthologs test using Fungi BUSCOs [18]. The CEGMA analysis indicated that 96.8% of the core eukaryotic genes were mapped to the G. incarnatum genome. The BUSCO analysis suggested that the annotation set was well completed, with 93.1% complete BUSCOs and 4.5% missing BUSCOs. Thus, our results indicate that the G. incarnatum genome assembly is high quality.
Table 1.
Comparison of genome assembly among representative edible mushrooms.
| Organism | Accession | Genome Size (Mbp) | Genome | Scaffold | N50 (Kbp) | GC Content (%) | Protein-Coding Genes | Sequencing Method |
|---|---|---|---|---|---|---|---|---|
| Gloeostereum incarnatum | 38.7 | 94× | 20 | 3500 | 49.0 | 15,251 | PacBio Sequel | |
| Lentinula edodes | LSDU00000000 | 46.1 | 60× | 31 | 3663 | 45.3 | 13,426 | PacBio RSII; Illumina HiSeq 2500 |
| Agrocybe aegerita | PRJEB21917 | 44.8 | 253× | 122 | 768 | 49.2 | 14,113 | PacBio RSII; Illumina HiSeq 2500 |
| Hericium erinaceus | PRJN361338 | 39.4 | 200× | 519 | 538 | 53.1 | 9895 | Illumina MiSeq; Hiseq 2500 |
| Antrodia cinnamomea | JNBV00000000 | 32.2 | 878× | 360 | 1035 | 50.6 | 9254 | Roche 454; Illumina GAIIx |
| Ganoderma lucidum | AGAX00000000 | 43.3 | 440× | 82 | 1388 | 55.9 | 16,113 | Roche 454; Illumina GAII |
| Wolfiporia cocos | AEHD00000000 | 50.5 | 40× | 348 | 2539 | 52.2 | 12,212 | Sanger; Roche 454 |
| Inonotus baumii | LNZH00000000 | 31.6 | 186× | 217 | 267 | 47.6 | 8455 | Illumina HiSeq |
| Agaricus bisporus var. bisporus | AEOK00000000 | 30.2 | 8.3× | 29 | 2300 | 46.6 | 10,438 | Sanger |
| Lignosus rhinocerotis | AXZM00000000 | 34.3 | 180× | 1338 | 90 | 53.7 | 10,742 | Illumina Hiseq 2000 |
| Sparassis latifolia | LWKX00000000 | 48.1 | 601× | 472 | 641 | 51.4 | 12,471 | Illumina HiSeq 2500 |
| Flammulina velutipes | BDAN00000000 | 35.3 | 132× | 5130 | 150 | 49.6 | 13,843 | Illumina HiSeq 2500 |
Figure 1.

The Gloeostereum incarnatum genome and comparative genomics analysis. (A) The G. incarnatum genome. Outside to inside of concentric circles show assembly scaffold number, gene density, non-coding RNA (ncRNA), GC count and GC skew. (B) Unique and homologous gene families. The number of unique and shared gene families is shown in each of the diagram components and the total number of gene families for each fungus is given in parentheses.
3.2. Gene and Repeat Sequence Prediction and Annotation
To most accurately predict the protein-coding genes in the G. incarnatum genome, we used a homology-based prediction strategy (against four representative mushroom genomes) combined with de novo gene prediction approaches. We predicted 15,251 protein-coding genes, accounting for 57.46% of the assembled G. incarnatum genome (Table S2). The predicted protein-coding genes had an average length of 1456.86 bp and contained 4.38 exons (each with an average length of 264.46 bp). The protein-coding genes were functionally annotated against several databases: NCBI nr, Swiss-Prot, InterPro, GO, COG, and KEGG. Of the 15,251 protein-coding genes predicted, 72.62% had homologs in one or more of the databases searched (Table S2).
We identified ~5.9 Mbp of repeat sequences in the G. incarnatum genome. Of these repeat sequences, 0.49% were predicted to be tandem repeats and 14.46% to be transposons (TEs) (Figure 1; Table S3). Most of the predicted TEs were long terminal repeats (LTRs), representing 13.78% of the genome (Table S3). Of the non-coding RNA species we identified in the G. incarnatum genome, 161 were tRNAs and 44 were rRNAs (Table S4). Nine of the identified tRNAs were possible pseudogenes, and the remaining 152 anti-codon tRNAs corresponded to the 20 common amino acids (Table S4). We also predicted 18 miRNAs and 18 snRNAs; the snRNAs comprised 15 spliceosomal RNAs and three C/D box small nucleolar RNAs (Table S4).
KOG functionally classified 4243 (27.82%) of the predicted proteins. Of these, 499 genes were associated with “amino acid transport and metabolism”, 476 genes were associated with “carbohydrate transport and metabolism”, and 337 with “secondary metabolite biosynthesis, transport, and catabolism”. This suggests that several G. incarnatum proteins are involved in nutrient absorption, transformation, and the synthesis of secondary metabolites. Similarly, KEGG classification indicated that both “amino acid metabolism” and “carbohydrate metabolism” were enriched in G. incarnatum genes (603 and 654 genes, respectively). KEGG analysis indicated that another 161 proteins were assigned to “biosynthesis of other secondary metabolites”, and 59 proteins were associated with the “metabolism of terpenoids and polyketides”. As the medicinal properties of edible mushrooms are closely related to the biosynthesis of secondary metabolites [49], these compounds were the focus of the remainder of our study.
3.3. Comparative Genomics and Evolutionary Analysis
With the exception of the G. incarnatum genome assembled in this study, no complete genomes are available for other fungi in Cyphellaceae. Thus, our evolutionary analysis compared whole genome sequences of seven representative species of the Agaricales: O. olearius, G. luxurians, L. bicolor, C. cinerea, A. ostoyae, L. edodes, and S. commune. We also included genomes of two additional agaricomycetid species having fossil calibrations—S. lacrymans and C. puteana [50]. We found that the G. incarnatum genome includes 7384 gene families, with 10,075 (66.1%) genes having homologs in at least one of the other nine fungal species (Figure 1, Table S5). Interestingly, 5,176 (33.9%) unclustered genes and 469 unique gene families (containing 1369 genes) were G. incarnatum specific (Table S5). These G. incarnatum-specific genes were associated with diverse biological processes, including steroid biosynthesis, terpenoid backbone biosynthesis, and polysaccharide biosynthesis.
We then constructed an ML phylogeny for G. incarnatum and the nine additional fungal species, based on 1822 shared single-copy orthologous genes (Figure 2). These data indicate that G. incarnatum is phylogenetically closer to A. ostoyae, diverging ~174 million years ago (Figure 2). We also identified 325 significantly expanded gene families in the G. incarnatum genome (p ≤ 0.01) (Figure 2); these families were primarily associated with carbohydrate metabolism (starch/sucrose metabolism and glycolysis/gluconeogenesis), amino acid and lipid metabolism, and genetic and environmental information processing. However, caution is warranted when interpreting species’ evolutionary time estimates and gene family expansions and contractions based on genomes generated with different sequencing platforms, assembly methods, and selection of comparative analysis groups. Nevertheless, our analysis provides new insights into the phylogeny of G. incarnatum and other mushroom species based on whole-genome data.
Figure 2.

The Gloeostereum incarnatum genome evolutionary analysis. The number of expanded (green) and contracted (red) gene families is shown at each branch. The estimated divergence time (MYA: million years ago) is shown at the bottom. MRCA: most recent common ancestor.
3.4. The Decomposition of Wood by CAZymes
To further classify the proteins associated with lignin digestion during carbohydrate metabolism, we mapped the protein sequences of G. incarnatum to the CAZy database [36]. We identified 311 non-overlapping CAZymes in six families in G. incarnatum (Table S6); the CAZymes in G. incarnatum were more diverse and abundant than those of brown rot fungi [51]. The G. incarnatum CAZymes consisted of 164 GHs, 66 proteins with AAs, 42 CBMs, 41 GTs, 18 PLs, and 10 CEs (Figure 3).
Figure 3.

(A) Annotation of carbohydrate-related genes in the G. incarnatum genome; (B) secondary metabolite-related gene clusters in the G. incarnatum genome. T1PK: Type I polyketide synthases; NRPS: nonribosomal peptide synthetase.
As the GHs include many cellulase families (such as GH16, GH5, GH3, GH6, and GH7) [36], the remarkably higher number of GHs in G. incarnatum was not unexpected. As a saprotrophic mushroom, G. incarnatum is likely to require many GHs to decompose cellulose from its woody hosts. AAs were the next most abundant family of CAZymes in G. incarnatum; AAs identified in this species included 21 AA9, 18 AA1, and 14 AA3 enzyme families. These three AA families are also the most abundant AAs in other fungi [10,11,12,13]. However, only three AA2 family proteins, the lignin-modifying fungal peroxidases (PODs), were identified in G. incarnatum. PODs are the primary lignin decomposers in the model white rot fungus Phanerochaete chrysosporium and other fungal species [52]. As G. incarnatum is restricted to elm tree hosts, the few AA2s identified in this fungus may be sufficient to decompose lignins produced by elm. We also identified 21 genes encoding enzymes for pectin digestion in G. incarnatum. Thus, the wood-decaying mushroom, G. incarnatum, may utilize complex strategies to decompose plant cell walls.
3.5. Secondary Metabolites and Terpene Pathway
The pharmacological properties of medicinal mushrooms are largely conferred by secondary metabolites; these metabolites have received intense research attention [1,49,53]. Here, we used antiSMASH to search for gene clusters encoding secondary metabolites in G. incarnatum [38]. We identified 65 gene clusters: one saccharide, 15 terpene synthases (TSs), one fatty acid, three polyketide synthases (PKSs), two siderophores, one non-ribosomal peptide-synthetase (NRPS), and 37 putative gene clusters of unknown type (Figure 3).
Terpenoid biosynthesis is of particular interest as terpenoids are important pharmacologically active compounds in G. incarnatum [14,54]. G. incarnatum contains two unique sesquiterpene compounds, gloeosteretriol and incarnal [14,54]. Both compounds demonstrate antibacterial activity against the Gram-positive bacteria Staphylococcus aureus and Bacillus subtilis, but not against any of the Gram-negative bacteria tested to date [14,54]. Incarnal extracted from G. incarnatum and another fungus in Cyphellaceae (Chondrostereum sp.) also shows potent cytotoxicity against several cancer cell lines [8,55]. To further investigate the biosynthesis of terpenoids in G. incarnatum, we mapped 35 proteins to 17 enzymes in the “terpenoid backbone biosynthesis” (KEGG: ko00900) pathway, and 10 proteins to four enzymes in the “sesquiterpenoid and triterpenoid biosynthesis” (KEGG: ko00909) pathway (Figure 4). The pathway mapping results suggested that the G. incarnatum terpenoids are likely to be synthesized through the mevalonate (MVA) pathway, not the 2-C-methyl-D-erythritol 4-phosphate/1-deoxy-d-xylulose 5-phosphate (MEP/DOXP) pathway. This is in line with the results for other mushrooms, such as G. lucidum and H. erinaceus [11,12].
Figure 4.
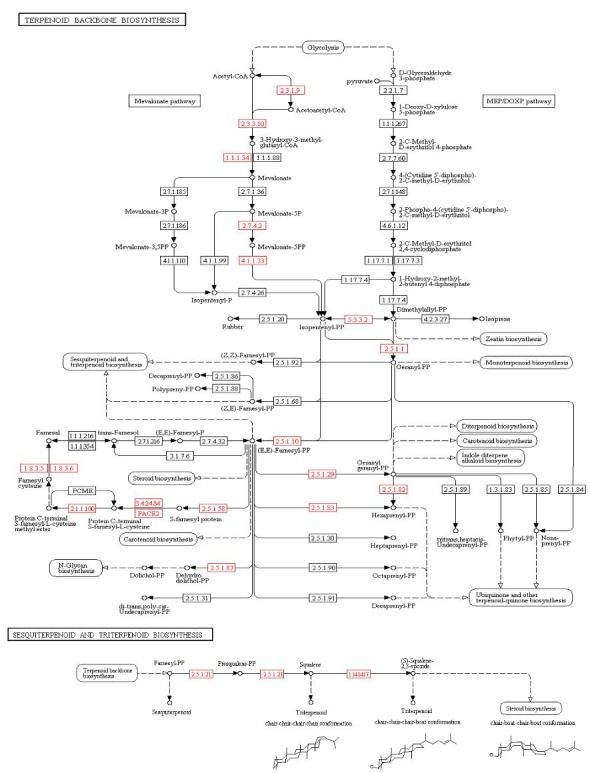
“Terpenoid backbone biosynthesis” (KEGG: ko00900) and “sesquiterpenoid and triterpenoid biosynthesis” (KEGG: ko00909) pathways of G. incarnatum. Red boxes indicate the presence of the enzymes, whereas white boxes indicate enzyme is not present.
Sesquiterpenoids are synthesized from farnesyl diphosphate (FPP) by various sesquiterpene synthases [56]. We located five genes encoding sesquiterpene synthases in G. incarnatum—three encoding trichodiene synthase (EC 4.2.3.6) and two encoding aristolochene synthase (EC 4.2.3.9; Figure 4). Interestingly, trichodiene is a precursor for the biosynthesis of the mycotoxin nivalenol, which is widely found in Fusarium species, and the biosynthesis of aristolochene, which is a precursor for the PR toxin found in Penicillium species [57,58]. To the best of our knowledge, neither of these compounds are produced by G. incarnatum. It would be interesting to know if trichodiene or aristolochene was a substrate for the synthesis of gloeosteretriol or incarnal in G. incarnatum. Based on structural similarity, incarnal might potentially be synthesized from trichodiene in conjunction with certain cyclization, bond-shift rearrangement, oxidation, and hydroxylation reactions. Further experiments are thus necessary to confirm the production of trichodiene and aristolochene, as well as their association with the biosynthesis of gloeosteretriol or incarnal, in G. incarnatum.
Regarding triterpenoid, two farnesyltransferases (EC 2.5.1.21), three squalene monooxygenases (EC 1.14.14.17), and one lanosterol synthase (EC 5.4.99.7) were encoded in the G. incarnatum genome. This suggested that G. incarnatum synthesizes squalene, (S)-2,3-epoxysqualene, and lanosterol, all of which are intermediates in the synthesis of triterpenoid and sterol [59]. Notably, lanosterane-type triterpenoids are produced by several medical mushrooms, including species of Ganoderma, Innonotus, and Antrodia (reviewed in [60]), although the relevant biosynthesis pathways are unknown. The existence of these triterpenoid-related proteins in G. incarnatum suggests that this species may produce previously uncharacterized triterpenoids.
3.6. The CYP Family
Although the pathway for terpenoid backbone biosynthesis in fungi is relatively well studied [61,62], the steps following terpenoid cyclization are largely unknown. The structural diversity of terpenoids depends on post-modification of many specific chemical groups. These modifications involve a series of hydroxylation, reduction, oxidation, and acylation reactions, largely mediated by CYPs (cytochrome P450s) [63,64,65]. In fungi, CYPs are especially important for xenobiotic degradation and the biosynthesis of several secondary metabolites, including terpenoids and polyketides [63]. Based on a comparative search of the Fungal Cytochrome P450 Database [66], 145 CYP proteins were identified in G. incarnatum. These proteins were classified into 57 families following Nelson’s nomenclature [37]. The family CYP5144 included the greatest number of G. incarnatum CYPs (16); CYP5144 also included the most CYPs in another medicinal mushroom, Lignosus rhinocerotis [46] (Table 2). It is thus likely that CYP5144 family proteins play key roles in the biosynthesis of terpenoids in G. incarnatum.
Table 2.
Summary of the CYP genes in the G. incarnatum genome.
| Family | Subfamily | Corresponding Gene Number | Total Gene Number | Family | Subfamily | Corresponding Gene Number | Total Gene Number |
|---|---|---|---|---|---|---|---|
| CYP5144 | C,F | 15,1 | 16 | CYP675 | A | 3 | 3 |
| CYP620 | A,B,E,H | 1,1,4,2 | 8 | CYP682 | B | 3 | 3 |
| CYP5015 | C | 6 | 6 | CYP504 | A | 3 | 3 |
| CYP5014 | F,H | 2,3 | 5 | CYP51 | F | 3 | 3 |
| CYP5068 | B | 5 | 5 | CYP55 | A | 3 | 3 |
| CYP5080 | B,D | 3,2 | 5 | CYP65 | J,X | 1,1 | 2 |
| CYP5093 | A | 5 | 5 | CYP5070 | A | 2 | 2 |
| CYP505 | C,D | 3,1 | 4 | CYP5074 | A | 2 | 2 |
| CYP535 | A | 4 | 4 | CYP5078 | A | 2 | 2 |
| CYP536 | A | 4 | 4 | CYP5081 | A | 2 | 2 |
| CYP617 | A,B | 1,2 | 3 | CYP5125 | A | 2 | 2 |
| CYP5037 | B | 3 | 3 | CYP540 | B | 2 | 2 |
| CYP5110 | A | 3 | 3 | CYP630 | B | 2 | 2 |
| CYP530 | A | 3 | 3 | Others | - | - | 30 |
As previously noted, the G. incarnatum genome encoded two sesterterpenoid synthases—aristolochene synthase and trichodiene synthase. Interestingly, the trichodiene synthase genes (GI_10004653, GI_10004654, and GI_10004694), but not the aristolochene synthase gene, (GI_10003231) were identified in gene clusters containing several CYPs (i.e., CYP65X, CYP530A, and CYP617B; Figure 5). Based on the logic in the Fungal Cytochrome P450 Database (FDPD) pipeline [66], CYP530A and CYP617B were assigned to the families CYP512 and CYP5144, respectively. Both of these families may be involved in the biosynthesis of bioactive terpenoids in G. lucidum and L. rhinocerotis [12]. These results further support our hypothesis that incarnal, the bioactive sesterterpenoid produced by G. incarnatum, might be synthesized from trichodiene, mediated by CYPs. The second-largest family of CYPs identified in G. incarnatum was CYP620 (Table 2), which is a relatively rare family in other medicinal mushrooms (absent in G. lucidum, one in A. cinnamomea, and three in L. rhinocerotis) [11,12,46]. This indicates that CYP distributions and functions vary among medicinal mushrooms. The exact roles of the identified CYPs in terpenoid post-modification or other biological functions remain to be experimentally validated.
Figure 5.

Genetic structures of sesterterpenoid synthase genes and their neighboring genes. Each gene is represented by an arrow. The aristolochene synthase gene (GI_10003231) is indicated by green color; the trichodiene synthase genes (GI_10004653, GI_10004654 and GI_10004694) are indicated by light blue color; the cytochrome P450 (CYP) genes are indicated by red color; choline dehydrogenase genes are indicated by yellow color; The Sec1-like protein genes are indicated by purple color.
3.7. Polysaccharide Biosynthesis
Composition of G. incarnatum polysaccharides also had immunomodulatory and immuno-enhancing effects in a mice model [6]. Some of the most potent immunomodulatory polysaccharides produced by medical mushrooms are water soluble 1,3-β- and 1,6-β-glucans [67]. In G. incarnatum, we identified four 1,3-β-glucan synthases (K00706 and K01180), three UTP–glucose-1-phosphate uridylyltransferases (K00963), 12 GTPase-activating-associated proteins (K12492, K12493, K19838, K19844, K19845, K14319, K17265, K18470, K20315, and K19839), two hexokinases (K00844), and two phosphoglucomutases (K01835) (Table 3). We also identified 15 β-glucan biosynthesis-associated proteins (PF03935; Table 3); β-glucan biosynthesis-associated proteins were shown to be involved in the biosynthesis of 1,6-β-glucans in Saccharomyces cerevisiae [68]. The polysaccharide biosynthesis-related proteins identified in G. incarnatum are summarized in Table S5. Compared with five other species of medicinal mushrooms (Auricularia heimuer [69], A. cinnamomea [11], Sparassis latifolia [47], L. rhinocerotis [46], and G. lucidum [12]), G. incarnatum produced more 1,3-β-glucan synthases, GTPase-activating-associated proteins, and β-glucan biosynthesis-associated proteins, as well as similar numbers of UTP–glucose-1-phosphate uridylyltransferases, hexokinases, and phosphoglucomutases (Table S7). This suggests that G. incarnatum might produce more 1,3-β- and 1,6-β-glucans. In-parallel quantifications of 1,3-β- and 1,6-β-glucan production among these medicinal mushrooms during different growth phases should be performed and compared.
Table 3.
Summary of the polysaccharide biosynthesis-related proteins in G. incarnatum.
| Enzyme Family | KO Term | EC Number | Gene Number | Gene Name |
|---|---|---|---|---|
| 1,3-β-glucan synthase | K01180 | EC:3.2.1.6 | 1 | GI_10004256 |
| K00706 | EC:2.4.1.34 | 3 | GI_10014134, GI_10014600, GI_10010064 | |
| UTP–glucose-1-phosphate uridylyltransferase | K00963 | EC:2.7.7.9 | 3 | GI_10009949, GI_10009950, GI_10009951 |
| Hexokinase | K00844 | EC:2.7.1.1 | 2 | GI_10010509, GI_10009252 |
| Phosphoglucomutase | K01835 | EC:5.4.2.2 | 2 | GI_10003989, GI_10014463 |
| GTPase-activating-associated protein | K12492 | - | 1 | GI_10009154 |
| K19838 | - | 1 | GI_10009280 | |
| K12493 | - | 1 | GI_10004440 | |
| K14319 | - | 1 | GI_10004658 | |
| K19845 | - | 2 | GI_10004984, GI_10007354 | |
| K19839 | - | 3 | GI_10003462, GI_10010380, GI_10012746 | |
| K19844 | - | 2 | GI_10014590, GI_10000357 | |
| K18470 | - | 1 | GI_10014667 |
3.8. Transcriptomic Analysis
As the expression levels of target genes encoding pharmacologically relevant proteins in G. incarnatum might differ across developmental stages, we profiled the transcriptomes of two major developmental stages of G. incarnatum—the mycelium and the fruiting body. We generated 70,634,952 raw reads from the cDNA libraries of the two stages. After data filtering and trimming, 69,716,944 high-quality clean reads remained. Of these clean reads, 92% were successfully mapped to the G. incarnatum genome. Across both stages, 11,015 genes were expressed, with 944 genes expressed only in the mycelium, and 718 genes only expressed in the fruiting body (Figure 6). We identified 3524 differentially expressed genes (DEGs) in the fruiting body as compared to the mycelium (Figure 6). Of these 1822 were significantly upregulated in the fruiting body as compared to the mycelium, and 1702 were significantly downregulated (Figure 6).
Figure 6.

Comparative transcriptome profiling of the mycelium and the fruiting body of G. incarnatum: (A) Fruiting bodies of G. incarnatum; (B) Venn diagram of the genes expressed in the mycelium and/or the fruiting body; (C) number of genes being significantly downregulated or upregulated in the fruiting body compared with the mycelium; (D) heatmap of the genes associated with the biosynthesis of polysaccharides and terpenes.
The gene expression patterns in the mycelium and fruiting body of G. incarnatum varied depending on the type of secondary metabolite encoded. For example, 17 of 45 terpenoid biosynthesis-related genes (38%) were differentially expressed between the mycelium and the fruiting body (Figure 6, Table S8). Of these, 65% were upregulated in the mycelium as compared to the fruiting body (Figure 6, Table S8), indicating that the biosynthesis of terpenoid compounds might be greater in the mycelium of G. incarnatum. In contrast, 10 of the 23 genes associated with polysaccharide biosynthesis (43%) were differentially expressed between the mycelium and the fruiting body, with 70% of these being significantly upregulated in the fruiting body as compared to the mycelium (Figure 6). This indicates that the fruiting body of G. incarnatum might be a richer source of polysaccharides. These findings were consistent with those for terpenoid- and polysaccharide-related genes in H. erinaceus [13]. Therefore, different secondary metabolites might be more enriched at different fungal development stages. Due to the complexity of secondary metabolite biosynthesis, further studies should investigate the molecular mechanisms underlying the secondary metabolism of G. incarnatum.
4. Conclusions
In this study, we presented the first whole-genome sequence of G. incarnatum, which is the first sequenced genome for a fungus belonging to Cyphellaceae. The G. incarnatum genome is one of the most completely assembled edible mushroom genomes available to date, consisting of 20 scaffolds with an N50 of 3.5 Mbp. The remarkably higher number of GHs and AAs in G. incarnatum may contribute to the active decomposition of lignin and cellulose of its woody hosts. We identified 65 gene clusters involved in the biosynthesis of secondary metabolites in the G. incarnatum genome. We also investigated the functions of the proteins involved in terpenoid biosynthesis; terpenoids are one of the main types of pharmacologically active compounds produced by G. incarnatum. We found two sesquiterpenoid synthase genes, one encoding aristolochene synthase and the other encoding trichodiene synthase, in gene clusters enriched with CYP genes. This suggested that CYPs play an active role in the post-modification of aristolochene and trichodiene sesquiterpenoids. We also predicted 38 proteins involved in polysaccharides biosynthesis, another main class of bioactive compounds in G. incarnatum. Genes involved in terpenoid biosynthesis were generally upregulated in mycelium, while the polysaccharide biosynthesis-related genes were upregulated in the fruiting body. These results provide a foundation for future studies of the genetic basis underlying the medicinal properties of G. incarnatum.
Abbreviations
| SMRT | Single-Molecule, Real-Time |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| CYP | cytochrome P450 |
| CAZymes | carbohydrate-active enzymes |
| DEG | differentially expressed genes |
| FPKM | fragments per kilobase of transcript per million mapped reads |
Supplementary Materials
Supplementary materials can be found at https://www.mdpi.com/2073-4425/10/3/188/s1.
Author Contributions
Conceptualization and supervision, Y.L. and Y.F.; sample preparation, W.C.; formal analysis and writing, J.P. and X.W.; software, J.P., L.S. and J.W.; critical review, G.B.
Funding
This research was funded by the Special Fund for Agro-Scientific Research in the Public Interest (No. 201503137); the Program of Creation and Utilization of Germplasm of Mushroom Crop of “111” Project (No. D17014); National-Level International Joint Research Center (2017B01011).
Conflicts of Interest
The authors declare no conflict of interest.
References
- 1.Wasser S.P. Medicinal mushroom science: Current perspectives, advances, evidences, and challenges. Biomed. J. 2014;37:345–356. doi: 10.4103/2319-4170.138318. [DOI] [PubMed] [Google Scholar]
- 2.Song C., Liu Y., Song A., Dong G., Zhao H., Sun W., Ramakrishnan S., Wang Y., Wang S., Li T., et al. The Chrysanthemum nankingense genome provides insights into the evolution and diversification of chrysanthemum flowers and medicinal traits. Mol. Plant. 2018;11:1482–1491. doi: 10.1016/j.molp.2018.10.003. [DOI] [PubMed] [Google Scholar]
- 3.Guggenheim A.G., Wright K.M., Zwickey H.L. Immune modulation from five major mushrooms: Application to integrative oncology. Integr. Med. 2014;13:32–44. [PMC free article] [PubMed] [Google Scholar]
- 4.Petersen R.H., Parmasto E. A redescription of Gloeostereum incarnatum. Mycol. Res. 1993;97:1213–1216. doi: 10.1016/S0953-7562(09)81287-2. [DOI] [Google Scholar]
- 5.Zhang Z.-F., Lv G.-Y., Jiang X., Cheng J.-H., Fan L.-F. Extraction optimization and biological properties of a polysaccharide isolated from Gleoestereum incarnatum. Carbohydr. Polym. 2015;117:185–191. doi: 10.1016/j.carbpol.2014.09.059. [DOI] [PubMed] [Google Scholar]
- 6.Wang D., Li Q., Qu Y., Wang M., Li L., Liu Y., Li Y. The investigation of immunomodulatory activities of Gloeostereum incaratum polysaccharides in cyclophosphamide-induced immunosuppression mice. Exp. Ther. Med. 2018;15:3633–3638. doi: 10.3892/etm.2018.5810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lull C., Wichers H.J., Savelkoul H.F. Antiinflammatory and immunomodulating properties of fungal metabolites. Mediat. Inflamm. 2005;2005:63–80. doi: 10.1155/MI.2005.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Asai R., Mitsuhashi S., Shigetomi K., Miyamoto T., Ubukata M. Absolute configurations of (−)-hirsutanol A and (−)-hirsutanol C produced by Gloeostereum incarnatum. J. Antibiot. 2011;64:693–696. doi: 10.1038/ja.2011.73. [DOI] [PubMed] [Google Scholar]
- 9.Liu W., Chen L., Cai Y., Zhang Q., Bian Y. Opposite polarity monospore genome de novo sequencing and comparative analysis rreveal the possible heterothallic life cycle of Morchella importuna. Int. J. Mol. Sci. 2018;19:2525. doi: 10.3390/ijms19092525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dai Y., Su W., Yang C., Song B., Li Y., Fu Y. Development of novel polymorphic EST-SSR markers in Bailinggu (Pleurotus tuoliensis) for crossbreeding. Genes. 2017;8:325. doi: 10.3390/genes8110325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lu M.Y., Fan W.L., Wang W.F., Chen T., Tang Y.C., Chu F.H., Chang T.T., Wang S.Y., Li M.Y., Chen Y.H., et al. Genomic and transcriptomic analyses of the medicinal fungus Antrodia cinnamomea for its metabolite biosynthesis and sexual development. Proc. Natl. Acad. Sci. USA. 2014;111:E4743–E4752. doi: 10.1073/pnas.1417570111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen S., Xu J., Liu C., Zhu Y., Nelson D.R., Zhou S., Li C., Wang L., Guo X., Sun Y., et al. Genome sequence of the model medicinal mushroom Ganoderma lucidum. Nat. Commun. 2012;3:913. doi: 10.1038/ncomms1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen J., Zeng X., Yang Y.L., Xing Y.M., Zhang Q., Li J.M., Ma K., Liu H.W., Guo S.X. Genomic and transcriptomic analyses reveal differential regulation of diverse terpenoid and polyketides secondary metabolites in Hericium erinaceus. Sci. Rep. 2017;7:10151. doi: 10.1038/s41598-017-10376-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Takazawa H., Kashino S. Incarnal. A new antibacterial sesquiterpene from Basidiomycetes. Chem. Pharm. Bull. 1991;39:555–557. doi: 10.1248/cpb.39.555. [DOI] [PubMed] [Google Scholar]
- 15.Li C., Lin F., An D., Wang W., Huang R. Genome sequencing and assembly by long reads in plants. Genes. 2017;9:6. doi: 10.3390/genes9010006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fu Y., Dai Y., Yang C., Wei P., Song B., Yang Y., Sun L., Zhang Z.W., Li Y. Comparative transcriptome analysis identified candidate genes related to Bailinggu mushroom formation and genetic markers for genetic analyses and breeding. Sci. Rep. 2017;7:9266. doi: 10.1038/s41598-017-08049-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Parra G., Bradnam K., Korf I. CEGMA: A pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics. 2007;23:1061–1067. doi: 10.1093/bioinformatics/btm071. [DOI] [PubMed] [Google Scholar]
- 18.Simao F.A., Waterhouse R.M., Ioannidis P., Kriventseva E.V., Zdobnov E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 2015;31:3210–3212. doi: 10.1093/bioinformatics/btv351. [DOI] [PubMed] [Google Scholar]
- 19.Stanke M., Keller O., Gunduz I., Hayes A., Waack S., Morgenstern B. AUGUSTUS: Ab initio prediction of alternative transcripts. Nucleic Acids Res. 2006;34:W435–W439. doi: 10.1093/nar/gkl200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Burge C., Karlin S. Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 1997;268:78–94. doi: 10.1006/jmbi.1997.0951. [DOI] [PubMed] [Google Scholar]
- 21.Allen J.E., Majoros W.H., Pertea M., Salzberg S.L. JIGSAW, GeneZilla, and GlimmerHMM: Puzzling out the features of human genes in the ENCODE regions. Genome Biol. 2006;7(Suppl. 1):S9. doi: 10.1186/gb-2006-7-s1-s9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Korf I. Gene finding in novel genomes. BMC Bioinform. 2004;5:59. doi: 10.1186/1471-2105-5-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Elsik C.G., Mackey A.J., Reese J.T., Milshina N.V., Roos D.S., Weinstock G.M. Creating a honey bee consensus gene set. Genome Biol. 2007;8:R13. doi: 10.1186/gb-2007-8-1-r13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ashburner M., Ball C.A., Blake J.A., Botstein D., Butler H., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T., et al. Gene ontology: Tool for the unification of biology. The gene ontology consortium. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tatusov R.L., Galperin M.Y., Natale D.A., Koonin E.V. The COG database: A tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000;28:33–36. doi: 10.1093/nar/28.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kanehisa M., Goto S., Kawashima S., Okuno Y., Hattori M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004;32:D277–D280. doi: 10.1093/nar/gkh063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jurka J., Kapitonov V.V., Pavlicek A., Klonowski P., Kohany O., Walichiewicz J. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 2005;110:462–467. doi: 10.1159/000084979. [DOI] [PubMed] [Google Scholar]
- 28.Tarailo-Graovac M., Chen N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinform. 2009 doi: 10.1002/0471250953.bi0410s25. [DOI] [PubMed] [Google Scholar]
- 29.Benson G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999;27:573–580. doi: 10.1093/nar/27.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lagesen K., Hallin P., Rodland E.A., Staerfeldt H.H., Rognes T., Ussery D.W. RNAmmer: Consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007;35:3100–3108. doi: 10.1093/nar/gkm160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lowe T.M., Eddy S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–964. doi: 10.1093/nar/25.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gardner P.P., Daub J., Tate J.G., Nawrocki E.P., Kolbe D.L., Lindgreen S., Wilkinson A.C., Finn R.D., Griffiths-Jones S., Eddy S.R., et al. Rfam: Updates to the RNA families database. Nucleic Acids Res. 2009;37:D136–D140. doi: 10.1093/nar/gkn766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Edgar R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stamatakis A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007;24:1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- 36.Cantarel B.L., Coutinho P.M., Rancurel C., Bernard T., Lombard V., Henrissat B. The Carbohydrate-Active EnZymes database (CAZy): An expert resource for Glycogenomics. Nucleic Acids Res. 2009;37:D233–D238. doi: 10.1093/nar/gkn663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nelson D.R. The cytochrome p450 homepage. Hum. Genom. 2009;4:59–65. doi: 10.1186/1479-7364-4-1-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Weber T., Blin K., Duddela S., Krug D., Kim H.U., Bruccoleri R., Lee S.Y., Fischbach M.A., Muller R., Wohlleben W., et al. antiSMASH 3.0—A comprehensive resource for the genome mining of biosynthetic gene clusters. Nucleic Acids Res. 2015;43:W237–W243. doi: 10.1093/nar/gkv437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Trapnell C., Pachter L., Salzberg S.L. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Anders S., Pyl P.T., Huber W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Anders S., Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shim D., Park S.G., Kim K., Bae W., Lee G.W., Ha B.S., Ro H.S., Kim M., Ryoo R., Rhee S.K., et al. Whole genome de novo sequencing and genome annotation of the world popular cultivated edible mushroom, Lentinula edodes. J. Biotechnol. 2016;223:24–25. doi: 10.1016/j.jbiotec.2016.02.032. [DOI] [PubMed] [Google Scholar]
- 43.Gupta D.K., Ruhl M., Mishra B., Kleofas V., Hofrichter M., Herzog R., Pecyna M.J., Sharma R., Kellner H., Hennicke F., et al. The genome sequence of the commercially cultivated mushroom Agrocybe aegerita reveals a conserved repertoire of fruiting-related genes and a versatile suite of biopolymer-degrading enzymes. BMC Genom. 2018;19:48. doi: 10.1186/s12864-017-4430-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shu S., Chen B., Zhou M., Zhao X., Xia H., Wang M. De novo sequencing and transcriptome analysis of Wolfiporia cocos to reveal genes related to biosynthesis of triterpenoids. PLoS ONE. 2013;8:e71350. doi: 10.1371/journal.pone.0071350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Morin E., Kohler A., Baker A.R., Foulongne-Oriol M., Lombard V., Nagye L.G., Ohm R.A., Patyshakuliyeva A., Brun A., Aerts A.L., et al. Genome sequence of the button mushroom Agaricus bisporus reveals mechanisms governing adaptation to a humic-rich ecological niche. Proc. Natl. Acad. Sci. USA. 2012;109:17501–17506. doi: 10.1073/pnas.1206847109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yap H.Y., Chooi Y.H., Firdaus-Raih M., Fung S.Y., Ng S.T., Tan C.S., Tan N.H. The genome of the Tiger Milk mushroom, Lignosus rhinocerotis, provides insights into the genetic basis of its medicinal properties. BMC Genom. 2014;15:635. doi: 10.1186/1471-2164-15-635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xiao D., Ma L., Yang C., Ying Z., Jiang X., Lin Y.Q. De novo sequencing of a Sparassis latifolia genome and its associated comparative analyses. Can. J. Infect. Dis. Med. Microbiol. 2018;2018:1857170. doi: 10.1155/2018/1857170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kurata A., Fukuta Y., Mori M., Kishimoto N., Shirasaka N. Draft genome sequence of the basidiomycetous fungus Flammulina velutipes TR19. Genome Announc. 2016;4 doi: 10.1128/genomeA.00505-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhong J.J., Xiao J.H. Secondary metabolites from higher fungi: Discovery, bioactivity, and bioproduction. Adv. Biochem. Eng. Biotechnol. 2009;113:79–150. doi: 10.1007/10_2008_26. [DOI] [PubMed] [Google Scholar]
- 50.Floudas D., Binder M., Riley R., Barry K., Blanchette R.A., Henrissat B., Martinez A.T., Otillar R., Spatafora J.W., Yadav J.S., et al. The Paleozoic origin of enzymatic lignin decomposition reconstructed from 31 fungal genomes. Science. 2012;336:1715–1719. doi: 10.1126/science.1221748. [DOI] [PubMed] [Google Scholar]
- 51.Sista Kameshwar A.K., Qin W. Comparative study of genome-wide plant biomass-degrading CAZymes in white rot, brown rot and soft rot fungi. Mycology. 2018;9:93–105. doi: 10.1080/21501203.2017.1419296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Martinez A.T., Ruiz-Duenas F.J., Martinez M.J., Del Rio J.C., Gutierrez A. Enzymatic delignification of plant cell wall: From nature to mill. Curr. Opin. Biotechnol. 2009;20:348–357. doi: 10.1016/j.copbio.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 53.Lee H.Y., Moon S., Shim D., Hong C.P., Lee Y., Koo C.D., Chung J.W., Ryu H. Development of 44 novel polymorphic SSR markers for determination of shiitake mushroom (Lentinula edodes) cultivars. Genes. 2017;8:109. doi: 10.3390/genes8040109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gao J., Yue D.C., Cheng K.D., Wang S.C., Yu K.B., Zheng Q.T., Yang J.S. Gloeosteretriol, a new sesquiterpene from the fermentation products of Gloeostereum incarnatum S. Ito et Imai. Yao Xue Xue Bao = Acta Pharm. Sin. 1992;27:33–36. [PubMed] [Google Scholar]
- 55.Li H.J., Chen T., Xie Y.L., Chen W.D., Zhu X.F., Lan W.J. Isolation and structural elucidation of chondrosterins F-H from the marine fungus Chondrostereum sp. Mar. Drugs. 2013;11:551–558. doi: 10.3390/md11020551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Christianson D.W. Unearthing the roots of the terpenome. Curr. Opin. Chem. Biol. 2008;12:141–150. doi: 10.1016/j.cbpa.2007.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hidalgo P.I., Ullan R.V., Albillos S.M., Montero O., Fernandez-Bodega M.A., Garcia-Estrada C., Fernandez-Aguado M., Martin J.F. Molecular characterization of the PR-toxin gene cluster in Penicillium roqueforti and Penicillium chrysogenum: Cross talk of secondary metabolite pathways. Fungal Genet. Biol. FG B. 2014;62:11–24. doi: 10.1016/j.fgb.2013.10.009. [DOI] [PubMed] [Google Scholar]
- 58.Schothorst R.C., van Egmond H.P. Report from SCOOP task 3.2.10 “collection of occurrence data of Fusarium toxins in food and assessment of dietary intake by the population of EU member states”. Subtask: Trichothecenes. Toxicol. Lett. 2004;153:133–143. doi: 10.1016/j.toxlet.2004.04.045. [DOI] [PubMed] [Google Scholar]
- 59.Benveniste P. Biosynthesis and accumulation of sterols. Annu. Rev. Plant Biol. 2004;55:429–457. doi: 10.1146/annurev.arplant.55.031903.141616. [DOI] [PubMed] [Google Scholar]
- 60.Rios J.L., Andujar I., Recio M.C., Giner R.M. Lanostanoids from fungi: A group of potential anticancer compounds. J. Nat. Prod. 2012;75:2016–2044. doi: 10.1021/np300412h. [DOI] [PubMed] [Google Scholar]
- 61.Schmidt-Dannert C. Biosynthesis of terpenoid natural products in fungi. Adv. Biochem. Eng. Biotechnol. 2015;148:19–61. doi: 10.1007/10_2014_283. [DOI] [PubMed] [Google Scholar]
- 62.Kanehisa M., Goto S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cresnar B., Petric S. Cytochrome P450 enzymes in the fungal kingdom. Biochim. Biophys. Acta. 2011;1814:29–35. doi: 10.1016/j.bbapap.2010.06.020. [DOI] [PubMed] [Google Scholar]
- 64.Sanglard D., Loper J.C. Characterization of the alkane-inducible cytochrome P450 (P450alk) gene from the yeast Candida tropicalis: Identification of a new P450 gene family. Gene. 1989;76:121–136. doi: 10.1016/0378-1119(89)90014-0. [DOI] [PubMed] [Google Scholar]
- 65.Mansuy D. The great diversity of reactions catalyzed by cytochromes P450. Comp. Biochem. Physiol. Part C Pharmacol. Toxicol. Endocrinol. 1998;121:5–14. doi: 10.1016/S0742-8413(98)10026-9. [DOI] [PubMed] [Google Scholar]
- 66.Park J., Lee S., Choi J., Ahn K., Park B., Park J., Kang S., Lee Y.H. Fungal cytochrome P450 database. BMC Genom. 2008;9:402. doi: 10.1186/1471-2164-9-402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Xu Z., Chen X., Zhong Z., Chen L., Wang Y. Ganoderma lucidum polysaccharides: Immunomodulation and potential anti-tumor activities. Am. J. Chin. Med. 2011;39:15–27. doi: 10.1142/S0192415X11008610. [DOI] [PubMed] [Google Scholar]
- 68.Montijn R.C., Vink E., Muller W.H., Verkleij A.J., Van Den Ende H., Henrissat B., Klis F.M. Localization of synthesis of beta1,6-glucan in Saccharomyces cerevisiae. J. Bacteriol. 1999;181:7414–7420. doi: 10.1128/jb.181.24.7414-7420.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yuan Y., Wu F., Si J., Zhao Y.F., Dai Y.C. Whole genome sequence of Auricularia heimuer (Basidiomycota, Fungi), the third most important cultivated mushroom worldwide. Genomics. 2017 doi: 10.1016/j.ygeno.2017.12.013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.