

Abstract
Spider venom-derived cysteine knot peptides are a mega-diverse class of molecules that exhibit unique pharmacological properties to modulate key membrane protein targets. Voltage-gated sodium channels (NaV) are often targeted by these peptides to allosterically promote opening or closing of the channel by binding to structural domains outside the channel pore. These effects can result in modified pain responses, muscle paralysis, cardiac arrest, priapism, and numbness. Although such effects are often deleterious, subtype selective spider venom peptides are showing potential to treat a range of neurological disorders, including chronic pain and epilepsy. This review examines the structure–activity relationships of cysteine knot peptides from spider venoms that modulate NaV and discusses their potential as leads to novel therapies for neurological disorders.
Keywords: spider venoms, ICK peptides, voltage-gated ion channels, structure–activity relationship, novel drugs
Introduction
Animal venoms are an extraordinary source of bio-active peptides that modulate membrane proteins to facilitate prey capture and defense. Venomous spiders, cone snails, fish, sea anemones, wasps, scorpions, snakes and dinoflagellates produce small molecules and/or peptides exhibiting pharmacological properties of singular value for the research in pharmacological tools and novel drugs (Ziegman and Alewood, 2015; Dongol et al., 2016; Cardoso and Lewis, 2017; Kessler et al., 2017; Abraham and Lewis, 2018; Cardoso et al., 2018). These toxins modulate a range of receptors and channels, including VGIC, TRP, muscarinic and nicotinic acetylcholine receptors (mAChR and nAChR), ASIC, NET and G protein coupled receptors (GPCRs). Based on the number of species and venom complexity, spider venoms provide a mega-diverse source of bio-active cysteine knot peptides, many of which modulate NaV with high potency and selectivity (Cardoso and Lewis, 2017).
Voltage-gated sodium channels (NaV1.1–1.9), in particular, are key players in the transmission of electrical signals in excitable cells and also involved in the pathophysiology of neurological disorders, including poorly treated conditions as chronic pain and epilepsy (Luiz and Wood, 2016; Klein-Weigel et al., 2018; Sloan et al., 2018; Szepetowski, 2018). The NaV channels α-subunit comprises four domains (DI–DIV), each formed by six transmembrane segments (S1–S6), including S4 which contributes transmembrane voltage sensitivity, the tip of S5–S6 which contributes to sodium ion selectivity, and the intracellular loop connecting S6 of DIII and S1 of DIV which contributes to fast inactivation (Figure 1). The co-associated auxiliary β-subunits (β1–β4) are positioned above the VSD explaining their ability to influence channel gating (Figure 1A,B). Interestingly, human genetic studies disclosed mutations in NaV1.7 and NaV1.9 channels leading to congenital insensitivity to pain, a rare condition characterized by lack of physical pain (Cox et al., 2006; Phatarakijnirund et al., 2016), while gain-of-function mutations in NaV1.6, NaV1.7, NaV1.8, and NaV1.9 lead to painful neuropathies such as trigeminal neuralgia and erythromelalgia (Drenth and Waxman, 2007; Faber et al., 2012; Huang et al., 2014; Grasso et al., 2016). Genetic mutations in the NaV1.1 and NaV1.2 channels result in functional defects linked to epileptic syndromes (Meisler and Kearney, 2005; Thompson et al., 2011). Furthermore, altered NaV channels function and expression are prominent in chronic inflammatory and neuropathic pains, with localization remodeling, altered expression and sensitization often observed in the subtypes NaV1.3, NaV1.6, NaV1.7, NaV1.8, and NaV1.9 (Cardoso and Lewis, 2017). NaV1.4 and NaV1.5 have restricted expression in the skeletal and cardiac muscle, respectively, which are important off-target pharmacologies to be considered when developing NaV channel therapeutics.
FIGURE 1.
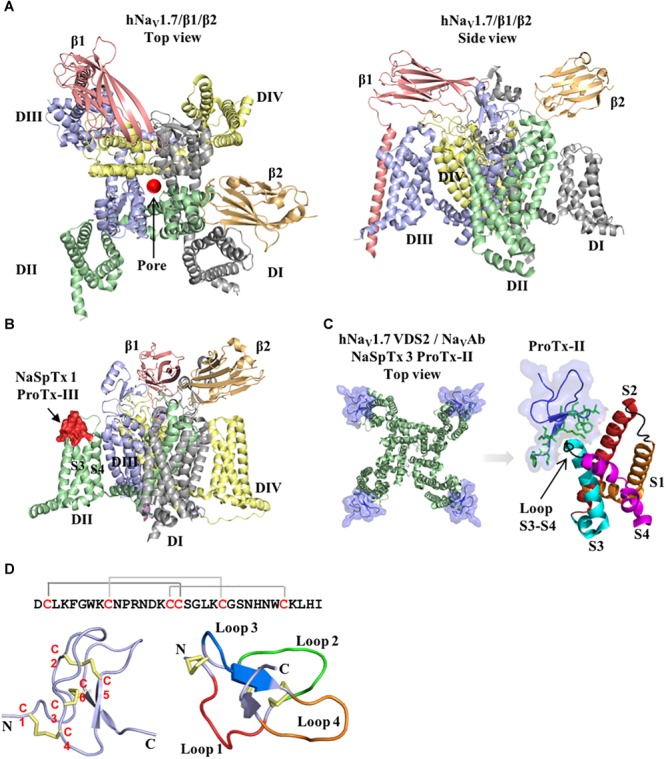
Structure of the voltage-gated sodium channel and the inhibitory cysteine knot (ICK) spider peptide. (A) Three-dimensional structure of the human NaV1.7 in the presence of the auxiliary subunits β1 and β2 determined by cryo-EM (PDB 6J8J) (Shen et al., 2019). Top and side views are presented. The domain I (DI) is colored in gray, domain II (DII) is colored in green, domain III (DIII) is colored in blue and domain IV (DIV) is colored in yellow. The auxiliary subunits β1 and β2 are colored in salmon and orange, respectively. (B) Representative binding site of a NaSpTx 1 in the DII S3-S4 loop of the hNaV1.7 channel (PDB 6J8J) (Shen et al., 2019). Domains and auxiliary subunits are colored as in (A), and the NaSpTx1 peptide ProTx-III (PDB 2MXM) (Cardoso et al., 2015) is colored in red. (C) Detailed binding site of the NaSpTx 3 ProTx-II over the hNaV1.7 voltage–sensor domain 2 (VSD2)-NaVAb chimeric channel (PBD 6N4I) (Xu et al., 2019). Top view of the hNaV1.7 VSD2-NaVAb chimeric channel colored in green showing ProTx-II colored in blue bound to the voltage sensor domain 2 in close proximity with the S3–S4 loop. In the structure on the left, the segments S1 to S4 are colored in orange, red, cyan and magenta, respectively. The loops S1–S2 and S3–S4 are colored in black and the residues in the loop 4 and C-terminal of ProTx-II are represented by green sticks. (D) Typical structure of an inhibitory cysteine knot (ICK) peptide from spider. Primary and three-dimensional structure of ProTx-III (PDB 2MXM) (Cardoso et al., 2015) showing cysteine connectivity (C1 – C4, C2 – C5, and C3 – C6) and loops 1–4 colored in red, green, blue and orange, respectively.
The role of NaV channels in both health and pathological pain have been further elucidated with the support of potent and selective NaV channels modulators isolated from spider venoms. By using a subtype selective NaV1.1 activator isolated from the tarantula Heteroscodra maculata, the key role played by NaV1.1 in physiological mechanical pain (Osteen et al., 2016) and chronic visceral pain (Salvatierra et al., 2018) was established. More interestingly, pain relief is achieved in pre-clinical models of inflammatory and neuropathic pain administrated with NaV channels inhibitors isolated from other spiders venoms, as for the ICK peptides ProTx-II (Tanaka et al., 2015; Flinspach et al., 2017), HnTX-IV (Liu et al., 2014a), Hl1a (Meng et al., 2016), HwTx-IV (Liu et al., 2014b), and Pn3a (Deuis et al., 2017).
The unique properties of spider ICK peptides in modulating ion channels give rise to opportunities for developing better and safer therapies targeting NaV channels. This, in association to the current need for effective drugs to treat challenging neurological disorders and to overcome severe side-effects by opioid analgesic drugs foster the use of these bio-active spider peptides in therapeutics development. In this review, we examine recent advances in the SAR of cysteine knot peptides from spider venoms that inhibit NaV and discuss their potential for the development of novel therapies.
Structure–Activity Relationships of Spider ICK Peptides
Spiders are the largest group of venomous animals, with more the 40,000 species described to date (Platnick, 2014). Their venoms are rich in peptides that inhibit or activate NaV channels by binding to domains outside the channel pore to allosterically promote opening or closing of the channel (Figure 1A–C) (Cardoso and Lewis, 2017). These binding sites include the VSD associated with domains II and IV that bind site 4 and 3 NaV channel toxins, respectively. Interestingly, these peptides have a conserved ICK scaffold that confers high stability and resistance to high temperatures, low pH and digestion by proteases. A typical spider peptide ICK scaffold, with few exceptions, comprises three disulphide bridges C1–C4, C2–C5, and C3–C6 that fold these peptides into a globular structure with four distinct loops and an extended C-terminal tail (Figure 1D). These peptides were classified into distinct families of NaV modulators named NaSpTx 1–12 based on their amino acids sequence and cysteine position (Klint et al., 2012), with extensive SAR studies reported for NaSpTx 1 and 3, and to a lesser extent for NaSpTx 7. Modern high throughput screening technologies using fluorescence-imaging and automated patch-clamp cell-based assays have facilitated the identification of peptides that display high potency and selectivity to modulate NaV subtypes (Cardoso et al., 2015; Klint et al., 2015b). These peptides have been the focus of studies to unravel their pharmacological properties and potential for the development of novel and more effective drugs, studies which have been considerably advanced through investigations of the SAR of spider ICK peptides over NaV channel subtypes. By using state-of-art methods for peptide production and detailed pharmacology characterization through patch-clamp electrophysiology in primary isolated neurons (e.g., DRG) or mammalian cells expressing NaV channels subtypes, and X-ray and nuclear magnetic resonance (NMR) for determination of three-dimensional structure, these SAR studies have unraveled key features associated to modulation NaV channels. Our current understanding of the SAR of each of the main ICK family peptides at NaV channels are outlined below.
NaSpTx 1
GpTx-1
The ICK peptide GpTx-1 (μ/ω-TRTX-Gr2a) isolated from the tarantula Grammostola rosea was first described as a nanomolar inhibitor of CaV (Ono et al., 2011) and later as a nanomolar NaV inhibitor from Grammostola porter (Murray et al., 2015). Its high potency for the sodium channel subtype NaV1.7 (IC50 of 10 nM) made it an attractive lead for SAR studies (Murray et al., 2015, 2016) (Table 1). Alanine scanning revealed the residues W29, K31 and F34 are essential for the NaV1.7 inhibition (Murray et al., 2015). In its native form, GpTx-1 is 20-fold and 1000-fold selective over NaV1.4 and NaV1.5, respectively, with the F5A mutant enhancing to 300-fold selectivity over NaV1.4. In the same study, additional positional substitutions using natural and non-natural amino acids other than alanine in the positions 5, 6, 26, and 28 put forward the rational design of an optimized GpTx-1 containing the substitutions F5A, M6F, T26L, K28R. This new GpTx-1 analog displayed 6-fold enhanced potency for NaV1.7 and 1000-fold selectivity over NaV1.4 and NaV1.5.
Table 1.
Structure–activity relationship of spider ICK peptides and NaV channels.
| Peptide | Amino acids sequences | In vitro properties | References |
|---|---|---|---|
| NaSpTx 1 | |||
| GpTX-I | wt-DCLGFMRKCIPDNDKCCRPNLVCSRTHKWCKYVF | IC50 value of 4 nM for NaV1.7 and 68- and 950-fold selective over NaV1.4 and NaV1.5, respectively. | Murray et al., 2015 |
| DCLGAMRKCIPDNDKCCRPNLVCSRTHKWCKYVF | Improved 300-fold selectivity over NaV1.4. | ||
| DCLGFFRKCIPDNDKCCRPNLVCSRLHRWCKYVF | Improved 6-fold potency for NaV1.7 and 1000-fold selective over NaV1.4 and NaV1.5. | Murray et al., 2016 | |
| HwTx-IV | wt-ECLEIFKACNPSNDQCCKSSKLVCSRKTRWCKYQI∗ | IC50 value of 26 nM for NaV1.7 and 6-, 13- and 15-fold selective over NaV1.2, NaV1.3, and NaV1.4, respectively. | Xiao et al., 2008b; Deng et al., 2013; Minassian et al., 2013 |
| (Pyro)ECLEIFKACNPSNDQCCKSSKLVCSRKTRWCKYQI∗ | Confer irreversible inhibition for TTX-S NaV currents in DRG. | Rong et al., 2013 | |
| ACLEIFKACNPSNDQCCKSSKLVCSRKTRWCKYQI | Improved 2 to 4-fold potency for NaV1.7 and NaV1.2. | Minassian et al., 2013 | |
| ECLAIFKACNPSNDQCCKSSKLVCSRKTRWCKYQI | |||
| GCLGIFKACNPSNDQCCKSSKLVCSRKTRWCKWQI∗ | Improved potency at 42-fold for NaV1.7. | Revell et al., 2013 | |
| GCLGIFKACNPSNDQCCKSSKLVCSRKTRWCKWQI | Improved potency at 15-fold for NaV1.7 and 4-fold for NaV1.2#. | Rahnama et al., 2017 | |
| ECLEIFKACNPSNDQCCKSSKLVCSRKTRWCKYQI∗ | Altered lipid binding in the cell membrane. | Henriques et al., 2016 | |
| CcoTx1 | wt-DCLGWFKSCDPKNDKCCKNYTCSRRDRWCKYDL | IC50 value of 3 nM for NaV1.2, 75 nM for Nav1.7 and >300 nM for other NaV subtypes (NaV1.6 was not tested). | Bosmans et al., 2006; Shcherbatko et al., 2016 |
| DCLGMFKSCDPENDKCCKRLVCSRSHRWCKWKL | IC50 value of 25 nM for NaV1.7 and selectivity of 6-fold over NaV1.2 and 4-fold over NaV1.6. | Shcherbatko et al., 2016 | |
| ICLGMFKSCDPENDKCCKRLVCSRSHRWCKWKL | IC50 value of 11 (C-terminal carboxi) and 2 nM (C-terminal amide) for NaV1.7, and selectivity of 15-fold over NaV1.2 and 6- fold over NaV1.6. | ||
| ICLGMFKSCDPENDKCCKRLVCSRSHRWCKWKL∗ | |||
| (Pyro)ECLGIFKSCDPENDKCCYRLVCSKSHRWCKWKL∗ | IC50 value of 2.5 nM for NaV1.7, and selectivity of 80-fold over NaV1.2 and 20-fold over Nav1.6, and extra 1.5-fold of irreversible binding for NaV1.7. | ||
| HNTX-I | wt-ECKGFGKSCVPGKNECCSGYACNSRDKWCKVLL | No activity over NaV channels | Klint et al., 2015a; Zhang et al., 2018b |
| ECKGFWKSCVPGKNECCSGYACSSRDKWCKVLL | IC50 value of 440 nM for NaV1.7 | Klint et al., 2015a | |
| GCKGFGKSCVPGKNECCSGYACSSRHKWCKVWL | IC50 value of 36 nM for NaV1.7 | Zhang et al., 2018b | |
| HNTX-III | wt-GCKGFGDSCTPGKNECCPNYACSSKHKWCKVYL | IC50 value of 150 nM for NaV1.7 | Zhang et al., 2015 |
| HNTX-IV | wt-ECLGFGKGCNPSNDQCCKSSNLVCSRKHRWCKYEI∗ | IC50 value of 34 nM for NaV1.7 | Li et al., 2004 |
| NaSpTx 3 | |||
| ProTx-II | wt-YCQKWMWTCDSERKCCEGMVCRLWCKKKLW | IC50 value of 0.3–1 nM for NaV1.7 and 26-146 nM for the other NaV subtypes (NaV1.1 was not tested). | Priest et al., 2007; Smith et al., 2007; Schmalhofer et al., 2008; Park et al., 2014 |
| YCQKWMWTCDSERKCCEGMVCRLWCKKKLW-NHCH3 | IC50 value of 42 pMol for NaV1.7, selectivity of 83-fold over NaV1.2. | Park et al., 2014 | |
| GPYCQKWMQTCDSERKCCEGMVCRLWCKKKLL | Improved selectivity over NaV1.4 and NaV1.5. | Flinspach et al., 2017 | |
| YCQKWMWTCDSERKCCEGMVCRLWCKKKLW | Altered lipid binding in the cell membrane. | Agwa et al., 2017 | |
| JZTX-V | wt-YCQKWMWTCDSKRACCEGLRCKLWCRKII∗ | IC50 value of 0.6 nM for NaV1.7, and selectivity of 4- and 4000-fold over NaV1.4 and NaV1.5, respectively. | Moyer et al., 2018 |
| YCQKWMWTCDSKRACCEGLRCKLWCRKEI∗ | Improved selectivity over NaV1.4 to 500-fold (AM-8145). | ||
| (Pra)YCQKWMWTCDSKRACCEGLRCKLWCRKEI∗ | Improved selectivity to 300- and 6000-fold over NaV1.4 and NaV1.5, respectively. | ||
| (CyA)YCQKWMWTCDSKRACCEGLRCKLWCRKEI∗ (Pra) | Improved selectivity to 128- and 1200-fold over NaV1.4 and NaV1.5, respectively. | ||
| NaSpTx 7 | |||
| JZTX-III | wt-DGECGGFWWKCGRGKPPCCKGYACSKTWGWCAVEAP | IC50 value of 348 nM for NaV1.5, not active over other NaV subtypes | Rong et al., 2011 |
| DGECGGFWWKCGEGKPPCCKGYACSKTWGWCAVEAP | Improved potency by 11-fold for NaV1.5. |
Key amino acids residues identified in SAR studies of GpTx-1, HwTx-IV, CcoTx1, ProTx-II, JZTX-V, and JZTX-III which are involved in the potency and/or selectivity of ICK peptides for the NaV channels family are described, along with the NaSpTx family of these peptides. Cysteines are colored in gray. Amino acids residues colored in red play a key role in potency over NaV channels. Amino acids substitutions colored in blue lead to improvement in potency over key NaV channels subtypes, and colored in green lead to improvement in selectivity over key NaV off-targets. Amino acids residues colored in orange play a key role in lipid cell membrane binding. Asterisk (∗) denotes C-terminal amidation. #Compared to Minassian et al. (2013). wt, wild-type.
Later, another positional scan using glutamic acid, arginine, lysin, tryptophan and 1-naphthylalanine substitutions supported previous alanine scanning observations and revealed a number of new positions in GpTx-1 essential for the NaV1.7 activity, all together these key residues are F5, M6, S24, H27, W29, K31, Y32, and F34 (Murray et al., 2016). Contrary to alanine substitutions, most of the substitutions with 1-naphthylalanine didn’t produce folded peptides, with few similar cases for substitutions with tryptophan, followed by lysine, arginine and glutamic acid. Furthermore, the importance of S24 was observed only in the S24E mutant, being all other substitutions at position 24 unable to produce folded peptides. In the three-dimensional structure of GpTx-1, these identified active residues form a cluster on the surface allowing a direct interaction with NaV1.7 (Murray et al., 2016). These potential interactions were further investigated using a homology model of the hNaV1.7 channel docked with the NMR structure of GpTx-1, which predicted interactions of the residues F5 and K31 of GpTx-1 with I767 and E811 located in the domain II voltage sensor region, respectively. Other potential electrostatic interactions were predicted between R25 and R7 of GpTx-1 and E759 and E818 of the ion channel, respectively. Overall, the optimization of GpTx-1 toward an increase in potency for NaV1.7 and selectivity over NaV1.4 and NaV1.5 was possible with the addition of a hydrophobic aromatic residue at position 6 (substitution M6F), and a hydrophobic residue at position 26 (substitution T26L). A summary of the SAR for GpTx-1 is shown in Table 1.
HwTx-IV
Huwentoxin-IV (μ-TRTX-Hs2a), isolated from the venom of the Chinese bird spider Selenocosmia huwena, was first identified as a potent (IC50 of 30 nM) NaV inhibitor of TTX-S currents in DRG neurons (Peng et al., 2002). This inhibitor has been the subject of extensive SAR studies (Table 1 and Figure 2). Initial SAR observations for HwTx-IV described a naturally occurring toxin variant with an N-terminal pyroglutamate instead of glutamate that had unchanged potency despite its irreversibility at strong depolarizing potentials (e.g., +200 mV) in DRG neurons and in NaV1.7 expressed in HEK293 cells (Rong et al., 2013). More detailed SAR studies of HwTx-IV revealed that C-terminal mutations T28D, R29A or Q34D reduced NaV potency in DRG neurons (Deng et al., 2013), while an alanine scan of HwTX-IV additionally revealed W30 and K32 also critical for high affinity interactions at NaV1.2 and NaV1.7 (Minassian et al., 2013) (Figure 2A). In this latter study, molecular dynamics simulations revealed substitutions displaying loss of potency for NaV1.2 and NaV1.7 also led to an increase in the flexibility of the loops 2 (substitutions P11A and D14A), in loop 3 (substitutions F6A, L22A, W30A, and Y33A) and in loop 4 (substitutions R26A and K27A), while the substitutions S25A, K32A, and I35A reduced HwTx-IV potency for NaV1.2 and NaV1.7 without affecting its structure and loops flexibility (Minassian et al., 2013).
FIGURE 2.
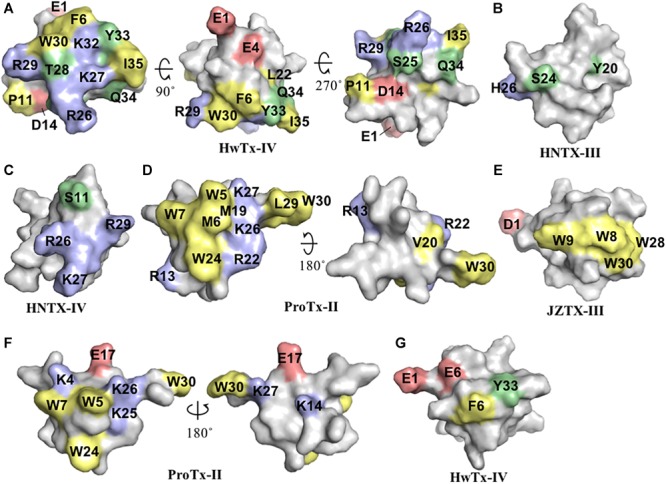
Three-dimensional structure of spider ICK peptides displaying key residues involved in the inhibition of NaV channels and cell membrane binding. (A) Three-dimensional structure of HwTx-IV determined by NMR (PBD 2m4x) (Minassian et al., 2013), (B) HNTX-III determined by NMR (PBD 2jtb), (C) HNTX-IV determined by NMR (PBD 1niy) (Li et al., 2004), (D) ProTx-II determined by X-Ray (PBD 5o0u) (Wright et al., 2017), and (E) JZTX-III determined by NMR (PBD 2i1t). The labeled amino acids residues have key role in potency over NaV channels and lead to loss in activity as described in the text and Table 1. (F) Structure of ProTx-II determined by X-Ray (PDB 5o0u) (Wright et al., 2017) and (G) Structure of HwTx-IV (PDB 2m4x) (Minassian et al., 2013) determined by NMR. The labeled amino acids residues have key role in cell membrane binding as described in the text and Table 1. Amino acids residues are colored as follow: yellow for hydrophobic, red for acid, blue for basic and green for neutral. All three-dimensional structures were prepared in PyMOL (DeLano, 2002).
Further alanine scanning of HwTx-IV identified E1, E4, F6, and Y33 as important contributors for NaV1.7 affinity (Revell et al., 2013). These findings afforded the rational design and optimization of HwTx-IV with increased potency for NaV1.7 and maintaining low affinity for the off-target NaV1.5 (Figure 3). Among these new HwTx-IV analogs, the mutant HwTx-IV-G1/G4/W33 demonstrated the highest increase in activity, with 42-fold enhanced potency for NaV1.7 inhibition, followed by HwTx-G1/G4 and HwTx-IV-A1/A4/W33 (Revell et al., 2013). More recently, the activity of HwTx-IV-G1/G4/W33 was tested over other members of the NaV family, showing an increase in inhibition of 12-fold for NaV1.2 and 47-fold for NaV1.3 compared to wild-type HwTx-IV, but maintained the low potency for the off-target NaV1.4 and NaV1.5 (Xiao et al., 2008a; Rahnama et al., 2017). Altogether, the residues F6, P11, D14, L22, T28, R29, W30, K32, Y33, and Q34, when mutated, led to a loss of affinity of HwTx-IV for NaV channels (Table 1 and Figure 3A). Furthermore, the optimization of HwTx-IV activity for NaV1.7 was possible with the removal of acid negative residues at the N-terminal (substitutions E1 to A or G), and increase in hydrophobicity at the C-terminal (substitution Y33W) (Table 1 and Figure 3A). These produced a new surface in the HwTx-IV three-dimensional structure with increased polarity and hydrophobicity.
FIGURE 3.
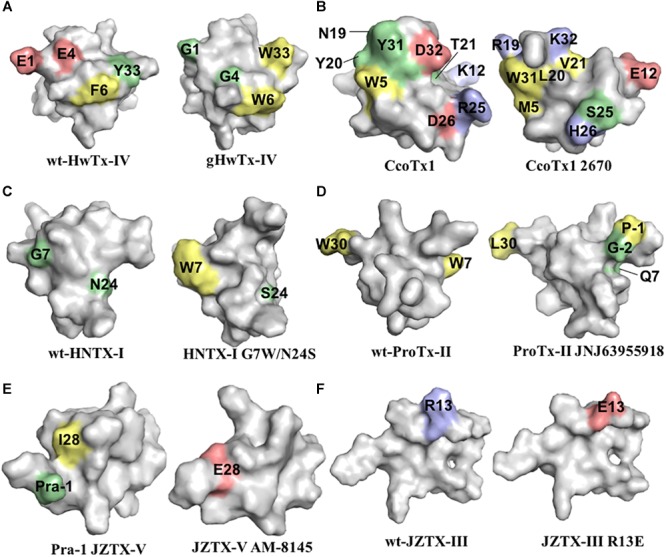
Three-dimensional structure of spider ICK peptides displaying key residues involved in the enhancement of activity for NaV channels, or enhancement of selectivity for NaV channels off-targets. (A) Structures of HwTx-IV (PDB 2m4x) (Minassian et al., 2013) and gHwTx-IV (PDB 5tlr) (Agwa et al., 2017) determined by NMR. (B) Structures of CcoTx1 determined by NMR (PDB 6br0) (Agwa et al., 2018) and the derived analog 2670 determined by X-ray (PDB 5epm) (Shcherbatko et al., 2016). (C) Structures of HNTX-I (PDB 2mqf) and the derived analog G7W/N24S (PDB 2mxo) determined by NMR (Klint et al., 2015a). (D) Structures of ProTx-II determined by X-Ray (PDB 5o0u) (Wright et al., 2017) and derived analog JNJ63955918 determined by NMR (PDB 5tcz) (Flinspach et al., 2017). (E) Structure of analogs Pra-1 JZTX-V (PDB 6chc) and AM-8145 (6cgw) determined by NMR (Moyer et al., 2018). (F) Structure of JZTX-III determined by NMR (PDB 2i1t) (Liao et al., 2006) and modeling of the derived analog R13A using SWISS-MODEL (Arnold et al., 2006). Amino acids residues are colored as follow: yellow for hydrophobic, red for acid, blue for basic and green for neutral. Amino acids substitutions or additions labeled in the respective analogs are involved in the improvement of NaV inhibitory activity or selectivity as described in the text and Table 1. All three-dimensional structures alignments were prepared in PyMOL (DeLano, 2002).
CcoTx1
CcoTx1 (β-TRTX-Cm1a) is a potent NaV inhibitor isolated from the tarantula Ceratogyrus cornuatus (Bosmans et al., 2006). It has strong preference for the subtype NaV1.2 and lower affinity for the off-targets NaV1.4 and NaV1.5 (Table 1). The SAR of CcoTx1 was investigated using a combination of direct evolution, saturation mutagenesis, chemical modifications and rational drug design to unravel key residues involved in potency and selectivity of this peptide over the NaV family (Shcherbatko et al., 2016). Using direct evolution, CcoTx1 was optimized to improve its potency for the subtype NaV1.7, and off-target selectivity for NaV1.2 and NaV1.6, producing the variant named 2670. This mutant contained the substitutions W5M, K12E, N19R, Y20L, T21V, R25S, D26H, Y31W, and D32K (Table 1 and Figure 3B).
Further SAR investigations aiming to improve selectivity and potency for NaV1.7 were performed using saturation mutagenesis, revealing that substitutions at the N- and C-terminal regions, along with positions 20 and 21 of 2670, were not well tolerated. Combining this SAR information, the NaV1.7 pharmacophore of 2670 was defined by the hydrophobic residues M5, F6, W28, W31, and L33, and polar or positively charged residues R19, H26, K30, and K32. Saturation mutagenesis substitutions that conferred improved selectivity over off-targets and didn’t alter the NaV1.7 inhibitory potency were K18Y, R24K, and R27N. Still using this approach, a new mutant was identified with one single substitution D1I that conferred improved activity and selectivity (Table 1). C-terminal amidation of 2670 (2670a) and D1I (D1Ia) improved potency for NaV1.7 which was accompanied by an increase in potency for NaV1.6, but not for NaV1.2. In addition, a D1Ia variant containing a terminal pyroglutamate (D1Za) improved binding to NaV1.7 by 27% at strong depolarizing potentials, and maintained the same potency as D1Ia. This observation resembles the SAR properties of HwTx-IV (Rong et al., 2013). Overall, the optimization of CcoTx-1 activity toward NaV1.7 was possible with the replacement of acid negative residues at both N- and C-terminal to residues with increased hydrophobicity and positive charges, and with modifications toward the C-terminal that included substitutions for aromatic residues H and W. This led to a considerable change in the CcoTx-1 surface to include more hydrophobic and positively charged residues (Figure 3B).
Hainantoxin
Hainantoxins (HNTXs) are ICK peptides comprised in the venom of the Chinese bird spider Selenocosmia hainana. Among these, the HNTX-I (μ-TRTX-Hhn2b) has weak to no activity over NaV channels. SAR studies to restore the NaV inhibitory activity of HNTX-I were performed using rational design based on other members of the NaSpTx 1 (Klint et al., 2015a; Zhang et al., 2018b) (Table 1 and Figure 3C). The substitutions G6W/N23S/W28F and G6W/N23S produced analogs with IC50 values of 1 and 0.44 μM for NaV1.7, respectively (Klint et al., 2015a). Later, the analogs N23S/D26H, N23S/D26H/L32W and E1G/N23S/D26H/L32W showed IC50 values of 79, 71, and 36 nM for NaV1.7, respectively. In this study, the motif X1X2SWCKX3 was identified as critical for the inhibitory activity for NaV1.7. Altogether, the HNTX-I activity for NaV1.7 was restored by removal of acid negative residues at both N- and C-terminal, and the addition of residues with increased hydrophobicity and positive charges, including the aromatic residues H and W.
Another hainantoxin, HNTX-III (μ-TRTX-Hhn2a), is an inhibitor of NaV1.7 with IC50 value of 232 nM (Liu et al., 2013). The SAR of this peptide was performed consequent to the discovery of several isoforms variants present in the transcriptome of the venom gland of this spider (Zhang et al., 2015) (Table 1). The pharmacological properties of these variants were tested, revealing that the substitutions Y20H, S24N, H26D and Y20H/S24N were all detrimental to the NaV1.7 activity (Figure 2B). The last hainantoxin discussed is HNTX-IV (μ-TRTX-Hhn1b), a potent inhibitor of TTX-S currents in DRG neurons (IC50 of 34 nM; Li et al., 2004). In this study, the SAR of HNTX-IV revealed K27 and R29 residues positions involved in the NaV activity (Table 1 and Figure 2C). More specifically, the mutants S12A and R26A produced IC50 values of 58 and 96 nM, respectively, while K27A and R29A produced IC50 values of 3.2 and 7 μM, respectively. This work reinforces the essential role of positively charged residues located in the surface of spider ICK peptides for NaV1.7 inhibition.
NaSpTx 3
ProTx-II
ProTx-II (β/ω-TRTX-Tp2a) is amongst the most potent NaV inhibitors described to date, with reported IC50 value of 0.3 nM for inhibition of NaV1.7 (Schmalhofer et al., 2008). This toxin was isolated from the spider Thrixopelma pruriens using rNav1.8 assay guided fraction that evaluated the inhibitory properties for the NaV1.8 channel of individual venom fractions separated by cation exchange liquid chromatography (Middleton et al., 2002). Despite of its exquisite inhibitory potency for NaV1.7, it also potently inhibits other members of the NaV family, including the off-targets NaV1.4 and NaV1.5. The SAR of ProTx-II was initially investigated over the NaV1.5 channel (Table 1 and Figure 2D). Analogs of ProTx-II produced by recombinant expression and chemical synthesis containing alanine or glutamine substitutions revealed a peptide active face composed of hydrophobic and cationic residues (Smith et al., 2007). More specifically, ten of these analogs showed losses in potency from 10- to 125-fold, with major losses associated with W5A and K26A substitutions. The positively charged residues substitutions K27Q, R13Q, and R22A led a significant loss in potency, while neutralization of negatively charged residues didn’t affect the NaV1.5 inhibition. Furthermore, the substitutions M6A, W7A, M19L, V20A, W24L, L29A, and W30A each produced a > 10-fold loss in potency. Similarly, another SAR study of ProTx-II identified the residues in the hydrophobic face essential for NaV1.5 activity, including W5, M6, W7, W24, while residues identified as not critical for inhibitory activity included Y1, Q3, T8, N10, S11, E12, E17, and L23 (Priest et al., 2007).
SAR of ProTx-II has also been investigated over the channels NaV1.2 and NaV1.7 (Park et al., 2014) (Table 1 and Figure 2D). In this study, besides the identification of residues mutations detrimental to NaV activity such as substitution of C-terminal KLW to II, an optimized analog ProTx-II-NHCH3 with ∼23-fold greater potency for NaV1.7 was identified. In this same work, the ICK peptide Phrixotoxin I, also known as PaTx I, isolated from the tarantula Phrixotrichus auratus, and a potent KV inhibitor with IC50 value of 28 nM was submitted to a SAR study over NaV channels (Diochot et al., 1999). It differs from ProTx-II by only 4 amino acids residues, and has moderate potency for NaV channels, with IC50 value of 423 nM for NaV1.7, and no activity for NaV1.2 (Park et al., 2014). These naturally occurring differences were useful for the understand of the ProTx-II SAR over NaV channels. The PaTx I C-terminal substitutions I28K and I29K, and addition of L30 and W31 (actual ProTx-II C-terminal) lead to an increase of 85-fold in the NaV1.7 potency, and an IC50 value of 45 nM for NaV1.2. This work unraveled a key role of the C-terminal for activity of NaSpTx 3 peptides, and describes interesting approaches for the improvement of NaV activity through C-terminal modifications.
More recently, a detailed SAR of ProTx-II over Nav channels was investigated by 1500 toxin-derived peptides to identify a double mutant with extended N-terminal that maintained the inhibitory properties for NaV1.7 but lost affinity for NaV1.4 and NaV1.5 (Flinspach et al., 2017) (Table 1 and Figure 3D). This mutant, named JNJ63955918, contained additional G-2 and P-1 at the N-terminal and the substitutions W7Q and W30L. The interactions of ProTx-II with lipid membranes as well as with the hNav1.7 channels were investigated, revealing ProTx-II does interact with the cell membrane as part of its strategy to inhibit the NaV1.7 channel (Henriques et al., 2016) (Figure 3). In this study, substitutions of residues K to R and W to Y lead to a reduction in the potency of ProTx-II for hNaV1.7. The ProTx-II lipid interactions will be discussed in more details later in this review. Altogether, the residues W5, M6, W7, R13, M19, V20, R22, W24, K26, K27, L29, and W30, when mutated, led to a loss of affinity of ProTx-II for NaV channels (Table 1 and Figure 2D). Interestingly, the optimization of ProTx-II toward a potent and selective NaV1.7 inhibitor was possible through the extension of the N-terminal with neutral and positively charged residues, and introduction of a positively charged group in the C-terminal.
JZTX-V
Isolated from the tarantula Chilobrachys jingzhao, JZTX-V (β-TRTX-Cg2a) is a potent NaV1.7 inhibitor with IC50 value of 0.6 nM (Moyer et al., 2018). The SAR of JZTX-V over the subtypes NaV1.4, NaV1.5, and NaV1.7 was investigated using alanine or glutamic acid positional scanning to unravel key residues involved in NaV inhibition, and to produce new mutants with improved potency and selectivity (Table 1 and Figure 3E). Substitutions that contribute to loss of the NaV1.7 inhibitory activity observed by alanine substitutions were W5, L19, W24, and R26, and by glutamic acid substitutions were M6, T8, D10, R13, and L23. Interestingly, a key substitution that improved off-target selectivity of JZTX-V over NaV1.4 was I28E. This mutation was able to induce a change in the conformation of JZTX-V to produce 500-fold selectivity and maintain the potency for NaV1.7.
In the same study, further investigation of the SAR of JZTX-V was performed through the introduction of side-chains containing residues such as Pra and β-cyanoalanine (CyA). Positions showing minimum involvement in NaV1.7 inhibitory activity disclosed by the glutamic acid scanning were Y1, S11, A14, and E17, and therefore selected to test this approach. The addition of Pra at the N-terminus of I28E created a new analog named AM-8145 with improved selectivity for NaV1.4 (300-fold) and NaV1.5 (6000-fold) and maintained potency for NaV1.7. Similar results were found for the addition of CyA at the N-terminus of I28E (analog AM-0422) and substitutions in the other selected positions. These properties were not present in the wild-type JZTX-V containing the Pra additions, but only in the I28E mutant. In summary, the optimization of JZTX-V toward a more selective NaV1.7 inhibitor was possible through the extension of the N-terminal with neutral or hydrophobic residues containing side-chains and addition of negative charge at the C-terminal. Interestingly, JZTX-V share similar key residues with ProTx-II essential for the inhibition of NaV1.7, as for W5, M6, R26, and W24 in JZTX-V, and selectivity optimization for NaV1.4 and NaV1.5 through N-terminal extensions.
NaSpTx 7
JZTX-III
The spider peptide JZTX-III (β/κ-TRTX-Cg1a), also isolated from the tarantula Chilobrachys jingzhao, was first characterized as a potent inhibitor of TTX-R currents in rat cardiac myocytes with IC50 value of 380 nM (Xiao et al., 2004), and later found to also inhibit KV2.1 channels (Yuan et al., 2007). The SAR of JZTX-III with NaV1.5 channels using alanine substitutions revealed residues D1, E3 and W8, W9, W28, and W30 as key players for its NaV1.5 inhibition (Rong et al., 2011) (Table 1 and Figure 2E). In addition, the substitution R13E enhanced the NaV1.5 inhibition by 11-fold (Figure 3F). Interestingly, JZTX-III does not inhibit the NaV1.7 channel, and this lack of affinity was associated to the residue D816 in the NaV1.7. The substitution D816R (corresponding to the R800 in the NaV1.5 channel) enhanced significantly the inhibitory properties of JZTX-III for NaV1.7.
Interactions With the Cell Membrane
Spider ICK peptides are known to interact with the lipids in the cell membrane. Early studies showed these peptides are water-soluble and bind to the aqueous-exposed extracellular surface of ion channels, and surprisingly reach the target by partitioning into the lipid membrane (Lee and MacKinnon, 2004). This strategy allows the peptide to reach the voltage-sensor and enhance high-affinity inhibition. The ability to bind to lipids seems exclusive of gating modifiers ICK peptides binding to site 4 of NaV channels (Smith et al., 2005). Furthermore, the NaV VSD s are affected by the cell membrane lipid composition, with changes from native lipids to sphingomyelin altering G-V relations and affinity of ProTx-I for the domain II and domain IV S3-S4 loops (Milescu et al., 2009).
Detailed studies of these interactions using NMR revealed that ICK peptides can interact with the headgroup region of lipid membrane to induce a thinning of the bilayer (Mihailescu et al., 2014). In this interaction, many basic residues are positioned toward the aqueous phase, the W residues adopt an interfacial position and hydrophobic residues are in direct contact with the membrane. The SAR of ProTx-II, membrane lipids and NaV1.7 confirmed the previous observations, and revealed the ProTx-II analog E17K had increased on-rate for NaV1.7 compared to wild-type, but had no changes in the NaV1.7 potency (Henriques et al., 2016) (Table 1 and Figure 2F). Interestingly, all mutations at residues W to Y and K to R lead to a lower affinity for cell membranes. Changes in the structure of ICK peptides can also occur in the presence of lipids. This was observed in structural studies of ω-Aga-IVA (a CaV channel inhibitor ICK peptide) that undergoes changes in the presence of micelles (Ryu et al., 2017). More specifically, the C-terminal tail of ω-Aga-IVA assume a β-turn like conformation which is disordered in water.
The HwTx-IV mutant E1G/E4G/F6W/Y30W had increased affinity for the lipid membrane and higher potency for the inhibition of hNaV1.7 compared to wild-type (Agwa et al., 2017) (Table 1 and Figure 2G). Interestingly, the introduction of a PyroE in the N-terminal for HwTx-IV didn’t alter the membrane interactions and potency for NaV1.7. This confirmed previous observations for the naturally occurring PyroE-HwTx-IV that binds irreversibly to the NaV channel voltage-sensor but at same potency of wild-type HwTx-IV (Rong et al., 2013). Although membrane interactions are key for the high affinity binding of ICK peptides over ion channels, such interactions have to-date had little influence on the selectivity for the NaV channel family (Agwa et al., 2018).
Binding Sites on NaV Channels
The binding sites of spider ICK peptides at mammalian NaV channels are starting to be characterized (Cardoso and Lewis, 2017). These gating-modifier toxins are known for their ability to change the voltage-dependence of activation and inactivation of NaV channels to either inhibit or activate Na+ currents. Overall, they have preference for binding to the site 4 in domain II of the NaV channel to trap the voltage–sensor and inhibit Na2+ currents, and to site 3 in domain IV to slow channel inactivation and maintain Na2+ currents. These sites have been reported for a range of spider ICK peptides, including HwTx-IV, ProTx-I, ProTx-II, PaurTx3, CcoTx-1, Hd1a and Df1a that inhibit NaV channels (Bosmans et al., 2008; Xiao et al., 2008a; Klint et al., 2015b; Shcherbatko et al., 2016; Cardoso et al., 2017) and to SGTx1 and Hm1a that slow NaV channels inactivation (Bosmans et al., 2008; Osteen et al., 2016).
The crystal structure of the related double-ICK spider peptide Dc1a bound to the NaVPaS was determined (Shen et al., 2018). Dc1a binds to a cleft between the VSD II and the pore loop of domain III of the NaVPaS and induce minimal changes in the channel structure. On the other hand, the Dc1a peptide undergoes considerable rearrangement to achieve its fit into the cleft. This information brings new insights into the docking and binding of other ICK peptides into the mammalian channel, which although widely reported to bind to domain II and domain IV S3-S4 loops of mammalian NaV channels, could also be interacting with other proximal regions of the channel to reaches its final fit and maximal activities.
More recently, the cryo-EM structure of the human NaV1.7 channel was described in association of the auxiliary β1 and β2 subunits (Figure 1A,B) and the spider peptides HwTx-IV and ProTx-II (Shen et al., 2019). This study confirmed previous observations that HwTx-IV binds to the voltage-sensor of DII site 4 (Xiao et al., 2008a, 2010) and that ProTx-II binds to the voltage-sensors of DII and DIII sites 4 and 3 (Bosmans et al., 2008; Xiao et al., 2010). The cryo-EM structure of a chimeric hNaV1.7-NaVAb channel bound to ProTx-II was also described recently (Figure 1C) (Xu et al., 2019), which confirmed ProTx-II binds to the voltage-sensor of DII site 4 of the NaV1.7 channel (Xiao et al., 2010). Although the determination of the three-dimensional structure of spider ICK peptides using X-ray or NMR are essential for the understanding of the SAR of these peptides, additional conformational changes are likely to happen for most of these ICK peptides when they achieved their final bound conformation with the NaV channel and associated membrane lipids.
Sar Integration of Spider ICK Peptides
Prevalent features in the SAR of spider ICK peptides and NaV channels have been disclosed (Figure 4A and Table 1). Amongst the remarkable positions associated to NaV activity in the NaSpTx 1 toxins are the residues 5 and 6, which are typically occupied by the hydrophobic residue phenylalanine. Similarly, NaSpTx 3 toxins typically have positions 5 and 6 occupied by hydrophobic residues W and M, which are also critical for NaV inhibitory activity. Another interesting aspect of these SAR studies is apparent in optimized analogs with increased NaV potency and selectivity that often display a decrease in negative charge at the N-terminal compared to wild-type toxins. This is supported by the introduction of Pyro, Pra, CyA and the residues G and P at the N-terminal in optimized peptides belonging to NaSpTx families 1 and 3 (Rong et al., 2013; Shcherbatko et al., 2016; Flinspach et al., 2017; Moyer et al., 2018), and for the substitutions E1A, E1G and D1I in optimized peptides from NaSpTx 1 (Minassian et al., 2013; Revell et al., 2013; Shcherbatko et al., 2016; Rahnama et al., 2017; Zhang et al., 2018b). A decrease in negative charge at the N-terminal also enhances the lipid affinity of NaSpTx 1 toxins such as HwTx-IV (Henriques et al., 2016), suggesting these optimized peptides may have enhanced lipid binding in addition to any enhanced interactions directly with NaV channels. This is relevant when alterations to NaV channel selectivity are sought, since enhanced lipid binding is unlikely to have less ability to influence NaV channel subtype selectivity (Agwa et al., 2018).
FIGURE 4.
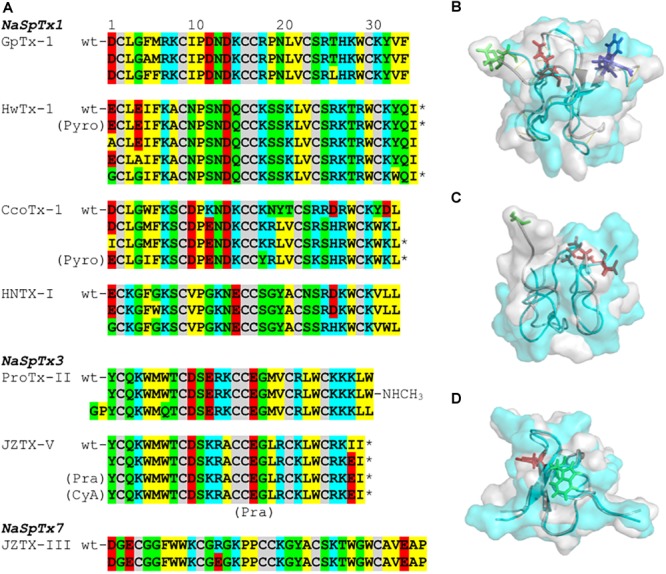
SAR integration of spider ICK peptides. Primary and three-dimensional structures comparison of spider ICK peptides belonging to NaSpTx families 1, 3, and 7. (A) Primary structure alignment of wild-type peptides and respective optimized analogs showing enhancement of activity and/or selectivity of NaV channels. The amino acids residues are highlighted as follow: yellow for hydrophobic, red for acid, blue for basic, green for neutral, and gray for the cysteines. (B) Alignment of the three-dimensional structures of HwTx-IV (PDB 2m4x) (Minassian et al., 2013) colored in gray and gHwTx-IV (PDB 5tlr) (Agwa et al., 2017) colored in cyan. Structures are represented by cartoon and surface, and substitutions E1G, E4G and Y32W are represented by sticks colored in green, red, and blue, respectively. (C) Alignment of the three-dimensional structures of Pra-1 JZTX-V (PDB 6chc) colored in gray and AM-8145 (6cgw) colored in cyan (Moyer et al., 2018). Structures are represented by cartoon and surface, and the Pra-1 addition and substitution I28E are represented by sticks colored in green and red, respectively. (D) Alignment of the three-dimensional structures of HNTX-I (PDB 2mqf) colored in gray and the derived analog G7W/N24S (PDB 2mxo) colored in cyan (Klint et al., 2015a). Structures are represented by cartoon and surface, and substitutions G7W and N24S are represented by sticks colored in green and red, respectively. All three-dimensional structures alignments were performed in PyMOL (DeLano, 2002). Asterisk (∗) denotes C-terminal amidation.
Modifications in the C-terminal region that enhance inhibition of NaV channels often involve the introduction of positive charges (Figure 4A and Table 1). This is observed for ICK peptides belonging to NaSpTx 1 and 3, as for the C-terminal amidation of HwTx-IV and CcoTx-1, the introduction of a NHCH3 group in ProTx-II and the substitution D32K in CcoTx-1 (Minassian et al., 2013; Revell et al., 2013; Park et al., 2014; Shcherbatko et al., 2016). An increase in hydrophobicity in the C-terminal region also enhanced the NaV inhibition properties of peptides in NaSpTx 1, especially Y33W in HwTx-IV and Y31W in CcoTx-1 (Revell et al., 2013; Shcherbatko et al., 2016; Rahnama et al., 2017). Interestingly, the reduction in hydrophobicity and introduction of negative charges in the C-terminal of the NaSpTx 3 peptide JZTX-V led to a decrease in NaV affinity (analog I28E), but only at NaV1.4 (Moyer et al., 2018). A similar reduction in hydrophobicity in the N-terminal region of the NaSpTx 1 analog F5A-GpTx-1 also decreased NaV1.4 inhibition (Murray et al., 2015). As our SAR knowledge of NaSpTx 1 and 3 deepens, we anticipate further opportunities to develop more selective and/or potent inhibitors are expected to emerge.
Structural changes in optimized molecules suggest gain of affinity can be associated with regions of the ICK peptide beyond those associated with the local effects of the substitution. For example, structural rearrangements are observed in the HwTx-IV, JZTX-V and HNTX-I optimized analogs (Figure 4B–D) that alter the surface structure of wild-type vs. optimized peptide, with pronounced changes in the N-termini of JZVTX-V with the introduction of Pra (Figure 4C) and following the introduction of W7 in HNTX-I (Figure 4D). However, the three-dimensional structure of a NaSpTx inhibitor bound to a NaV channel embedded in the cell membrane is still to be elucidated.
The Phylogeny of NaSpTx Targeting NaV1.7
In addition to the SAR studies of the nine NaSpTx discussed above, a number of other NaSpTx display interesting NaV1.7 modulatory properties (Figure 5). To date, twenty-three other peptides belonging to NaSpTx 1, 2, 3, and 7 are described with potencies ranging from 0.3 nM to 7.4 μM for half-maximal activity for NaV1.7 (Figure 5A). These include μ-TRTX-Pho1a, μ-TRTX-Pho1b and μ-TRTX-Pho2a (Chow et al., 2015), κ/μ-TRTX-Gr2c, κ/μ-TRTX-Gr3a, μ-TRTX-Gr1a, β-TRTX-Gr1d and β-TRTX-Gr1a (Redaelli et al., 2010), μ-TRTX-Pn3a (Deuis et al., 2017), μ-TRTX-Df1a (Cardoso et al., 2017), β/ω-TRTX-Tp2a (Priest et al., 2007), δ-TRTX-Cg1a (Xiao et al., 2005), μ-TRTX-Tp1a (Cardoso et al., 2015), β-TRTX-Cm1b and β-TRTX-Cd1a (Bosmans et al., 2006; Sousa et al., 2017) and μ-TRTX-Hd1a (Klint et al., 2015b). Amongst these, only δ-TRTX-Cg1a activates NaV1.7, while the other β- and μ-toxins are NaV1.7 inhibitors. Although detailed SAR studies on these toxins have not been described, their sequences similarities make a detailed analysis of their primary structure using phylogenetic approaches useful.
FIGURE 5.
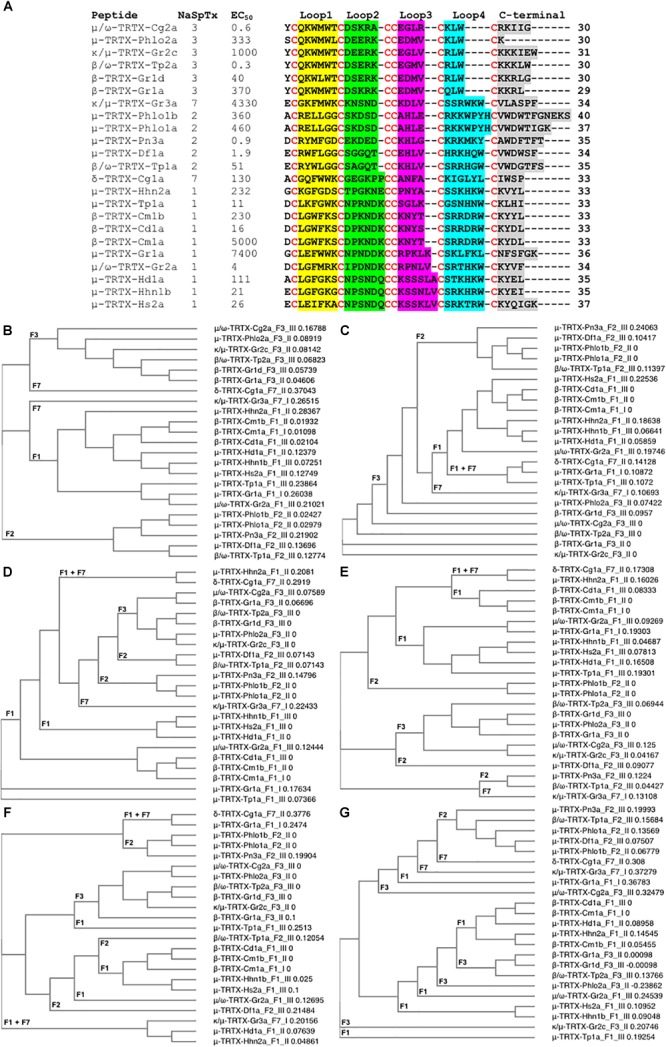
Primary amino acids sequence alignment and phylogenetic analysis of NaSpTx peptides with described NaV1.7 modulatory activity. (A) Primary sequences alignment of members of NaSpTx1, 2, 3, and 7, with EC50 values represented in nM, Loops, 1, 2, 3, 4, and C-terminal shaded in yellow, green, pink, (blue and gray, respectively, and cysteines colored in red. (B) Phylogenetic analysis of the full primary sequences of NaSpTx NaSpTx1, 2, 3, and 7, followed by Loop 1 (C), Loop 2 (D), Loop 3 (E), Loop 4 (F), and C-terminals (G). For the phylogenetic trees, the peptides names are followed by their respective NaSpTx families represented by F1, F2, F3, and F7, and their NaV1.7 potency represented by I (EC50 > 1 μM), II (EC50 between 1 and 0.1 μM), and III (EC50 < 0.1 μM). These analyses were performed using Clustal Omega (Sievers et al., 2011) and Simple Phylogeny (Saitou and Nei, 1987). The loops were flanked by cysteine for these analyses.)
Not surprisingly, NaSpTxs targeting NaV1.7 show independent phylogenetic origins and generate well defined clades for NaSpTx 1, 2, and 3, while NaSpTx 7, with only two representatives, clustered as outliers (Figure 5B). These results support previous findings underpinning the classification of the NaSpTx into 12 families (Klint et al., 2012). However, examining relatedness within loops revealed some interesting differences. For example, loop 1 showed similar clustering as the complete primary sequences except NaSpTx 7 clustered with NaSpTx 1 (Figure 5C), while loop 2 had more complex phylogenetic origins, with a primitive ancestor possibly arising from NaSpTx 1 (Figure 5D). Loop 3 also showed complex phylogenetic origins. It was divided into a major clade including the NaspTx1, two members of NaSpTx 2 and one member of NaSpTx 7, a second clade containing NaSpTx 3 and one member of the NaSpTx 2 as outlier, and a small clade including NaSpTx 2 and 7 (Figure 5E). These observations suggest a less purifying selection in loop 3, with NaSpTx 3 remaining the most conservative loop 3 group. The complexity of the phylogenetic origins of the loop 4 and C-terminal regions are considerably higher compared to loops 1, 2, and 3 (Figure 5F,G). Loop 4 diverged into four clades containing a mix of NaSpTx members, and again the NaSpTx 3 clustered further in a single clade, but now with one member of the NaSpTx 1 as an outlier. Finally, the C-terminal was diverged into three major clades that also contained a mix of NaSpTx members. Interestingly, the C-terminals of NaSpTx 2 clustered further into a single clade, indicating that a more conservative evolutionary pressure is occurring in this region of the NaSpTx 2 members.
Overall, we observed that the NaSpTx 1, 2 and 3 families have independent phylogenetic origins. For the NaSpTx 7, more representatives of this family are essential for an appropriated phylogenetic analysis. Remarkably, the loops 2, 3, and 4 forming the peptides in NaSpTx 3 are under a more conservative pressure compared to NaSpTx 1 and 2. This agrees with the SAR studies for ProTx-II (β/ω-TRTX-Tp2a), where optimization was possible only with modifications located in less conserved regions, the C-terminal and in the loop 1, while modifications in loops 2, 3, and 4 were often deleterious for NaV inhibition (Priest et al., 2007; Smith et al., 2007; Schmalhofer et al., 2008; Park et al., 2014; Flinspach et al., 2017). For NaSpTx 1, loop 2 was more conserved and appears to be in agreement with the SAR of CcoTx1 (β-TRTX-Cm1a) where modifications to loop 2 were not well tolerated, while loops 3, 4 and C-terminal tolerated multiple modification (Shcherbatko et al., 2016). Similarly, the C-terminal of NaSpTx 2 was also more conserved compared to the other NaSpTx families, but studies of SAR aiming to unravel the role this C-terminal region in NaV inhibition are yet to be pursued. These patterns of evolutionary pressure provide clues to further explore the SAR of loops and C-terminal residues to that can help further expand the SAR of these NaV channel toxins. However, a simple correlation between phylogenetic origins and potency at NaV channels remains to be established.
Perspectives in Therapeutic Development
Advances in understanding disease mechanisms and associated novel therapeutic targets position spider ICK peptides as novel drug leads of sufficient size and pharmacophore complexity to restrict their in vivo distribution and limit off-target effects. To fully exploit this potential, a deeper understanding of ICK peptide SAR and bio-engineering is required to address specific therapeutic needs. This undoubtedly involves bio-activity and three-dimensional structural determinations for a detailed view of the SAR features. Pre-clinical studies in rodent pain models showing reversal of different types of pain have been reported for spider toxins, including several lacking published SAR data (Table 2).
Table 2.
Therapeutical potential of NaSpTx peptides evaluated in pre-clinical rodent pain models.
| Peptide | NaSpTx family | Spider species | Preferred NaV subtypes | Therapeutic potential | References |
|---|---|---|---|---|---|
| Gr1b (GsAFI) Gr2c (GsAFII) | 3 | Grammostola rosea | NaV1.7 > 1.4 > 1.1 > 1.2 NaV1.7 > 1.4 > 1.1 | Acute and inflammatory pain | Lampe, 1998 |
| HwTx-IV | 1 | Ornithoctonus huwena | NaV1.7 > 1.2 > 1.3 > 1.4 | Inflammatory and SNI-induced neuropathic pain | Liu et al., 2014b |
| HnTx-IV | 1 | Haplopelma hainanum | NaV1.2 > 1.3 > 1.7 | SNI-induced neuropathic and formalin-induced inflammatory pain. | Liu et al., 2014a |
| ProTx-II | 3 | Thrixopelma Pruriens | NaV1.7 > 1.6 > 1.2 > 1.5 > 1.3 > 1.8 | Painful diabetic neuropathy Inflammatory pain | Tanaka et al., 2015; Flinspach et al., 2017 |
| Hl1a | 7 | Haplopelma lividum | NaV1.8 | Inflammatory and neuropathic pain | Meng et al., 2016 |
| Pn3a | 2 | Pamphobeteus nigricolor | NaV1.7 > 1.1 | Inflammatory (with opioid co-administration) and post-surgical pain | Deuis et al., 2017; Mueller et al., 2019 |
| Ca1a | Unknown | Cyriopagopus albostriatus | NaV1.7 > 1.2 > 1.6 > 1.4 > 1.3 | Inflammatory and thermal pain | Zhang et al., 2018c |
| Ca2a | 1 | Cyriopagopus albostriatus | NaV1.7 > 1.2 > 1.6 > 1.3 | Inflammatory and thermal pain | Zhang et al., 2018a |
| Cyriotoxin-1a | 1 | Cyriopagopus schioedtei | NaV1.1 > 1.2 > 1.6 > 1.7 > 1.3 | Thermal pain | Goncalves et al., 2019 |
The NaSpTx peptide with respective family, spider species, preferred voltage-gated sodium channels (NaV) subtypes and the pre-clinical pain model showing therapeutic efficacy are described. The preferred NaV subtypes for some of these NaSpTx peptides were described in: Gr1b and Gr2c (Redaelli et al., 2010), HwTx-IV (Minassian et al., 2013), HnTx-IV (Cai et al., 2015), and ProTx-II (Middleton et al., 2002).
The NaSpTx3 peptides Gr1b and Gr2c reversed acute and inflammatory pain intrathecally (Lampe, 1998), while ProTx-II and its optimized analog JNJ63955918 reversed neuropathic and inflammatory pain when administered intrathecally or locally (Tanaka et al., 2015; Flinspach et al., 2017). The NaSpTx1 peptides HwTx-IV and HNTX-IV also reversed neuropathic and inflammatory pains intraperitonially (Liu et al., 2014a,b), while ides Ca2a and cyriotoxin-1a reversed inflammatory and thermal pain following intraperitoneal or intraplantar administration, respectively (Zhang et al., 2018a; Goncalves et al., 2019).
In contrast, the NaSpTx2 peptide Pn3a reversed inflammatory pain but only when co-administered with an intraperitoneal opioid (Deuis et al., 2017), despite reversing post-surgical pain following intraperitoneal or local administration (Mueller et al., 2019). Finally, the unclassified spider peptide Ca1a reversed inflammatory and thermal pain following intraperitoneal or intraplantar administrations, respectively (Zhang et al., 2018c). This review of the continuing advancements in the SAR of spider venom ICK peptides will hopefully facilitate efforts to optimize NaV channel modulators for the treatment of complex channelopathies, including different forms of chronic pain.
Author Contributions
FC performed the phylogenetic analysis, bibliography research, wrote the manuscript, and prepared the figures. RL provided scope, guidance and critically reviewed the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
- ASIC
acid sensing channels
- CaV
voltage-gated calcium channel
- CyA
β-cyanoalanine
- DRG
dorsal root ganglion
- ICK
inhibitory cysteine knot
- KV
voltage-gated potassium channel
- mAChR
muscarinic acetylcholine receptor
- nAchR
nicotinic acetylcholine receptor
- NaV
voltage-gated sodium channels
- NaVPaS
insect NaV channel
- NET
norepinephrine transporter
- Pra
propargylglycine
- PyroE
pyroglutamate
- SAR
structure-activity relationship
- TRP
transient receptor potential channels
- TTX-R
tetrodotoxin-resistant
- TTX-S
tetrodotoxin-sensitive
- VGIC
voltage-gated ion channels
- VSD
voltage-sensor domain
Footnotes
Funding. The National Health and Medical Research Council of Australia APP1072113 provided research funding to RL that produced background to the review and APP1119056 provided a Fellowship to RL.
References
- Abraham N., Lewis R. J. (2018). Neuronal nicotinic acetylcholine receptor modulators from cone snails. Mar. Drugs 16:E208. 10.3390/md16060208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agwa A. J., Lawrence N., Deplazes E., Cheneval O., Chen R., Craik D. J., et al. (2017). Spider peptide toxin HwTx-IV engineered to bind to lipid membranes has an increased inhibitory potency at human voltage-gated sodium channel hNaV1.7. Biochim. Biophys. Acta 1859 835–844. 10.1016/j.bbamem.2017.01.020 [DOI] [PubMed] [Google Scholar]
- Agwa A. J., Peigneur S., Chow C. Y., Lawrence N., Craik D. J., Tytgat J., et al. (2018). Gating modifier toxins isolated from spider venom: modulation of voltage-gated sodium channels and the role of lipid membranes. J. Biol. Chem. 293 9041–9052. 10.1074/jbc.RA118.002553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold K., Bordoli L., Kopp J., Schwede T. (2006). The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics 22 195–201. 10.1093/bioinformatics/bti770 [DOI] [PubMed] [Google Scholar]
- Bosmans F., Martin-Eauclaire M. F., Swartz K. J. (2008). Deconstructing voltage sensor function and pharmacology in sodium channels. Nature 456 202–208. 10.1038/nature07473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosmans F., Rash L., Zhu S., Diochot S., Lazdunski M., Escoubas P., et al. (2006). Four novel tarantula toxins as selective modulators of voltage-gated sodium channel subtypes. Mol. Pharmacol. 69 419–429. 10.1124/mol.105.015941 [DOI] [PubMed] [Google Scholar]
- Cai T., Luo J., Meng E., Ding J., Liang S., Wang S., et al. (2015). Mapping the interaction site for the tarantula toxin hainantoxin-IV (β-TRTX-Hn2a) in the voltage sensor module of domain II of voltage-gated sodium channels. Peptides 68 148–156. 10.1016/j.peptides.2014.09.005 [DOI] [PubMed] [Google Scholar]
- Cardoso F. C., Dekan Z., Rosengren K. J., Erickson A., Vetter I., Deuis J. R., et al. (2015). Identification and characterization of ProTx-III [μ-TRTX-Tp1a], a new voltage-gated sodium channel inhibitor from the venom of the tarantula Thrixopelma pruriens. Mol. Pharmacol. 88 291–303. 10.1124/mol.115.098178 [DOI] [PubMed] [Google Scholar]
- Cardoso F. C., Dekan Z., Smith J. J., Deuis J. R., Vetter I., Herzig V., et al. (2017). Modulatory features of the novel spider toxin μ-TRTX-Df1a isolated from the venom of the spider Davus fasciatus. Br. J. Pharmacol. 174 2528–2544. 10.1111/bph.13865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardoso F. C., Hasan M., Zhao T., Lewis R. J. (2018). Toxins in pain. Curr. Opin. Support. Palliat. Care 12 132–141. 10.1097/SPC.0000000000000335 [DOI] [PubMed] [Google Scholar]
- Cardoso F. C., Lewis R. J. (2017). Sodium channels and pain: from toxins to therapies. Br. J. Pharmacol. 175 2138–2157. 10.1111/bph.13962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow C. Y., Cristofori-Armstrong B., Undheim E. A., King G. F., Rash L. D. (2015). Three peptide modulators of the human voltage-gated sodium channel 1.7 an important analgesic target, from the venom of an Australian tarantula. Toxins 7 2494–2513. 10.3390/toxins7072494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J. J., Reimann F., Nicholas A. K., Thornton G., Roberts E., Springell K., et al. (2006). An SCN9A channelopathy causes congenital inability to experience pain. Nature 444 894–898. 10.1038/nature05413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLano W. L. (2002). The PyMOL Molecular Graphics System, Version 1.5.0.4 Schrödinger. Scotts Valley, CA: LLC. [Google Scholar]
- Deng M., Luo X., Jiang L., Chen H., Wang J., He H., et al. (2013). Synthesis and biological characterization of synthetic analogs of Huwentoxin-IV (μ-theraphotoxin-Hh2a), a neuronal tetrodotoxin-sensitive sodium channel inhibitor. Toxicon 71 57–65. 10.1016/j.toxicon.2013.05.015 [DOI] [PubMed] [Google Scholar]
- Deuis J. R., Dekan Z., Wingerd J. S., Smith J. J., Munasinghe N. R., Bhola R. F., et al. (2017). Pharmacological characterisation of the highly NaV1.7 selective spider venom peptide pN3a. Sci. Rep. 7:40883. 10.1038/srep40883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diochot S., Drici M. D., Moinier D., Fink M., Lazdunski M. (1999). Effects of phrixotoxins on the KV4 family of potassium channels and implications for the role of Ito1 in cardiac electrogenesis. Br. J. Pharmacol. 126 251–263. 10.1038/sj.bjp.0702283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dongol Y., Dhananjaya B. L., Shrestha R. K., Aryal G. (2016). Wasp venom toxins as a potential therapeutic agent. Protein Pept. Lett. 23 688–698. 10.2174/0929866523666160511151039 [DOI] [PubMed] [Google Scholar]
- Drenth J. P., Waxman S. G. (2007). Mutations in sodium-channel gene SCN9A cause a spectrum of human genetic pain disorders. J. Clin. Invest. 117 3603–3609. 10.1172/JCI33297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faber C. G., Lauria G., Merkies I. S., Cheng X., Han C., Ahn H. S., et al. (2012). Gain-of-function NaV1.8 mutations in painful neuropathy. Proc. Natl. Acad. Sci. U.S.A. 109 19444–19449. 10.1073/pnas.1216080109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flinspach M., Xu Q., Piekarz A. D., Fellows R., Hagan R., Gibbs A., et al. (2017). Insensitivity to pain induced by a potent selective closed-state NaV1.7 inhibitor. Sci. Rep. 7:39662. 10.1038/srep39662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goncalves T. C., Benoit E., Kurz M., Lucarain L., Fouconnier S., Combemale S., et al. (2019). From identification to functional characterization of cyriotoxin-1a, an antinociceptive toxin from Cyriopagopus schioedtei spider. Br. J. Pharmacol. 10.1111/bph.14628 [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grasso G., Landi A., Alafaci C. (2016). A novel pathophysiological mechanism contributing to trigeminal neuralgia. Mol. Med. 22 452–454. 10.2119/molmed.2016.00172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henriques S. T., Deplazes E., Lawrence N., Cheneval O., Chaousis S., Inserra M., et al. (2016). Interaction of tarantula venom peptide ProTx-II with lipid membranes is a prerequisite for its inhibition of human voltage-gated sodium channel NaV1.7. J. Biol. Chem. 291 17049–17065. 10.1074/jbc.M116.729095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J., Han C., Estacion M., Vasylyev D., Hoeijmakers J. G., Gerrits M. M., et al. (2014). Gain-of-function mutations in sodium channel NaV1.9 in painful neuropathy. Brain 137 1627–1642. 10.1093/brain/awu079 [DOI] [PubMed] [Google Scholar]
- Kessler P., Marchot P., Silva M., Servent D. (2017). The three-finger toxin fold: a multifunctional structural scaffold able to modulate cholinergic functions. J. Neurochem. 142(Suppl. 2) 7–18. 10.1111/jnc.13975 [DOI] [PubMed] [Google Scholar]
- Klein-Weigel P. F., Volz T. S., Richter J. G. (2018). Erythromelalgia. Vasa 47 91–97. 10.1024/0301-1526/a000675 [DOI] [PubMed] [Google Scholar]
- Klint J. K., Chin Y. K., Mobli M. (2015a). Rational engineering defines a molecular switch that is essential for activity of spider-venom peptides against the analgesics target NaV1.7. Mol. Pharmacol. 88 1002–1010. 10.1124/mol.115.100784 [DOI] [PubMed] [Google Scholar]
- Klint J. K., Smith J. J., Vetter I., Rupasinghe D. B., Er S. Y., Senff S., et al. (2015b). Seven novel modulators of the analgesic target NaV1.7 uncovered using a high-throughput venom-based discovery approach. Br. J. Pharmacol. 172 2445–2458. 10.1111/bph.13081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klint J. K., Senff S., Rupasinghe D. B., Er S. Y., Herzig V., Nicholson G. M., et al. (2012). Spider-venom peptides that target voltage-gated sodium channels: pharmacological tools and potential therapeutic leads. Toxicon 60 478–491. 10.1016/j.toxicon.2012.04.337 [DOI] [PubMed] [Google Scholar]
- Lampe R. A. (1998). Analgesic peptides from venom of Grammostola spatulata and use thereof. U.S. Patent No 5807821A. [Google Scholar]
- Lee S. Y., MacKinnon R. (2004). A membrane-access mechanism of ion channel inhibition by voltage sensor toxins from spider venom. Nature 430 232–235. 10.1038/nature02632 [DOI] [PubMed] [Google Scholar]
- Li D., Xiao Y., Xu X., Xiong X., Lu S., Liu Z., et al. (2004). Structure-activity relationships of hainantoxin-IV and structure determination of active and inactive sodium channel blockers. J. Biol. Chem. 279 37734–37740. 10.1074/jbc.M405765200 [DOI] [PubMed] [Google Scholar]
- Liao Z., Yuan C., Deng M., Li J., Chen J., Yang Y., et al. (2006). Solution structure and functional characterization of jingzhaotoxin-XI: a novel gating modifier of both potassium and sodium channels. Biochemistry 45 15591–15600. 10.1021/bi061457+ [DOI] [PubMed] [Google Scholar]
- Liu Y., Tang J., Zhang Y., Xun X., Tang D., Peng D., et al. (2014a). Synthesis and analgesic effects of μ-TRTX-Hhn1b on models of inflammatory and neuropathic pain. Toxins 6 2363–2378. 10.3390/toxins6082363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y., Wu Z., Tang D., Xun X., Liu L., Li X., et al. (2014b). Analgesic effects of Huwentoxin-IV on animal models of inflammatory and neuropathic pain. Protein Pept. Lett. 21 153–158. [DOI] [PubMed] [Google Scholar]
- Liu Z., Cai T., Zhu Q., Deng M., Li J., Zhou X., et al. (2013). Structure and function of hainantoxin-III, a selective antagonist of neuronal tetrodotoxin-sensitive voltage-gated sodium channels isolated from the Chinese bird spider Ornithoctonus hainana. J. Biol. Chem. 288 20392–20403. 10.1074/jbc.M112.426627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luiz A. P., Wood J. N. (2016). Sodium channels in pain and cancer: new therapeutic opportunities. Adv. Pharmacol. 75 153–178. 10.1016/bs.apha.2015.12.006 [DOI] [PubMed] [Google Scholar]
- Meisler M. H., Kearney J. A. (2005). Sodium channel mutations in epilepsy and other neurological disorders. J. Clin. Invest. 115 2010–2017. 10.1172/JCI25466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng P., Huang H., Wang G., Yang S., Lu Q., Liu J., et al. (2016). A novel toxin from Haplopelma lividum selectively inhibits the NaV1.8 channel and possesses potent analgesic efficacy. Toxins 9:7. 10.3390/toxins9010007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Middleton R. E., Warren V. A., Kraus R. L., Hwang J. C., Liu C. J., Dai G., et al. (2002). Two tarantula peptides inhibit activation of multiple sodium channels. Biochemistry 41 14734–14747. 10.1021/bi026546a [DOI] [PubMed] [Google Scholar]
- Mihailescu M., Krepkiy D., Milescu M., Gawrisch K., Swartz K. J., White S. (2014). Structural interactions of a voltage sensor toxin with lipid membranes. Proc. Natl. Acad. Sci. U.S.A. 111 E5463–E5470. 10.1073/pnas.1415324111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milescu M., Bosmans F., Lee S., Alabi A. A., Kim J. I., Swartz K. J. (2009). Interactions between lipids and voltage sensor paddles detected with tarantula toxins. Nat. Struct. Mol. Biol. 16 1080–1085. 10.1038/nsmb.1679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minassian N. A., Gibbs A., Shih A. Y., Liu Y., Neff R. A., Sutton S. W., et al. (2013). Analysis of the structural and molecular basis of voltage-sensitive sodium channel inhibition by the spider toxin huwentoxin-IV (μ-TRTX-Hh2a). J. Biol. Chem. 288 22707–22720. 10.1074/jbc.M113.461392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyer B. D., Murray J. K., Ligutti J., Andrews K., Favreau P., Jordan J. B., et al. (2018). Pharmacological characterization of potent and selective NaV1.7 inhibitors engineered from Chilobrachys jingzhao tarantula venom peptide JzTx-V. PLoS One 13:e0196791. 10.1371/journal.pone.0196791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller A., Starobova H., Morgan M., Dekan Z., Cheneval O., Schroeder C. I., et al. (2019). Anti-allodynic effects of the selective NaV1.7 inhibitor Pn3a in a mouse model of acute post-surgical pain: evidence for analgesic synergy with opioids and baclofen. Pain [Epub ahead of print]. 10.1097/j.pain.0000000000001567 [DOI] [PubMed] [Google Scholar]
- Murray J. K., Ligutti J., Liu D., Zou A., Poppe L., Li H., et al. (2015). Engineering potent and selective analogues of GpTx-1, a tarantula venom peptide antagonist of the NaV1.7 sodium channel. J. Med. Chem. 58 2299–2314. 10.1021/jm501765v [DOI] [PubMed] [Google Scholar]
- Murray J. K., Long J., Zou A., Ligutti J., Andrews K. L., Poppe L., et al. (2016). Single residue substitutions that confer voltage-gated sodium ion channel subtype selectivity in the NaV1.7 inhibitory peptide GpTx-1. J. Med. Chem. 59 2704–2717. 10.1021/acs.jmedchem.5b01947 [DOI] [PubMed] [Google Scholar]
- Ono S., Kimura T., Kubo T. (2011). Characterization of voltage-dependent calcium channel blocking peptides from the venom of the tarantula Grammostola rosea. Toxicon 58 265–276. 10.1016/j.toxicon.2011.06.006 [DOI] [PubMed] [Google Scholar]
- Osteen J. D., Herzig V., Gilchrist J., Emrick J. J., Zhang C., Wang X., et al. (2016). Selective spider toxins reveal a role for the NaV1.1 channel in mechanical pain. Nature 534 494–499. 10.1038/nature17976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J. H., Carlin K. P., Wu G., Ilyin V. I., Musza L. L., Blake P. R., et al. (2014). Studies examining the relationship between the chemical structure of protoxin II and its activity on voltage gated sodium channels. J. Med. Chem. 57 6623–6631. 10.1021/jm500687u [DOI] [PubMed] [Google Scholar]
- Peng K., Shu Q., Liu Z., Liang S. (2002). Function and solution structure of huwentoxin-IV, a potent neuronal tetrodotoxin (TTX)-sensitive sodium channel antagonist from Chinese bird spider Selenocosmia huwena. J. Biol. Chem. 277 47564–47571. 10.1074/jbc.M204063200 [DOI] [PubMed] [Google Scholar]
- Phatarakijnirund V., Mumm S., Mcalister W. H., Novack D. V., Wenkert D., Clements K. L., et al. (2016). Congenital insensitivity to pain: fracturing without apparent skeletal pathobiology caused by an autosomal dominant, second mutation in SCN11A encoding voltage-gated sodium channel 1.9. Bone 84 289–298. 10.1016/j.bone.2015.11.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platnick N. I. (2014). The World Spider Catalog, version 15. New York, NY: American Museum of Natural History. [Google Scholar]
- Priest B. T., Blumenthal K. M., Smith J. J., Warren V. A., Smith M. M. (2007). ProTx-I and ProTx-II: gating modifiers of voltage-gated sodium channels. Toxicon 49 194–201. 10.1016/j.toxicon.2006.09.014 [DOI] [PubMed] [Google Scholar]
- Rahnama S., Deuis J. R., Cardoso F. C., Ramanujam V., Lewis R. J., Rash L. D., et al. (2017). The structure, dynamics and selectivity profile of a NaV1.7 potency-optimised huwentoxin-IV variant. PLoS One 12:e0173551. 10.1371/journal.pone.0173551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redaelli E., Cassulini R. R., Silva D. F., Clement H., Schiavon E., Zamudio F. Z., et al. (2010). Target promiscuity and heterogeneous effects of tarantula venom peptides affecting Na+ and K+ ion channels. J. Biol. Chem. 285 4130–4142. 10.1074/jbc.M109.054718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revell J. D., Lund P. E., Linley J. E., Metcalfe J., Burmeister N., Sridharan S., et al. (2013). Potency optimization of Huwentoxin-IV on hNaV1.7: a neurotoxin TTX-S sodium-channel antagonist from the venom of the Chinese bird-eating spider Selenocosmia huwena. Peptides 44 40–46. 10.1016/j.peptides.2013.03.011 [DOI] [PubMed] [Google Scholar]
- Rong M., Chen J., Tao H., Wu Y., Jiang P., Lu M., et al. (2011). Molecular basis of the tarantula toxin jingzhaotoxin-III (β-TRTX-Cj1α) interacting with voltage sensors in sodium channel subtype Nav1.5. FASEB J. 25 3177–3185. 10.1096/fj.10-178848 [DOI] [PubMed] [Google Scholar]
- Rong M., Duan Z., Chen J., Li J., Xiao Y., Liang S. (2013). Native pyroglutamation of huwentoxin-IV: a post-translational modification that increases the trapping ability to the sodium channel. PLoS One 8:e65984. 10.1371/journal.pone.0065984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu J. H., Jung H. J., Konishi S., Kim H. H., Park Z. Y., Kim J. I. (2017). Structure-activity relationships of ω-Agatoxin IVA in lipid membranes. Biochem. Biophys. Res. Commun. 482 170–175. 10.1016/j.bbrc.2016.11.025 [DOI] [PubMed] [Google Scholar]
- Saitou N., Nei M. (1987). The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4 406–425. [DOI] [PubMed] [Google Scholar]
- Salvatierra J., Castro J., Erickson A., Li Q., Braz J., Gilchrist J., et al. (2018). NaV1.1 inhibition can reduce visceral hypersensitivity. JCI Insight 3:121000. 10.1172/jci.insight.121000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmalhofer W. A., Calhoun J., Burrows R., Bailey T., Kohler M. G., Weinglass A. B., et al. (2008). ProTx-II, a selective inhibitor of NaV1.7 sodium channels, blocks action potential propagation in nociceptors. Mol. Pharmacol. 74 1476–1484. 10.1124/mol.108.047670 [DOI] [PubMed] [Google Scholar]
- Shcherbatko A., Rossi A., Foletti D., Zhu G., Bogin O., Galindo Casas M., et al. (2016). Engineering highly potent and selective microproteins against NaV1.7 sodium channel for treatment of pain. J. Biol. Chem. 291 13974–13986. 10.1074/jbc.M116.725978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen H., Li Z., Jiang Y., Pan X., Wu J., Cristofori-Armstrong B., et al. (2018). Structural basis for the modulation of voltage-gated sodium channels by animal toxins. Science 362:eaau2596. 10.1126/science.aau2596 [DOI] [PubMed] [Google Scholar]
- Shen H., Liu D., Wu K., Lei J., Yan N. (2019). Structures of human NaV1.7 channel in complex with auxiliary subunits and animal toxins. Science 363 1303–1308. 10.1126/science.aaw2493 [DOI] [PubMed] [Google Scholar]
- Sievers F., Wilm A., Dineen D., Gibson T. J., Karplus K., Li W., et al. (2011). Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7:539. 10.1038/msb.2011.75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloan G., Shillo P., Selvarajah D., Wu J., Wilkinson I. D., Tracey I., et al. (2018). A new look at painful diabetic neuropathy. Diabetes Res. Clin. Pract. 144 177–191. 10.1016/j.diabres.2018.08.020 [DOI] [PubMed] [Google Scholar]
- Smith J. J., Alphy S., Seibert A. L., Blumenthal K. M. (2005). Differential phospholipid binding by site 3 and site 4 toxins. Implications for structural variability between voltage-sensitive sodium channel domains. J. Biol. Chem. 280 11127–11133. 10.1074/jbc.M412552200 [DOI] [PubMed] [Google Scholar]
- Smith J. J., Cummins T. R., Alphy S., Blumenthal K. M. (2007). Molecular interactions of the gating modifier toxin ProTx-II with NaV1.5: implied existence of a novel toxin binding site coupled to activation. J. Biol. Chem. 282 12687–12697. 10.1074/jbc.M610462200 [DOI] [PubMed] [Google Scholar]
- Sousa S. R., Wingerd J. S., Brust A., Bladen C., Ragnarsson L., Herzig V., et al. (2017). Discovery and mode of action of a novel analgesic β-toxin from the African spider Ceratogyrus darlingi. PLoS One 12:e0182848. 10.1371/journal.pone.0182848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szepetowski P. (2018). Genetics of human epilepsies: continuing progress. Presse Med. 47 218–226. 10.1016/j.lpm.2017.10.020 [DOI] [PubMed] [Google Scholar]
- Tanaka K., Sekino S., Ikegami M., Ikeda H., Kamei J. (2015). Antihyperalgesic effects of ProTx-II, a NaV1.7 antagonist, and A803467, a NaV1.8 antagonist, in diabetic mice. J. Exp. Pharmacol. 7 11–16. 10.2147/JEP.S79973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson C. H., Kahlig K. M., George A. L., Jr. (2011). SCN1A splice variants exhibit divergent sensitivity to commonly used antiepileptic drugs. Epilepsia 52 1000–1009. 10.1111/j.1528-1167.2011.03040.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright Z. V. F., Mccarthy S., Dickman R., Reyes F. E., Sanchez-Martinez S., Cryar A., et al. (2017). The role of disulfide bond replacements in analogues of the tarantula toxin ProTx-II and their effects on inhibition of the voltage-gated sodium ion channel NaV1.7. J. Am. Chem. Soc. 139 13063–13075. 10.1021/jacs.7b06506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Y., Bingham J. P., Zhu W., Moczydlowski E., Liang S., Cummins T. R. (2008a). Tarantula huwentoxin-IV inhibits neuronal sodium channels by binding to receptor site 4 and trapping the domain II voltage sensor in the closed configuration. J. Biol. Chem. 283 27300–27313. 10.1074/jbc.M708447200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Y., Luo X., Kuang F., Deng M., Wang M., Zeng X., et al. (2008b). Synthesis and characterization of huwentoxin-IV, a neurotoxin inhibiting central neuronal sodium channels. Toxicon 51 230–239. [DOI] [PubMed] [Google Scholar]
- Xiao Y., Blumenthal K., Jackson J. O., II, Liang S., Cummins T. R. (2010). The tarantula toxins ProTx-II and huwentoxin-IV differentially interact with human NaV1.7 voltage sensors to inhibit channel activation and inactivation. Mol. Pharmacol. 78 1124–1134. 10.1124/mol.110.066332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Y., Tang J., Hu W., Xie J., Maertens C., Tytgat J., et al. (2005). Jingzhaotoxin-I, a novel spider neurotoxin preferentially inhibiting cardiac sodium channel inactivation. J. Biol. Chem. 280 12069–12076. 10.1074/jbc.M411651200 [DOI] [PubMed] [Google Scholar]
- Xiao Y., Tang J., Yang Y., Wang M., Hu W., Xie J., et al. (2004). Jingzhaotoxin-III, a novel spider toxin inhibiting activation of voltage-gated sodium channel in rat cardiac myocytes. J. Biol. Chem. 279 26220–26226. 10.1074/jbc.M401387200 [DOI] [PubMed] [Google Scholar]
- Xu H., Li T., Rohou A., Arthur C. P., Tzakoniati F., Wong E., et al. (2019). Structural basis of NaV1.7 inhibition by a gating-modifier spider toxin. Cell 176 1238–1239. 10.1016/j.cell.2019.01.047 [DOI] [PubMed] [Google Scholar]
- Yuan C., Yang S., Liao Z., Liang S. (2007). Effects and mechanism of Chinese tarantula toxins on the KV2.1 potassium channels. Biochem. Biophys. Res. Commun. 352 799–804. 10.1016/j.bbrc.2006.11.086 [DOI] [PubMed] [Google Scholar]
- Zhang F., Liu Y., Zhang C., Li J., Yang Z., Gong X., et al. (2015). Natural mutations change the affinity of μ-theraphotoxin-Hhn2a to voltage-gated sodium channels. Toxicon 93 24–30. 10.1016/j.toxicon.2014.11.220 [DOI] [PubMed] [Google Scholar]
- Zhang Y., Peng D., Huang B., Yang Q., Zhang Q., Chen M., et al. (2018a). Discovery of a novel NaV1.7 inhibitor from Cyriopagopus albostriatus venom with potent analgesic efficacy. Front. Pharmacol. 9:1158. 10.3389/fphar.2018.01158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Yang Q., Zhang Q., Peng D., Chen M., Liang S., et al. (2018b). Engineering gain-of-function analogues of the spider venom peptide HNTX-I, a potent blocker of the hNaV1.7 sodium channel. Toxins 10:E358. 10.3390/toxins10090358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y. X., Peng D. Z., Zhang Q. F., Huang B., Yang Q. C., Tang D. F., et al. (2018c). μ-TRTX-Ca1a: a novel neurotoxin from Cyriopagopus albostriatus with analgesic effects. Acta Pharmacol. Sin. 10.1038/s41401-018-0181-9 [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegman R., Alewood P. (2015). Bioactive components in fish venoms. Toxins 7 1497–1531. 10.3390/toxins7051497 [DOI] [PMC free article] [PubMed] [Google Scholar]