

Abstract
This work takes advantage of one of the hallmarks of cancer, that is, the presence of tumor infiltrating cells of the immune system and leukocyte‐secreted enzymes, to promote the activation of an anticancer drug at the tumor site. The peptidomimetic integrin ligand cyclo(DKP‐RGD) was found to accumulate on the surface of αvβ3 integrin‐expressing human renal cell carcinoma 786‐O cells. The ligand was conjugated to the anticancer drug paclitaxel through a Asn‐Pro‐Val (NPV) tripeptide linker, which is a substrate of neutrophil‐secreted elastase. In vitro linker cleavage assays and cell antiproliferative experiments demonstrate the efficacy of this tumor‐targeting conjugate, opening the way to potential therapeutic applications.
Keywords: antitumor agents, drug delivery, elastase, inflammation, neutrophils
The conjugation of anticancer drugs to specific ligands, capable of selective binding to tumor‐associated receptors, represents a widely explored strategy to improve the accumulation of cytotoxic agents at the tumor site, sparing healthy tissues and resulting in better therapeutic outcomes. Antibody–drug conjugates (ADCs) represent the first‐in‐class in this oncology area.1 To date, four ADC products have gained marketing authorization (Adcetris, Kadcyla, Mylotarg, and Besponsa), while more than 50 are presently under clinical investigation.2 Similarly, anticancer agents have also been coupled to small ligands (e.g., peptides/peptidomimetics,3 vitamins,4 steroids,5 and enzyme inhibitors),6 targeting specific receptors overexpressed by cancer cells. An increasing body of evidences suggests that these so‐called small molecule–drug conjugates (SMDCs)7 may accumulate in the tumor mass homogeneously and with high tumor/organ and tumor/blood ratios, potentially showing better anticancer efficacy than conventional cytotoxic agents and ADC products.8 In general, these conjugates have been designed to bind the target receptor on the surface of cancer cells and to release the cytotoxic cargo in intracellular compartments (e.g., lysosomes), upon receptor‐mediated internalization and selective cleavage of a linker (e.g., a short peptide sequence or a reducible disulfide bond) connecting the targeting unit to the payload.9
Among the protein antigens that have been explored for tumor targeting applications, integrin αvβ3 is a heterodimeric transmembrane receptor overexpressed in a variety of cancer types (such as melanoma, glioblastoma, renal cell carcinoma, and tumors of lung, ovary, breast, prostate, and colon), where it is involved in disease progression.10 Upon the observation that cyclic peptides bearing the Arg‐Gly‐Asp (RGD) sequence are potential αvβ3 integrin ligands, a large number of RGD‐bearing peptides and peptidomimetics have been explored for SMDC development.11
Within this frame, our group synthesized a cyclic peptidomimetic compound bearing the RGD integrin recognition motif and a diketopiperazine (DKP) scaffold, as a low nanomolar αvβ3 integrin ligand (compound 1, Figure 1).12 The ligand was successfully linked to different cytotoxic payloads (i.e., paclitaxel,13 camptothecin,14 and α‐amanitin)15 and the resulting conjugates maintained high affinity for the integrin receptor. In line with literature data that reported evidence of internalization of integrin ligands equipped with fluorescent dyes,16 our cyclo(DKP‐RGD)‐drug conjugates were endowed with suitable linkers for selective payload release in intracellular compartments of the cancer cells. The ability of these SMDCs to selectively hit αvβ3‐positive cells was quantified through in vitro cell antiproliferative assays performed using cancer cell lines expressing the integrin receptor at different levels (i.e., expressing vs. non‐expressing cells). To our delight, some of these SMDCs proved to be highly selective for the αvβ3‐expressing cells,17 indicating that 1 effectively recognizes the integrin receptor on the cancer cell membrane. However, the potency of all our conjugates against αvβ3‐positive cells proved to be significantly lower (typically one order of magnitude) than that of the free drug. This observation is in contrast with literature data reporting the high potency of internalizing ADCs and SMDCs,18, 19 and it may indicate a suboptimal endocytosis of our ligand–drug conjugates. In order to confirm this hypothesis and evaluate the internalization properties of the cyclo(DKP‐RGD) integrin ligand, we labeled it with the near‐infrared (NIR) dye sulfo‐cyanine5 (sCy5, Figure 1). The ability of the resulting conjugate cyclo(DKP‐RGD)–sCy5 (2) to compete with biotinylated vitronectin for the binding to the isolated αvβ3 receptor was measured. The low‐nanomolar IC50 value obtained confirmed that integrin recognition was not affected by the conjugation with the probe (2, Figure 1). Moreover, we devised the preparation of a negative control, possessing almost identical physical‐chemical properties, while showing negligible affinity for the receptor. Inspired by the literature,20 the Arg‐Gly‐Asp tripeptide was mutated into a Arg‐[βAla]‐Asp sequence, giving rise to the cyclo(DKP‐RβAD) ligand 3 (Figure 1). In comparison with the parent ligand 1, the new compound showed a dramatically lower integrin binding affinity (micromolar IC50 value) and a similar result was obtained for the fluorescent conjugate cyclo(DKP‐RβAD)‐sCy5 4 (Figure 1).
Figure 1.
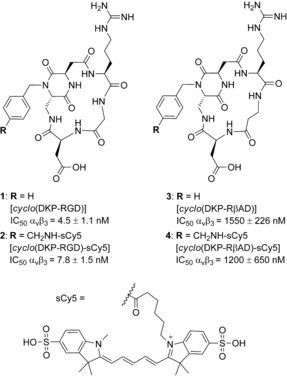
Molecular structure of the αvβ3 integrin ligand cyclo(DKP‐RGD) (1) and its analog cyclo(DKP‐RβAD) (3), alongside with their relative sulfo‐cyanine5 (sCy5) conjugates cyclo(DKP‐RGD)‐sCy5 (2) and cyclo(DKP‐RβAD)‐sCy5 (4). Data of inhibition of biotinylated vitronectin binding to the αvβ3 receptor are shown.
Confocal microscopy experiments were carried out with live human cancer cells 786‐O (renal cell carcinoma), expressing αvβ3 integrin (as detected by Western blot analysis; see Figure S1 in the Supporting Information), in the presence of conjugates 2 and 4. The uncoupled sulfo‐cyanine5 probe was also included in the experiment as additional control. As shown in Figure 2, a fair accumulation of the cyclo(DKP‐RGD)‐sCy5 conjugate (2) on the membrane of cancer cells was detected, whereas no significant intracellular uptake was observed. On the other hand, no accumulation of control compounds cyclo(DKP‐RβAD)‐sCy5 (4) and free sulfo‐cyanine5 was detected neither in intracellular compartments, nor on the cell surface.
Figure 2.
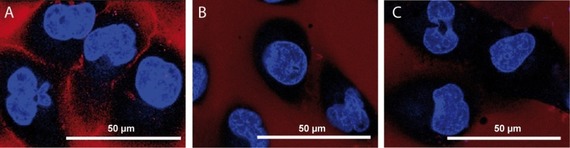
Confocal microscopy images of live αvβ3‐expressing human renal cell carcinoma 786‐O cells after exposure to conjugates 2 (A), 4 (B) and free sCy5 (C) (1 μmol L−1, exposure time=2 h). Accumulation at the cell membrane was detected only upon exposure to conjugate 2. No fluorescent signal was detected in the intracellular compartments for all compounds tested. Blue: Hoechst; Red: NIR dye.
Overall, this analysis confirmed that the integrin ligand cyclo(DKP‐RGD) 1 accumulates on the cell membrane of αvβ3 integrin‐expressing cancer cells, while it is poorly internalized by receptor‐mediated endocytosis. This finding may be related to the antagonist behavior of 1, which has been found to display inhibitory effects on the FAK/Akt integrin‐activated transduction pathway and on integrin‐mediated cell infiltration processes.21 The elucidation of the link between the agonist/antagonist behavior of integrin ligands and the receptor‐mediated internalization is currently a hot topic in this field.22
Interestingly, the importance of conjugate internalization has been recently challenged.23 Indeed, ADCs and SMDCs specific to non‐internalizing receptors (e.g., collagen IV,24 carbonic anhydrase IX,25 fibrin,26 and splice variants of fibronectin27 and tenascin‐C)28 were found to elicit strong antitumor responses in vivo, while proving significantly less potent than the parent drug in in vitro cell antiproliferative assays.29 For these reasons, we envisioned the preparation of a non‐internalizing, αvβ3 integrin‐targeted conjugate, featuring a specific linker for the extracellular release of the payload.
In 2002, researchers at Bayer developed a new SMDC, whose linker consisted in the tripeptide Asn‐Pro‐Val (NPV), a specific substrate of neutrophil elastase.30 The latter is a serine protease stored in azurophilic granules of neutrophils and released into the extracellular space upon infections or inflammation stimuli. High levels of elastase have been reported in primary tumors and metastasis, where it promotes oncogenic signaling and inhibits tumor suppressors. As a result, elevated neutrophil elastase levels correlate with poor prognosis in different types of solid tumors.31 To the best of our knowledge, no further investigations of this promising NPV peptide linker have been reported in the literature. For these reasons, we designed an extracellularly cleavable conjugate (5, Figure 3) in which the αvβ3 integrin ligand cyclo(DKP‐RGD) is connected to paclitaxel (PTX) via a self‐immolative spacer, the NPV linker and a hydrophilic PEG4 spacer. Two additional conjugates were prepared as negative controls: the cyclo(DKP‐RGD)‐NPv‐PTX conjugate (6), featuring the non‐proteinogenic amino acid d‐valine, and the cyclo(DKP‐RGD)‐unc‐PTX conjugate (7), in which the tripeptide moiety is replaced by a proteolytically stable (or “uncleavable”) tertiary amide bond. This panel of compounds was designed to gain insights into the specificity of elastase cleavage and the stability of the whole linker system. All synthetic details and analytics are included in the Supporting Information.
Figure 3.
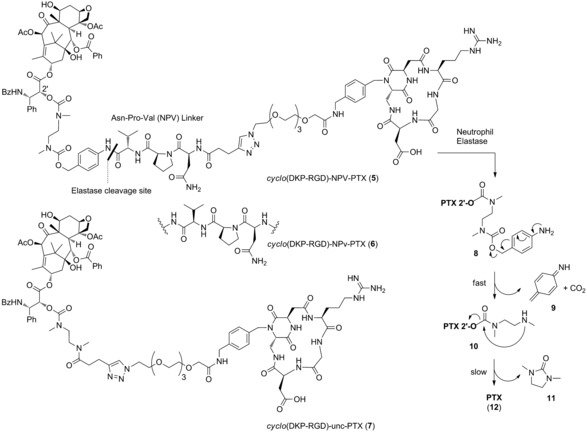
Molecular structures of the αvβ3 integrin‐targeted conjugate cyclo(DKP‐RGD)‐NPV‐PTX (5), of its diastereoisomer cyclo(DKP‐RGD)‐NPv‐PTX (6), whose linker bears d‐valine (v), and of the uncleavable conjugate cyclo(DKP‐RGD)‐unc‐PTX (7). Mechanism of PTX release upon linker cleavage is depicted, consisting in the fast 1,6‐elimination of the p‐aminobenzyl carbamate 8, followed by amine cyclization in compound 10, which results in the formation of cyclic urea 11 and free Paclitaxel (12).
The affinity of compounds 5–7 for αvβ3 integrin was estimated as described previously and low‐nanomolar values were obtained throughout the series (Table 1).
Table 1.
Inhibition of biotinylated vitronectin binding to the αvβ3 receptor.
| Conjugate | Structure | αvβ3 IC50 [nm][a] |
|---|---|---|
| 5 | cyclo(DKP‐RGD)‐NPV‐PTX | 12.9±1.4 |
| 6 | cyclo(DKP‐RGD)‐NPv‐PTX | 24.9±2.1 |
| 7 | cyclo(DKP‐RGD)‐unc‐PTX | 5.8±1.2 |
[a] IC50 values were determined as the concentration of compound required for 50 % inhibition of biotinylated vitronectin binding as estimated by GraphPad Prism software. All values are the arithmetic mean±the standard deviation (SD) of duplicate determinations.
To evaluate the cleavage of the tripeptide linker and the subsequent paclitaxel release in the presence of neutrophil elastase, conjugate 5 was treated with the enzyme in PBS solution at 37 °C, and metabolites were detected by HPLC‐MS analysis. The enzymatic cleavage of the tripeptide linker was observed over a 2 h period, resulting in the complete formation of metabolite 10 32 (Figure 3), whereas the use of inactivated enzyme did not lead to metabolite formation (Figure S2 in the Supporting Information). The selectivity of linker cleavage was analyzed by treating 5 with rat liver‐derived lysosome extract, composed of a mixture of proteolytic enzymes. The broad‐scope cysteine proteases inhibitor E‐64 was used in this experiment to gain insight into the effector enzymes involved in the cleavage. Upon 2 h exposure to lysosome extract, conjugate 5 was digested only partially (Figure S4 in the Supporting Information), possibly indicating the presence of elastase in the lysosome extract.33 This hypothesis was supported by the partial formation of metabolite 10 also in the presence of the E‐64 inhibitor, (Figure S4 in the Supporting Information) indicating that cysteine proteases are not responsible for the cleavage of the NPV linker.
Additionally, conjugate 5 was treated with mouse plasma at 37 °C, showing an excellent stability under these conditions (t 1/2=35.3 h, Figure S5 in the Supporting Information).
Following the protocol reported by Bayer,30 the in vitro cell antiproliferative activity of conjugates 5–7 was tested against 786‐O cancer cells, in the presence or absence of elastase. Importantly, Western blot analysis showed no significant elastase expression in these cancer cells (see Figure S1 in the Supporting Information). This model assay aimed at evaluating the extracellular cleavage of the NPV linker upon activation of neutrophils and release of elastase in the extracellular tumor environment, followed by the PTX internalization into cancer cells by passive diffusion through the cell membrane. In the absence of elastase, conjugates 5–7 did not exhibit a significant cytotoxic activity (IC50>5 μm, Table 2), whereas free PTX inhibited cell proliferation at nanomolar concentrations (IC50=35.8±16.7 nm). Interestingly, conjugate 5 displayed a >250‐fold increased activity (IC50=19.6±4.1 nm) upon addition of elastase (50 nm), and the observed potency was comparable to the one exhibited by free PTX under the same conditions (IC50=29.5±7.6 nm). On the other hand, the presence of elastase did not modify the original cell antiproliferative activity of conjugates 6 and 7. This result indicates not only that the PTX payload is inactive when it is not released from the targeting vehicle, but also that the use of the natural amino acid l‐Val at the linker C‐terminus is crucial for the recognition of the tripeptide sequence by elastase.
Table 2.
Antiproliferative activity of conjugates 5–7 and free PTX in αvβ3‐expressing human renal cell carcinoma 786‐O cells after 96 h treatment, in the presence (or absence) of elastase from human leukocytes.
| IC50 [nm][a] | ||
|---|---|---|
| Added elastase | ||
| Structure | NO | YES |
| Paclitaxel (PTX) (12) | 35.8±16.7 | 29.5±7.6 |
| cyclo(DKP‐RGD)‐NPV‐PTX (5) | >5000 | 19.6±4.1 |
| cyclo(DKP‐RGD)‐NPv‐PTX (6) | >5000 | >5000 |
| cyclo(DKP‐RGD)‐unc‐PTX (7) | >5000 | >5000 |
[a] IC50 values were determined as the concentration of compound required for 50 % inhibition of cell viability in presence or absence of 50 nm elastase (ELANE, 324681, Millipore). Samples were measured in triplicate.
In conclusion, integrin ligands represent promising vehicles for the selective release of anticancer drugs at the tumor site. However, it is still not clear whether different RGD‐based ligands may have different effects on the receptor internalization and recycling in cancer cells. Upon the observation that the integrin ligand cyclo(DKP‐RGD) is not efficiently internalized by αvβ3‐expressing cancer cells, we focused on suitable strategies for the delivery of anticancer agents in the extracellular tumor environment. In this work, the peptide sequence NPV, substrate of the serine protease elastase, has been investigated as trigger for the release of paclitaxel from an αvβ3‐targeted conjugate. Ideally, the integrin recognition unit would drive the conjugate accumulation at the tumor site, where the pro‐inflammatory stimuli result in the recruitment of tumor‐infiltrating leukocytes, such as neutrophils. The activation of the latter promotes the release of elastase, which triggers the payload release in the tumor microenvironment. As described elsewhere,23 this mode of activation may possess potential therapeutic benefits, since the free payload would diffuse in the tumor mass, and act against a large variety of cells (e.g., antigen‐negative cancer cells, endothelial and other cancer‐associated host cells) leading to a localized damage. It has been reported that lipophilic payloads are most suited for this strategy, as the membrane permeability would facilitate the cytotoxic activity by the so‐called “bystander effect.”34 Our in vitro data indicate that each individual unit of our SMDC (i.e., ligand, linker, and drug) may efficiently act according to this ideal mechanism of action. Moreover, the pro‐inflammatory environment and the presence of infiltrating cells of the immune system are well‐established hallmarks of cancers.35 It is therefore conceivable that elastase‐activatable prodrugs may be therapeutically active against a large variety of tumor types.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank the European Commission (Marie Skłodowska‐Curie ITN MAGICBULLET 642004) for a Ph.D. fellowship (to A.R.M.D.) and financial support. We also thank the University of Milan for a Ph.D. fellowship (to A.P.) and Ministero dell′Università e della Ricerca (PRIN 2015 project 20157WW5EH) for financial support. Amelia Dean (Cardiff University, UK) thanks the Erasmus+ Programme for a fellowship. Precious discussions on the biological assays with Dr. Beatrix Stelte‐Ludwig (Bayer AG) are thankfully acknowledged. We thank Dr. Nicoletta Colombo (Nerviano Medical Sciences) for the plasma stability experiments.
A. Raposo Moreira Dias, A. Pina, A. Dean, H.-G. Lerchen, M. Caruso, F. Gasparri, I. Fraietta, S. Troiani, D. Arosio, L. Belvisi, L. Pignataro, A. Dal Corso, C. Gennari, Chem. Eur. J. 2019, 25, 1696.
Contributor Information
André Raposo Moreira Dias, http://sites.unimi.it/gennarigroup/.
Prof. Dr. Cesare Gennari, Email: cesare.gennari@unimi.it.
References
- 1.
- 1a. Chari R. V. J., Miller M. L., Widdison W. C., Angew. Chem. Int. Ed. 2014, 53, 3796–3827; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 3872–3904; [Google Scholar]
- 1b. Beck A., Goetsch L., Dumontet C., Corvaïa N., Nat. Rev. Drug Discovery 2017, 16, 315–337. [DOI] [PubMed] [Google Scholar]
- 2. Dott J., Abila B., Wuerthner J. U., Pharm. Med. 2018, 32, 259–273. [Google Scholar]
- 3.
- 3a. Chatzisideri T., Leonidis G., Sarli V., Future Med. Chem. 2018, 10, 2201–2226; [DOI] [PubMed] [Google Scholar]
- 3b. Sartori A., Portioli E., Battistini L., Calorini L., Pupi A., Vacondio F., Arosio D., Bianchini F., Zanardi F., J. Med. Chem. 2017, 60, 248–262; [DOI] [PubMed] [Google Scholar]
- 3c. Bennett G., Harrison H., Campbell S., Teufel D., Langford G., Watt A., Bonny C., Eur. J. Cancer 2016, 69, S21. [Google Scholar]
- 4.
- 4a. Santos F. M. F., Matos A. I., Ventura A. E., Gonçalves J., Veiros L. F., Florindo H. F., Gois P. M. P., Angew. Chem. Int. Ed. 2017, 56, 9346–9350; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 9474–9478; [Google Scholar]
- 4b. Fernández M., Javaid F., Chudasama V., Chem. Sci. 2018, 9, 790–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Dao K.-L., Hanson R. N., Bioconjugate Chem. 2012, 23, 2139–2158. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Roy J., Nguyen T. X., Kanduluru A. K., Venkatesh C., Lv W., Reddy P. V. N., Low P. S., Cushman M., J. Med. Chem. 2015, 58, 3094–3103; [DOI] [PubMed] [Google Scholar]
- 6b. Kularatne S. A., Wang K., Santhapuram H. K. R., Low P. S., Mol. Pharm. 2009, 6, 780–789; [DOI] [PubMed] [Google Scholar]
- 6c. Krall N., Pretto F., Decurtins W., Bernardes G. J. L., Supuran C. T., Neri D., Angew. Chem. Int. Ed. 2014, 53, 4231–4235; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 4315–4320. [Google Scholar]
- 7.
- 7a. Krall N., Scheuermann J., Neri D., Angew. Chem. Int. Ed. 2013, 52, 1384–1402; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 1424–1443; [Google Scholar]
- 7b. Srinivasarao M., Galliford C. V., Low P. S., Nat. Rev. Drug Discovery 2015, 14, 203–219; [DOI] [PubMed] [Google Scholar]
- 7c. Srinivasarao M., Low P. S., Chem. Rev. 2017, 117, 12133–12164. [DOI] [PubMed] [Google Scholar]
- 8. Cazzamalli S., Dal Corso A., Widmayer F., Neri D., J. Am. Chem. Soc. 2018, 140, 1617–1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Joubert N., Denevault-Sabourin C., Bryden F., Viaud-Massuard M.-C., Eur. J. Med. Chem. 2017, 142, 393–415. [DOI] [PubMed] [Google Scholar]
- 10. Desgrosellier J. S., Cheresh D. A., Nat. Rev. Cancer 2010, 10, 9–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dal Corso A., Pignataro L., Belvisi L., Gennari C., Curr. Top. Med. Chem. 2016, 16, 314–329. [DOI] [PubMed] [Google Scholar]
- 12. Marchini M., Mingozzi M., Colombo R., Guzzetti I., Belvisi L., Vasile F., Potenza D., Piarulli U., Arosio D., Gennari C., Chem. Eur. J. 2012, 18, 6195–6207. [DOI] [PubMed] [Google Scholar]
- 13.
- 13a. Colombo R., Mingozzi M., Belvisi L., Arosio D., Piarulli U., Carenini N., Perego P., Zaffaroni N., De Cesare M., Castiglioni V., Scanziani E., Gennari C., J. Med. Chem. 2012, 55, 10460–10474; [DOI] [PubMed] [Google Scholar]
- 13b. Dal Corso A., Caruso M., Belvisi L., Arosio D., Piarulli U., Albanese C., Gasparri F., Marsiglio A., Sola F., Troiani S., Valsasina B., Pignataro L., Donati D., Gennari C., Chem. Eur. J. 2015, 21, 6921–6929; [DOI] [PubMed] [Google Scholar]
- 13c. Dias A. R. M., Pina A., Dal Corso A., Arosio D., Belvisi L., Pignataro L., Caruso M., Gennari C., Chem. Eur. J. 2017, 23, 14410–14415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pina A., Dal Corso A., Caruso M., Belvisi L., Arosio D., Zanella S., Gasparri F., Albanese C., Cucchi U., Fraietta I., Marsiglio A., Pignataro L., Donati D., Gennari C., ChemistrySelect 2017, 2, 4759–4766. [Google Scholar]
- 15. Bodero L., Rivas P. L., Korsak B., Hechler T., Pahl A., Müller C., Arosio D., Pignataro L., Gennari C., Piarulli U., Beilstein J. Org. Chem. 2018, 14, 407–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.
- 16a. Lee M. H., Kim J. Y., Han J. H., Bhuniya S., Sessler J. L., Kang C., Kim J. S., J. Am. Chem. Soc. 2012, 134, 12668–12674; [DOI] [PubMed] [Google Scholar]
- 16b. Nahrwold M., Weiß C., Bogner T., Mertink F., Conradi J., Sammet B., Palmisano R., Gracia S. R., Preuße T., Sewald N., J. Med. Chem. 2013, 56, 1853–1864. [DOI] [PubMed] [Google Scholar]
- 17. Rivas P. L., Ranđelović I., Dias A. R. M., Pina A., Arosio D., Tóvári J., Mező G., Dal Corso A., Pignataro L., Gennari C., Eur. J. Org. Chem. 2018, 2902–2909. [Google Scholar]
- 18. Wang X., Ma D., Olson W. C., Heston W. D. W., Mol. Cancer Ther. 2011, 10, 1728–1739. [DOI] [PubMed] [Google Scholar]
- 19.
- 19a. Legigan T., Clarhaut J., Tranoy-Opalinski I., Monvoisin A., Renoux B., Thomas M., Le Pape A., Lerondel S., Papot S., Angew. Chem. Int. Ed. 2012, 51, 11606–11610; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 11774–11778; [Google Scholar]
- 19b. Vineberg J. G., Wang T., Zuniga E. S., Ojima I., J. Med. Chem. 2015, 58, 2406–2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Garanger E., Boturyn D., Jin Z., Dumy P., Favrot M. C., Coll J. L., Mol. Ther. 2005, 12, 1168–1175. [DOI] [PubMed] [Google Scholar]
- 21. Panzeri S., Zanella S., Arosio D., Vahdati L., Dal Corso A., Pignataro L., Paolillo M., Schinelli S., Belvisi L., Gennari C., Piarulli U., Chem. Eur. J. 2015, 21, 6265–6271. [DOI] [PubMed] [Google Scholar]
- 22.
- 22a. Tolomelli A., Galletti P., Baiula M., Giacomini D., Cancers 2017, 9, 78; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22b. Nardelli F., Paissoni C., Quilici G., Gori A., Traversari C., Valentinis B., Sacchi A., Corti A., Curnis F., Ghitti M., Musco G., J. Med. Chem. 2018, 61, 7474–7485. [DOI] [PubMed] [Google Scholar]
- 23.
- 23a. Dal Corso A., Cazzamalli S., Neri D. in Innovations for Next-Generation Antibody–Drug Conjugates (Ed.: M. Damelin), Humana, New York, 2018, pp. 299–319; [Google Scholar]
- 23b. Gerber H. P., Senter P. D., Grewal I. S., MAbs 2009, 1, 247–253; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23c. Staudacher A. H., Brown M. P., Br. J. Cancer 2017, 117, 1736–1742; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23d. Gébleux R., Casi G., Pharmacol. Ther. 2016, 167, 48–59. [DOI] [PubMed] [Google Scholar]
- 24. Yasunaga M., Manabe S., Tarin D., Matsumura Y., Bioconjugate Chem. 2011, 22, 1776–1783. [DOI] [PubMed] [Google Scholar]
- 25.
- 25a. Cazzamalli S., Dal Corso A., Neri D., Mol. Cancer Ther. 2016, 15, 2926–2935; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25b. Cazzamalli S., Ziffels B., Widmayer F., Murer P., Pellegrini G., Pretto F., Wulhfard S., Neri D., Clin. Cancer Res. 2018, 24, 3656–3667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fuchigami H., Manabe S., Yasunaga M., Matsumura Y., Sci. Rep. 2018, 8, 14211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.
- 27a. Bernardes G. J., Casi G., Trussel S., Hartmann I., Schwager K., Scheuermann J., Neri D., Angew. Chem. Int. Ed. 2012, 51, 941–944; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 965–968; [Google Scholar]
- 27b. Perrino E., Steiner M., Krall N., Bernardes G. J., Pretto F., Casi G., Neri D., Cancer Res. 2014, 74, 2569–2578; [DOI] [PubMed] [Google Scholar]
- 27c. Gébleux R., Wulhfard S., Casi G., Neri D., Mol. Cancer Ther. 2015, 14, 2606–2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.
- 28a. Gébleux R., Stringhini M., Casanova R., Soltermann A., Neri D., Int. J. Cancer 2017, 140, 1670–1679; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28b. Dal Corso A., Cazzamalli S., Gébleux R., Mattarella M., Neri D., Bioconjugate Chem. 2017, 28, 1826–1833; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28c. Dal Corso A., Gébleux R., Murer P., Soltermann A., Neri D., J. Controlled Release 2017, 264, 211–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.
- 29a. Cazzamalli S., Dal Corso A., Neri D., J. Controlled Release 2017, 246, 39–45; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29b. Renoux B., Raes F., Legigan T., Peraudeau E., Eddhif B., Poinot P., Tranoy-Opalinski I., Alsarraf J., Koniev O., Kolodych S., Lerondel S., Le Pape A., Clarhautad J., Papot S., Chem. Sci. 2017, 8, 3427–3433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lerchen H. G., Baumgarten J., Schoop A., Albers M., (Bayer AG), PCT/EP2002/002501, WO 2002/072151, 2002.
- 31. Lerman I., Hammes S. R., Steroids 2018, 133, 96–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Compound 10 was synthesized from PTX (see the Supporting Information). The cyclization rate of the ethylenediamine self-immolative spacer in compound 10 was found to be pH dependent: though a half-life of 4.7 h was measured in a DMSO:PBS mixture at pH 7.5 and 37 °C (see Figure S3 A in the Supporting Information), no release of free PTX was detected in a DMSO:acetate buffer mixture at pH 5.5 over a 24 h period. The antiproliferative activity of compound 10 was evaluated in αvβ3-expressing human renal cell carcinoma 786-O cells after 110 h treatment, in comparison with free PTX (12). Under these experimental conditions, compound 10 proved moderately less potent than PTX (see Figure S3B in the Supporting Information).
- 33. Starkey P. M., Barrett A. J., Biochem. J. 1976, 155, 265–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Li F., Emmerton K. K., Jonas M., Zhang X., Miyamoto J. B., Setter J. R., Nicholas N. D., Okeley N. M., Lyon R. P., Benjamin D. R., Law C. L., Cancer Res. 2016, 76, 2710–2719. [DOI] [PubMed] [Google Scholar]
- 35. Hanahan D., Weinberg R. A., Cell 2011, 144, 646–674. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary