

Abstract
In contrast to organic cages which are formed by exploiting dynamic covalent chemistry, such as boronic ester cages, imine cages, or disulfide cages, those with a fully carbonaceous backbone are rarer. With the exception of alkyne metathesis based approaches, the vast majority of hydrocarbon cages need to be synthesized by kinetically controlled bond formation. This strategy implies a multiple step synthesis and no correction mechanism in the final macrocyclization step, both of which are responsible for low overall yields. Whereas for smaller cages the intrinsic drawbacks are not always obvious, larger cages are seldom synthesized in yields beyond a few tenths of a percent. Presented herein is a three‐step method to convert imine cages into hydrocarbon cages. The method has been successfully applied to even larger structures such as derivatives of C72H72 , an unknown cage suggested by Fritz Vögtle more than 20 years ago.
Keywords: cage compounds, hydrocarbons, imines, macrocycles, reduction
The interest in organic cages has risen in recent years not the least because some of them became accessible in good yields from rather simple building blocks by applying dynamic covalent chemistry (DCC) reactions, such as the formation of imines from amines and aldehydes,1 boronic esters from boronic acids and diols,2 or disulfides from thiols,3 to name a few.4 All these cages have in common that the constructive elements are typically based on bonds with at least one heteroatom. In contrast, cages with pure carbon atom backbones are rarer and, with a few exceptions by exploiting the reversibility of alkyne metathesis,5 need to be synthesized by irreversible formation of C−C bonds.6 One of the earliest examples of a hydrocarbon cage synthesis is the approach of Olof Wennerström et al.:7 In a one‐pot reaction 1,3,5‐triformyl benzene underwent a sixfold Wittig reaction with 1,3‐bisbenzylphosphonium bromide, giving the helical olefin cage 1 in less than 2 % yield upon isolation (Figure 1 a, top). The follow‐up reductive transformation into the ethylene‐bridged compound 2 was nearly quantitative. Ten years later, Vögtle et al. presented the seven‐step synthesis of a tetrahedral hydrocarbon cage C36H36, where four benzene rings are connected by ethylene units (Figure 1 a, bottom).8 The overall yield of this route was about 3 %, because the limiting step is the final macrocyclization or cage formation, which was achieved in only 11 % despite using high‐dilution conditions. Nevertheless, as Vögtle et al. have pointed out, the scaffold of this hydrocarbon cage is a cut‐out structure of fullerene C60 and therefore attractive as a potential precursor to synthesize it (Figure 1 b). In follow‐up contributions larger related structures were proposed, and some of them (a C54H48 and a C60H60) synthesized,9 again in ten to fourteen consecutive steps with overall yields of less than 0.1 % in both cases. Among the suggested structures was also a larger “cubic” C72H72 with eight benzene rings connected by ethylene bridges (Figure 1 a), the synthesis of which has not been reported to date. Similar to how the tetrahedral C36H36 is a cut‐out structure of fullerene C60, the C72H72 is related to a cubic C120 derivative containing six cyclooctatetraene rings (Figure 1 c).10
Figure 1.
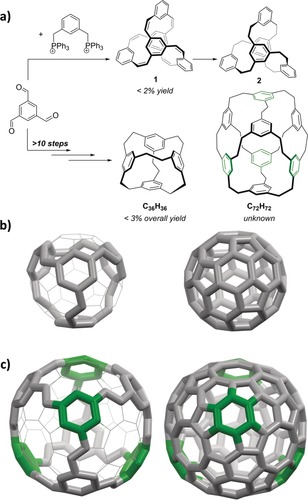
a) Reported syntheses of the helical cages 1 and 2 7 and tetrahedral cage C36H36,8 and the unknown structure C72H72, suggested by Vögtle et al.9 b) Scaffold of the tetrahedral cage C36H36 and fullerene C60 for comparison. c) Scaffold of cubic cage C72H72 and fullerene C120 for comparison.
Although the synthetic transformation of C72H72 into C120 is very challenging, the latter is an attractive compound, making it worthwhile to work on new methods for generating such ethylene‐bridged hydrocarbon cages in shorter and thus more efficient synthetic routes, which will be presented herein. In classical cyclophane chemistry an often‐used strategy is the late‐stage ring contraction under either elimination or extrusion of small heteroatomic molecules to generate the ethylene bridges, even for making strained cyclophanes. Among the possibilities to realize the C−C‐bond formation is the extrusion of nitrosamines, developed by Overberger et al.,11 as has been reported by Takemura et al. to form [2.2]‐meta or [2.2]‐para cyclophanes.12 We investigated this reaction to convert imine cages via nitrosamine cages into hydrocarbon cages.
The first target was the transformation of [2+3] imine cage 5 a 11 into the Wennerström‐type hydrocarbon cage 8 a (Figure 2). The imine cage 5 a was synthesized in 83 % from the reaction of triamine 3 and isophthaldehyde 4 a.11a The follow‐up reduction to the amine cage 6 a occurred in 93 % yield by using NaBH4 as a reducing agent.11a Exhaustive nitrosylation of the secondary amine functions could either be achieved with sodium nitrite and hydrochloric acid or neat isoamyl nitrite, giving 48 or 68 % yield, respectively, of the nitrosylated cage 7 a. The analytics of the transformation into 7 a is restricted to IR spectroscopy, DOSY NMR spectroscopy, mass spectrometry, and elemental analysis. Although 1H and 13C NMR spectra of various batches of the product looked always nearly identical, such as fingerprints (see Figure 2 b and the Supporting Information), no reasonable peak assignment can be done because for each nitrosamine group there exist two thermally stable E/Z isomers resulting from a partial N=N double bond character.12 Therefore, by nitrosamine formation there are 26=64 different possible isomers for 7 a, which explains the complicated 1H and 13C NMR spectra. Investigations by DOSY NMR spectroscopy revealed for all peaks the same diffusion coefficient (D=9.16 m2 s−1), thus all isomers have a very similar solvodynamic radius (r S=0.58 nm), which is expected (see the Supporting Information). Nitrosamine formation could be clearly found by two bands at 1443 and 1134 cm−1 in the IR spectrum.13 By mass spectrometry a peak at m/z=1001.512 was detected, matching the expected m/z ratio of 1001.513 for a sixfold nitrosylation (see the Supporting Information). Furthermore, the elemental analysis is in the expected range of specification. The final step of the transformation of the imine cage into a hydrocarbon cage is the reductive elimination of the nitrosylamine unit by ring contraction, and it was performed by using the original conditions (EtOH, NaOHaq, Na2S2O4, reflux) of Overberger et al.14 Besides the desired hydrocarbon cage 8 a (25 % yield), the mono‐nitrosamine cage 9 was isolated in 20 % yield. Any attempts to perform the ring contraction on this compound by applying either the same or similar reaction conditions gave only minor conversion (about 20 %, see Figure S98 in the Supporting Information). This observation implies that the absolute E/Z configuration of the nitrosamine unit plays a yet underestimated role in the reaction mechanism, which was postulated to occur by the formation of a diazene (or N‐nitrene) as an intermediate and therefore would not be affected by the stereochemistry of the nitrosamine groups.14 However, more detailed studies need to be done to obtain further mechanistic insights. The overall yield of the three‐step method is 13 %, thus approximately ten times higher than for the reported two‐step approach of Wennerström et al.7 Other [2+3] imine cages, such as the pyridine cage 5 b 11b can be transformed within three steps in the same way to the corresponding hydrocarbon cage 8 b in yields of 53 % (overall 24 %). And even a cage with sterically more demanding methoxy groups on the interior (5 c) was transformed by ring contraction in 20 % in the last step into 8 c (Figure 2 a). The much higher yields of the Overberger rearrangement of 7 b provides a hint that the electronic demand of the aromatic rings may stabilize the intermediates. To further elucidate this, more diverse imine cages with additional substituents need to be investigated.
Figure 2.
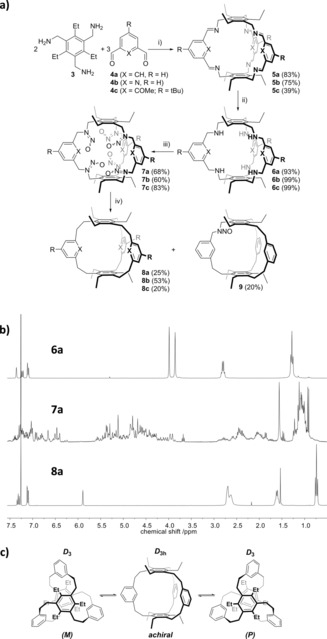
a) Synthesis of the Wennerström‐type carbon cages 8 a–c. i) MeOH, room temp., 2 d (8 a, 8 b) or 12 h (8 c); ii) NaBH4, MeOH, 12 h; iii) isoamyl nitrite, 50 °C (8 a,8 b), or 60 °C (8 c) 12 h; iv) Na2S2O4, NaOHaq (20 %), EtOH, reflux 12 h. b) Comparison of the 1H NMR spectra of 6 a, 7 a, and 8 a in CDCl3 (300 MHz, room temp.). c) Interconversion of D 3‐symmetric helical conformers of 8 a via the achiral D 3h transition.
All small hydrocarbon cages, 8 a–8c, and the mono nitrosamine cage 9 have been characterized by single‐crystal X‐ray diffraction (Figure 3; for crystallographic details see the Supporting Information). The hydrocarbon cage 8 a crystallizes as a racemate of two helical conformers along with two molecules of CHCl3 in the unit cell . All ethyl chains point outside the cage, which is expected because of the high rotation barrier of these found in an alternating fashion.15 The ethylene bridges are arranged in a gauche conformation with torsional angles between 76.7° and 88.1°. The distance between the two coplanar benzene rings on top and bottom is 6.1 Å, and the distances between the inner aromatic hydrogen atoms are about 3.2 Å, which is too small to host larger guest molecules. The cage 9 also crystallized as racemic mixture of the two helical conformers. It is worth mentioning that the NNO, which is conformationally fixed, points with the oxygen towards the threefold ethyl‐substituted ring. The two other cages 8 b and 8 c are of lower symmetry (C 1) in the solid state (Figure 3 c, d).
Figure 3.
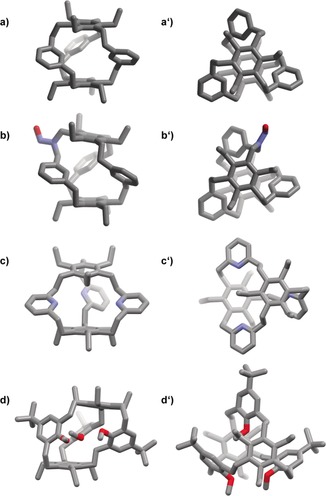
Solid‐state structures of 8 a (a: side view and a′: top view), 9 (b and b′), 8 b (c and c′), and 8 c (d and d′), determined by single‐crystal X‐ray diffraction. Note: hydrogen atoms and solvate molecules are omitted for clarity. Depicted is only one enantiomer of the racemic mixture, each. For crystallographic details, see the Supporting Information.
Using temperature‐dependent 1H NMR spectroscopy, the inversion barrier for 8 a between the helical D 3h‐symmetric conformers via the D 3‐symmetric achiral transition state (Figure 2 c) was determined, by monitoring the coalescence of the two peaks of the ethylene unit. The coalescence occurs at T c=263 K, which corresponds to an inversion barrier of ΔG=51 kJ mol−1 and switching rates of k c=242 and 769 Hz. In comparison to the original measurements of Wennerström′s unsubstituted cage,16 the barrier is 14 kJ mol−1 higher, and can be explained by a further restriction of movement by the additional ethyl chains. The barrier of the pyridyl cage 8 b is somewhat lower at ΔG=40 kJ mol−1 (T c=208 K; k c=195 and 804 Hz), whereas the methoxy cage 8 c does not invert in the temperature range between 223 and 323 K.
We were interested if this method allows the synthesis of even larger structures, such as derivatives of the unknown C72H72 depicted in Figure 1.9b Recently we published the formation of truncated tetrahedral [4+4] imine cages, such as 10, which already contain the eight aromatic rings necessary for this compound.17 Reduction of 10 to the amine 11 with NaBH4 was quantitative as well as the nitrosylation to 12 (Figure 4 a). After performing the Overberger rearrangement, the hydrocarbon cage 13, which is a dodecakis‐ethyl‐substituted derivative of previously unknown C72H72, was isolated in 5 % yield. Considering that this is a 12‐fold reaction, the average yield per C−C bond formation is 77 %, exactly what was reported by Overberger in his original contribution for a single reaction.14 Including the imine cage formation, 13 was accessible from rather simple precursors in only four steps with an overall yield of 1.4 % yield. Although no direct comparison to a reported route exists, one can assume that if it would have been synthesized in a similar way to that of C54H54 it would have been many more steps and most likely with overall yields less than tenth of a thousandth.
Figure 4.
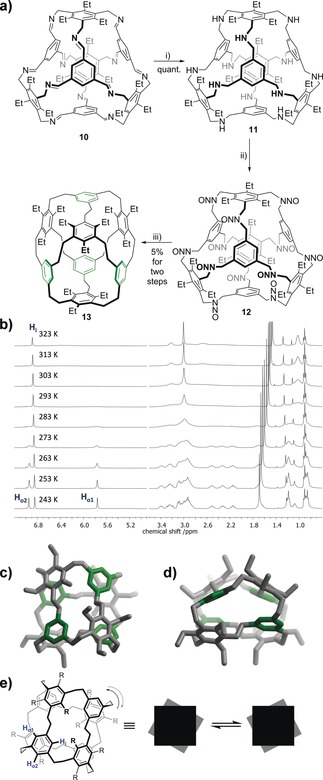
a) Synthesis of the “cubic” cage derivative 13. i) NaBH4, MeOH, reflux, 72 h; ii) tert‐butyl nitrite, 50 °C, 72 h; iii) Na2S2O4, NaOHaq (20 %), EtOH, reflux 72 h. b) 1H NMR spectra of 13 at various temperatures (CDCl3, 400 MHz). c,d) Single‐crystal structures by X‐ray diffraction. Shown is only one conformer each. Hydrogen atoms and solvate molecules are omitted for clarity. To highlight the connectivity, alternating rings are colored in green. For detailed crystallographic information, see the Supporting Information). e) Interconversion of helical conformers and assignment of aromatic protons as detected at 253 K.
The cage 13 crystallizes in the tetragonal unit cell with a flattened molecule of S 4 symmetry. As for the smaller cages 8 a–c and 9, all ethyl groups are pointing outwards the molecular structure. Here, three different conformational orientations of the ethylene bridges can be found, one that is anti‐periplanar with 172.7°, and two that are synclinal oriented with torsional angles of 48.6° and 86.2°. The 1H NMR spectrum of 13 shows a number of broad peaks at room temperature, however when cooled to 253 K only sharp peaks can be detected (Figure 4 b). In the aromatic region, three distinct peaks with the same integrals are resonating at δ=6.95, 6.85, and 5.76 ppm, exactly the number of different peaks that are expected for the S 4‐symmetric molecular structure found in the X‐ray crystal structure (Figures 4 c and d). A closer look at the structure reveals one inner aromatic proton (Hi) pointing towards the ethyl groups and two outer ones, whereas one (Ho1) is located over the center of the adjacent ethyl‐substituted ring at a distance of approximately 3.6 Å, thus experiencing the anisotropic ring current, and thus explaining the up‐field shift to δ=5.76 ppm (Figure 4 b). Upon heating the NMR sample, only the two aromatic peaks Ho1 and Ho2 start to broaden and coalesce at 303 K (k c=1057 Hz, ΔG=57 kJ mol−1), and can be explained by a twisting movement of the two disc‐like parts of the molecule against each other (Figure 4 e).
Finally, the [4+6] tetrahedral imine cage 14 18 was converted within three steps into an even larger C84H84 derivative (17; Figure 5 a) with 4 % yield in the 12‐fold Overberger reaction, which is nearly the same as for 13. The 1H NMR spectrum of 17 at room temperature shows only a few sharp distinct signals, and is a hint of a faster dynamic interconversion of conformers. Indeed, by measurements at variable temperatures a coalescence temperature, T c, of 238 K was found, and correlates to an inversion frequency of k c=120 Hz and an energy of ΔG=48 kJ mol−1. The single‐crystal structure of 17 is a C 2‐symmetric conformer. With the assumption that the para‐substituted phenylene rings (highlighted in blue) can freely rotate in solution along the 1,4‐axis, for the fixed conformer four different NMR signals for the aromatic protons are expected, and is in fact the case at 218 K (δ=7.32, 6.85, 6.10 and 5.95 ppm; see the Supporting Information).
Figure 5.
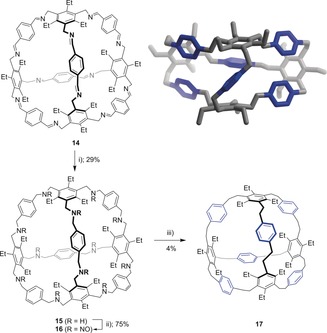
a) Synthesis of the C84H84 derivative 17. i) NaBH4, MeOH, reflux, 2 d; ii) isoamyl nitrite, 50 °C, 12 h; iii) Na2S2O4, NaOHaq (20 %), EtOH, reflux 12 h. b) Single‐crystal structures of 17 by X‐ray diffraction. Shown is only one conformer each. Hydrogen atoms and solvate molecules are omitted for clarity. To highlight the connectivity, alternating rings are colored in blue. For detailed crystallographic information, see the Supporting Information.
In conclusion, we have developed a straightforward synthetic method to transform imine cages into hydrocarbon cages in only three steps, exploiting the high‐yielding imine bond formation in the first step to generate the cage scaffold. To transform the imine cages into hydrocarbon cages, these needed to be reduced and nitrosylated to apply the Overberger reaction to finally form the C−C bonds. By this method, the Wennerström‐type cages 8 a–c were synthesized in 13–24 % overall yield, which is ten to twenty times higher than by the original one‐step method reported before. The real strength of this method is demonstrated in the four‐step synthesis of a C72H72 and C84H84 derivative both of which were previously unknown. The overall yields for the four‐step synthesis of the C72H72 derivative 13, for instance, is with 1.4 % already higher than those reported for smaller C60H60 (0.8 %, 20 steps!).9b Doubtless, carbon cages based on alkyne metathesis can be achieved in much higher yields,5c but it does not allow the synthesis of less symmetric compounds from more than one building block as it is the case with our method, thus providing a protocol that gives the opportunity to access a larger variety of structures. Currently we are re‐investigating the Overberger conditions to finally improve the yields of the carbon cage formation, as well as looking in more detail at the substrate scope.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
The authors would like to thank the European Research Council (ERC) for funding this project in the frame of the consolidators grant CaTs n DOCs (grant agreement no.725765). We thank Alexandra Rowse for proof reading.
T. H. G. Schick, J. C. Lauer, F. Rominger, M. Mastalerz, Angew. Chem. Int. Ed. 2019, 58, 1768.
References
- 1.
- 1a. MacDowell D., Nelson J., Tetrahedron Lett. 1988, 29, 385–386; [Google Scholar]
- 1b. Skowronek P., Gawronski J., Org. Lett. 2008, 10, 4755–4758; [DOI] [PubMed] [Google Scholar]
- 1c. Liu X., Liu Y., Li G., Warmuth R., Angew. Chem. Int. Ed. 2006, 45, 901–904; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 915–918; [Google Scholar]
- 1d. Mastalerz M., Chem. Commun. 2008, 4756–4758. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Christinat N., Scopelliti R., Severin K., Angew. Chem. Int. Ed. 2008, 47, 1848–1852; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 1874–1878; [Google Scholar]
- 2b. Zhang G., Presly O., White F., Oppel I. M., Mastalerz M., Angew. Chem. Int. Ed. 2014, 53, 1516–1520; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 1542–1546; [Google Scholar]
- 2c. Hutin M., Bernardinelli G., Nitschke J. R., Chem. Eur. J. 2008, 14, 4585–4593; [DOI] [PubMed] [Google Scholar]
- 2d. Klotzbach S., Scherpf T., Beuerle F., Chem. Commun. 2014, 50, 12454–12457. [DOI] [PubMed] [Google Scholar]
- 3. Collins M. S., Carnes M. E., Nell B. P., Zakharov L. N., Johnson D. W., Nat. Commun. 2016, 7, 11052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.
- 4a. Mastalerz M., Angew. Chem. Int. Ed. 2010, 49, 5042–5053; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 5164–5175; [Google Scholar]
- 4b. Zhang G., Mastalerz M., Chem. Soc. Rev. 2014, 43, 1934–1947; [DOI] [PubMed] [Google Scholar]
- 4c. Beuerle F., Gole B., Angew. Chem. Int. Ed. 2018, 57, 4850–4878; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 4942–4972; [Google Scholar]
- 4d. Hasell T., Cooper A. I., Nat. Rev. Mater. 2016, 1, 16053; [Google Scholar]
- 4e. Mastalerz M., Acc. Chem. Res. 2018, 51, 2411–2422. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Wang Q., Zhang C., Noll B. C., Long H., Jin Y., Zhang W., Angew. Chem. Int. Ed. 2014, 53, 10663–10667; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 10839–10843; [Google Scholar]
- 5b. Zhang C., Wang Q., Long H., Zhang W., J. Am. Chem. Soc. 2011, 133, 20995–21001; [DOI] [PubMed] [Google Scholar]
- 5c. Lee S., Yang A., Moneypenny T. P., Moore J. S., J. Am. Chem. Soc. 2016, 138, 2182–2185. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Högberg H.-E., Thulin B., Wennerström O., Tetrahedron Lett. 1977, 18, 931–934; [Google Scholar]
- 6b. Wu Z., Moore J. S., Angew. Chem. Int. Ed. Engl. 1996, 35, 297–299; [Google Scholar]; Angew. Chem. 1996, 108, 320–322; [Google Scholar]
- 6c. Wu Z., Lee S., Moore J. S., J. Am. Chem. Soc. 1992, 114, 8730–8732; [Google Scholar]
- 6d. Rubin Y., Parker T. C., Khan S. I., Holliman C. L., McElvany S. W., J. Am. Chem. Soc. 1996, 118, 5308–5309; [Google Scholar]
- 6e. Matsui K., Segawa Y., Itami K., J. Am. Chem. Soc. 2014, 136, 16452–16458; [DOI] [PubMed] [Google Scholar]
- 6f. Matsui K., Segawa Y., Namikawa T., Kamada K., Itami K., Chem. Sci. 2013, 4, 84–88; [Google Scholar]
- 6g. Tobe Y., Nakagawa N., − J. y. Kishi, M. Sonoda, K. Naemura, T. Wakabayashi, T. Shida, Y. Achiba, Tetrahedron 2001, 57, 3629–3636; [Google Scholar]
- 6h. Tobe Y., Nakagawa N., Naemura K., Wakabayashi T., Shida T., Achiba Y., J. Am. Chem. Soc. 1998, 120, 4544–4545; [Google Scholar]
- 6i. Rubin Y., Parker T. C., Pastor S. J., Jalisatgi S., Boulle C., Wilkins C. L., Angew. Chem. Int. Ed. 1998, 37, 1226–1229; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 1998, 110, 1353–1356; [Google Scholar]
- 6j. Kayahara E., Iwamoto T., Takaya H., Suzuki T., Fujitsuka M., Majima T., Yasuda N., Matsuyama N., Seki S., Yamago S., Nat. Commun. 2013, 4, 2694; [DOI] [PubMed] [Google Scholar]
- 6k. Cremers J., Haver R., Rickhaus M., Gong J. Q., Favereau L., Peeks M. D., Claridge T. D. W., Herz L. M., Anderson H. L., J. Am. Chem. Soc. 2018, 140, 5352–5355; [DOI] [PubMed] [Google Scholar]
- 6l. Sato H., Bender J. A., Roberts S. T., Krische M. J., J. Am. Chem. Soc. 2018, 140, 2455–2459; [DOI] [PubMed] [Google Scholar]
- 6m. Tomoya M., Shoko K., Isao A., Soichiro W., ChemistryOpen 2018, 7, 278–281;29657913 [Google Scholar]
- 6n. Gu X., Gopalakrishna T. Y., Phan H., Ni Y., Herng T. S., Ding J., Wu J., Angew. Chem. Int. Ed. 2017, 56, 15383–15387; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 15585–15589; [Google Scholar]
- 6o. Avellaneda A., Valente P., Burgun A., Evans J. D., Markwell-Heys A. W., Rankine D., Nielsen D. J., Hill M. R., Sumby C. J., Doonan C. J., Angew. Chem. Int. Ed. 2013, 52, 3746–3749; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 3834–3837; [Google Scholar]
- 6p. Burgun A., Valente P., Evans J. D., Huang D. M., Sumby C. J., Doonan C. J., Chem. Commun. 2016, 52, 8850–8853; [DOI] [PubMed] [Google Scholar]
- 6q. Avellaneda A., Valente P., Burgun A., Evans J. D., Markwell-Heys A. W., Rankine D., Nielsen D. J., Hill M. R., Sumby C. J., Doonan C. J., Angew. Chem. Int. Ed. 2013, 52, 3746–3749; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 3834–3837; [Google Scholar]
- 6r. Zhang C., Chen C.-F., J. Org. Chem. 2007, 72, 9339–9341. [DOI] [PubMed] [Google Scholar]
- 7. Högberg H.-E., Wennerstroem O., Acta Chem. Scand. Ser. B 1982, 36, 661–667. [Google Scholar]
- 8. Vögtle F., Groß J., Seel C., Nieger M., Angew. Chem. Int. Ed. Engl. 1992, 31, 1069–1071; [Google Scholar]; Angew. Chem. 1992, 104, 1112–1113. [Google Scholar]
- 9.
- 9a. Gross J., Harder G., Vögtle F., Stephan H., Gloe K., Angew. Chem. Int. Ed. Engl. 1995, 34, 481–484; [Google Scholar]; Angew. Chem. 1995, 107, 523–526; [Google Scholar]
- 9b. Gross J., Harder G., Siepen A., Harren J., Vögtle F., Stephan H., Gloe K., Ahlers B., Cammann K., Rissanen K., Chem. Eur. J. 1996, 2, 1585–1595. [Google Scholar]
- 10. Szefler B., Diudea M., Molecules 2014, 19, 15468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.
- 11a. Mateus P., Delgado R., Brandão P., Carvalho S., Félix V., Org. Biomol. Chem. 2009, 7, 4661–4673; [DOI] [PubMed] [Google Scholar]
- 11b. Mateus P., Delgado R., Brandão P., Félix V., J. Org. Chem. 2009, 74, 8638–8646; [DOI] [PubMed] [Google Scholar]
- 11c. De Rycke N., Marrot J., Couty F., David O. R. P., Tetrahedron Lett. 2010, 51, 6521–6525. [Google Scholar]
- 12. Looney C. E., Phillips W. D., Reilly E. L., J. Am. Chem. Soc. 1957, 79, 6136–6142. [Google Scholar]
- 13. Williams R. L., Pace R. J., Jeacocke G. J., Spectrochim. Acta 1964, 20, 225–236. [Google Scholar]
- 14. Overberger C. G., Lombardino J. G., Hiskey R. G., J. Am. Chem. Soc. 1958, 80, 3009–3012. [Google Scholar]
- 15. Wang X., Hof F., Beilstein J. Org. Chem. 2012, 8, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Olsson T., Tanner D., Thulin B., Wennerstrōm O., Tetrahedron 1981, 37, 3485–3490. [Google Scholar]
- 17. Lauer J. C., Zhang W.-S., Rominger F., Schroeder R. R., Mastalerz M., Chem. Eur. J. 2018, 24, 1816–1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Greenaway R. L., Santolini V., Bennison M. J., Alston B. M., Pugh C. J., Little M. A., Miklitz M., Eden-Rump E. G. B., Clowes R., Shakil A., Cuthbertson H. J., Armstrong H., Briggs M. E., Jelfs K. E., Cooper A. I., Nat. Commun. 2018, 9, 2849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.CCDC 1858590, 1858591, 1858592, 1858593, 1858594, 1858595, 1858596, 1858597, 1858598 (13, 13, 11, 5 a, 8 a, 9, 8 b, 17, 8 c) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre. All other data supporting the findings of this study are available within the Article and its Supplementary Information, or from the corresponding author upon reasonable request.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary