

Abstract
An in‐situ laboratory‐based X‐ray Absorption Near Edge Structure (XANES) Spectroscopy set‐up is presented, which allows performing long‐term experiments on a solid catalyst at relevant reaction conditions of temperature and pressure. Complementary to research performed at synchrotron radiation facilities the approach is showcased for a Co/TiO2 Fischer‐Tropsch Synthesis (FTS) catalyst. Supported cobalt metal nanoparticles next to a (very small) fraction of cobalt(II) titanate, which is an inactive phase for FTS, were detected, with no signs of re‐oxidation of the supported cobalt metal nanoparticles during FTS at 523 K, 5 bar and 200 h, indicating that cobalt metal is maintained as the main active phase during FTS.
Keywords: X-ray spectroscopy, heterogeneous catalysis, hydrogenation reaction, Fischer-Tropsch Synthesis, Cobalt
The developments in synchrotron radiation‐based X‐ray absorption (XAS) and X‐ray emission (XES) techniques accomplished during the last decades, have positioned them as prominent approaches for in‐depth characterization of materials and complex reactions systems.1 Specifically, for the characterization of solid catalysts the development of in‐situ and operando spectroscopy as well as methods offering high spatial and temporal resolution has established X‐rays techniques as crucial for studying the functionality of these materials.2,3 Among those techniques, X‐ray Absorption Near Edge Structure (XANES) spectroscopy has become a powerful, highly versatile and useful characterization technique for the study of different catalytic systems of practical relevance.4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14
However, one of the disadvantages of synchrotron‐based techniques is their limited availability and accessibility, i. e. the number of hours of beamtime access granted, which exclude certain characterization studies and developments. For instance, the restricted beamtime access typically confines in‐situ studies of the deactivation of catalysts to a few days, which is often insufficient to conduct a conclusive study about long‐term deactivation processes that can take place over much longer time frames, e. g. the long‐term deactivation process of Fischer‐Tropsch Synthesis (FTS) catalyst, which happens over days/weeks, and is a major challenge for FTS technology.15,16 Additional difficulties include the transport of the in‐situ set‐up and samples (e. g. air sensitive samples) to the synchrotron radiation facilities.17 To overcome this availability gap, during the last years research on the development of laboratory‐based X‐rays facilities has been intensified.17, 18, 19, 20, 21, 22, 23, 24, 25, 26 By using laboratory‐based set‐ups, different successful ex‐situ XANES studies have been performed on diverse systems.27, 28, 29, 30, 31, 32 Interestingly, one can consider these developments as a revival of a somewhat older idea. For example, during the 1980s, Koningsberger et al. developed a laboratory‐based EXAFS set‐up, and successfully performed ex‐situ experiments.33 However, limitations in the energy resolution of the monochromator crystal (∼14 eV) limited the applicability of the set‐up. We refer the reader for this older work to a book chapter by Koningsberger.34
In the present study, we introduce the first in‐situ XANES laboratory‐based experiments of a solid catalyst, measured in transmission mode, under relevant conditions of temperature and pressure. We have performed such experiments for an industrial relevant Co/TiO2 FTS catalyst. The experiments were compared and complemented by synchrotron‐based measurements carried out on the same catalyst material. By using both set‐ups, namely a laboratory‐based and synchrotron‐based system, it was possible to obtain fundamental physicochemical insights about the activation and deactivation process of a Co/TiO2 FTS catalyst and compare the capabilities of both measurement approaches for collecting in‐situ XANES data for this solid catalyst as well as relevant reference compounds, i. e. Co3O4, CoO, Co, and CoTiO3.
Results and Discussion
The in‐situ XANES laboratory‐based set‐up used in the experiments consists of an Ag‐tube as the X‐ray source, 40 mA and 20 kV, a spherically bent crystal monochromator utilizing the Johann geometry employing a Si (533) crystal with a bending radius R=0.5 m that was used for Co K‐edge (7709 eV) measurements, and a NaI scintillator detector. The spectrometer uses the Bremstrahlung spectrum and is designed to work in the 4–20 keV photon energy range. The detector, monochromator and X‐ray source are located at a Rowland circle. The X‐ray source is kept fixed and the monochromator and detector are moved during scanning across the Co K‐edge. Details about the laboratory‐based X‐ray absorption spectrometer can be found in the work of Honkanen et al.31 The in‐situ XANES set‐up is illustrated in Figure 1.30
Figure 1.

Schematic of the in‐situ XANES set‐up, principles and configuration.
i. Validation of the Laboratory‐Based XANES Set‐Up
The first set of experiments completed with the in‐situ XANES laboratory‐scale set‐up were the measurements of the different cobalt reference materials. These data were then used to identify the different cobalt species present during the different stages of reaction, and to elucidate the possible formation of CoO and CoTiO3 during FTS. The second stage in our study was to test the Co/TiO2 FTS catalyst under reaction conditions for 35 h at 523 K, 1 bar, and a H2/CO ratio of 0.5. In order to validate the research accomplished by the in‐situ XANES laboratory‐scale set‐up, analogous experiments were performed at the BM26 beamline at the European Synchrotron Radiation Facility (ESRF), wherein the different cobalt references were measured and the Co/TiO2 catalyst was tested under FTS reaction conditions for 15 h at 523 K, 1 bar, and a H2/CO ratio of 0.5. The ex‐situ XANES measurements of the cobalt references performed using the laboratory‐based set‐up and at the BM26 beamline of ESRF are presented in Figure 2. The analysis of these ex‐situ spectra shows that the laboratory‐based set‐up produces high quality data comparable to synchrotron measurements, although at reduced time‐resolution (∼0.5 h for the synchrotron‐based measurements and ∼3 h for the laboratory‐based measurements). This is illustrated in Figure S1. The spectra collected during FTS at different times of reaction using the laboratory‐based set‐up, which shows a lower signal‐to‐noise ratio because of the relative small amount of cobalt in the sample and the high absorbance rate of the support (TiO2), and at the BM26 beamline are presented in Figure 3. By analyzing the XANES data from the laboratory‐based set‐up, it was concluded that the catalyst consisted of a mixture of metallic Co (active phase for the FTS reaction), and CoTiO3 (inactive phase for the FTS reaction), formed during the activation step of the catalyst at 673 K and 1 bar, in a pure H2 flow, this inactive phase was formed because of the presence of hot‐spot in the bed of the capillary reactor. By least squares linear combination fitting of the cobalt reference spectra to the unknown spectra collected during FTS reaction (Figure 4.I), it was established that ∼33 % of the cobalt was converted into CoTiO3. After 35 h of FTS reaction, a small fraction of CoO was identified by the fit (Figure S2.1.4.a). However, this fraction was found within measurement uncertainty, i. e. at/below the detection limit and therefore does not provide conclusive evidence of a re‐oxidation process during FTS reaction.
Figure 2.
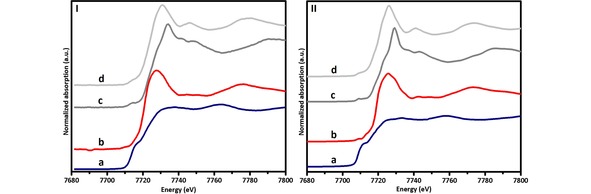
XANES of the cobalt reference materials measured: I) using the laboratory‐based set‐up; a) Co, b) CoTiO3, c) Co3O4 and d) CoO, and II) at the BM26 beamline; a) Co, b) CoTiO3, c) Co3O4 and d) CoO.
Figure 3.
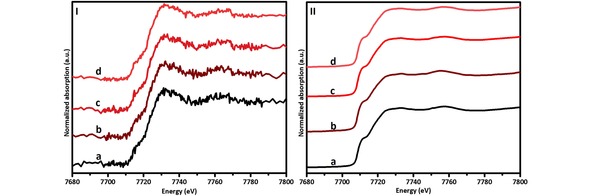
In‐situ XANES spectra of the FTS reaction over a Co/TiO2 catalyst at a reaction temperature of 523 K, a reaction pressure of 1 bar and a H2/CO ratio of 0.5 at different reaction times: I) measured with a laboratory‐based set‐up at a) 8.75 h, b) 17.5 h, c) 26.25 h and d) 35 h, and II) measured at the BM26 beamline of ESRF at a) 6 h, b) 10 h, c) 12 h and d) 15 h. A glitch due to the monochromator is visible at 7764 eV in some of the spectra.
Figure 4.

Least Squares Linear Combination fitting of the data recorded during the experiments perform: I) using the in‐situ XANES laboratory‐scale set‐up at different reaction times; a) 8.75 h, b) 17.5 h, c) 26.25 h and d) 35 h, and II) at the BM26 beamline of the ESRF after a) 6 h, b) 10 h, c) 12 h and d) 15 h of FTS. Experimental data is plotted in grey, fits are plotted in red.
In the case of the synchrotron measurements, a similar procedure was applied (Figure 4.II) and no substantial amount of CoTiO3 was detected during the activation stage of the catalyst. Additionally, no statistically relevant re‐oxidation of the cobalt could be observed after 15 h of FTS reaction.
ii. Long‐Term FTS Reaction Experiments
A second set of in‐situ experiments was completed with the XANES laboratory‐scale set‐up using FTS reaction conditions of 523 K and 5 bar, using a H2/CO ratio of 0.5 and 2. These experiments were performed for 200 h each. The different spectra collected at different times for both long‐term reactions are presented in Figure 5.
Figure 5.
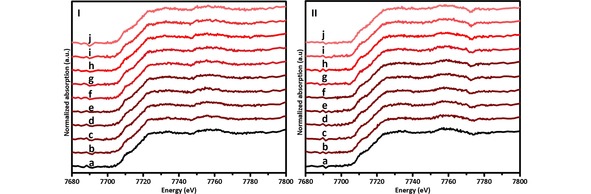
In‐situ XANES spectra of the FTS reaction over a Co/TiO2 catalyst at a reaction temperature of 523 K, and a reaction pressure of 5 bar: I) using a H2/CO ratio of 0.5 at different reaction times; a) 20 h, b) 40 h, c) 60 h, d) 80 h, e) 100 h, f) 120 h, g) 140 h, h) 160 h, i) 180 h and j) 200 h; and II) using a H2/CO ratio of 2 at different reaction times; a) 20 h, b) 40 h, c) 60 h, d) 80 h, e) 100 h, f) 120 h, g) 140 h, h) 160 h, i) 180 h and j) 200 h.
By least squares linear combination analysis of the long‐term XANES data using the same procedure as presented in the previous section, it was established that a small fraction of CoTiO3 was identified by the fit in the different steps of both reactions, as illustrated in Figure 6.
Figure 6.
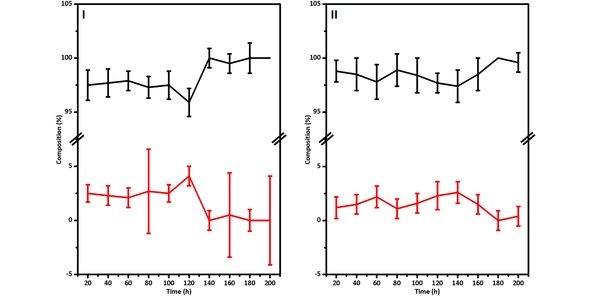
Composition of the different cobalt species obtained from Least Squares Linear Combination fitting of the data recorded during the experiments perform: I) using a H2/CO ratio of 0.5 at different reaction times, and II) using a H2/CO ratio of 2 at different reaction time, fitting values for Co in black and for CoTiO3 in red.
For all the spectra measured the combination of the reference spectra of Co and CoTiO3 gives the best fitting, except for one case (see section S2.2.12). However, as in the case of the data from the BM26 beamline, CoTiO3 and CoO fractions were found within measurement uncertainty and does not provide conclusive evidence about their formation during the activation step and the FTS. The complete fitting data is presented in section S2.2.
In addition, no re‐oxidation of the cobalt metal nanoparticles was observed in any of the FTS experiments performed at different pressures (1 and 5 bar), and H2/CO ratios (0.5 and 2), which agrees with previous studies from our research group that indicate that re‐oxidation is not observed during the activation or deactivation stages of a Co/TiO2 FTS catalyst.35, 36, 37
Conclusions
A laboratory‐scale set‐up is presented that allows in‐situ XANES measurements of a solid catalyst. This instrument provides the opportunity to carry out unique experiments, which are complimentary to the research performed at synchrotron radiation facilities. Thanks to the stability of the set‐up, as demonstrated in Figure S3, this technical development shows large potential for long‐term experiments, e. g. when activation and/or deactivation studies of complex reaction systems have to be conducted over time frames of days or weeks. Such experiments are difficult to perform at synchrotrons due to the limited time available during granted beamtimes. By using this in‐situ laboratory‐scale XANES set‐up it was not only possible to establish Co K‐edge XANES of different Co reference compounds, namely Co3O4, CoO, Co and CoTiO3, but also to follow the activation and expected long‐term deactivation15 of a Co/TiO2 catalyst during realistic Fischer‐Tropsch Synthesis (FTS) conditions; i. e., at elevated pressure and temperature and while flowing hydrogen and carbon monoxide. From the performed laboratory‐scale in‐situ FTS experiments, it is observed that at a temperature higher than 573 K, a small fraction of the cobalt can be converted into CoTiO3 during the reduction process, which agrees with the concept of strong metal‐support interaction.16 Furthermore, during FTS at different conditions (pressure and H2/CO ratio) no cobalt re‐oxidation was observed, which is in line with earlier reports35, 36, 37 from our research group, confirming that oxidation of the cobalt is not a main deactivation route for FTS, leaving sintering and coke formation as main reasons for the detrimental activity over time for Co‐based FTS catalyst.15
Experimental Section
The Co/TiO2 catalyst under study was synthesized by incipient wetness impregnation (IWI) using TiO2 P90 as support (Evonik, 90 % anatase and 10 % rutile) and Co(NO3)2x6H2O (Acros Organics, 99+%) as precursor. The cobalt loading obtained was 14.1 wt %, which was confirmed by Inductively Coupled Plasma‐Atomic Emission Spectrometry (ICP‐AES). The calcined catalyst consists of Co3O4 nano‐crystallites of ∼14 nm in diameter. The crystal size was estimated by X‐Ray Diffraction (XRD) data analysis and confirmed by Scanning Transmission Electron Microscopy‐Energy Dispersion X‐ray Spectroscopy (STEM‐EDX). These data can be found in Figures S4 and S5. The in‐situ reactor set‐up, used in the experiments, is made of a quartz capillary plug‐flow reactor with an inner diameter of 0.98 mm and outer diameter of 1 mm.38,39 The catalyst was placed and fixed in the isothermal zone (Figures S6.1 and S6.2) of the capillary by quartz wool and the plug‐flow reactor was heated by two IR heaters. The temperature was controlled by a thermocouple positioned in the bed of the capillary. During the experiments, two different measurements were performed: a) ex‐situ measurements of cobalt references; Co3O4 (Sigma‐Aldrich, 99.5 %), CoO (Acros Organics, 99+%), CoTiO3 (Alfa Aesar, 99.8 %) and Co (Co foil, 99.9 %); the powders were diluted with potato starch to increase the transmittance of the sample, and b) in‐situ measurements by using the FTS reaction on Co/TiO2 (14.1 wt %) catalyst at 523 K, 1 and 5 bar. The FTS reaction was divided into two different stages: i) activation (reduction of the Co3O4 nano‐crystallites to Co0) at 673 K and 1 bar, in a H2 flow of 1 ml/min (AGA, H2 >99.999 %) and ii) the FTS reaction, feeding a mixture of H2/CO (AGA, CO >99.97 %) of 0.5 (0.3 ml/min of H2 and 0.6 ml/min of CO) and 2 (0.6 ml/min of H2 and 0.3 ml/min of CO). Before starting the activation step, a Co K‐edge spectrum was collected to identify the oxidation state of the cobalt in the sample. The different spectra obtained during the execution of the experiments were collected continuously. The spectra were collected every ∼38 min and were summed up every 8.75 h (∼14 scans per spectrum) for the experiment performed at 1 bar, and every 20 h (∼32 scans per spectrum) for the experiments at 5 bar, which thereby determined the effective time resolution. During the activation stage, the temperature was raised from room temperature (RT) to 673 K with a heating rate of 5 K/min, and kept at 673 K for 2 h. Once the activation stage was concluded, the temperature was decreased to 523 K with a rate of 5 K/min and the gases were switched to a mixture to H2/CO (0.5) in the case of the experiment at 1 bar. The FTS reaction was performed for 35 h. For the experiments performed at 5 bar, after the activation step was finished the temperature was reduced to RT, the system was pressurized in a mixture of H2/CO (0.5 and 2 in each case), and the temperature was raised to 523 K with a heating rate of 5 K/min. The FTS reactions were performed for 200 h. A new set of experiments was performed at the Dutch‐Belgian Beamline BM26 (DUBBLE) at the ESRF following an equivalent experimental procedure. These experiments were planned to complement and compare with the data obtained from the laboratory‐based set‐up. In this second set of experiments the different cobalt references (i. e., Co3O4, CoO, CoTiO3 and Co) were measured and the FTS reaction was performed at 1 bar and for 15 h. The Athena XAS data processing software was used for the analysis of all sets of data. The Co/TiO2 catalyst under study has been previously tested to check its catalytic activity by using a fixed‐bed reactor. The FTS products were analyzed every hour by an on‐line Gas Chromatograph (GC, Varian 430).40
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
Pasi Paalanen and Ramon Oord (Utrecht University) are thanked for their technical feedback. Merja Blomberg (University of Helsinki) is thanked for her help during the setting‐up of the configuration. Shell Global Solutions and the Netherlands Organization for Scientific Research (NWO) are acknowledged for financial support via a CHIPP grant. Simo Huotari and Ari‐Pekka Honkanen were supported by the Academy of Finland (grant no. 1295696), the University of Helsinki Doctoral Program in Materials Research and Nanosciences (MATRENA), and the Jenny and Antti Wihuri Foundation.
J. G. Moya-Cancino, A.-P. Honkanen, A. M. J. van der Eerden, H. Schaink, L. Folkertsma, M. Ghiasi, A. Longo, F. M. F. de Groot, F. Meirer, S. Huotari, B. M. Weckhuysen, ChemCatChem 2019, 11, 1039.
Contributor Information
Prof. Dr. Simo Huotari, Email: simo.huotari@helsinki.fi
Prof. Dr. Bert M. Weckhuysen, Email: b.m.weckhuysen@uu.nl.
References
- 1. Bokhoven J. A. van, Lamberti C., Eds., X-Ray Absorption and X-Ray Emission Spectroscopy, John Wiley & Sons, West Sussex, 2016. [Google Scholar]
- 2. Meirer F., Weckhuysen B. M., Nat. Rev. Mater. 2018, 3, 324–340. [Google Scholar]
- 3. Beale A. M., Jacques S. D. M., Weckhuysen B. M., Chem. Soc. Rev. 2010, 39, 4656–4672. [DOI] [PubMed] [Google Scholar]
- 4. Bordiga S., Groppo E., Agostini G., Bokhoven J. A. van, Lamberti C., Chem. Rev. 2013, 113, 1736–1850. [DOI] [PubMed] [Google Scholar]
- 5. Jacobs G., Patterson P. M., Zhang Y., Das T., Li J., Davis B. H., Appl. Catal. A 2002, 233, 215–226. [Google Scholar]
- 6. Jacobs G., Das T. K., Patterson P. M., Li J., Sanchez L., Davis B. H., Appl. Catal. A 2003, 247, 335–343. [Google Scholar]
- 7. Das T. K., Jacobs G., Patterson P. M., Conner W. A., Li J., Davis B. H., Fuel 2003, 82, 805–815. [Google Scholar]
- 8. Jacobs G., Chaney J. A., Patterson P. M., Das T. K., Davis B. H., Appl. Catal. A 2004, 264, 203–212. [Google Scholar]
- 9. Jacobs G., Ji Y., Davis B. H., Cronauer D., Kropf J., Marshall C. L., Appl. Catal. A 2007, 333, 177–191. [Google Scholar]
- 10. Saib A. M., Borgna A., van de Loosdrecht J., Berge P. J. van, Niemantsverdriet J. W., Appl. Catal. A 2006, 312, 12–19. [Google Scholar]
- 11. Girardon J.-S., Lermontov A. S., Gengembre L., Chernavskii P. A., Griboval-Constant A., Khodakov A. Y., J. Catal. 2005, 230, 339–352. [Google Scholar]
- 12. Chu W., Chernavskii P. A., Gengembre L., Pankina G. A., Fongarland P., Khodakov A. Y., J. Catal. 2007, 252, 215–230. [Google Scholar]
- 13. Rønning M., Tsakoumis N. E., Voronov A., Johnsen R. E., Norby P., Beek W. van, Borg Ø., Rytter E., Holmen A., Catal. Today 2010, 155, 289–295. [Google Scholar]
- 14. Bezemer G. L., Bitter J. H., Kuipers H. P. C. E., Oosterbeek H., Holewijn J. E., Xu X., Kapteijn F., Dillen J. van, Jong K. P. de, J. Am. Chem. Soc. 2006, 128, 3956–3964. [DOI] [PubMed] [Google Scholar]
- 15. Tsakoumis N. E., Rønning M., Borg Ø., Rytter E., Holmen A., Catal. Today 2010, 154, 162–182. [Google Scholar]
- 16. Khodakov A. Y., Chu W., Fongarland P., Chem. Rev. 2007, 107, 1692–1744. [DOI] [PubMed] [Google Scholar]
- 17. Seidler G. T., Mortensen D. R., Remesnik A. J., Pacold J. I., Ball N. A., Barry N., Styczinski M., Hoidn O. R., Rev. Sci. Instrum. 2014, 85, 113906–113918. [DOI] [PubMed] [Google Scholar]
- 18. Seidler G. T., Mortensen D. R., Ditter A. S., Ball N. A., Remesnik A. J., J. Phys. Conf. Ser. 2016, 712, 012015. [Google Scholar]
- 19. Holden W. M., Hoidn O. R., Ditter A. S., Seidler G. T., Kas J., Stein J. L., Cossairt B. M., Kozimor S. A., Guo J., Ye Y., Marcus M. A., Fakra S., Rev. Sci. Instrum. 2017, 88, 073904–073914. [DOI] [PubMed] [Google Scholar]
- 20. Mortensen D. R., Seidler G. T., J. Electron Spectrosc. Relat. Phenom. 2017, 215, 8–15. [Google Scholar]
- 21. Mortensen D. R., Seidler G. T., Ditter A. S., Glatzel P., J. Phys. Conf. Ser. 2016, 712, 012036. [Google Scholar]
- 22. Németh Z., Szlachetko J., Bajnóczi É. G., Vankó G., Rev. Sci. Instrum. 2016, 87, 103105–103114. [DOI] [PubMed] [Google Scholar]
- 23. Bajnóczi É. G., Németh Z., Vankó G., Inorg. Chem. 2017, 56, 14220–14226. [DOI] [PubMed] [Google Scholar]
- 24. Szlachetko M., Berset M., Dousse J.-Cl., Hoszowska J., Szlachetko J., Rev. Sci. Instrum. 2013, 84, 093104–093116. [DOI] [PubMed] [Google Scholar]
- 25. Yuryev Y. N., Lee H.-J., Park H.-M., Cho Y.-K., Lee M.-K., Pogrebitsky K. J., Rev. Sci. Instrum. 2007, 78, 025108–025113. [DOI] [PubMed] [Google Scholar]
- 26. Schlesiger C., Anklamm L., Stiel H., Malzera W., Kanngießer B., J. Anal. At. Spectrom. 2015, 30, 1080–1085. [Google Scholar]
- 27. Anklamm L., Schlesiger C., Malzer W., Grötzsch D., Neitzel M., Kanngießer B., Rev. Sci. Instrum. 2014, 85, 053110–053115. [DOI] [PubMed] [Google Scholar]
- 28. Mantouvalou I., Witte K., Martyanov W., Jonas A., Grötzsch D., Streeck C., Löchel H., Rudolph I., Erko A., Stiel H., Kanngießer B., Appl. Phys. Lett. 2016, 108, 201106–201110. [Google Scholar]
- 29. Wang W., Kuai L., Cao W., Huttula M., Ollikkala S., Ahopelto T., Honkanen A.-P., Huotari S., Yu M., Geng B., Angew. Chem. 2017, 129, 15173–15177; [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2017, 56, 14977–14981. [DOI] [PubMed] [Google Scholar]
- 30. Bes R., Ahopelto T., Honkanen A.-P., Huotari S., Leinders G., Pakarinen J., Kvashnina K., J. Nucl. Mater. 2018, 507, 50–53. [Google Scholar]
- 31.A.-P. Honkanen, S. Ollikkala, T. Ahopelto, A.-J. Kallio, M. Blomberg, S. Huotari, arXiv:1812.01075. [DOI] [PubMed]
- 32.M. Tromp, https://www.c2w.nl/achtergrond/chemicus-bouwt-eigen-spectrometer/item19387, 2017.
- 33. Kampers F. W. H., Duivenvoorden F. B. M., van Zon J. B. A. D., Brinkgreve P., Viegers M. P. A., Koningsberger D. C., Solid State Ion. 1985, 16, 55–64. [Google Scholar]
- 34. Koningsberger D. C., in X-ray Absorption: Principles, Applications, Techniques of EXAFS, SEXAFS and XANES, D. C. Koningsberger, R. Prins (Eds.), John Wiley & Sons, New York, 1988, p. 163. [Google Scholar]
- 35. Cats K. H., Gonzalez-Jimenez I. D., Liu Y., Nelson J., van Campen D., Meirer F., van der Eerden A. M. J., De Groot F. M. F., Andrews J. C., Weckhuysen B. M., Chem. Commun. 2013, 49, 4622–4624. [DOI] [PubMed] [Google Scholar]
- 36. Cats K. H., Weckhuysen B. M., ChemCatChem 2016, 8, 1531–1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cats K. H., Andrews J. C., Stéphan O., March K., Karunakaran C., Meirer F., Groot F. M. F. de, Weckhuysen B. M., Catal. Sci. Technol. 2016, 6, 4438–4449. [Google Scholar]
- 38. Fischer N., Clapham B., Feltes T., Claeys M., ACS Catal. 2015, 5, 113–121. [Google Scholar]
- 39. Fischer N., Clapham B., Feltes T., Steen E. van, Claeys M., Angew. Chem. Int. Ed. 2014, 53, 1342–1345. [DOI] [PubMed] [Google Scholar]
- 40. Ravenhorst I. K. van, Vogt C., Oosterbeek H., Bossers K. W., Moya-Cancino J. G., van Bavel A. P., Eerden A. M. J. van der, Vine D., Groot F. M. F. de, Meirer F., Weckhuysen B. M., Angew. Chem. Int. Ed. 2018, 57, 11957–11962. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary