

Abstract
Fluorogenic oligonucleotide probes allow mRNA imaging in living cells. A key challenge is the cellular delivery of probes. Most delivery agents, such as cell‐penetrating peptides (CPPs) and pore‐forming proteins, require interactions with the membrane. Charges play an important role. To explore the influence of charge on fluorogenic properties and delivery efficiency, we compared peptide nucleic acid (PNA)‐ with DNA‐based forced intercalation (FIT) probes. Perhaps counterintuitively, fluorescence signaling by charged DNA FIT probes proved tolerant to CPP conjugation, whereas CPP–FIT PNA conjugates were affected. Live‐cell imaging was performed with a genetically engineered HEK293 cell line to allow the inducible expression of a specific mRNA target. Blob‐like features and high background were recurring nuisances of the tested CPP and lipid conjugates. By contrast, delivery by streptolysin‐O provided high enhancements of the fluorescence of the FIT probe upon target induction. Notably, DNA‐based FIT probes were brighter and more responsive than PNA‐based FIT probes. Optimized conditions enabled live‐cell multicolor imaging of three different mRNA target sequences.
Keywords: bioconjugation, fluorescence, imaging agents, oligonucleotides, RNA
Introduction
Labeled oligonucleotide probes that fluoresce upon hybridization provide the opportunity to detect and visualize mRNA molecules within their native environment.1 Live‐cell measurements enable analysis of dynamic processes and avoid artefacts caused by cell fixation.2 Useful imaging probes should provide strong enhancements of fluorescence upon specific recognition of the complementary target. In addition, the emission should have sufficient brightness to exceed cellular autofluorescence at the selected wavelength.
The most frequently used hybridization probes capitalize on the molecular beacon approach, which involves the spatial separation of two dyes upon target‐induced opening of a hairpin structure.3 To reduce the risk of false‐positive signaling caused by unintentional hairpin opening, we introduced forced intercalation (FIT) probes (Figure 1).4 These single‐labeled hybridization probes lack a hairpin structure, but instead rely on the responsiveness of a TO dye, which is linked to a DNA or PNA scaffold as a nucleobase surrogate.5 We, and others, used FIT probes for live‐cell imaging of viral mRNA,6 cellular mRNA,7 and noncoding RNA.8 Alternative RNA imaging methods include binary probes,9 ECHO probes,10 and reactive probes,11 which typically rely on the dye separation principle for the activation of fluorescence.12
Figure 1.
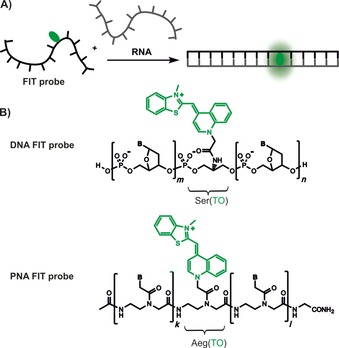
A) A dye nucleotide (green) within the FIT probe fluoresces upon hybridization with a complementary RNA target. B) Chemical structures of DNA and peptide nucleic acid (PNA) FIT probes containing a serinol (Ser)‐ or aminoethylglycine (Aeg)‐linked thiazole orange (TO) dye (green).
Regardless of the mechanism of fluorescence activation applied in RNA imaging technologies, the cellular delivery of probes is the major challenge. Microinjection is a very reliable approach that provides full control over the amount of intracellularly delivered probes. However, microinjection is tedious and practical only for large cells. Owing to its ease, delivery through microporation is a standard method to introduce plasmids into cells.13 This method has also been used for the cellular delivery of molecular beacons.14 However, the electric fields applied during microporation can affect cell viability.
Alternative delivery methods reported to allow live‐cell mRNA imaging involve the addition of chemical or biological agents, such as pore‐forming proteins (e.g., streptolysin‐O (SLO)),15 lipofection,16 lipidation,17 or cell‐penetrating peptides (CPPs).18 A characteristic hallmark is the strong interaction of the reagents with the hybridization probe and/or the cell membrane. Charge interactions play an important role. As a result, the charged nature of the hybridization probe should have a major influence on the efficiency of delivery. For example, the CPPs used in nucleic acid delivery are net positively charged.19 Although CPP‐mediated cellular delivery has been reported to succeed for both polyanionic DNA probes and charge‐neutral PNA probes,20 it seems plausible to anticipate major differences in delivery efficiency. Indeed, it has been reasoned that charge neutrality would be more appropriate for achieving delivery of therapeutic oligonucleotide analogues.19 However, we wish to emphasize that reports describing the delivery of perturbation probes, such as antisense molecules, provide little guidance for the delivery of imaging probes. Minor amounts of perturbation probes within the cytosol can exert the sought‐after biological effect, despite enrichment of the probe in endosomal compartments. On the contrary, imaging probes should provide spatial information and, therefore, must not impose a localization bias.
Once we considered the multitude of delivery modalities, we noticed that the lack of knowledge as to which backbone, DNA or PNA, better suited the demands of mRNA imaging extended beyond CPP‐mediated delivery to methods based on passive diffusion. Owing to the lack of comparative studies, we set out to evaluate both PNA‐ and DNA‐type fluorogenic hybridization probes in live‐cell mRNA imaging. For this purpose, we designed and constructed sequence‐identical FIT probes that were comprised of either DNA or PNA backbones. To enable control measurements, we established a genetically engineered HEK293 cell line that allowed the inducible expression of a mRNA target sequence. By using this cell line, we investigated different transfection methods/conditions and assessed the magnitude of fluorescence increase upon induction of target mRNA expression. This knowledge was used for the development of conditions that allowed the facile cellular delivery of FIT probes for the live‐cell multicolor imaging of three different mRNA targets.
Results and Discussion
Design of probes and cell line
Most endogenous mRNA molecules in cells are expressed at <1000 copy numbers. According to a typical approach, dozens of hybridization probes are directed against different segments of the mRNA molecule to increase the sensitivity of mRNA imaging.21 Alternatively, the mRNA target is equipped with repeated sequence tags at the 5′‐ or 3′‐untranslated region.22 For the design of a sequence tag, we considered the following criteria: 1) the hybridization probe should be GC‐rich to permit the use of short probes, 2) cross reactions with cell‐endogenous transcripts should be avoided by using the basic local alignment search tool (BLAST), and 3) an individual sequence repeat should have sufficient length to avoid self‐quenching of adjacently aligned probes.
The 22 nt long segment (F‐tag) shown in Figure 2 satisfied these criteria. Next, we constructed the 11 nt long FIT‐DNA1 probe. This probe was comprised predominantly of 2′‐OMe units to resist degradation by nucleases inside cells. The probe furthermore contained a single LNA building block,23 which was known to increase the brightness of emission from the adjacent TO nucleotide.7b Hybridization with the synthetic RNA target at 37 °C was accompanied by a 29‐fold enhancement of fluorescence intensity. The value of T M (51.4 °C) was considered to be sufficiently low to avoid base‐mismatched hybridization, yet high enough to enable live‐cell mRNA imaging. Sequence‐identical FIT‐PNA1 did not require the incorporation of special building blocks to confer nuclease resistance. The target affinity of the FIT‐PNA1 probe is comparable to that of the DNA‐based probe FIT‐DNA1 (T M=52.3 °C). A comparison revealed that the extinction of light by TO in the FIT‐PNA1 probe was lower than that in the DNA probe FIT‐DNA1 (Figure 2). This is probably due to more efficient intercalation of TO adjacent to the rigidifying LNA unit in DNA–RNA duplexes.7b Hybridization of FIT‐PNA1 with the RNA target resulted in eightfold enhancement of fluorescence at λ=535 nm. As a result, the brightness of TO emission is higher in the DNA–RNA duplex formed upon hybridization of FIT‐DNA than that in the PNA–RNA duplex formed with FIT‐PNA. This also is in agreement with previous studies, which have shown that the nonconjugated TO dye is a rather modest stain of PNA–DNA duplexes.5b
Figure 2.
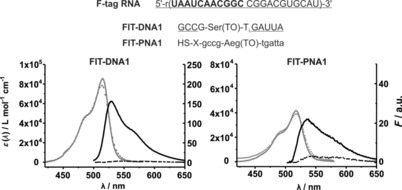
The 22 nt long F‐tag RNA sequence and complementary FIT probes, and the corresponding absorption and emission spectra of probes in the absence (‐ ‐ ‐ ‐) and presence (—) of F‐tag RNA. Small letters indicate PNA monomers; capital letters represent nucleotides. Underlined nucleotides mark 2′‐OMe‐modified nucleotides. Subscript L highlights a locked nucleic acid (LNA) building block. X: (CH2)2CO−NH(CH2)5−CO. Conditions: 0.5 μm probe with target RNA (5 equiv) in phosphate‐buffered saline (PBS; 100 mm NaCl, 10 mm Na2HPO4, pH 7) at 37 °C. λ ex=485 nm, λ em=500–650 nm, slitex=5 nm, slitem=5 nm.
Next, we appended the 22 nt long sequence tag in multiple repeats to the 3′‐untranslated region (3′‐UTR) of mRNA coding for the fluorescent protein mCherry. For this purpose, we synthesized two 51‐nt long oligonucleotides (Figure S7 in the Supporting Information) that, after hybridization, comprised two copies of the sequence tag, as well as overhangs compatible with BamHI and BglII restriction enzymes, and allowed T4 ligase mediated polymerization in the presence of both restriction enzymes (Figure 3). Products of 300–500 bp in length were purified by gel electrophoresis and cloned into a custom Gateway vector. Sequencing showed a product containing nine repeats. With the help of a shuttle vector, this product was repeatedly reinserted by making use of the terminal BamHI and BglII restriction sites, until we obtained a vector with 45 repeats of the sequence tag. For the construction of an expression plasmid, three vectors containing the doxycyclin (Dox)‐responsive CMV‐TO promotor, the mCherry open reading frame (ORF), and the F‐tag were used as donors in a three‐fragment recombination with a custom destination vector containing R4 and R3 recombination sites for the Multiside‐Gateway system, as well as an FRT site for Flp‐In recombination. Cotransfection of Flp‐In 293 T‐Rex cells with the plasmid encoding the tagged mCherry and a plasmid expressing Flp recombinase afforded a stable cell line that produced mCherry upon stimulation with Dox. Measurements by quantitative real‐time PCR with primers specific for the mCherry ORF suggested that incubation with 2 μg cm−1 Dox increased the expression of F‐tagged mCherry mRNA by 8.5‐fold within 1 h (Figure S11). Considering the number of sequence repeats and given the typical volume of HEK293 cells, the mRNA target occurs at an average concentration of 30 nm after 1 h of incubation in the presence of Dox.
Figure 3.
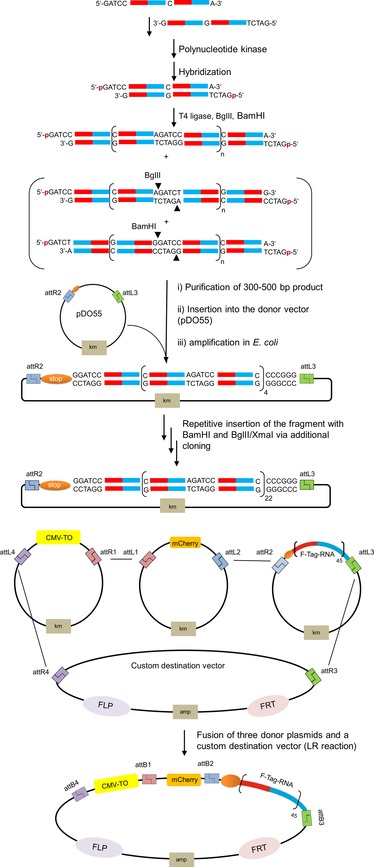
Schematic description of the cloning procedure used for the construction of a repeat motif (=F‐tag) appended to the 3′‐UTR of mCherry mRNA. BglII and BamHI: cut sites for restriction enzymes; attR1, attR2, attL1, attL2, attL3, attL4, attB1, attB2, attB3, and attB4: recombination sites; km: kanamycin resistance gene; amp: ampicillin resistance gene; CMV‐TO: doxycycline inducible CMV promoter (Tet‐ON); mCherry: fluorescent protein; FLP: recombinase flippase; FRT: FLP recognition target.
Design and synthesis of FIT probe conjugates
To assess the cellular delivery of the FIT probes, we considered a range of chemical delivery agents. PNA FIT probes were equipped with an N‐terminal mercaptopropionyl linker to allow conjugation with the maleimide‐modified penetratin peptide in Pen_FIT‐PNA1 (Figure 4). An identical peptide was used for conjugation with the 5′‐thiol‐modified DNA FIT probe Pen_FIT‐DNA1. Penetratin, similar to most CPPs, is net positively charged.24 The polyanionic nature of the DNA backbone might affect the cell‐delivery properties of the cationic CPPs. Exploration of additional CPP conjugates, which included Pep2,25 transportan,26 and MPG (comprising a fusion sequence of HIV gp41 and a hydrophilic segment of the nuclear localization signal),27 was therefore restricted to PNA‐based payloads in Pep2_FIT‐PNA1, Transp_FIT‐PNA1, and MPG_FIT‐PNA1. The conjugates featured a disulfide linkage, which might allow a reduction‐triggered removal of the long CPPs inside the cytosol.
Figure 4.
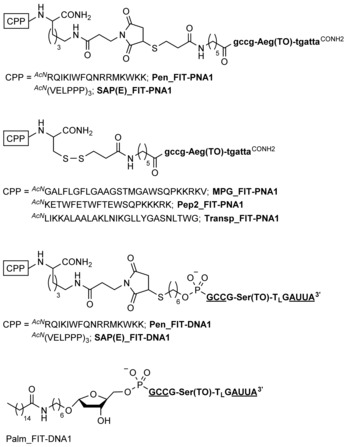
FIT probe conjugates used for live‐cell imaging of mRNA in HEK293 cells. Amino acids are represented with one‐letter codes. Bold small letters indicate PNA monomers; bold capital letters represent nucleotides. Underlined nucleotides mark 2′‐OMe‐modified nucleotides. Subscript L highlights a LNA building block.
In a next set of conjugates, we avoided the introduction of positive charges. Recently, a net negatively charged version of the sweet arrow CPP (SAP(E)) was introduced.28 This peptide was prepared as a maleimido conjugate and fused to both PNA and DNA FIT probes (SAP(E)_FIT‐DNA1 and SAP(E)_FIT‐PNA1).
Successful cellular delivery has also been described for lipid‐modified DNA and RNA.29 We used commercially available 5′‐palmitate to connect a lipid chain to the 5′‐end of DNA FIT probe Palm_FIT‐DNA1. Lipidation of the PNA FIT probe proved problematic, owing to the low solubility of the FIT‐PNA‐lipid conjugates.
Fluorescence with synthetic RNA targets
The conjugation of FIT probes with CPPs may change the fluorescence properties. We therefore measured the absorption and fluorescence emission spectra of conjugated FIT probes before and after hybridization with synthetic target RNA (Figures S2 and S3). We observed that conjugation with penetratin did not alter the fluorescence properties of the FIT‐DNA (compare entries 1 and 2 in Table 1; see also Figure S2). Likewise, the target affinity remained unaffected. In contrast, the TO emissions and T m values were enhanced if penetratin was conjugated with the PNA‐based probe (compare entries 6 and 7 in Table 1). However, the single‐stranded probe experienced higher emission enhancements than those of the probe–target duplex, leading to a decreased responsiveness of Pen_FIT‐PNA1. This effect was also observed with negatively charged SAP(E) (entry 8 in Table 1). Again, the emission increase was higher for the single‐stranded form of PNA conjugate SAP(E)_FIT‐PNA1 than that for the probe‐bound form. As observed for penetratin conjugation, fluorescence of the DNA FIT probe showed rather modest changes if SAP(E) was attached.
Table 1.
Optical properties of FIT probes and FIT probe conjugates.
| Probe | F 0 [a] | F [b] | F/F 0 | ϵ max [c] | T m [d] | |
|---|---|---|---|---|---|---|
| 1 | FIT‐DNA1 | 4.9 | 142 | 29 | 85 640 | 49 |
| 2 | Pen_FIT‐DNA1 | 4.7 | 140 | 30 | 75 780 | 49 |
| 3 | SAP(E)_FIT‐DNA1 | 3.8 | 110 | 29 | 64 780 | 44 |
| 4 | R8+FIT‐DNA1 | 5.7 | 161 | 28 | 85 640 | 60 |
| 5 | Palm_FIT‐DNA1 | 9.7 | 115 | 12 | 57 170 | 49 |
| 6 | FIT‐PNA1 | 2.7 | 22 | 8 | 41 790 | 52 |
| 7 | Pen_FIT‐PNA1 | 15 | 68 | 4.5 | 64 310 | 58 |
| 8 | SAP(E)_FIT‐PNA1 | 9.9 | 45 | 4.5 | 45 460 | 56 |
| 9 | Pep2_FIT‐PNA1 | 11 | 68 | 6 | 62 320 | 53 |
| 10 | Transp_FIT‐PNA1 | 3.2 | 11 | 3.5 | 43 460 | 49 |
| 11 | MPG_FIT‐PNA1 | 2.5 | 12 | 5 | 40 620 | 52 |
[a] Fluorescence intensity of a single strand. [b] Fluorescence intensity after hybridization with F‐tag‐RNA (Figure 2). [c] Extinction coefficient at the TO absorption maximum. [d] Melting temperature of a double strand at 0.5 μm. Conditions: 0.5 μm probes, complementary RNA (5 equiv, if added) in PBS (100 mm NaCl, 10 mm Na2HPO4, pH 7) at 37 °C, λ ex=488 nm, λ em=535 nm.
Given the potential for intra‐ or intermolecular charge–charge interactions in CPP–DNA conjugates, it seems counterintuitive that the PNA‐based probe responded to peptide conjugation, whereas the DNA probe proved to be inert. At this stage, we cannot explain the different sensitivities. However, it should be noted that both CPPs and PNA have hydrophobic properties, and we speculate that hydrophobic interactions may affect the structure of PNA in both single‐stranded and probe‐bound forms. Regardless of the mechanistic origin of the undesirable consequences for RNA imaging, we observed that each of the CPPs tested led to a decrease in the hybridization‐induced fluorescence response of PNA FIT probes. Interestingly, although Pep2 provided brightness enhancements (which were stronger for the single strand than that for the double strand), transportan and MPG (>23 AS) conferred decreases of fluorescence emission (which in this case were stronger for the double strand than for the single strand).
It has been reported that positively charged peptides enable the cellular delivery of DNA without conjugation.30 To assess whether uptake of noncovalent CPP–DNA complexes enabled mRNA imaging, we incubated FIT‐DNA1 with octaarginine at various charge ratios. The ideal charge ratio (positive charge, P, to negative charge, N) for the aggregation of peptide and oligonucleotide was determined by gel analysis (Figure S13). The fluorescence spectra measured once octaarginine was added to the DNA FIT probe, R8+FIT‐DNA1, at the optimal charge ratio (16:1) showed an increased intensity of TO emission, for both single‐stranded and target‐bound forms (compare entries 1 and 4 in Table 1). This and the increase in duplex stability induced by octaarginine (T m=50 vs. 60 °C) suggest that the polycationic additive induces a more compact state of the probe–target duplex. Gratifyingly, the fluorescence response factor of the DNA FIT probe remained unchanged.
Motivated by reports describing cellular delivery of lipidated oligonucleotides,29 we analyzed the palmitoylated DNA FIT probe Palm_FIT‐DNA1 (Table 1, entry 5). The weak fluorescence of the single‐stranded probe was red‐shifted. The broad emission band (Figure S2) is indicative of dye–dye interactions, which may be due to the formation of aggregates.
Fluorescence of FIT probe conjugates/complexes in cells
In the initial experiments, unconjugated FIT probes were added to HEK293 cells. After incubation for 30 min over a concentration range of 1–2 μm, neither the PNA nor the DNA FIT probes were delivered to cells, as evidenced by the lack of fluorescence signals after induction of target expression (Figure 5). We then turned our attention to CPP–probe conjugates. The DNA–penetratin conjugate Pen_FIT‐DNA1 provided weak fluorescence signals, which remained low, regardless of whether the mRNA target expression was induced or not. In contrast, the PNA–penetratin conjugate was successfully delivered into the cells. A diffuse fluorescence in the cytosol, which was stronger in cells induced with Dox than that in untreated cells, suggests target recognition (Figure S12). However, massive fluorescent spots occurred in both Dox‐induced and noninduced cells. The large size of the spots probably results from aggregation. The size of the fluorescent spots was smaller if the cells were incubated with SAP(E) conjugates. Visual inspection suggested that the PNA conjugate afforded higher increases in fluorescence in Dox‐treated cells than that in the DNA conjugate, but, again, fluorescent spots appeared in the presence and absence of target. The situation remained unchanged with the other CPP–PNA conjugates. It has been reported that CPP–nucleic acid conjugates form large aggregates in PBS.31 We performed delivery experiments in Dulbecco's modified eagle medium (DMEM). In our hands, however, spot‐like features appeared again in induced and noninduced cells (Figure S15). Next, we delivered DNA FIT probes into cells by means of octaarginine complexation. Again, large fluorescent spots caused artefacts, which rendered RNA imaging impossible.
Figure 5.
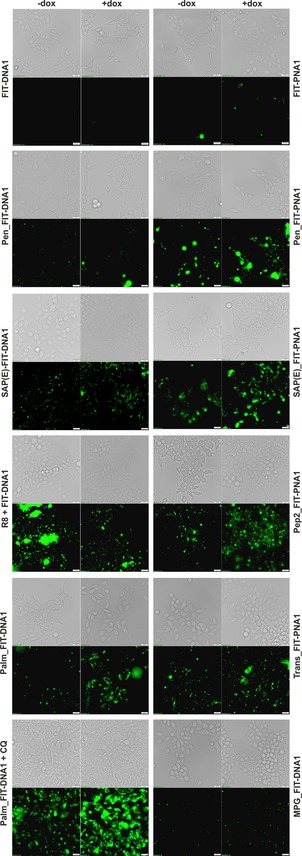
Fluorescence and bright‐field (gray) microscopy images of living Flp‐In 293 T‐REx cells incubated with DNA (2 μm) or PNA FIT probes (1 μm) for 30 min in PBS at 37 °C and 5 % CO2. Green shows the signal from TO emission with a λ=500/24 nm filter; −dox and +dox: without and with, respectively, the addition of Dox (2 μg mL−1) 1 h before incubation with probes. Scale bar: 20 μm.
We also examined cellular delivery aided by lipidation. Fluorescence microscopy images suggested a noteworthy gain of fluorescence upon induction of the target if the cells were incubated with lipidated Palm_FIT‐DNA1. It has been reported that chloroquine promotes endosomal escape.32 In fact, magnified fluorescence images (Figure S14) suggested that chloroquine provided substantial enhancements of cytosolic fluorescence in cells treated with Dox, which appeared to be more homogenous than that observed with the other conjugates. However, the background level in cells expressing low levels of the mRNA target remained high.
Fluorescence of FIT probes delivered by SLO
Delivery experiments with FIT probe conjugates revealed major sources of artefacts. Large fluorescent spots can occur if the conjugates form aggregates in or on cells. Furthermore, conjugated or probe‐complexed delivery agents that rely on interactions with the cell membrane typically form fluorescent spots, which are known to result from aggregation on the membrane or endosomal entrapment of probes. To avoid interactions between the probes and the cell membrane, we assessed delivery aided by the protein SLO; a channel forming protein, which grants entry into cells.33 The duration of permeabilization can be controlled because the membranes reseal upon exchange of cell media. We compared mRNA imaging with PNA and DNA FIT probes at different concentrations (Figure 6). No aggregates or interactions of probes with the cell membrane were observed. With 2 μm FIT‐DNA1, Doxinduced cells showed a fourfold higher median fluorescence than noninduced cells (Figure 6 A, see also Figure S17 for data from the analysis of three independent cell cultures). The fluorescence intensification was lower if the DNA FIT probe was applied at lower or higher concentrations. Notably, the FIT‐PNA1 probe required a lower concentration of 600 nm for an optimal fluorescence response (Figure 6 B). Analysis of three different cell cultures (Figure S17) suggests that the magnitude of the fluorescence enhancement was higher if FIT‐DNA1 was used, which was probably due to its higher responsiveness.
Figure 6.
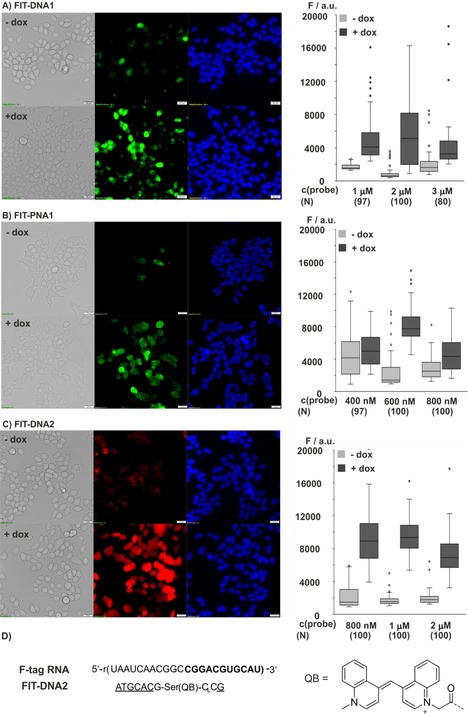
Fluorescence and bright‐field (gray) microscopy images of Flp‐In 293 T‐REx cells after SLO‐induced delivery of A) FIT‐DNA1, B) FIT‐PNA1, and C) FIT‐DNA2 without (−dox) and with (+dox) induction of F‐tagged mCherry mRNA. The Box‐Whisker plots (1.5 IQR) show the signal intensity of Dox‐induced cells (dark gray) and noninduced cells (light gray) measured by means of image analysis (Figure S16) at varied probe concentrations. The number, N, of cells analyzed is given in brackets. Conditions: 2 μg mL−1 Dox (if added); after 1 h cells are permeabilized with 150 U mL−1 SLO for 10 min in PBS+1 mm MgCl2 at 37 °C and 5 % CO2 in the presence of FIT probes. Green: TO emission; red: QB emission; blue: Hoechst 33342 nucleus stain. Filter sets: TO λ ex=500/24 nm, TO λ em=545/40 nm; QB λ ex=575/25, QB λ em=628/40 nm; Hoechst 33342=350/50 nm. Scale bar: 20 μm.
The fluorescence microscopy image exposed a few noninduced cells (3 out of 143 in Figure 6 A) that showed rather bright signals. We also observed a substantial variation of fluorescence intensity of induced cells. We ascribe this to cell‐to‐cell variations of SLO activity and differences in cell states. The observation that optimal staining required lower concentrations of PNA probes than that of DNA probes might suggest that delivery of the uncharged PNA probe is easier than delivery of the negatively charged DNA probe.
The FIT‐DNA2 probe contains a QB fluorophore34 and targets a segment that is adjacent to a sequence recognized by the TO probe (Figure 6 D). This probe provided a remarkable 114‐fold enhancement of fluorescence at λ=605 nm upon hybridization with synthetic target (Figure S2). At 1 μm concentration in the medium, this DNA FIT probe showed a 450 % higher fluorescence in Dox‐induced cells than in noninduced cells (Figure 6 C; see also Figure S17). This fluorescence increase is in the order of the eightfold change of target expression determined by means of quantitative PCR.
Cell viability
We assessed the viability of the genetically engineered HEK293 cell line under conditions of live‐cell imaging. Among the compounds studied, lipidated probe Pen_FIT‐DNA1 and complexes formed upon incubation of the R8+FIT‐DNA1 probe with octaarginine proved most harmful by inducing 3–4 % cell death (Figure S18), as estimated by propidium iodide staining. Incubation with 1 μm CPP–PNA conjugates and 2 μm DNA‐CPP conjugates was well tolerated, as previously reported. Cell death was reduced to 2 %. Notably, a similarly high rate of cell survival was obtained if the HEK293 cells were treated with SLO. This and the superior properties in the mRNA imaging experiments recommend SLO as a suitable delivery agent.
Multicolor imaging
We have previously shown a set of three differently labeled FIT DNA probes and demonstrated the simultaneous detection of three different targets in fixed cells.34 We combined the TO‐containing probe FIT‐DNA1 with two different probes, which included the known fluorescent dyes BO (see Figure S2 for a structure)34 and QB to cover the cyan and red ranges, respectively, of the light spectrum. The QB probe FIT‐DNA2 and the TO probe FIT‐DNA1 were designed to align in adjacent positions. In this way, the colocalization of TO and QB emission signals provides a stringent control for the specificity of mRNA target recognition inside live cells. The BO‐containing probe FIT‐DNA3 was directed against the poly‐A tail of mRNAs and it was, therefore, expected that fluorescence microscopy images based on BO emission would reveal different features than that of images based on TO and QB emission. Fluorescence spectrometric analysis confirmed that fluorescence signals from the three different dyes could be resolved (Figure 7 A). The BO‐oligo U probe FIT‐DNA3 had lower responsiveness than that of the TO‐ and QB‐containing probes and afforded a fivefold emission enhancement upon addition of A18 RNA, relative to 29‐ and 114‐fold enhancements provided by probes FIT‐DNA1 and FIT‐DNA2. However, given the high levels of oligo‐A sequences in living cells, this degree of fluorescence response was deemed to be sufficient for live‐cell mRNA imaging.
Figure 7.
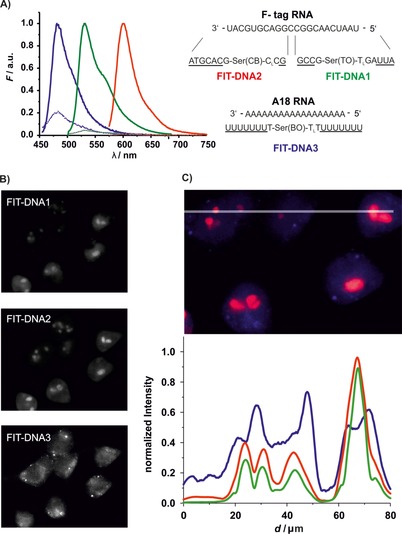
A) FIT probes for multicolor live‐cell imaging and their emission spectra before (‐ ‐ ‐ ‐) and after (—) addition of target RNA. Conditions: 0.5 μm probe with RNA target (5 equiv) in PBS (100 mm NaCl, 10 mm Na2HPO4, pH 7) at 37 °C. BO: λ ex=440 nm, λ em=485 nm, TO: λ ex=485 nm, λ em=535 nm, QB: λ ex=560 nm, λ em=605 nm, slitex=5 nm, slitem=5 nm. B) Gray‐scale fluorescence microscopy images of Flp‐In 293 T‐REx cells after SLO‐induced delivery of FIT‐DNA1, FIT‐DNA2, and FIT‐DNA3 with induction of F‐tagged mCherry mRNA in separate channels. C) Line scan and normalized intensities for emission from FIT‐DNA1 (green), FIT‐DNA2 (red), and FIT‐DNA3 (blue). Conditions: 2 μg mL−1 Dox; after 1 h incubation with 150 U mL−1 SLO for 10 min in PBS+1 mm MgCl2 and FIT‐DNA1 (2 μm), FIT‐DNA2 (1 μm), and FIT‐DNA3 (4 μm) at 37 °C and 5 % CO2. Filter sets: BO λ ex=438/24 nm, BO λ em=483/32 nm; TO λ ex=500/24 nm, TO λ em=545/40 nm; QB λ ex=575/25, QB λ em=628/40 nm.
The three different probes were delivered to the genetically engineered HEK cells by means of SLO‐mediated permeabilization. The TO and QB probes FIT‐DNA1 and FIT‐DNA2 were mixed in 2 and 1 μm concentrations, respectively, as described in the optimization experiments (Figure 6). A higher concentration (4 μm) was required for the BO probe FIT‐DNA3. As anticipated, the features resolved by the TO probe colocalized with the features exposed by the QB probe (Figure 7 B). The line scan (Figure 7 C) shows a strong correlation between intensities in the TO and QB detection channels. In contrast (and as expected), there was little overlap between patterns exposed by the poly‐A‐specific BO probe and patterns from the TO and QB probes. The BO probe produced a rather diffuse stain, thus suggesting that the majority of poly‐A‐containing mRNAs were rather regularly distributed over the cytosol. In addition, a few bright spots appeared, which were not obtained with FIT‐DNA1 and FIT‐DNA2. At present, we cannot tell whether the spots reflect high local concentrations of polyadenylated mRNA or result from local enrichment of unbound probes. A distinction between these two scenarios would be possible with qFIT probes, which carry an additional nonresponsive dye to report local probe concentration.7c
Conclusion
Robust methods that allow the imaging of mRNA in live cells are urgently needed to provide spatial and temporal resolution of the events that control intracellular RNA trafficking and degradation. Fluorogenic hybridization probes offer the unique opportunity to analyze unmodified RNA molecules without perturbations that may be caused by the genetic modifications required to create “vector‐based” imaging systems. However, delivery of the synthetic hybridization probes across the cell membrane is the central hurdle. A plethora of reports describe cellular delivery aided by CPPs. CPPs have frequently been used to introduce molecules that perturb biological processes. Typically, a minor amount of such perturbation probes suffices to induce a distinct phenotype and the subcellular localization of such perturbation probes is less important. The situation is entirely different with imaging probes for which a localization bias leads to artefacts. Therefore, the delivery of imaging probes is more challenging than the delivery of perturbation probes. To critically evaluate reagent‐based delivery of RNA imaging probes, we established a genetically engineered HEK293 cell line that expressed mCherry under the control of Dox. The 3′‐UTR of the mCherry RNA was tagged with a 22 nt long sequence tag in 45 repeats. Our data obtained with the inducible expression system attested to the challenges associated with CPP‐based delivery systems. The CPP penetratin was rather inefficient in conferring delivery of DNA‐based FIT probes. Better results were obtained with the net negatively charged SAP(E) peptide. However, spot‐like features appearing in the presence and absence of the target indicate the formation of aggregates as a key problem. Initially, we had hoped that aggregation problems would be lessened with the charge‐neutral PNA backbone. However, we also observed spot‐like features with the PNA–CPP conjugates. Among the different CPPs tested (penetratin, SAP(E), transportan, Pep2, MPG), again the best results were obtained with the negatively charged SAP(E)‐CPP. However, spots remained in noninduced cells that had low target expression levels. This finding questions the previous idea that charge neutrality will facilitate cellular delivery. We inferred that the formation of aggregates might be a hallmark of CPPs. Unfortunately, similar characteristics were observed in imaging experiments with lipidated probes.
The use of SLO provided a solution to the aggregation problem in HEK cells. SLO acts on cell membranes and probably does not interact with the probes. Therefore, the probes can freely diffuse through the pores transiently opened upon treatment of cells with SLO. With DNA‐based FIT probes, concentrations of 1–2 μm were required to afford a 500 % signal increase upon a Dox‐induced increase of target expression. This is in the order of the theoretically possible maximum, which is governed by the eightfold increase in target concentration. With the PNA‐based probe, the concentration could be reduced to 600 nm; however, at the cost of an attenuated signal increase. A viability test revealed that the HEK293 cells tolerated treatment with SLO remarkably well. We are, however, aware that other cells may be less tolerant. In such cases, CPPs may provide delivery options. However, it is conceivable that the formation of aggregates we have observed with the CPPs penetratin, transportan, MPG, Pep2, or SAP(E) is independent of the cell. For cells that do not tolerate SLO, microporation or cell squeezing may provide viable alternatives. We wish to note that SLO and other membrane‐disrupting techniques induce stress responses.35 This argues against long‐term imaging of stress‐associated processes.
A comparison of DNA and PNA scaffolds suggests that DNA‐based FIT probes are typically brighter and more responsive than PNA‐based FIT probes. This, and the higher solubility of DNA, motivated us to explore DNA FIT probes in live‐cell multicolor mRNA imaging. Differently colored probes targeting adjacent sequences within the mCherry sequence tag, in combination with a probe directed against a different target (poly‐A tail), provided unambiguous proof of the target specificity. Based on these results, we consider SLO‐mediated delivery of DNA FIT probes as a powerful tool for mRNA imaging.
Experimental Section
Cell culture: Permanently transfected Flp‐In 293 T‐REx cells capable of expressing mCherry mRNA tagged with a 45× repeat sequence (5′‐TAATC AACGG CCGGA CGTGC AT‐3′) were grown in DMEM from Thermo Fisher Scientific supplemented with 10 % fetal bovine serum purchased from Merck, 1 % penicillin/streptomycin and three different antibiotics: hygromycin (0.2 μg mL−1), blasticidin (0.045 μg mL−1), and zeocin (0.1 μg mL−1) from Invitrogen at 37 °C and 5 % CO2. Prior to imaging, the cells were subcultured and plated in μ‐slide 8‐well Ibidi from Ibidi (Munich, Germany) treated with 0.01 % poly(d‐lysine) acquired from Sigma–Aldrich with a confluency of 80 %.
Optical spectroscopy in vitro: Fluorescence and absorption spectra were measured at a concentration of 0.5 μm probe in 10 mm quartz cuvettes with phosphate buffer. The probes were measured in the single‐stranded state, without the target nucleic acid; BO: λ ex=440 nm, λ em=450–650 nm; TO: λ ex=485 nm, λ em=500–700 nm; QB: λ ex=560 nm, λ em=575–750 nm. Next, the absorption spectrum (λ=700–220 nm, 1 nm steps) of the probe was measured in the same cuvette. RNA (5 equiv) was added and the fluorescence measurements were repeated for the double‐stranded probe. The average of three fluorescence measurements (corrected with the fluorescence of the blank and normalized by the absorption at λ=260 nm) was calculated for both single‐ and double‐stranded states of the probes.
Transfection with FIT probes conjugates: Dox (2 μg mL−1) was added to the cells 1 h before transfection, then cells were washed once with Dulbecco's phosphate‐buffered saline (DPBS)+1 mm MgCl2 and incubated with 1 μm PNA probes and 2 μm DNA probes in DPBS for 30 min at 37 °C and 5 % CO2. The cells were washed twice with DPBS with MgCl2 (1 mm), and imaged in DPBS or for longer time in phenol red free DMEM.
Transfection with SLO: SLO from Sigma–Aldrich was aliquoted in about 350 U/10 μL RNAse‐free water and supplemented with tris(2‐carboxyethyl)phosphine (TCEP; 10 μm) and incubated for 30 min at 37 °C. Dox (2 μg mL−1) was added to the positive cells. After 1 h, the cells were washed once with DPBS with MgCl2 (1 mm), and then incubated with 30 U SLO and different concentrations of FIT probes in DPBS with MgCl2 (1 mm) for 10 min at 37 °C. The cells were washed twice with DPBS with MgCl2 (1 mm), and incubated with recovery medium (DMEM) with adenosine triphosphate (ATP; 2 mm) and guanosine‐5′‐triphosphate (GTP, 2 mm) for 2 h at 37 °C. Subsequently, cells were washed once more with DPBS with MgCl2 (1 mm), and supplemented with Hoechst 33342 dye in PBS for 5 min, and washed with DPBS with MgCl2 (1 mm) to continue with fluorescence microscopy in DPBS or for longer time in phenol red free DMEM.
Fluorescence microscopy: Images were acquired with an inverted Olympus IX83 microscope (Hamburg, Germany) with a 60×/1.35 UPLSAPO oil objective and an Orca Flash 4.0 V2 camera from Hamamatsu (Ammersee, Germany). A JC12V100W halogen lamp from Traydon (Frechen, Germany) for bright‐field images and a light‐emitting diode (LED) lamp (pE‐4000) acquired from Cool LED (Andover, UK) with a λ=500/24 nm filter for the TO probes, a λ=575/25 nm filter for QB probes, and a λ=350/50 nm filter for BO or 4′,6‐diamidino‐2‐phenylindole (DAPI) from AHF (Pfrondorf, Germany) was used. The images were analyzed by using the Olympus IX83 software with the region of interest (ROI) tool to surround the whole cells (25–50 cells were analyzed per measurement).
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We acknowledge support from the Einstein‐Stiftung Berlin and from the European Research Commission (ERC AdG TRIGGDRUG).
J. Chamiolo, G.-m. Fang, F. Hövelmann, D. Friedrich, A. Knoll, A. Loewer, O. Seitz, ChemBioChem 2019, 20, 595.
References
- 1.
- 1a. Bao G., Rhee W. J., Tsourkas A., Annu. Rev. Biomed. Eng. 2009, 11, 25–47; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1b. Tyagi S., Nat. Methods 2009, 6, 331–338; [DOI] [PubMed] [Google Scholar]
- 1c. Buxbaum A. R., Haimovich G., Singer R. H., Nat. Rev. Mol. Cell Biol. 2015, 16, 95–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.
- 2a. Tutucci E., Livingston N. M., Singer R. H., Wu B., Annu. Rev. Biophys. 2018, 47, 85–106; [DOI] [PubMed] [Google Scholar]
- 2b. Yamauchi K., Yang M., Jiang P., Xu M., Yamamoto N., Tsuchiya H., Tomita K., Moossa A. R., Bouvet M., Hoffman R. M., Cancer Res. 2006, 66, 4208–4214. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Tyagi S., Kramer F. R., Nat. Biotechnol. 1996, 14, 303–308; [DOI] [PubMed] [Google Scholar]
- 3b. Tan W. H., Wang K. M., Drake T. J., Curr. Opin. Chem. Biol. 2004, 8, 547–553; [DOI] [PubMed] [Google Scholar]
- 3c. Wang K. M., Tang Z. W., Yang C. Y. J., Kim Y. M., Fang X. H., Li W., Wu Y. R., Medley C. D., Cao Z. H., Li J., Colon P., Lin H., Tan W. H., Angew. Chem. Int. Ed. 2009, 48, 856–870; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 870–885. [Google Scholar]
- 4.
- 4a. Köhler O., Jarikote D. V., Seitz O., ChemBioChem 2005, 6, 69–77; [DOI] [PubMed] [Google Scholar]
- 4b. Seitz O., Bergmann F., Heindl D., Angew. Chem. Int. Ed. 1999, 38, 2203–2206; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 1999, 111, 2340–2343; [Google Scholar]
- 4c. Hövelmann F., Bethge L., Seitz O., ChemBioChem 2012, 13, 2072–2081. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Karunakaran V., Lustres J. L. F., Zhao L. J., Ernsting N. P., Seitz O., J. Am. Chem. Soc. 2006, 128, 2954–2962; [DOI] [PubMed] [Google Scholar]
- 5b. Jarikote D. V., Krebs N., Tannert S., Röder B., Seitz O., Chem. Eur. J. 2007, 13, 300–310; [DOI] [PubMed] [Google Scholar]
- 5c. Hövelmann F., Seitz O., Acc. Chem. Res. 2016, 49, 714–723. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Kummer S., Knoll A., Socher E., Bethge L., Herrmann A., Seitz O., Angew. Chem. Int. Ed. 2011, 50, 1931–1934; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 1972–1975; [Google Scholar]
- 6b. Kummer S., Knoll A., Sucher E., Bethge L., Herrmann A., Seitz O., Bioconjugate Chem. 2012, 23, 2051–2060; [DOI] [PubMed] [Google Scholar]
- 6c. Haralampiev I., Schade M., Chamiolo J., Jolmes F., Prisner S., Witkowski P. T., Behrent M., Hovelmann F., Wolff T., Seitz O., Herrmann A., ChemBioChem 2017, 18, 1589–1592. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Hövelmann F., Gaspar I., Ephrussi A., Seitz O., J. Am. Chem. Soc. 2013, 135, 19025–19032; [DOI] [PubMed] [Google Scholar]
- 7b. Hövelmann F., Gaspar I., Loibl S., Ermilov E. A., Röder B., Wengel J., Ephrussi A., Seitz O., Angew. Chem. Int. Ed. 2014, 53, 11370–11375; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 11553–11558; [Google Scholar]
- 7c. Gaspar I., Hövelmann F., Chamiolo J., Ephrussi A., Seitz O., ACS Chem. Biol. 2018, 13, 742–749; [DOI] [PubMed] [Google Scholar]
- 7d. Kam Y., Rubinstein A., Nissan A., Halle D., Yavin E., Mol. Pharm. 2012, 9, 685–693; [DOI] [PubMed] [Google Scholar]
- 7e. Kolevzon N., Hashoul D., Naik S., Rubinstein A., Yavin E., Chem. Commun. 2016, 52, 2405–2407; [DOI] [PubMed] [Google Scholar]
- 7f. Sonar M. V., Wampole M. E., Jin Y. Y., Chen C. P., Thakur M. L., Wickstrom E., Bioconjugate Chem. 2014, 25, 1697–1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.
- 8a. Kam Y., Rubinstein A., Naik S., Djavsarov I., Halle D., Ariel I., Gure A. O., Stojadinovic A., Pan H. G., Tsivin V., Nissan A., Yavin E., Cancer Lett. 2014, 352, 90–96; [DOI] [PubMed] [Google Scholar]
- 8b. Torres A. G., Fabani M. M., Vigorito E., Williams D., Al-Obaidi N., Wojciechowski F., Hudson R. H. E., Seitz O., Gait M. J., Nucleic Acids Res. 2012, 40, 2152–2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.
- 9a. Kolpashchikov D. M., Chem. Rev. 2010, 110, 4709–4723; [DOI] [PubMed] [Google Scholar]
- 9b. Martí A. A., Jockusch S., Stevens N., Ju J. Y., Turro N. J., Acc. Chem. Res. 2007, 40, 402–409. [DOI] [PubMed] [Google Scholar]
- 10. Okamoto A., Chem. Soc. Rev. 2011, 40, 5815–5828. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Abe H., Kool E. T., Proc. Natl. Acad. Sci. USA 2006, 103, 263–268; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11b. Pianowski Z., Gorska K., Oswald L., Merten C. A., Winssinger N., J. Am. Chem. Soc. 2009, 131, 6492–6497; [DOI] [PubMed] [Google Scholar]
- 11c. Gorska K., Keklikoglou I., Tschulena U., Winssinger N., Chem. Sci. 2011, 2, 1969–1975; [Google Scholar]
- 11d. Sadhu K. K., Winssinger N., Chem. Eur. J. 2013, 19, 8182–8189; [DOI] [PubMed] [Google Scholar]
- 11e. Wu H. X., Alexander S. C., Jin S. J., Devaraj N. K., J. Am. Chem. Soc. 2016, 138, 11429–11432. [DOI] [PubMed] [Google Scholar]
- 12. Holzhauser C., Liebl R., Goepferich A., Wagenknecht H. A., Breunig M., ACS Chem. Biol. 2013, 8, 890–894. [DOI] [PubMed] [Google Scholar]
- 13.
- 13a. Neumann E., Schaefer-Ridder M., Wang Y., Hofschneider P. H., EMBO J. 1982, 1, 841–845; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13b. Teruel M. N., Blanpied T. A., Shen K., Augustine G. J., Meyer T., J. Neurosci. Methods 1999, 93, 37–48. [DOI] [PubMed] [Google Scholar]
- 14. Chen A. K., Behlke M. A., Tsourkas A., Nucleic Acids Res. 2008, 36, e69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Santangelo P. J., Nix B., Tsourkas A., Bao G., Nucleic Acids Res. 2004, 32, e57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wile B. M., Ban K., Yoon Y. S., Bao G., Nat. Protoc. 2014, 9, 2411–2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Seo Y. J., Jeong H. S., Bang E. K., Hwang G. T., Jung J. H., Jang S. K., Kim B. H., Bioconjugate Chem. 2006, 17, 1151–1155. [DOI] [PubMed] [Google Scholar]
- 18.
- 18a. Nitin N., Santangelo P. J., Kim G., Nie S. M., Bao G., Nucleic Acids Res. 2004, 32, e58; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18b. Yeh H. Y., Yates M. V., Mulchandani A., Chen W., Proc. Natl. Acad. Sci. USA 2008, 105, 17522–17525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Boisguérin P., Deshayes S., Gait M. J., O'Donovan L., Godfrey C., Betts C. A., Wood M. J. A., Lebleu B., Adv. Drug Delivery Rev. 2015, 87, 52–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nielsen P. E., Egholm M., Berg R. H., Buchardt O., Science 1991, 254, 1497–1500. [DOI] [PubMed] [Google Scholar]
- 21. Raj A., van den Bogaard P., Rifkin S. A., van Oudenaarden A., Tyagi S., Nat. Methods 2008, 5, 877–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.
- 22a. Bertrand E., Chartrand P., Schaefer M., Shenoy S. M., Singer R. H., Long R. M., Mol. Cell 1998, 2, 437–445; [DOI] [PubMed] [Google Scholar]
- 22b. Tutucci E., Vera M., Biswas J., Garcia J., Parker R., Singer R. H., Nat. Methods 2018, 15, 81–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Singh S. K., Nielsen P., Koshkin A. A., Wengel J., Chem. Commun. 1998, 455–456. [Google Scholar]
- 24. Dupont E., Prochiantz A., Joliot A., Methods Mol. Biol. 2011, 683, 21–29. [DOI] [PubMed] [Google Scholar]
- 25. Morris M. C., Chaloin L., Choob M., Archdeacon J., Heitz F., Divita G., Gene Ther. 2004, 11, 757–764. [DOI] [PubMed] [Google Scholar]
- 26.
- 26a. Pooga M., Hallbrink M., Zorko M., Langel U., FASEB J. 1998, 12, 67–77; [DOI] [PubMed] [Google Scholar]
- 26b. Pooga M., Soomets U., Hällbrink M., Valkna A., Saar K., Rezaei K., Kahl U., Hao J. X., Xu X. J., Wiesenfeld-Hallin Z., Hökfelt T., Bartfai T., Langel Ü., Nat. Biotechnol. 1998, 16, 857–861. [DOI] [PubMed] [Google Scholar]
- 27. Morris M. C., Vidal P., Chaloin L., Heitz F., Divita G., Nucleic Acids Res. 1997, 25, 2730–2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Martín I., Teixidó M., Giralt E., ChemBioChem 2011, 12, 896–903. [DOI] [PubMed] [Google Scholar]
- 29.
- 29a. Raouane M., Desmaële D., Urbinati G., Massaad-Massade L., Couvreur P., Bioconjugate Chem. 2012, 23, 1091–1104; [DOI] [PubMed] [Google Scholar]
- 29b. Lorenz C., Hadwiger P., John M., Vornlocher H. P., Unverzagt C., Bioorg. Med. Chem. Lett. 2004, 14, 4975–4977. [DOI] [PubMed] [Google Scholar]
- 30. Emi N., Kidoaki S., Yoshikawa K., Saito H., Biochem. Biophys. Res. Commun. 1997, 231, 421–424. [DOI] [PubMed] [Google Scholar]
- 31.
- 31a. Jafari M., Xu W., Pan R., Sweeting C. M., Karunaratne D. N., Chen P., PLoS One 2014, 9, e97797; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31b. Konate K., Lindberg M. F., Vaissiere A., Jourdan C., Aldrian G., Margeat E., Deshayes S., Boisguerin P., Int. J. Pharm. 2016, 509, 71–84. [DOI] [PubMed] [Google Scholar]
- 32. Guy J., Drabek D., Antoniou M., Mol. Biotechnol. 1995, 3, 237–248. [DOI] [PubMed] [Google Scholar]
- 33. Ahnert-Hilger G., Bader M. F., Bhakdi S., Gratzl M., J. Neurochem. 1989, 52, 1751–1758. [DOI] [PubMed] [Google Scholar]
- 34. Hövelmann F., Gaspar I., Chamiolo J., Kasper M., Steffen J., Ephrussi A., Seitz O., Chem. Sci. 2016, 7, 128–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Stewart M. P., Langer R., Jensen K. F., Chem. Rev. 2018, 118, 7409–7531. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary