

Abstract
Herein, we communicate a selective and efficient protocol for oxidative arylating carbocyclization of enallenynes using O2 as the oxidant. The key to success for this aerobic transformation is the application of a specific electron transfer mediator (ETM), a bifunctional catalyst consisting of a metal‐macrocycle and quinone moieties. This catalyst significantly facilitates the reoxidation of Pd0 to PdII under atmospheric pressure of O2. Diverse functionalized enallenynes react with aryl boronic acids to afford the corresponding cyclic tetraenes in moderate to good yields.
Keywords: aerobic oxidation, carbocyclization, electron transfer mediator, enallenyne, palladium
The use of molecular oxygen (O2) in enzyme‐catalyzed oxidation reactions is highly widespread in nature.1 In the perspective of synthetic organic chemistry, molecular oxygen is an inexpensive, abundant, and highly atom‐efficient oxidant, which generates no toxic byproducts, thus fulfilling the requirements of “green chemistry”.2 Over the past decades, palladium‐catalyzed aerobic oxidations have provided the basis for streamlined conversion of various feedstocks into valuable products.3 Illustrative examples include Wacker oxidations,4 alcohol oxidations,5 alkene functionalizations,6 and oxidative C−H activations.7 In spite of the significant synthetic progress offered by these reactions, the relative low catalyst efficiency due to palladium deactivation still remains challenging in most cases.8 This oxidation problem can be explained by the slow electron transfer directly between Pd0 and O2, compared to the rapid precipitation of palladium black from active palladium species (Scheme 1 a).
Scheme 1.

a) The “Oxidation Problem” in palladium‐catalyzed aerobic reactions and the solutions. b) The cobalt‐based bifunctional catalyst (Co(salophen)‐HQ) as an electron transfer mediator in this study.
To circumvent the problem of getting Pd black in reoxidation of Pd0 by O2, considerable attention has been focused on the development of ancillary air‐stable ligands such as amines, pyridines, sulfoxides, and carbene derivatives that can inhibit precipitation of Pd black during the catalytic cycle.9 On the other hand, inspired by nature, a coupled catalyst system with electron transfer mediators (ETMs) can facilitate the transport of the electrons from the reduced palladium catalyst to O2, thereby increasing the efficiency of the reoxidation of Pd0 to PdII.10 After the original work by Bäckvall in the late 1980s,11 several groups have explored this concept by developing mild and efficient PdII‐catalyzed aerobic oxidative reactions.12 The key to success for these transformations is the use of macrocyclic metal complexes and quinones as ETMs under aerobic conditions. Additionally, improved coupled catalyst systems, in which a metal‐macrocycle and quinones are merged into one molecule, have been reported by our group.13 These bifunctional catalysts led to an increased efficiency of the electron transfer compared to that observed for the system with the quinone and metal‐macrocycle as separate molecules.13 Based on these state‐of‐the‐art methods, we present herein the work on the use of such a bifunctional catalyst (Co(salophen)‐HQ; HQ=hydroquinone, Scheme 1 b) as an efficient electron transfer mediator in Pd‐catalyzed aerobic oxidative carbocyclizations of enallenynes.
Allenes constitute an important class of synthons in organic synthesis, which can be applied to construct a variety of valuable molecules.14 Our group has a long‐standing interest in Pd‐catalyzed C−C bond‐forming reactions from allene‐substituted unsaturated compounds, such as enallenes, allenynes, and bisallenes.15 In the present work, we report for the first time an example of Pd‐catalyzed oxidative carbocyclization of rationally designed enallenynes. These interesting molecules contain three different C−C π‐bond functionalities (allene, olefin, and alkyne groups), all of which can be potential reaction sites.16 However, the presence of these three π systems provides a challenge concerning control of regioselectivity in the reaction. As shown in Scheme 2, there are three possible pathways for the transformation of vinylpalladium species Int‐A, which is generated from an initial allenic C−H cleavage15 of enallenyne 1 by PdII: 1) According to our previous work,17 the close‐by olefin insertion of Int‐A could lead to a highly strained cyclobutene complex Int‐B, followed by arylation to give a four‐membered carbocycle 3′ (Path I). 2) Intermediate Int‐A can also be directly trapped by a nucleophile such as ArB(OH)2 to give an acyclic arylated product 3′′ (Path II).18 3) A ligand exchange of the olefin for alkyne in Int‐A would give Int‐C, which on subsequent alkyne insertion can generate six‐membered ring species Int‐D. Trapping of Int‐D by ArB(OH)2 would lead to cyclic tetraene 3 (Path III).
Scheme 2.

Possible pathways for palladium‐catalyzed oxidative functionalization (arylation) of enallenynes.
With these possibilities in mind, our goal was to develop a general and efficient catalyst system for oxidative functionalization of enallenynes in a selective manner. More specifically, in light of recent growing interest in green and sustainable chemistry, we have undertaken preliminary investigations to use molecular oxygen as terminal oxidant.
At the outset of our investigations, the palladium‐catalyzed aerobic oxidation of a readily accessible enallenyne 1 a and phenylboronic acid 2 a in H2O/acetone was chosen as the benchmark reaction (Table 1).19 In the absence of ETMs, we did not observe any formation of the arylated product under aerobic conditions and the starting material 1 a could be recovered in 93 % yield (entry 1). When catalytic amounts of BQ (p‐benzoquinone) was added, a highly unsaturated six‐membered carbocycle 3 a was selectively formed albeit in low yield (15 %, entry 2).20 It is noteworthy that this unexpected cyclization takes place between the allene moiety and distal triple bond of enallenyne 1 a, while the olefin group remains intact.21 This interesting result motivated us to further explore various established metal‐based electron transfer mediators to improve the reaction efficiency. The application of VO(acac)2, Fe(Pc), and Co(Pc) (Pc=phthalocyanine) as ETMs did not lead to any significant improvement of the transformation of 1 a to 3 a. (21–24 % yields, entries 3–5). To our delight, the use of cobalt‐based salen and salophen complexes led to good yields of 3 a (71 and 74 % yields, entries 6 and 7). As to the reduced state of BQ, HQ (hydroquinone) also furnished the cyclization product 3 a in 63 % yield (entry 8). Interestingly, a bifunctional cobalt catalyst (Co(salophen)‐HQ), combining a metal‐macrocycle and quinones moieties, was found to be the best performing catalyst and afforded the desired product 3 a in 79 % yield (entry 9). Notably, even under lower catalyst loading (2 mol % Pd(OAc)2 and 5 mol % Co(salophen)‐HQ), no significant decrease in yield was observed (76 % yield, entry 10 vs. 79 % yield, entry 9).
Table 1.
Evaluation of different electron transfer mediators (ETMs) for oxidative carbocyclization of enallenyne 1 a.[a]

| Entry | ETM1 (10 mol %) | ETM2 (20 mol %) | Yield of 3 a [%] | Recovery of 1 a [%] |
|---|---|---|---|---|
| 1 | – | – | 0 | 93 |
| 2 | – | BQ | 15 | 70 |
| 3 | VO(acac)2 | BQ | 24 | 64 |
| 4 | Fe(Pc) | BQ | 23 | 55 |
| 5 | Co(Pc) | BQ | 21 | 56 |
| 6 | Co(salen) | BQ | 71 | 8 |
| 7 | Co(salophen) | BQ | 74 | 6 |
| 8 | Co(salophen) | HQ | 63 | 18 |
| 9[b] | Co(salophen)‐HQ | – | 79 | <1 |
| 10[c] | Co(salophen)‐HQ | – | 76 | 3 |

| ||||
[a] Unless otherwise noted, the following reaction condition were employed: 1 a (0.1 mmol, 1.0 equiv), 2 a (0.15 mmol, 1.5 equiv), Pd(OAc)2 (5 mol %), ETM1 (10 mol %), ETM2 (20 mol %), H2O (1.0 equiv) in 0.1 m acetone, O2 (1 atm) at room temperature (23 °C) for 48 h. Yield and conversion are determined by 1H NMR using anisole as internal standard. [b] 10 mol % Co(salophen)‐HQ was added. [c] 5 mol % Co(salophen)‐HQ with 2 mol % Pd(OAc)2 was added.
Next, the catalytic reaction progress with different electron transfer mediators (ETMs) was examined using 1 mol % of Pd(OAc)2, 2.5 mol % Co(salophen)‐HQ or 2.5 mol % Co(salophen), and 5 mol % quinone, at 30 °C under 1 atm of air. As shown in Figure 1, the use of Co(salophen)‐HQ resulted in a higher reaction rate than that with Co(salophen) and quinone as separate ETMs. This result indicates that the intramolecular electron transfer between the hydroquinone unit and the oxidized metal‐macrocycle of this bifunctional catalyst leads to a more efficient overall reaction under aerobic conditions.22
Figure 1.

Reaction progress with different ETMs.
After optimizing the reaction conditions, we continued to explore the scope of this transformation with various arylboronic acids (Scheme 3). First, when applying PhB(OH)2 as the arylating partner, 77 % isolated yield of cyclization product 3 a was achieved. Importantly, this aerobic catalytic system appeared to be equally effective in comparison to the application of stoichiometric BQ as oxidant (76 % yield in Scheme 3), therefore highlighting the advancement of this efficient and sustainable method. In addition to PhB(OH)2, arylboronic acids bearing an electron neutral group (3‐Me), donating group (3‐OMe), and withdrawing group (4‐Acyl) all reacted well and produced the corresponding cyclic tetraenes (3 b–3 d) in good yields (61–77 %, respectively). Various halogen‐containing substrates (3‐F, 3‐Cl, and 4‐Br) were well compatible with this methodology and gave the desired products (3 e–3 g) in 54–74 % yields. 2‐Naphthylboronic acid also reacted smoothly and a moderate yield (53 %) of 3 h could be achieved. Moreover, when 4‐vinylphenylboronic acid was used as the substrate, the olefin bond remained intact and 52 % yield of the desired product 3 i was obtained. However, 2‐thienylboronic acid, 4‐pyridinylboronic acid, and trans‐2‐phenylvinylboronic acid did not give the corresponding tetraenes under the optimal reaction conditions.
Scheme 3.
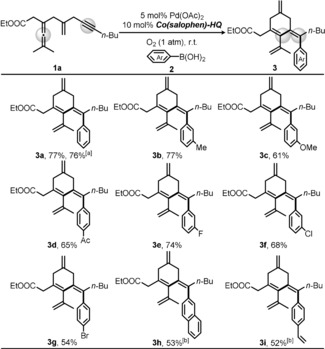
Substrate scope of different arylboronic acids 2. Reaction conditions: 1 a (1.0 equiv), ArB(OH)2 2 (1.5 equiv), Pd(OAc)2 (5 mol %), Co(salophen)‐HQ (10 mol %), H2O (1.0 equiv) in 0.1 m acetone, O2 (1 atm) at room temperature (23 °C) for 48 h. Yield of isolated product. [a] Conditions employing 1.1 equiv BQ instead of 10 mol % Co(salophen)‐HQ under Ar. [b] Reaction time of 72 h.
We next investigated the reactivity of structurally diverse enallenynes 1 in this aerobic arylating carbocyclization (Scheme 4). Various substitutions on the allene moiety of 1, such as phenyl (3 j), cyclopentyl (3 k), and cyclohexyl (3 l) groups were well tolerated, furnishing products in 61–81 % yields. Moreover, functional groups on the alkyne moiety, bearing ‐OAc (3 m), ‐OBn (3 n), and ‐CN (3 o) were compatible with the aerobic conditions and gave the corresponding carbocycles in moderate to good yields (65–79 %). Under the optimized aerobic condition, cyclohexylacetylene‐substituted enallenyne substrate 1 p afforded 3 p in only 36 % yield. We attribute this diminished reactivity to the increased steric bulk of the alkyne moiety. In addition to an ester group, enallenynes containing butyl (3 q), benzyl (3 r), hydroxyl (3 s), silyl (3 t), sulfonamide (3 u), and imide (3 v) functionalities underwent this transformation smoothly highlighting the broad substrate scope of this protocol. Late‐stage oxidative reaction is a powerful approach for the streamlining and diversification of complex natural products and medicinal compounds. Here, an estrone‐derived substrate, participated in this reaction efficiently to afford a useful yield (75 %) of a functionalized complex molecule 3 w.
Scheme 4.

Substrate scope of different enallenyne 1. Reaction conditions: enallenyne 1 (1.0 equiv), PhB(OH)2 2 a (1.5 equiv) Pd(OAc)2 (5 mol %), Co(salophen)‐HQ (10 mol %), H2O (1.0 equiv) in 0.1 m acetone, O2 (1 atm) at room temperature (23 °C) for 48 h. Yield of isolated product.
Furthermore, to demonstrate the necessity of the olefin group (X is C=CH2) in substrate 1 m, control experiments were performed with substrates lacking the olefin group (Table 2). At first, allenyne 1 m′, without the olefin group in the β‐position (X is CH2) of the allene moiety, was applied under the standard reaction conditions. Notably, the six‐membered carbocycle 3 m′ was not observed (entry 2 vs. entry 1). Allenynes containing heteroatom linkers such as O (1 x) and NTs (1 y) were evaluated in this aerobic arylative carbocyclization; however, neither of them was found to be effective (entries 3 and 4). In addition to an olefin group, we recently showed that a hydroxyl functionality, can trigger allene attack by weak coordination to the PdII center, enabling efficient oxidative carbocyclizations of enallenols.23 We therefore examined the reactivity of an allenynol 1 z, with a hydroxyl group at the β‐position of the allene moiety. However, the desired oxidative carbocyclization did not take place in this case (entry 5). These experiments show that the initial step of allenic C(sp3)−H cleavage by PdII requires the coordination of a pending olefin bond.18 A recent computational study by the Liu group supports that the assisting olefin group plays an indispensable role in the formation of the vinyl‐palladium intermediate.24
Table 2.
Investigation of different linkers in allenynes for Pd‐catalyzed aerobic arylating carbocyclization.[a]

| Entry | Substrate 1 | Product 3 | Yield of 3 [%] |
|---|---|---|---|
| 1 | 1 m, X=C=CH2 | 3 m | 65 |
| 2 | 1 m′, X=CH2 | 3 m′ | 0 |
| 3 | 1 x, X=O | 3 x | 0 |
| 4 | 1 y, X=NTs | 3 y | 0 |
| 5 | 1 z, X=CH‐OH | 3 z | 0 |
[a] Reaction conditions: allene 1 (1.0 equiv), PhB(OH)2 2 a (1.5 equiv), Pd(OAc)2 (5 mol %), Co(salophen)‐HQ (10 mol %), H2O (1.0 equiv) in 0.1 m acetone, O2 (1 atm) at room temperature (23 °C) for 48 h.
On the basis of these experimental findings, the mechanism given in Scheme 5 is proposed for this oxidative arylating carbocyclization. Initially, the coordination of allene and olefin units to the PdII center leads to a chelate palladium complex Int‐II. This special coordination of the close‐by olefin to PdII is essential for triggering the allenic C(sp3)−H cleavage and generating a vinylpalladium intermediate Int‐III.18 Next, instead of direct arylation to give 3′′ or olefin insertion to form a cyclobutene complex Int‐B (see Scheme 2), the envisioned ligand exchange of olefin25 by the distant alkyne moiety takes place to give Int‐IV. Subsequent carbocyclization of Int‐IV by alkyne insertion gives a cyclic vinylpalladium Int‐V. Transmetallation of Int‐V with arylboronic acid 2,26 followed by reductive elimination provides the target product 3 and Pd0 species Int‐VI, respectively. Finally, with the assistance of the cobalt hybrid catalyst as electron transfer mediator, aerobic oxidation of Pd0 regenerates PdII to close the catalytic cycle.27
Scheme 5.
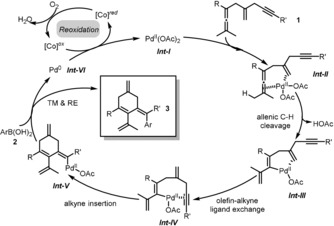
Proposed mechanism.
In summary, we have developed an efficient PdII‐catalyzed oxidative carbocyclization of enallenynes using molecular oxygen as the oxidant. By applying a bifunctional catalyst (Co(salophen)‐HQ) as an efficient electron transfer mediator, a wide range of enallenynes and arylboronic acids were transformed into the corresponding six‐membered carbocycles in moderate to good yields. In view of the reaction efficiency and good selectivity, this protocol is expected to complement the current approach for oxidative functionalization in homogeneous catalysis and organic synthesis.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We are grateful for the financial support from the European Research Council (ERC AdG 247014), Swedish Research Council (2016‐03897, 2018‐00830), and Carl Tryggers Foundation. We thank Dr. Zoltán Bacsik for the characterization of the catalyst.
J. Liu, A. Ricke, B. Yang, J.-E. Bäckvall, Angew. Chem. Int. Ed. 2018, 57, 16842.
Dedicated to Professor Elias J. Corey and Professor Xiyan Lu on the occasion of their 90th birthday
Contributor Information
Dr. Jie Liu, http://www.organ.su.se/jeb/
Dr. Bin Yang, Email: binyang@organ.su.se
Prof. Dr. Jan‐E. Bäckvall, Email: jeb@organ.su.se.
References
- 1.
- 1a. Tang M.-C., Zou Y., Watanabe K., Walsh C. T., Tang Y., Chem. Rev. 2017, 117, 5226–5333; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1b. Huang X., Groves J. T., Chem. Rev. 2018, 118, 2491–2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bäckvall J.-E., Modern Oxidation Methods, Second Edition, Wiley-VCH, Weinheim, 2011. [Google Scholar]
- 3. Stahl S. S., Science 2005, 309, 1824. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Jira R., Angew. Chem. Int. Ed. 2009, 48, 9034–9037; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 9196–9199; [Google Scholar]
- 4b. Keith J. A., Henry P. M., Angew. Chem. Int. Ed. 2009, 48, 9038–9049; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 9200–9212. [Google Scholar]
- 5.
- 5a. Sheldon R. A., Arends I. W. C. E., ten Brink G.-J., Dijksman A., Acc. Chem. Res. 2002, 35, 774–781; [DOI] [PubMed] [Google Scholar]
- 5b. Sigman M. S., Jensen D. R., Acc. Chem. Res. 2006, 39, 221–229; [DOI] [PubMed] [Google Scholar]
- 5c. Horn E. J., Rosen B. R., Baran P. S., ACS Cent. Sci. 2016, 2, 302–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.For recent reviews, see:
- 6a. McDonald R. I., Liu G., Stahl S. S., Chem. Rev. 2011, 111, 2981–3019; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6b. Sigman M. S., Werner E. W., Acc. Chem. Res. 2012, 45, 874–884; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6c. Wu W., Jiang H., Acc. Chem. Res. 2012, 45, 1736–1748; [DOI] [PubMed] [Google Scholar]
- 6d. Wu X.-F., Fang X., Wu L., Jackstell R., Neumann H., Beller M., Acc. Chem. Res. 2014, 47, 1041–1053; [DOI] [PubMed] [Google Scholar]
- 6e. Dong J. J., Browne W. R., Feringa B. L., Angew. Chem. Int. Ed. 2015, 54, 734–744; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 744–755; [Google Scholar]
- 6f. Mann S. E., Benhamou L., Sheppard T. D., Synthesis 2015, 47, 3079–3117; [Google Scholar]
- 6g. Yin G., Mu X., Liu G., Acc. Chem. Res. 2016, 49, 2413–2423; [DOI] [PubMed] [Google Scholar]
- 6h. Dong Z., Ren Z., Thompson S. J., Xu Y., Dong G., Chem. Rev. 2017, 117, 9333–9403; [DOI] [PubMed] [Google Scholar]
- 6i. Zhang D., Liu J., Córdova A., Liao W.-W., ACS Catal. 2017, 7, 7051–7063; [Google Scholar]
- 6j. Sommer H., Juliá-Hernández F., Martin R., Marek I., ACS Cent. Sci. 2018, 4, 153–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.For recent reviews, see:
- 7a. Liu C., Yuan J., Gao M., Tang S., Li W., Shi R., Lei A., Chem. Rev. 2015, 115, 12138–12204; [DOI] [PubMed] [Google Scholar]
- 7b. Hartwig J. F., Larsen M. A., ACS Cent. Sci. 2016, 2, 281–292; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7c. Iosub A. V., Stahl S. S., ACS Catal. 2016, 6, 8201–8213; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7d. Davies H. M. L., Morton D., ACS Cent. Sci. 2017, 3, 936–943; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7e. He J., Wasa M., Chan K. S. L., Shao Q., Yu J.-Q., Chem. Rev. 2017, 117, 8754–8786; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7f. Liang Y.-F., Jiao N., Acc. Chem. Res. 2017, 50, 1640–1653; [DOI] [PubMed] [Google Scholar]
- 7g. Yang Y., Lan J., You J., Chem. Rev. 2017, 117, 8787–8863; [DOI] [PubMed] [Google Scholar]
- 7h. Sterckx H., Morel B., Maes B. U. W., Angew. Chem. Int. Ed. 2018, 10.1002/anie.201804946; [DOI] [Google Scholar]; Angew. Chem. 2018, 10.1002/ange.201804946. [DOI] [Google Scholar]
- 8.
- 8a. Campbell A. N., Stahl S. S., Acc. Chem. Res. 2012, 45, 851–863; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8b. Jin L., Lei A., Sci. China Chem. 2012, 55, 2027–2035; [Google Scholar]
- 8c. Shi Z., Zhang C., Tang C., Jiao N., Chem. Soc. Rev. 2012, 41, 3381–3430. [DOI] [PubMed] [Google Scholar]
- 9. Wang D., Weinstein A. B., White P. B., Stahl S. S., Chem. Rev. 2018, 118, 2636–2679. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Piera J., Bäckvall J.-E., Angew. Chem. Int. Ed. 2008, 47, 3506–3523; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 3558–3576; [Google Scholar]
- 10b. Vasseur A., Muzart J., Le Bras J., Eur. J. Org. Chem. 2015, 4053–4069; [Google Scholar]
- 10c. Wendlandt A. E., Stahl S. S., Angew. Chem. Int. Ed. 2015, 54, 14638–14658; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 14848–14868. [Google Scholar]
- 11.
- 11a. Bäckvall J.-E., Awasthi A. K., Renko Z. D., J. Am. Chem. Soc. 1987, 109, 4750–4752; [Google Scholar]
- 11b. Bäckvall J.-E., Hopkins R. B., Grennberg H., Mader M., Awasthi A. K., J. Am. Chem. Soc. 1990, 112, 5160–5166. [Google Scholar]
- 12.For recent examples, see:
- 12a. Piera J., Naerhi K., Bäckvall J.-E., Angew. Chem. Int. Ed. 2006, 45, 6914–6917; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 7068–7071; [Google Scholar]
- 12b. Piera J., Persson A., Caldentey X., Bäckvall J.-E., J. Am. Chem. Soc. 2007, 129, 14120–14121; [DOI] [PubMed] [Google Scholar]
- 12c. Morandi B., Wickens Z. K., Grubbs R. H., Angew. Chem. Int. Ed. 2013, 52, 2944–2948; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 3016–3020; [Google Scholar]
- 12d. Volla C. M. R., Bäckvall J.-E., Angew. Chem. Int. Ed. 2013, 52, 14209–14213; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 14459–14463; [Google Scholar]
- 12e. Pattillo C. C., Strambeanu I. I., Calleja P., Vermeulen N. A., Mizuno T., White M. C., J. Am. Chem. Soc. 2016, 138, 1265–1272; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12f. Ta L., Axelsson A., Sunden H., Green Chem. 2016, 18, 686–690. [Google Scholar]
- 13.
- 13a. Grennberg H., Faizon S., Bäckvall J.-E., Angew. Chem. Int. Ed. Engl. 1993, 32, 263–264; [Google Scholar]; Angew. Chem. 1993, 105, 269–271; [Google Scholar]
- 13b. Purse B. W., Tran L.-H., Piera J., Åkermark B., Bäckvall J.-E., Chem. Eur. J. 2008, 14, 7500–7503; [DOI] [PubMed] [Google Scholar]
- 13c. Johnston E. V., Karlsson E. A., Lindberg S. A., Åkermark B., Bäckvall J.-E., Chem. Eur. J. 2009, 15, 6799–6801; [DOI] [PubMed] [Google Scholar]
- 13d. Johnston E. V., Karlsson E. A., Tran L.-H., Aakermark B., Bäckvall J.-E., Eur. J. Org. Chem. 2009, 3973–3976. [Google Scholar]
- 14.
- 14a. Krause N., Hashmi A. S. K., Modern Allene Chemistry, Wiley-VCH, Weinheim, 2008; [Google Scholar]
- 14b. Aubert C., Fensterbank L., Garcia P., Malacria M., Simonneau A., Chem. Rev. 2011, 111, 1954–1993; [DOI] [PubMed] [Google Scholar]
- 14c. Inagaki F., Kitagaki S., Mukai C., Synlett 2011, 594–614; [Google Scholar]
- 14d. Krause N., Winter C., Chem. Rev. 2011, 111, 1994–2009; [DOI] [PubMed] [Google Scholar]
- 14e. López F., Mascareñas J. L., Chem. Eur. J. 2011, 17, 418–428; [DOI] [PubMed] [Google Scholar]
- 14f. Rivera-Fuentes P., Diederich F., Angew. Chem. Int. Ed. 2012, 51, 2818–2828; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 2872–2882; [Google Scholar]
- 14g. Yu S., Ma S., Angew. Chem. Int. Ed. 2012, 51, 3074–3112; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 3128–3167; [Google Scholar]
- 14h. Ye J., Ma S., Acc. Chem. Res. 2014, 47, 989–1000. [DOI] [PubMed] [Google Scholar]
- 15. Yang B., Qiu Y., Bäckvall J.-E., Acc. Chem. Res. 2018, 51, 1520–1531. [DOI] [PubMed] [Google Scholar]
- 16.
- 16a. Saito N., Ichimaru T., Sato Y., Chem. Asian J. 2012, 7, 1521–1523; [DOI] [PubMed] [Google Scholar]
- 16b. Ohta Y., Yasuda S., Yokogawa Y., Kurokawa K., Mukai C., Angew. Chem. Int. Ed. 2015, 54, 1240–1244; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 1256–1260; [Google Scholar]
- 16c. Raviola C., Protti S., Ravelli D., Fagnoni M., Chem. Soc. Rev. 2016, 45, 4364–4390; [DOI] [PubMed] [Google Scholar]
- 16d. Cassú D., Parella T., Solà M., Pla-Quintana A., Roglans A., Chem. Eur. J. 2017, 23, 14889–14899. [DOI] [PubMed] [Google Scholar]
- 17. Qiu Y., Yang B., Zhu C., Bäckvall J.-E., Angew. Chem. Int. Ed. 2016, 55, 6520–6524; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 6630–6634. [Google Scholar]
- 18. Zhu C., Yang B., Jiang T., Bäckvall J.-E., Angew. Chem. Int. Ed. 2015, 54, 9066–9069; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 9194–9197. [Google Scholar]
- 19.There is an equilibrium between PhB(OH)2 and its anhydride form (PhBO)3 We speculate that the addition of water can promote the equilibrium toward the formation of PhB(OH)2. Please see the Supporting Information for details.
- 20.The side product 3 a′ and 3 a′′ in Scheme 2 were not obtained. For a discussion on the selectivity, please see the Supporting Information.
- 21.The isomer of arene from aromatization of tetraene 3 a was not observed.
- 22.A proposed catalytic cycle for reoxidation of Pd0 by Co(salophen)-HQ is given in the Supporting Information. For a related mechanistic study, see: Anson C. W., Ghosh S., Hammes-Schiffer S., Stahl S. S., J. Am. Chem. Soc. 2016, 138, 4186–4193. [DOI] [PubMed] [Google Scholar]
- 23. Posevins D., Qiu Y., Bäckvall J.-E., J. Am. Chem. Soc. 2018, 140, 3210–3214. [DOI] [PubMed] [Google Scholar]
- 24. Han L., Liu T., Org. Biomol. Chem. 2017, 15, 5055–5061. [DOI] [PubMed] [Google Scholar]
- 25.
- 25a. Zhu C., Yang B., Qiu Y., Bäckvall J.-E., Angew. Chem. Int. Ed. 2016, 55, 14405–14408; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 14617–14620; [Google Scholar]
- 25b. Qiu Y., Yang B., Zhu C., Bäckvall J.-E., Chem. Sci. 2017, 8, 616–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.In the absence of ArB(OH)2, no product was obtained. The Int-V could not be detected because a fast trapping of Int-V by ArB(OH)2 occurs and generates the corresponding cyclic tetraene.
- 27.Additionally, to facilitate electron transfer from the reduced palladium catalyst to O2, we speculate that the quinone moiety of the oxidized Co(salophen)-HQ can act as a ligand that coordinates to the PdII intermediate during the catalysis. Quinone coordination to PdII could withdraw electron density from the PdII center, therefore promoting the subsequent reductive elimination step.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary