

Abstract
The ubiquitin system regulates countless physiological and disease‐associated processes and has emerged as an attractive entryway for therapeutic efforts. With over 600 members in the human proteome, ubiquitin ligases are the most diverse class of ubiquitylation enzymes and pivotal in encoding specificity in ubiquitin signaling. Although considerable progress has been made in the identification of small molecules targeting RING ligases, relatively little is known about the “druggability” of HECT (homologous to E6AP C terminus) ligases, many of which are critically implicated in human pathologies. A major obstacle to optimizing the few available ligands is our incomplete understanding of their inhibitory mechanisms and the structural basis of catalysis in HECT ligases. Here, we survey recent approaches to manipulate the activities of HECT ligases with small molecules to showcase the particular challenges and opportunities these enzymes hold as therapeutic targets.
Keywords: drug discovery, enzymes, inhibitors, reaction mechanisms, structure–activity relationships, ubiquitin
1. Introduction
1.1. Targeting the ubiquitin system
The modification of eukaryotic proteins with ubiquitin controls their lifetimes, abundance, localization, interactions, and activities, thereby regulating protein function at all levels. It is thus not surprising that the ubiquitin system is critically implicated in many human diseases and has become a prime focus of therapeutic efforts.1 The remarkable success of proteasome inhibitors, such as bortezomib, in the treatment of multiple myeloma2 fed the idea of manipulating components of the ubiquitylation machinery upstream of the proteasome for even more specific therapeutic effects.
The ubiquitylation machinery operates as a catalytic cascade and is counteracted by deubiquitinases (DUBs). First, a ubiquitin‐activating (E1) enzyme hydrolyses adenosine triphosphate (ATP) to generate an energy‐rich thioester linkage between a cysteine residue at its catalytic center and the C terminus of ubiquitin. In the second step, ubiquitin is transferred to the catalytic cysteine of a ubiquitin‐conjugating (E2) enzyme. Finally, a ubiquitin ligase (E3 enzyme) interacts with the ubiquitin‐loaded E2 enzyme to link ubiquitin to a substrate protein. Depending on their architectures, E3 enzymes follow different mechanisms: really interesting new gene (RING)‐type and U‐box ligases act as scaffolds for the colocalization of a ubiquitin‐loaded E2 enzyme and a substrate and promote the direct transfer of ubiquitin from the E2 to the substrate. In contrast, HECT (homologous to E6AP C terminus) and RBR (RING‐in‐between‐RING) ligases have their own catalytic cysteine and take over ubiquitin from the E2 enzyme before passing it on to the substrate. Regardless of their mechanism, however, most ubiquitin ligases catalyze the formation of an isopeptide or peptide bond between the C terminus of ubiquitin and a primary amino group of the substrate.
Multiple cycles of the catalytic cascade lead to the modification of substrates at different sites (multi‐monoubiquitylation) or the formation of ubiquitin chains (polyubiquitylation), if ubiquitin itself functions as a substrate. Because ubiquitin has eight primary amino groups (seven lysines and the N terminus), such chains can have different linkage types and adopt distinct conformations associated with specific functional outcomes.3 The specificity of ubiquitin as a molecular signal thus depends on the identity of the substrate, the pattern of modification sites, and the types of ubiquitin modifications formed. Importantly, all of these features are controlled by the dynamic interplay of ubiquitin ligases and DUBs. The availability of specific receptors that recognize distinct modifications provides another important layer in encoding specificity in ubiquitin signaling downstream of E3 enzymes.
With over 600 members in the human proteome,4 ubiquitin ligases are pivotal in conferring specificity in ubiquitylation and provide particularly interesting targets for therapeutic interventions. Progress in manipulating the activities of RING ligases, as reviewed by Dixit and Huang,5 includes the discovery that the immunomodulatory drug thalidomide targets a RING ligase, CRL4CRBN.6 Along with landmark work on the structural basis of thalidomide action,7 this finding has refueled efforts to generate protein‐targeting chimeric compounds (PROTACs), which direct the activities of RING ligases at specific disease‐associated proteins, thereby marking them for proteasomal degradation.8 In contrast, few studies have focused on targeting HECT ligases by small molecules.
1.2. Targeting HECT‐type ubiquitin ligases
The 28 human HECT ligases regulate a wide range of cellular signaling pathways, many of which are critically linked to human pathologies.9 For example, E6AP (UBE3A) is hijacked by the E6 protein from high‐risk human papilloma viruses (HPVs) to promote proteasomal degradation of the tumor suppressor p53, thereby driving HPV‐induced cervical carcinogenesis.9 Maternally inherited deletion or mutation of this ligase gene results in a neurodevelopmental disease known as Angelman syndrome; in contrast, genetic amplification or mutational upregulation of E6AP is linked to autism‐spectrum disorders.9 Genetic alterations of HUWE1 (MULE, ARF‐BP1, HECTH9, LASU1, UREB1) have been associated with X‐linked intellectual impairment.10, 11, 12, 13, 14 Moreover, various HECT ligases, including HUWE1, NEDD4‐1, WWP1 (TIUL1, AIP1), WWP2 (AIP2), ITCH (NAPP1, AIP4), SMURF2, UBR5 (EDD), and members of the HERC subfamily have been implicated in tumorigenesis and immune signaling, respectively.9, 15
HECT ligases share a C‐terminal catalytic HECT domain of about 45 kDa that consists of two lobes, connected by a short, flexible linker.16, 17 The regions preceding the HECT domain, which range from about 40 to 490 kDa, have variable domain compositions and are poorly characterized.18 Most of our structural and mechanistic knowledge of HECT ligases originates from studies of the NEDD4 subfamily (NEDD4‐1, NEDD4‐2, ITCH, WWP1, WWP2, SMURF1, SMURF2, HECW1, HECW2), which constitutes 30 % of the human HECT ligases.18, 19 However, it is largely unknown to what extent the mechanistic features described for NEDD4‐type enzymes are conserved in the HECT ligase family.
Similar to all types of E3 enzymes, HECT ligases mediate a series of protein–protein interactions (PPIs), including those with E2 enzymes, ubiquitin, substrates, and regulatory factors. How these dynamic interactions are orchestrated at a structural level is incompletely understood and appears to vary between individual ligases.18, 19, 20 A conserved E2‐binding site was shown to reside on the N lobe of the HECT domain, as illustrated by crystal structures of the E2‐bound HECT domains of E6AP and NEDD4‐1.16, 21 Recently, the HECT domains of E6AP and NEDD4‐2 were proposed to contain a second E2 binding site, based on kinetic analyses and modeling approaches.22, 23, 24
The interactions of HECT ligases with ubiquitin involve several functionally distinct ubiquitin moieties: 1) The “donor” ubiquitin is thioester‐linked through its C terminus to the E3 active site in an intermediate step of catalysis; 2) the “acceptor” ubiquitin provides a primary amino group for the nucleophilic attack on the donor ubiquitin during chain formation; and 3) a regulatory ubiquitin moiety controls the processivity of chain formation by interacting with a particular site on the N lobe of NEDD4‐type enzymes, known as the exosite.25, 26, 27, 28, 29 The interactions of the donor ubiquitin with the C lobe and the regulatory ubiquitin molecule with the exosite, respectively, were shown to be conserved across several NEDD4‐type enzymes.25, 27, 28, 29, 30, 31 In contrast, the structural underpinnings of acceptor ubiquitin recognition and linkage‐specific ubiquitin chain formation by HECT ligases have not been structurally elucidated. Biochemical studies have demonstrated, however, that linkage specificity is determined by the C lobe.30, 32
How substrates are presented to the catalytic center of HECT ligases is also largely unclear. To date, this question has only been structurally analyzed for RSP5, a NEDD4‐orthologue from yeast.33 In the NEDD4 family, substrate recognition is mediated by WW domains located in proximity to the HECT domain that bind particular proline‐rich motifs in substrates.19 In other HECT ligases, substrate‐binding regions are separated from the catalytic domain by hundreds or even thousands of residues;18, 34, 35 how these regions are oriented with respect to the catalytic center in order to enable the ubiquitylation of specific substrates is an unresolved question.
Consequently, the structural underpinnings of HECT‐mediated ubiquitin chain formation on substrates remain puzzling. Different models have been proposed: On one hand, data on NEDD4‐type HECT ligases have frequently been interpreted in the context of a sequential addition model, which posits that ubiquitin chains are assembled on substrates in a stepwise fashion.19 This mechanism was recently experimentally validated for WWP1.36 On the other hand, kinetic analyses of E6AP and NEDD4‐2 indicate an en bloc/proximal indexation mechanism, in which a ubiquitin chain is preassembled on the ligase active site with the help of two E2 enzymes before the chain is transferred to a substrate.22, 23, 24, 37 Together, these results are in line with the idea that different ligases follow distinct mechanisms.37
Emerging structural insights into the intra‐ and intermolecular interactions that regulate the activities of HECT ligases have revealed that this E3 enzyme family indeed harbors considerable mechanistic diversity.18 Members of the NEDD4 subfamily contain a membrane‐targeting C2 domain that can form autoinhibitory interactions with the HECT domain, as first demonstrated for SMURF2.38, 39 This autoinhibition is modulated by accessory proteins, phospholipids, calcium, and phosphorylation.38, 39, 40, 41, 42 Studies on WWP1 and ITCH revealed that a linker region connecting the substrate‐binding WW‐2 and WW‐3 domains of these enzymes could also mediate autoinhibition of the HECT domain in a phosphorylation‐dependent manner.43, 44 Regulatory functions have also been assigned to the WW domains themselves.40, 43, 44, 45, 46, 47, 48, 49 Moreover, oligomerization has emerged as a regulatory principle in various HECT ligases. For example, HUWE1 can form an autoinhibited dimer,50 and the regulation of NEDD4‐1 activity was shown to involve trimerization.51 E6AP activity is also influenced by oligomerization;52, 53, 54, 55 a phenomenon that has been associated with a particular trimeric state seen in the crystal structure of an N‐terminally truncated construct of the HECT domain of this ligase.16
This diversity in regulatory mechanisms of HECT ligases might provide entry points for specific therapeutic interventions. Small molecules could conceivably be used to stabilize or block particular intra‐ or intermolecular interactions of HECT ligases or modulate their conformational dynamics, either directly or allosterically. In addition, the catalytic cysteine may present a target site for covalent probes (Figure 1).
Figure 1.
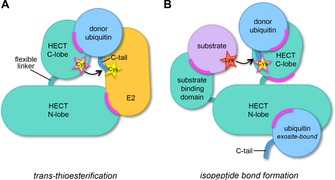
Possible target sites for the manipulation of HECT ligase activities. Cartoon representation of the minimal set of macromolecular interactions formed by the catalytic domain of HECT ligases during the two‐step catalytic cycle. The flexible, C‐terminal region of ubiquitin is abbreviated as C‐tail. The illustration highlights critical protein interfaces (magenta) that may present target sites for small‐molecule effectors. A) During the transthioesterification step, these interfaces include the N lobe–E2 enzyme interface and the C lobe–donor ubiquitin interface. B) During isopeptide bond formation, interfaces between the N and C lobes, substrate binding domains and substrates, and the N lobe and an exosite‐bound ubiquitin (as required for ubiquitin chain formation by NEDD4‐type enzymes) may be targeted. Additional interfaces that are expected to be relevant for HECT E3 function, but not displayed here include intramolecular interactions within the full‐length ligase, interactions between the ligase and acceptor ubiquitin, regulatory factors, or additional ligase subunits (in the context of an E3 oligomer). The catalytic cysteine residue of HECT ligases, highlighted as a star, may be targeted by covalent probes.
Herein, we summarize our current knowledge of small‐molecule effectors of HECT ligases. Key results, chemical structures, and details on screening methodologies for each study are listed in Table 1. Briefly, this survey shows that a variety of different ligand discovery approaches for HECT ligases have yielded diverse compounds, most of which are still moderate in potency and selectivity, but can serve as starting points for medicinal chemistry optimizations. How the available compounds act on their target ligases is, for the most part, unclear; to‐date only one crystal structure of a HECT domain in complex with a small‐molecule inhibitor has been reported.56 Structural and mutational elucidation of the mechanisms by which HECT ligases interact with small‐molecule effectors will therefore be an essential step towards developing high‐quality chemical probes. We envision that such probes will provide invaluable tools to illuminate the conformational landscape and catalytic requirements of HECT ligases and to assess how these enzymes may be exploited for therapeutic applications.
Table 1.
Overview of published small molecules targeting HECT ligases.[a]
| Target | Compound | Formula | Identification | Mechanism | Potency | Selectivity | Structure | Ref. |
|---|---|---|---|---|---|---|---|---|
| E3 | method | of action | of complex |
|||||
| NEDD4 type | ||||||||
| NEDD4‐1 | 1 |
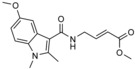
|
in vitro screen of 100 electrophilic fragments for covalent modification of NEDD4‐1 | covalent modification of exosite cysteine (Cys627) |
K
I=(250±50) μm for ubiquitin binding to the exosite (fluorescence polarization in vitro; [E3]=8 μm); k
inact=2.2 s−1; k inact/K I=0.089 m −1 s−1 |
n.a. | PDB ID: 5C91 | 56 |
| 2 |
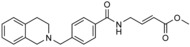
|
n.a. | n.a. | |||||
| 3 |
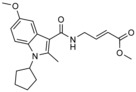
|
K
I=(29±6) μm for ubiquitin binding to the exosite (fluorescence polarization in vitro; [E3]=8 μm); k
inact=5.8 s−1; k inact/ K I=1.98 m −1 s−1 |
modifies NEDD4‐1/2, but not WWP1, E6AP, or other cysteine‐based enzymes | |||||
| I3C |
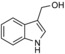
|
genetic analyses suggest NEDD4‐1 as a target of I3C | predicted to bind to the exosite of the HECT domain, based on docking | IC50=284 μm (in vitro autoubiquitylation assay); IC50=107 μm in cells (growth inhibition assay); K D=(88.1±13.0) μm (ITC) | n.a. | n.a. | 61 | |
| 1‐benzylI3C |
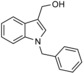
|
derivative of I3C | predicted to bind to the exosite of the HECT domain, based on docking | IC50=12.3 μm (in vitro autoubiquitylation assay); IC50=14.7 μm in cells (growth inhibition assay) | n.a. | n.a. | 62 | |
| NEDD4 subfamily | I |
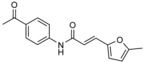
|
AlphaScreen technology‐based screen of ≈17 500 compounds for displacement of peptides from the SMURF2 HECT domain | n.a. | IC50 (SMURF2)=7.4 μm; IC50 (NEDD4)=7.1 μm; IC50 (WWP1)=8.7 μm; IC50 (UBE3C)=8.0 μm; IC50 (HUWE1)=23 μm (in vitro autoubiquitylation assay) | n.a. | n.a. | 64 |
| heclin |
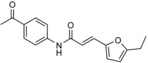
|
derivative of I | reversible; predicted to promote oxidation of the catalytic cysteine | IC50 (SMURF2)=6.8 μm; IC50 (NEDD4)=6.3 μm; IC50 (WWP1)=6.9 μm (in vitro autoubiquitylation assay); apparent IC50=9 μm (SMURF2) in cells | selective against RING ligase MDM2 | |||
| RSP5 | NAB |
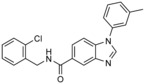
|
phenotypic screen of ≈190 000 compounds for rescue from α‐synuclein toxicity in yeast | n.a. | IC40=34 μm (cell‐based assay; rescue from α‐synuclein toxicity in yeast) | n.a. | n.a. | 67 |
| RSP5/NEDD4 | NAB2 |
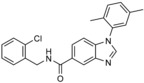
|
derivative of NAB | IC40=20.5 μm (cell‐based assay; rescue from α‐synuclein toxicity in yeast) | n.a. | |||
| ITCH | clomipramine |
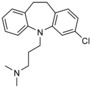
|
in vitro screen of ≈21 000 compounds, monitoring ITCH autoubiquitylation | n.a. | MIC 300 μm in vitro; 6‐30 μm in cells | inhibits E6AP, but not RING1B and DIAP | n.a. | 70 |
| 10 e |
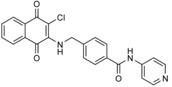
|
screen of a small synthetic library, designed based on known HECT E3 inhibitors, for antiproliferative activity | n.a. | not determined in vitro; GI50=0.4 to 4 μm, cell type‐dependent | inhibits several RING and RBR ligases | n.a. | 73 | |
| SMURF1 | A01 |
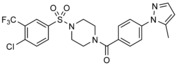
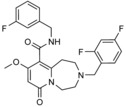
|
in silico screen of ≈106 compounds against the WW‐1 domain of SMURF1 | predicted to bind to the WW‐1 domain, based on docking | n.a. | n.a. | n.a. | 82 |
| A17 |
|
|||||||
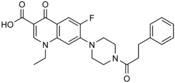
| B06 |
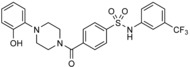
|
in silico screen of ≈106 compounds against a homology model of the HECT domain of SMURF1 | predicted to bind to the N lobe–C lobe interface, based on docking | n.a. | n.a. | n.a. | 83 | |
| B75 |
|
|||||||
| SVAK‐12 |
|
in silico screen of ≈70 000 compounds and a set of lead compounds predicted to potentiate BMP‐2 activity against a homology model of the WW‐2 domain of SMURF1 | predicted to bind to the WW‐2 domain, based on docking | EC50=2.6 μm (in vivo luciferase reporter assay monitoring BMP‐2 activity) | n.a. | n.a. | 79 | |
| Others | ||||||||
| E6AP | N‐acetylphenylalanine |
|
hypothesis‐based use of a phenylalanine derivative | impacts E6AP oligomerization; not E2‐competitive | K I=(12±3) mm | n.a. | n.a. | 52 |
| HUWE1 | BI8626 |
|
in vitro screen of ≈840 000 compounds, monitoring autoubiquitylation of the HUWE1 HECT domain | n.a. | IC50=0.9 μm (in vitro autoubiquitylation assay) | 9 other HECT ligases tested: IC50 >50 μm (in vitro autoubiquitylation assay) | n.a. | 92 |
| BI8622 |
|
|||||||
| IC50=3.1 μm (in vitro autoubiquitylation assay) | ||||||||
[a] n.a.: not available, ITC: isothermal titration calorimetry, MIC: minimum inhibitory concentration.
2. Case Studies on HECT Ligase‐Directed Ligand Discovery
2.1. Thiol‐reactive, irreversible inhibitors of NEDD4‐1/2
The presence of a catalytic cysteine in HECT ligases should render them susceptible to thiol‐reactive probes. Moreover, various HECT ligases contain a cysteine residue in a region that coincides with the functionally critical, ubiquitin‐binding exosite of NEDD4‐type enzymes. This residue may also provide a target site for ubiquitin‐competitive covalent probes. Based on this rationale, Statsyuk and co‐workers screened a library of 100 electrophilic fragments derived from methyl 4‐amino (E)‐crotonate, which they had previously developed to identify cysteine protease inhibitors,57 against the HECT domain of NEDD4‐1.56 This screen yielded two hits (an indole and a tetrahydroisoquinoline derivative, referred to as 1 and 2, respectively; Table 1) that selectively reacted with the (noncatalytic) cysteine residue, Cys627, in the exosite of the ligase (Scheme 1), as confirmed by mass spectrometry and mutational analyses. In contrast, a simpler compound, methyl 4‐acetamido (E)‐crotonate, was found to specifically target the generally more reactive, catalytic cysteine.
Scheme 1.
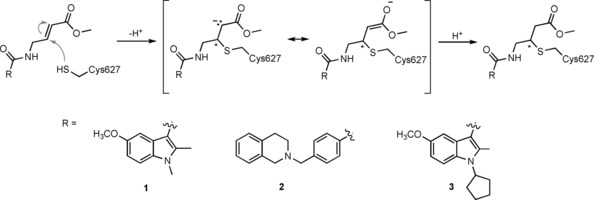
Conjugate addition (thiol‐ or sulfa‐Michael addition) of the side chain of Cys627 of NEDD4‐1 to compounds 1, 2, and 3.
The binding mode of compound 1 in a hydrophobic pocket adjacent to Cys627 was revealed by the crystal structure of the covalently modified HECT domain of NEDD4‐1 (Figure 2 A and B).56 In line with the identified binding mode, compound 1 and derivatives thereof block the interaction of ubiquitin with the exosite and inhibit processive ubiquitin chain formation. Moreover, inhibitor‐treated NEDD4‐1 is more efficiently counteracted by DUBs than the untreated ligase in vitro.
Figure 2.
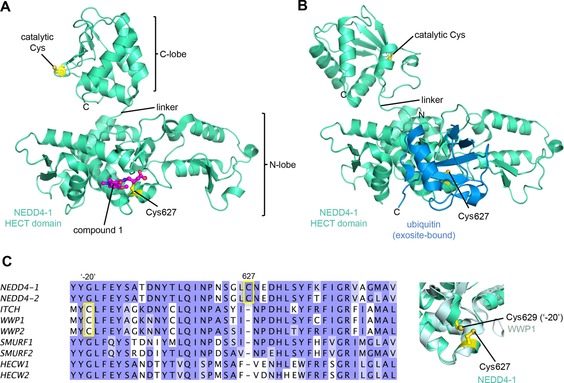
Covalent blockage of the NEDD4‐1 exosite. A) Crystal structure of the HECT domain of NEDD4‐1 with a small‐molecule inhibitor (1) covalently linked to Cys627 (PDB ID: 5C9156). B) Crystal structure of the HECT domain of NEDD4‐1 with ubiquitin bound noncovalently to the exosite on the N lobe (PDB ID: 2XBB27). The N lobes in A) and B) are shown in the same orientation. Notably, the C lobe adopts different orientations with respect to the N lobes in A) and B), reflecting interlobe flexibility. C) Left: sequence alignment of human NEDD4‐type enzymes for the region around Cys627 of NEDD4‐1. A cysteine at this particular position is only conserved in NEDD4‐1 and ‐2. However, in ITCH, WWP1, and WWP2, a cysteine residue is located 20 residues N‐terminal to this position (Cys−20). The alignment was rendered in JalView109 and colored according to the Blosum62 score.110 Right: detailed view of a structural superposition of the N lobe of NEDD4‐1 (PDB ID: 2XBB27) and WWP1 (PDB ID: 1ND727), which illustrates that the side chains of Cys627 of NEDD4‐1 and Cys629 (Cys−20) of WWP1 are in close spatial proximity to each other. Protein backbones in all structural representations are shown in cartoon representation; the side chains of relevant residues are displayed as balls and sticks.
Because compounds 1 and 2 are relatively weak binders of NEDD4‐1 and can be outcompeted by ubiquitin concentrations close to cellular levels, four N‐substituted analogues were prepared.56 One of these compounds, 3 (Table 1), features an N‐cyclopentyl substituent and is 22‐fold more potent than compound 1 with a rate of covalent modification (k inact/K I) of 1.98 m −1 s−1 at an enzyme concentration of 8 μm, as measured by fluorescence polarization. Compound 3 does not label the E1 enzyme UBE1, the E2 enzyme UBCH5A, the DUB USP8, and human rhinovirus 3C protease, all of which have catalytic cysteine residues. Moreover, although compound 3 reacted with the HECT domain of NEDD4‐2 (a close homologue of NEDD4‐1), no labeling of the HECT domains of E6AP and WWP1 was detected. In line with this observation, E6AP lacks a cysteine residue in the structurally homologous region to the exosite of NEDD4‐type enzymes. In contrast, WWP1 does contain a cysteine in spatial proximity to Cys627 of NEDD4‐1; however, this residue is not in an equivalent sequence position, but located 20 residues N‐terminally to it (Cys−20; Figure 2 C). The fact that this cysteine (Cys629) of WWP1 is not labeled underscores the specificity of compound 3. It will be interesting to investigate whether the activities of other HECT ligases that contain a cysteine residue homologous to Cys627 of NEDD4‐1 (e.g., HUWE1) rely on ubiquitin recognition through the exosite and could be modulated by small molecules in an analogous manner.
Taken together, this study provides a proof of concept for the accessibility of HECT ligases by irreversible thiol‐reactive probes. It will be important to test the identified compounds in cells. Because they contain relatively reactive ester‐derived Michael acceptor functionalities, cross‐reactions with glutathione and off‐target proteins are possible.58 Moreover, the ester group of the compounds may be susceptible to cleavage by esterases. However, these liabilities may be overcome by employing acrylamide‐derived Michael acceptors that typically have lower reactivity and increased stability in vivo. Alternatively, if high reactivity turns out to be required, covalent‐reversible targeting of the exosite cysteine, for example, by α‐cyanoacrylamides,59 may provide avenues towards optimized probes.
2.2. Exploiting phytochemicals to block NEDD4‐1 activity
Natural dietary phytochemicals have emerged as a rich source of compounds with potential as supplemental chemotherapeutic agents. One such compound is 1H‐indole‐3‐yl‐carbinol (I3C); a metabolite of glucobrassicin found in cruciferous vegetables that triggers a variety of antiproliferative responses in cancer cells (Table 1).60 Firestone and co‐workers dissected the effects of I3C in human melanoma cells, and concluded that the compound stabilized the tumor suppressor PTEN by inhibiting its ubiquitylation by NEDD4‐1.61 These effects were confirmed in vivo by using G‐361 cell‐derived xenograft models, albeit at high I3C doses (200 mg kg−1). I3C was also found to interact with NEDD4‐1 in vitro with an estimated dissociation constant in the micromolar range (88 μm, as determined by ITC). Based on computational docking using a crystal structure of the HECT domain of NEDD4‐2, it was predicted that I3C occupies a hydrophobic pocket on the N lobe in proximity to the exosite.61
To overcome the disadvantages of I3C, such as its low affinity for NEDD4‐1 and chemical instability, as caused by the formation of various condensation products, especially at low pH, a small library of N‐benzyl or N‐phenyl I3C analogues with different substitution patterns at the phenyl ring was prepared.62 The binding of these derivatives to NEDD4‐1 was confirmed by differential scanning fluorimetry, in which four out of six compounds caused significant shifts in melting temperature. The most potent compounds, among them 1‐benzyl‐I3C (Table 1), inhibit the autoubiquitylation activity of NEDD4‐1 in vitro and melanoma cell proliferation with IC50 values in the low micromolar range.
Guided by the crystal structure of the HECT domain of NEDD4‐1 in complex with the irreversible Cys‐reactive inhibitor 1 identified by Statsyuk and co‐workers (Figure 2 A, see Section 2.1), the I3C derivatives were docked onto the hydrophobic exosite region flanking Cys627,62 although this binding mode has not been validated experimentally, yet.
Given the small size of the I3C derivatives and their varying efficacies in different assay systems, it is conceivable that these compounds exert multitarget effects in cells. Notably, I3C derivatives can be activated by acid or the sulfotransferase SULT1A1 to form highly reactive carbocations, which can lead to cysteine modifications (Scheme 2).63 To prevent the formation of promiscuous reactive species, it may thus be necessary to replace the hydroxymethyl group with motifs that are less prone to elimination. Whether such modified compounds would retain the observed activity and could be rendered selective NEDD4‐1 inhibitors for in vivo use requires further investigation.
Scheme 2.
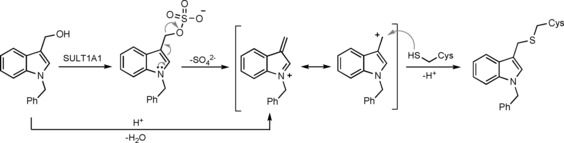
Activation of I3C derivatives, as exemplified by 1‐benzyl‐I3C, by sulfotransferase (SULT) 1A1 or acid.63 A highly electrophilic, resonance‐stabilized carbocation is formed that can be trapped by nucleophiles, such as cysteine residues.
2.3. Interfering with E2 recognition by NEDD4‐type ligases
To explore the possibility of harnessing macrocyclic compounds as HECT ligase inhibitors, Pelham and co‐workers screened a phage library that displayed bicyclic peptides for ligands of the HECT domain of SMURF2, NEDD4‐1, WWP1, and HUWE1, respectively.64 The best binders were used in subsequent selection rounds to evolve sequences that competed with E2 enzymes (UBCH7 in the case of SMURF2 and NEDD4‐1; UBCH5 in the case of WWP1 and HUWE1). After optimization, the peptides recognized their respective target enzyme specifically and inhibited ligase activity with IC50 values in the lowmicromolar range. That the compounds indeed interact with the E2 binding site on the HECT domain was verified by hydrogen–deuterium (H/D) exchange and mutational analyses.64
The most promising SMURF2 inhibitor was subjected to an AlphaScreen‐based assay (PerkinElmer) to identify competitive small‐molecule ligands with improved pharmacological properties.64 Based on a 17 500 compound library, 30 hits were identified, with a single one demonstrating promising inhibitory activity in vitro and in vivo (compound I; Table 1). Three closely related analogues of this compound were purchased, among them heclin (Table 1), which proved to be a single‐digit micromolar inhibitor of the autoubiquitylation activities of SMURF2, NEDD4‐1, and WWP1; no inhibition of the RING ligase MDM2 was observed. In cells, heclin reversibly suppresses the ubiquitylation of SMURF2 with an apparent IC50 value of 9 μm.
Unexpectedly, heclin was found to follow a different mechanism from that of the originally identified bicyclic peptide. H/D exchange studies and E2 competition experiments indicate that heclin does not obstruct the E2 binding site of SMURF2. Instead, the compound appears to render the catalytic cysteine of the ligase more susceptible to oxidation;64 however, the molecular underpinnings of this phenomenon are unclear. In this context, it is interesting to note that heclin acts on HECT ligases in a reversible manner;64 this suggests a noncovalent binding mode, despite the presence of an acrylamide‐derived Michael acceptor function, which may be trapped by cysteine residues (analogous to the mechanism depicted in Scheme 1). To understand precisely how heclin inhibits HECT ligases, structural and mutational studies will be required.
The stability of heclin in vivo also needs to be evaluated, particularly because this compound contains a metabolically unstable furan ring known to generate reactive oxidized metabolites,65 and a keto group that may be susceptible to reduction, for example, by aldo–keto reductases. Furthermore, it will be important to assess whether the broad target spectrum of heclin can be altered towards specific ligases. Alternatively, the promiscuity of this inhibitor could prove beneficial in particular disease settings. In a recent study identifying WWP1 as a biomarker for acute myeloid leukemia (AML), heclin was found to suppress the growth of leukemic blasts. This supports the idea that HECT ligases present attractive therapeutic targets in hematological malignancies.66
2.4. Phenotypic screening identifies small‐molecule activators of RSP5/NEDD4‐1 function
Lindquist and colleagues identified a small‐molecule effector of RSP5 in an unbiased, phenotypic high‐throughput screen directed at neurodegenerative processes.67 In this study, about 190 000 compounds were analyzed for their ability to restore the survival of yeast cells overexpressing TDP‐43, a cytotoxic protein linked to various neurodegenerative disorders. One hit, an N‐aryl benzimidazole (NAB) derivative (Table 1), also rescued yeast cells expressing toxic levels of α‐synuclein by restoring vesicular trafficking pathways, thus rendering this compound an attractive lead for the development of therapeutics against α‐synucleinopathies, such as Parkinson's and Alzheimer's disease.
In a small structure–activity relationship (SAR) study, 29 analogues of NAB were prepared.67 NAB2, a derivative with an additional ortho‐methyl group on the N‐phenyl ring, was identified as the most potent compound (Table 1). A favorable effect conferred by an ortho‐methyl group was also observed in other derivatives, which indicates that a conformational bias orienting the phenyl substituent out of the benzimidazole plane increases potency in this system. Moreover, the presented SAR suggests that the imidazole N3‐nitrogen atom, the amide carbonyl oxygen atom, the benzylic methylene spacer, and an appropriate substituent in the ortho position of the benzylic amide are required for activity.
Interestingly, genetic screens in yeast indicate that the cellular target of NAB/NAB2 is the HECT ligase RSP5, which modifies α‐synuclein and controls ubiquitin‐mediated endosomal trafficking.67, 68 A point mutation, G747E, in the C lobe of RSP5 causes cellular resistance to NAB2 treatment.67 However, NAB2 neither stimulates the ubiquitylation activity of RSP5 towards α‐synuclein in vitro nor does it affect α‐synuclein levels in cells. It therefore remains unclear whether NAB2 interacts with RSP5 directly and how it complements α‐synuclein‐induced dysfunctions in trafficking. Answering these questions will be an interesting area of future studies, particularly because the protective activity of NAB2 against α‐synuclein‐linked pathologies is conserved in human neurons, mediated by the RSP5‐orthologue NEDD4‐1.67, 69
2.5. High‐throughput screening against ITCH recovers an antidepressant
In a target‐based approach to identify HECT ligase inhibitors, Melino and co‐workers screened about 21 000 compounds for the inhibition of ITCH, a ligase best known for its critical functions in immune signaling.70 This screen recovered clomipramine, a tricyclic dibenzazepine‐derived antidepressant (Table 1). The compound specifically inhibits the transthioesterification reaction, in which ubiquitin is transferred from the E2 to the E3. The compound was found to inhibit E6AP in addition to ITCH, indicating that it has at least some promiscuity among HECT ligases. In contrast, clomipramine does not interfere with E1, E2 (UBCH7), and RING ligase (RING1B, DIAP2) activities in vitro.
Out of 17 tested chemical analogues of clomipramine, norclomipramine (the active metabolite of clomipramine) inhibits ITCH most potently; however, high micromolar concentrations of the compound are required for in vitro efficacy.70 In contrast, clomipramine and norclomipramine block autophagic flux and synergize with chemotherapeutics to kill cancer cells at low micromolar concentrations, thus highlighting ITCH‐independent effects.70, 71 Notably, both compounds target G protein–coupled receptors (GPCRs) in the low nanomolar range and bind to the serotonin transporter (clomipramine) and the noradrenalin transporter (norclomipramine) with picomolar affinities.72
Understanding the mechanism of nor‐/clomipramine action on HECT ligases awaits structural analyses. It may also be interesting to make use of the large number of clomipramine derivatives in industrial compound libraries to explore whether specific inhibitors of HECT ligases can be identified.
In another study on ITCH, Liou and co‐workers initiated a medicinal chemistry program based on the hypothesis that the minimal pharmacophore for an ITCH inhibitor was a rigid aromatic core substituted with a side chain of at least four carbon atoms in length.73 They used a naphthoquinone core bearing a chlorine atom (as identified in the study described above70) and a benzylamine linker as a structural template for the synthesis of 17 derivatives.73 One compound (10 e; Table 1) was found to inhibit ITCH in vitro at low micromolar concentrations and displayed antiproliferative activity in various cancer cell lines and xenograft models of multiple myeloma.73 At 10 μm concentration, the compound also inhibited several E2‐RING/U‐box/RBR ligase systems to various degrees (24 systems screened with Millipore UbiquitinProfiler Services; no HECT ligases were tested in this setup for direct comparison). The molecular mechanisms underlying these observations remain to be explored. However, covalent binding through a Michael‐type reaction of the naphthoquinone core of 10 e with cysteine residues is conceivable (analogous to the mechanism shown in Scheme 1, but with a concomitant elimination of chloride). Therefore, it will be important to interrogate target engagement and selectivity of this compound.
2.6. Blocking substrate binding to SMURF1
The NEDD4‐type ligase SMURF1 is critically involved in bone homeostasis through the BMP (TGF‐b/bone morphogenetic protein), WNT, and MAP kinase signaling pathways.74, 75, 76, 77 The fact that SMURF1 negatively regulates BMP signaling and osteogenesis sparked the idea of inhibiting this ligase to fight bone volume disorders, such as osteoporosis. Sangadala and co‐workers screened a virtual library, including FDA‐approved drugs and the MDL Available Chemicals Directory (ACD), for synthetic mimics of the LIM mineralization protein‐1 (LMP‐1); an enhancer of BMP activity known to bind to the WW‐2 domain of SMURF1. Docking of the compounds into a predicted hydrophobic pocket of the WW‐2 domain, based on a homology model of this domain, yielded a chemically unstable compound of undisclosed structure (SVAK‐3) that showed dose‐dependent enhancement of BMP‐2 activity in C2C12 cells.78
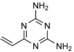
In a follow‐up study, a similar virtual screening approach returned 2,4‐diamino‐6‐vinyl‐1,3,5‐triazine (SVAK‐12; Table 1).79 This compound was found to potentiate BMP‐2 activity and induced differentiation of myoblastic C2C12 cells in a dose‐dependent manner. In animal models, SVAK‐12 enhanced rhBMP‐2‐induced bone formation and fracture repair.80 However, it is conceivable that the compound acts as a covalent modifier of cysteine residues, in analogy to 4‐vinylpyridines (Scheme 3).81 Therefore, target engagement and selectivity need to be assessed.
Scheme 3.
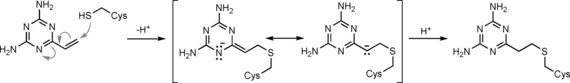
Mechanism suggested for the putative covalent modification of cysteine residues by SVAK‐12.
Recently, Zhang and co‐workers conducted a virtual screen directed at a hydrophobic pocket in the N‐terminal WW domain (WW‐1) of SMURF1.82 By employing an iterative docking workflow 19 compounds (from a virtual library of ≈1 000 000) were selected for experimental testing. Two of these compounds (A01 and A17; Table 1) displayed encouraging effects: they interfere with the interaction between the SMURF1 WW‐1 domain and the substrate SMAD1 (but not E2 enzymes) in vitro, stabilize SMURF1 substrates in cells, and enhance cellular responsiveness to BMP signaling and osteoblastic activity. However, neither target affinity nor selectivity have been experimentally assessed, yet.
An analogous virtual screen was performed against a predicted hydrophobic region on the HECT domain of SMURF1, based on a homology model derived from the crystal structure of the HECT domain of SMURF2.83 This region (defined by residues Asn431, Tyr439, Asn481, and Gln653 of SMURF1) is located at the interface between the two lobes of the HECT domain and adjacent to, but not overlapping with, the ubiquitin‐binding exosite on the N lobe of NEDD4‐type enzymes. Of 24 compounds selected from the virtual screen for experimental evaluation, two hits (B06 and B75; Table 1) were found to inhibit the ubiquitylation and turnover of substrates of SMURF1 in cells and enhanced osteoblast differentiation and proliferation. In vitro pull‐down experiments showed that these compounds disrupt the interaction of SMURF1 with ubiquitin, but not with E2 enzymes (UBCH5 and UBCH7). Furthermore, the compounds did not obstruct substrate (SMAD1/5) binding to SMURF1, as monitored by IP from cells, nor ubiquitin or substrate binding to SMURF2.
To fully interpret these results, structural analyses will be required. If the compounds bind to the proposed region at the interface between the two lobes of SMURF1, it is not clear how they would interfere with canonical ubiquitin binding to the exosite. Moreover, the proposed binding mode involves the last modeled residue (Glu724) in the C‐terminal region of the HECT domain, whereas the seven flanking residues that make up the functionally critical C‐terminal tail of the HECT domain (residues 725–731) are missing from the model used for docking. It is possible that these residues impact on the accessibility and other properties of the site onto which the compounds were docked. Therefore, structural and mutational studies (including the complete C‐terminal region of SMURF1) will be essential to define the mechanistic basis of inhibition by the identified compounds.
2.7. Targeting ligases outside of the NEDD4 subfamily: HUWE1
The 482 kDa HECT ligase HUWE1 functions in diverse cellular pathways, including protein quality control, transcription, DNA repair, apoptosis, neuronal differentiation, and mitophagy.9, 84, 85, 86, 87, 88, 89, 90, 91 It has been recognized as an important player in tumor biology;9, 92, 93, 94, 95, 96, 97, 98 its precise functions, however, are complex and likely to depend on tumor stage and/or entity.
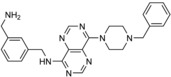
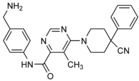
Eilers and co‐workers set out to block HUWE1 activity in colorectal cancer.92 They found that this ligase promotes MYC‐mediated transcriptional activation, thereby contributing to tumor maintenance. Together with Boehringer Ingelheim, a library of about 840 000 compounds was screened for inhibitors of the autoubiquitylation activity of the HECT domain in vitro.92 Two druglike hits from this screen (BI8662 and BI86262; Table 1) were found to inhibit HUWE1 with low micromolar IC50 values in vitro and displayed selectivity for HUWE1 over nine other HECT ligases tested, including members of the NEDD4 subfamily. In line with these data, the compounds reduce the ubiquitylation of cellular HUWE1 substrates and the growth of colorectal cancer cells, both with low micromolar IC50 values. The compounds also suppress transcriptional activation of MYC target genes in colorectal cancer cells, but not in normal colonic epithelial and HUWE1‐depleted cells. Due to their pharmacokinetic properties, the compounds are not suited for in vivo studies.92 However, encouragingly, the underlying scaffolds offer considerable room for medicinal chemistry optimization, so it will be interesting to unravel the structural mechanism by which these compounds inhibit HUWE1.
2.8. Manipulating oligomerization of HECT ligases
Oligomerization has emerged as a regulatory theme in various members of the HECT ligase family and may hold additional opportunities for therapeutic interference. For instance, the activity of E6AP is influenced by oligomerization;52, 53, 54, 55 the structural basis and functional consequences of this phenomenon, however, are incompletely understood. In particular, a trimeric arrangement seen in crystal structures of a truncated construct of the E6AP HECT domain has been analyzed with varied outcomes.16, 52, 53, 54, 55
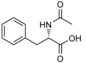
Haas and co‐workers observed that E6AP activity is inhibited by N‐acetylphenylalanine in a manner that is noncompetitive with binding of the E2 enzyme (K I=12 mm; Table 1).52 Addition of the compound leads to a reduction in the size of full‐length E6AP, as measured by light scattering, indicating a trimer‐to‐monomer transition. On the basis of the crystallographic trimer,16 it was proposed that N‐acetylphenylalanine disrupts this trimeric state by substituting for a phenylalanine contact at the subunit interface. Non‐E2‐competitive inhibition of full‐length E6AP was also observed upon the addition of a peptide that mimicks the N‐terminal region of the HECT domain missing in the crystallized fragment.52 To understand the structural basis of these observations and further explore the accessibility of oligomeric states of E6AP to small molecule or peptide‐based probes, structural analyses of extended constructs and their macromolecular complexes will be valuable.
3. Summary and Outlook
Identifying ubiquitin ligase inhibitors by conventional high‐throughput screening campaigns has proven challenging. This, a least in part, reflects the fact that traditional compound libraries are biased towards “classical” targets, such as kinases, proteases, GPCRs, ion channels, and nuclear receptors, which typically possess well‐defined small‐molecule binding pockets. In contrast, ubiquitin ligases act through the sequential formation and reorganization of weak PPIs, as mediated by rather large and flat surfaces that are notoriously difficult to target.99, 100 However, several strategies for modulating PPIs by small molecules have been developed99 and can applied for the design of HECT ligase inhibitors. For example, low‐molecular‐weight fragments (typically <250 Da) that bind PPI hotspot regions may be identified by biophysical assays, tethering or crosslinking and transformed into highly potent and specific inhibitors by fragment growing, linking, or merging approaches.101, 102 To identify starting points for the development of selective covalent inhibitors, the screening of electrophilic fragment libraries for compounds targeting cysteine residues at the exosite or the catalytic center of HECT ligases is a promising strategy.35 This review also highlights that virtual screening and the use of nontraditional libraries, including natural products, macrocycles, and peptides/peptide mimetics, can provide avenues towards novel ligands of HECT ligases.
However, another obstacle in identifying ubiquitin ligase inhibitors by high‐throughput screening lies in the complex nature of ubiquitylation reactions, which makes it tedious to quantify ligase activities specifically and distinguish their sensitivities towards compound‐mediated inhibition from effects on other members of the catalytic cascade. To overcome this issue, chemically modified ubiquitin probes have been developed that enable bypassing the E1 and E2 enzymes in a minimalist reaction setup.103, 104, 105 Yet, such approaches do not allow for the identification of compounds that act on intermediate states in the catalytic cycle of HECT ligases if stabilized, for instance, by interactions with E2 enzymes or E2–ubiquitin conjugates.
To uncover constitutive and transient allosteric pockets in HECT ligases, it will thus be essential to structurally characterize the macromolecular complexes in which these enzymes sequentially engage. Due to the low affinities of the underlying dynamic protein interactions, this is a challenging task. However, chemical modification and cross‐linking strategies, as successfully applied in several studies of NEDD4‐type ligases,30, 33 provide a powerful workaround and enable the visualization of relevant conformational states of HECT ligases in their macromolecular context. Additional insights into functionally sensitive sites in HECT ligases can be obtained from the characterization of engineered ubiquitin variants that modulate particular reaction steps.29
Structural insights into the catalytic intermediates of HECT ligases will also be required to evaluate whether the catalytic center can be targeted with specificity. Located on the globular C lobe, which does not display any clear small‐molecule binding grooves per se, the catalytic cysteine appears a challenging target for specific inhibitors, at least if considering the apo‐HECT domain, where the C lobe is flexible with respect the N lobe. It is possible, however, that inter‐ or intramolecular interactions, in the context of full‐length ligases and macromolecular complexes, generate distinct chemical environments at the catalytic center of individual ligases that allow for specific manipulations.
In light of the many open questions and technical challenges in understanding the structural mechanisms of HECT ligases, it is not surprising that the development of inhibitors targeting these enzymes is still in its infancy. We anticipate that the continued, joint efforts of structural biologists, cell biologists, and medicinal chemists will yield “high‐quality” chemical probes in the foreseeable future. The key criteria for such probes, as defined by Frye and others,106, 107, 108 include appropriate selectivity and dose‐dependent activity to link in vitro and cellular activity profiles, as well as defined SARs and mode of action, plus verifiable cellular target engagement and selectivity against off‐targets.
If innovative strategies, especially those for manipulating PPIs, prove successful in modulating ubiquitin ligase activities by small molecules, there is a chance for this class of enzymes to compete with kinases as the major drug targets of the 21st century in oncology. We envision HECT ligases to be particularly attractive targets, due to their key roles in various diseases, comparatively small number compared with RING ligases, and diverse domain structures that may facilitate specific manipulations.
Proof‐of‐principle work on small‐molecule effectors of HECT ligase activities may not only pave the way towards therapeutic applications. It will likely provide fundamental insights into the catalytic requirements, conformational dynamics, specificities, and cellular functions of HECT ligases, analogous to the defining impact of kinase inhibitors on our understanding of phosphorylation.
Conflict of interest
The authors declare no conflict of interest.
Biographical Information
Dan Chen obtained a diploma degree in biological sciences from Nanyang Technological University, Singapore, where she also received her PhD, working in Pär Nordlund's laboratory. At the start of 2018, she moved to Germany to pursue postdoctoral studies with Sonja Lorenz at the University of Würzburg. She is excited to explore the interaction patterns of ubiquitylation enzymes and eventually exploit these enzymes therapeutically.

Biographical Information
Matthias Gehringer studied chemistry at the Karlsruhe Institute of Technology, the École Nationale Supérieure de Chimie de Montpellier, and the University of Heidelberg, and obtained his PhD from Tübingen University working on kinase inhibitors. As a postdoctoral researcher at the Swiss Federal Institute of Technology Zürich, he worked on the total synthesis of mycolactones and on targeted antibiotic–protein conjugates. He recently returned to Tübingen with the aim of establishing an independent research group. His research interests include medicinal chemistry, chemical biology, natural product synthesis, and innovative drug targeting approaches.

Biographical Information
Sonja Lorenz studied biochemistry at the Universities of Regensburg and Berkeley, CA, and obtained a DPhil degree from the University of Oxford. As a postdoctoral researcher with John Kuriyan at UC Berkeley, she became fascinated by the ubiquitin system through collaborative work with Michael Rape. She is an independent group leader at the University of Würzburg, funded by the Emmy Noether Program of the German Research Foundation (DFG), and is the co‐speaker of a ubiquitin‐focused consortium (GRK2243, DFG). Her research combines structural and functional studies to understand the ubiquitylation machinery. In 2017, she was named an EMBO Young Investigator.

Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank the members of the Lorenz laboratory, particularly Bodo Sander, for helpful discussions and Aaron Cantor for insightful comments on the manuscript. M.G. acknowledges a “Recruitment of Excellent Junior Researchers” grant from the Institutional Strategy of the University of Tübingen (ZUK 63; DFG). S.L. is funded by the Emmy Noether Program of the German Research Foundation (LO 2003/1‐1; DFG), the Research Unit 2314 (“Targeting therapeutic windows in essential cellular processes for tumor therapy”, DFG), the Wilhelm‐Sander‐Stiftung (2015.147.1), and the EMBO Young Investigator Program.
D. Chen, M. Gehringer, S. Lorenz, ChemBioChem 2018, 19, 2123.
References
- 1. Popovic D., Vucic D., Dikic I., Nat. Med. 2014, 20, 1242–1253. [DOI] [PubMed] [Google Scholar]
- 2. Einsele H., Recent Results Cancer Res. 2014, 201, 325–345. [DOI] [PubMed] [Google Scholar]
- 3. Swatek K. N., Komander D., Cell Res. 2016, 26, 399–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Li W., Bengtson M. H., Ulbrich A., Matsuda A., Reddy V. A., Orth A., Chanda S. K., Batalov S., Joazeiro C. A. P., PLoS One 2008, 3, e1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Huang X., Dixit V. M., Cell Res. 2016, 26, 484–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ito T., Ando H., Suzuki T., Ogura T., Hotta K., Imamura Y., Yamaguchi Y., Handa H., Science 2010, 327, 1345–1350. [DOI] [PubMed] [Google Scholar]
- 7. Fischer E. S., Böhm K., Lydeard J. R., Yang H., Stadler M. B., Cavadini S., Nagel J., Serluca F., Acker V., Lingaraju G. M., Tichkule R. B., Schebesta M., Forrester W. C., Schirle M., Hassiepen U., Ottl J., Hild M., Beckwith R. E. J., Harper J. W., Jenkins J. L., Thomä N. H., Nature 2014, 512, 49–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sakamoto K. M., Kim K. B., Kumagai A., Mercurio F., Crews C. M., Deshaies R. J., Proc. Natl. Acad. Sci. USA 2001, 98, 8554–8559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Scheffner M., Kumar S., Biochim. Biophys. Acta Mol. Cell Res. 2014, 1843, 61–74. [DOI] [PubMed] [Google Scholar]
- 10. Bosshard M., Aprigliano R., Gattiker C., Palibrk V., Markkanen E., Backe P. H., Pellegrino S., Raymond F. L., Froyen G., Altmeyer M., Bjørås M., Dianov G. L., van Loon B., Sci. Rep. 2018, 8, 6010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Moortgat S., Berland S., Aukrust I., Maystadt I., Baker L., Benoit V., Caro-Llopis A., Cooper N. S., Debray F.-G., Faivre L., et al., Eur. J. Hum. Genet. 2018, 26, 64–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Friez M. J., Brooks S. S., Stevenson R. E., Field M., Basehore M. J., Adès L. C., Sebold C., McGee S., Saxon S., Skinner C., et al., BMJ Open 2016, 6, e009537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Froyen G., Corbett M., Vandewalle J., Jarvela I., Lawrence O., Meldrum C., Bauters M., Govaerts K., Vandeleur L., Van Esch H., et al., Am. J. Hum. Genet. 2008, 82, 432–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Froyen G., Belet S., Martinez F., Santos-Reboucas C. B., Declercq M., Verbeeck J., Donckers L., Berland S., Mayo S., Rosello M., et al., Am. J. Human Genet. 2012, 91, 252–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mao X., Sethi G., Zhang Z., Wang Q., Curr. Pharm. Des. 2018, 24, 1676–1681. [DOI] [PubMed] [Google Scholar]
- 16. Huang L., Kinnucan E., Wang G., Beuadenon S., Howley P. M., Huibregtse J. N., Pavletich N. P., Science 1999, 286, 1321–1326. [DOI] [PubMed] [Google Scholar]
- 17. Verdecia M. A., Joazeiro C. A. P., Wells N. J., Ferrer J.-L., Bowman M. E., Hunter T., Noel J. P., Mol. Cell 2003, 11, 249–259. [DOI] [PubMed] [Google Scholar]
- 18. Lorenz S., Biol. Chem. 2017, 399, 127–145. [DOI] [PubMed] [Google Scholar]
- 19. Fajner V., Maspero E., Polo S., FEBS Lett. 2017, 591, 2636–2647. [DOI] [PubMed] [Google Scholar]
- 20. Lorenz S., Cantor A. J., Rape M., Kuriyan J., BMC Biol. 2013, 11, 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kamadurai H. B., Souphron J., Scott D. C., Duda D. M., Miller D. J., Stringer D., Piper R. C., Schulman B. A., Mol. Cell 2009, 36, 1095–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ronchi V. P., Klein J. M., Haas A. L., J. Biol. Chem. 2013, 288, 10349–10360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ronchi V. P., Kim E. D., Summa C. M., Klein J. M., Haas A. L., J. Biol. Chem. 2017, 292, 18006–18023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Todaro D. R., Augustus-Wallace A. C., Klein J. M., Haas A. L., J. Biol. Chem. 2017, 292, 19521–19536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ogunjimi A. A., Wiesner S., Briant D. J., Varelas X., Sicheri F., Forman-Kay J., Wrana J. L., J. Biol. Chem. 2010, 285, 6308–6315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. French M. E., Kretzmann B. R., Hicke L., J. Biol. Chem. 2009, 284, 12071–12079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Maspero E., Mari S., Valentini E., Musacchio A., Fish A., Pasqualato S., Polo S., EMBO Rep. 2011, 12, 342–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kim H. C., Steffen A. M., Oldham M. L., Chen J., Huibregtse J. M., EMBO Rep. 2011, 12, 334–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhang W., Wu K.-P., Sartori M. A., Kamadurai H. B., Ordureau A., Jiang C., Mercredi P. Y., Murchie R., Hu J., Persaud A., et al., Mol. Cell 2016, 62, 121–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Maspero E., Valentini E., Mari S., Cecatiello V., Soffientini P., Pasqualato S., Polo S., Nat. Struct. Mol. Biol. 2013, 20, 696–701. [DOI] [PubMed] [Google Scholar]
- 31. Jäckl M., Stollmaier C., Strohäker T., Hyz K., Maspero E., Polo S., Wiesner S., J. Mol. Biol. 2018, 430, 3218–3233. [DOI] [PubMed] [Google Scholar]
- 32. Kim H. C., Huibregtse J. M., Mol. Cell. Biol. 2009, 29, 3307–3318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kamadurai H. B., Qiu Y., Deng A., Harrison J. S., MacDonald C., Actis M., Rodrigues P., Miller D. J., Souphron J., Lewis S. M., Kurinov I., Fujii N., Hammel M., Piper R., Kuhlman B., Schulman B. A, eLife 2013, 2, e00828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhong Q., Gao W., Du F., Wang X., Cell 2005, 121, 1085–1095. [DOI] [PubMed] [Google Scholar]
- 35. Warr M. R., Acoca S., Liu Z., Germain M., Watson M., Blanchette M., Wing S. S., Shore G. C., FEBS Lett. 2005, 579, 5603–5608. [DOI] [PubMed] [Google Scholar]
- 36. French M. E., Klosowiak J. L., Aslanian A., Reed S. I., Yates J. R., Hunter T., J. Biol. Chem. 2017, 292, 10398–10413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wang M., Pickart C. M., EMBO J. 2005, 24, 4324–4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wiesner S., Ogunjimi A. A., Wang H.-R., Rotin D., Sicheri F., Wrana J. L., Forman-Kay J. D., Cell 2007, 130, 651–662. [DOI] [PubMed] [Google Scholar]
- 39. Mari S., Ruetalo N., Maspero E., Stoffregen M. C., Pasqualato S., Polo S., Wiesner S., Structure 2014, 22, 1639–1649. [DOI] [PubMed] [Google Scholar]
- 40. Escobedo A., Gomes T., Aragón E., Martín-Malpartida P., Ruiz L., Macias M. J., Structure 2014, 22, 1446–1457. [DOI] [PubMed] [Google Scholar]
- 41. Wang J., Peng Q., Lin Q., Childress C., Carey D., Yang W., J. Biol. Chem. 2010, 285, 12279–12288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Persaud A., Alberts P., Mari S., Tong J., Murchie R., Maspero E., Safi F., Moran M. F., Polo S., Rotin D., Sci. Signaling 2014, 7, ra95. [DOI] [PubMed] [Google Scholar]
- 43. Chen Z., Jiang H., Xu W., Li X., Dempsey D. R., Zhang X., Devreotes P., Wolberger C., Amzel L. M., Gabelli S. B., Cole P. A., Mol. Cell 2017, 66, 345–357.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhu K., Shan Z., Chen X., Cai Y., Cui L., Yao W., Wang Z., Shi P., Tian C., Lou J., Xie Y., Wen W., EMBO Rep. 2017, 18, 1618–1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gallagher E., Gao M., Liu Y.-C., Karin M., Proc. Natl. Acad. Sci. USA 2006, 103, 1717–1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mund T., Pelham H. R. B., EMBO Rep. 2009, 10, 501–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mund T., Graeb M., Mieszczanek J., Gammons M., Pelham H. R. B., Bienz M., Open Biol. 2015, 5, 150185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Riling C., Kamadurai H., Kumar S., O'Leary C. E., Wu K.-P., Manion E. E., Ying M., Schulman B. A., Oliver P. M., J. Biol. Chem. 2015, 290, 23875–23887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bruce M. C., Kanelis V., Fouladkou F., Debonneville A., Staub O., Rotin D., Biochem. J. 2008, 415, 155–163. [DOI] [PubMed] [Google Scholar]
- 50. Sander B., Xu W., Eilers M., Popov N., Lorenz S., eLife 2017, 6, e21036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Attali I., Tobelaim W. S., Persaud A., Motamedchaboki K., Simpson-Lavy K. J., Mashahreh B., Levin-Kravets O., Keren-Kaplan T., Pilzer I., Kupiec M., Wiener R., Wolf D. A., Rotin D., Prag G., EMBO J. 2017, 36, 425–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ronchi V. P., Klein J. M., Edwards D. J., Haas A. L., J. Biol. Chem. 2014, 289, 1033–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Yi J. J., Berrios J., Newbern J. M., Snider W. D., Philpot B. D., Hahn K. M., Zylka M. J., Cell 2015, 162, 795–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Chan A.-L., Grossman T., Zuckerman V., Campigli Di Giammartino D., Moshel O., Scheffner M., Monahan B., Pilling P., Jiang Y.-H., Haupt S., Schueler-Furman O., Haupt Y., Biochemistry 2013, 52, 3119–3129. [DOI] [PubMed] [Google Scholar]
- 55. Mortensen F., Schneider D., Barbic T., Sladewska-Marquardt A., Kühnle S., Marx A., Scheffner M., Proc. Natl. Acad. Sci. USA 2015, 112, 9872–9877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kathman S. G., Span I., Smith A. T., Xu Z., Zhan J., Rosenzweig A. C., Statsyuk A. V., J. Am. Chem. Soc. 2015, 137, 12442–12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kathman S. G., Xu Z., Statsyuk A. V., J. Med. Chem. 2014, 57, 4969–4974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Jackson P. A., Widen J. C., Harki D. A., Brummond K. M., J. Med. Chem. 2017, 60, 839–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Serafimova I. M., Pufall M. A., Krishnan S., Duda K., Cohen M. S., Maglathlin R. L., McFarland J. M., Miller R. M., Frödin M., Taunton J., Nat. Chem. Biol. 2012, 8, 471–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ahmad A., Sakr W. A., Rahman K. M. W., Curr. Drug Targets 2010, 11, 652–666. [DOI] [PubMed] [Google Scholar]
- 61. Aronchik I., Kundu A., Quirit J. G., Firestone G. L., Mol. Cancer Res. 2014, 12, 1621–1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Quirit J. G., Lavrenov S. N., Poindexter K., Xu J., Kyauk C., Durkin K. A., Aronchik I., Tomasiak T., Solomatin Y. A., Preobrazhenskaya M. N., Firestonea G. L., Biochem. Pharmacol. 2017, 127, 13–27. [DOI] [PubMed] [Google Scholar]
- 63. Rothman D. M., Gao X., George E., Rasmusson T., Bhatia D., Alimov I., Wang L., Kamel A., Hatsis P., Feng Y., et al., Chem. Biol. 2015, 22, 1228–1237. [DOI] [PubMed] [Google Scholar]
- 64. Mund T., Lewis M. J., Maslen S., Pelham H. R., Proc. Natl. Acad. Sci. USA 2014, 111, 16736–16741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Peterson L. A., Chem. Res. Toxicol. 2013, 26, 6–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Sanarico A. G., Ronchini C., Croce A., Memmi E. M., Cammarata U. A., De Antoni A., Lavorgna S., Divona M., Giaco L., Melloni G. E. M., Brendolan A., Simonetti G., Martinelli G., Mancuso P., Bertolini F., Lo Coco F., Melino G., Pelicci P. G., Bernassola F., Leukemia 2018, 32, 911–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Tardiff D. F., Jui N. T., Khurana V., Tambe M. A., Thompson M. L., Chung C. Y., Kamadurai H. B., Kim H. T., Lancaster A. K., Caldwell K. A., Caldwell G. A., Rochet J. C., Buchwald S. L., Lindquist S., Science 2013, 342, 979–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Tofaris G. K., Kim H. T., Hourez R., Jung J.-W., Kim K. P., Goldberg A. L., Proc. Natl. Acad. Sci. USA 2011, 108, 17004–17009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Chung C. Y., Khurana V., Auluck P. K., Tardiff D. F., Mazzulli J. R., Soldner F., Baru V., Lou Y., Freyzon Y., Cho S., et al., Science 2013, 342, 983–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Rossi M., Rotblat B., Ansell K., Amelio I., Caraglia M., Misso G., Bernassola F., Cavasotto C. N., Knight R. A., Ciechanover A., Melino G., Cell Death Dis. 2014, 5, e1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Rossi M., Munarriz E. R., Bartesaghi S., Milanese M., Dinsdale D., Guerra-Martin M. A., Bampton E. T. W., Glynn P., Bonanno G., Knight R. A., Nicotera P., Melino G., J. Cell. Sci. 2009, 122, 3330–3339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Gillman P. K., Br. J. Pharmacol. 2007, 151, 737–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Liu Y.-M., HuangFu W.-C., Huang H.-L., Wu W.-C., Chen Y.-L., Yen Y., Huang H.-L., Nien C.-Y., Lai M.-J., Pan S.-L., Liou J. P., Eur. J. Med. Chem. 2017, 140, 84–91. [DOI] [PubMed] [Google Scholar]
- 74. Zhu H., Kavsak P., Abdollah S., Wrana J. L., Thomsen G. H., Nature 1999, 400, 687–693. [DOI] [PubMed] [Google Scholar]
- 75. Yamashita M., Ying S.-X., Zhang G.-M., Li C., Cheng S. Y., Deng C.-X., Zhang Y. E., Cell 2005, 121, 101–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Kavsak P., Rasmussen R. K., Causing C. G., Bonni S., Zhu H., Thomsen G. H., Wrana J. L., Mol. Cell 2000, 6, 1365–1375. [DOI] [PubMed] [Google Scholar]
- 77. Aragón E., Goerner N., Zaromytidou A.-I., Xi Q., Escobedo A., Massague J., Macias M. J., Genes Devel. 2011, 25, 1275–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Okada M., Sangadala S., Liu Y., Yoshida M., Reddy B. V. B., Titus L., Boden S. D., Cell Biochem. Funct. 2009, 27, 526–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Kato G., Kondo H., Aoki T., Hirono I., Dev. Comp. Immunol. 2012, 36, 349–358. [DOI] [PubMed] [Google Scholar]
- 80. Wong E., Sangadala S., Boden S. D., Yoshioka K., Hutton W. C., Oliver C., Titus L., J. Bone Jt. Surg. 2013, 95, 454–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Cavins J. F., Friedman M., Anal. Biochem. 1970, 35, 489–493. [DOI] [PubMed] [Google Scholar]
- 82. Cao Y., Wang C., Zhang X., Xing G., Lu K., Gu Y., He F., Zhang L., Sci. Rep. 2014, 4, 4965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Zhang Y., Wang C., Cao Y., Gu Y., Zhang L., Oncotarget 2017, 8, 50521–50533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Michel M. A., Swatek K. N., Hospenthal M. K., Komander D., Mol. Cell 2017, 68, 233–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Yau R. G., Doerner K., Castellanos E. R., Haakonsen D. L., Werner A., Wang N., Yang X. W., Martinez-Martin N., Matsumoto M. L., Dixit V. M., Rape M., Cell 2017, 171, 918–933.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Xu Y., Anderson D. E., Ye Y., Cell Discovery 2016, 2, 16040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Sung M.-K., Porras-Yakushi T. R., Reitsma J. M., Huber F. M., Sweredoski M. J., Hoelz A., Hess S., Deshaies R. J., eLife 2016, 5, e19105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Heidelberger J. B., Voigt A., Borisova M. E., Petrosino G., Ruf S., Wagner S. A., Beli P., EMBO Rep. 2018, 19, e44754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. King B., Boccalatte F., Moran-Crusio K., Wolf E., Wang J., Kayembe C., Lazaris C., Yu X., Aranda-Orgilles B., Lasorella A., Aifantis I., Nat. Immunol. 2016, 17, 1312–1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Zhao X., Heng J. I.-T., Guardavaccaro D., Jiang R., Pagano M., Guillemot F., Iavarone A., Lasorella A., Nat. Cell Biol. 2008, 10, 643–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. D'Arca D., Zhao X., Xu W., Ramirez-Martinez N. C., Iavarone A., Lasorella A., Proc. Natl. Acad. Sci. USA 2010, 107, 5875–5880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Peter S., Bultinck J., Myant K., Jaenicke L. A., Walz S., Müller J., Gmachl M., Treu M., Boehmelt G., Ade C. P., Schmitz W., Wiegering A., Otto C., Popov N., Sansom O., Kraut N., Eilers M., EMBO Mol. Med. 2014, 6, 1525–1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Inoue S., Hao Z., Elia A. J., Cescon D., Zhou L., Silvester J., Snow B., Harris I. S., Sasaki M., Li W. Y., et al., Genes Development 2013, 27, 1101–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Myant K. B., Cammareri P., Hodder M. C., Wills J., Von Kriegsheim A., Győrffy B., Rashid M., Polo S., Maspero E., Vaughan L., Gurung B., Barry E., Malliri A., Camargo F., Adams D. J., Iavarone A., Lasorella A., Sansom O. J., EMBO Mol. Med. 2016, 9, 181–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Yang D., Sun B., Zhang X., Cheng D., Yu X., Yan L., Li L., An S., Jiang H., Lasorella A., Iavarone A., Zhang S., Zou F., Zhao X., Cancer Res. 2017, 77, 4773–4784. [DOI] [PubMed] [Google Scholar]
- 96. Yang D., Cheng D., Tu Q., Yang H., Sun B., Yan L., Dai H., Luo J., Mao B., Cao Y., Yu X., Jiang H., Zhao X., Theranostics 2018, 8, 3517–3529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Adhikary S., Marinoni F., Hock A., Hulleman E., Popov N., Beier R., Bernard S., Quarto M., Capra M., Goettig S., Kogel U., Scheffner M., Helin K., Eilers M., Cell 2005, 123, 409–421. [DOI] [PubMed] [Google Scholar]
- 98. Chen D., Kon N., Li M., Zhang W., Qin J., Gu W., Cell 2005, 121, 1071–1083. [DOI] [PubMed] [Google Scholar]
- 99. Arkin M. R., Tang Y., Wells J. A., Chem. Biol. 2014, 21, 1102–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Cohen P., Tcherpakov M., Cell 2010, 143, 686–693. [DOI] [PubMed] [Google Scholar]
- 101. Valkov E., Sharpe T., Marsh M., Greive S., Hyvönen M., Top. Curr. Chem. 2012, 317, 145–179. [DOI] [PubMed] [Google Scholar]
- 102. Erlanson D. A., Wells J. A., Braisted A. C., Annu. Rev. Biophys. Biomol. Struct. 2004, 33, 199–223. [DOI] [PubMed] [Google Scholar]
- 103. Krist D. T., Foote P. K., Statsyuk A. V., Curr. Protoc. Chem. Biol. 2017, 9, 11–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Krist D. T., Park S., Boneh G. H., Rice S. E., Statsyuk A. V., Chem. Sci. 2016, 7, 5587–5595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Foote P. K., Krist D. T., Statsyuk A. V., Curr. Protoc. Chem. Biol. 2017, 9, 174–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Frye S. V., Nat. Chem. Biol. 2010, 6, 159–161. [DOI] [PubMed] [Google Scholar]
- 107. Arrowsmith C. H., Audia J. E., Austin C., Baell J., Bennett J., Blagg J., Bountra C., Brennan P. E., Brown P. J., Bunnage M. E., et al., Nat. Chem. Biol 2015, 11, 536–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Blagg J., Workman P., Cancer Cell 2017, 32, 9–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Waterhouse A. M., Procter J. B., Martin D. M. A., Clamp M., Barton G. J., Bioinformatics 2009, 25, 1189–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Henikoff S., Henikoff J. G., Proc. Natl. Acad. Sci. USA 1992, 89, 10915–10919. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary