

Abstract
Inverse‐electron‐demand Diels–Alder (iEDDA) cycloaddition between 1,2,4,5‐tetrazines and strained dienophiles belongs among the most popular bioconjugation reactions. In addition to its fast kinetics, this cycloaddition can be tailored to produce fluorescent products from non‐fluorescent starting materials. Here we show that even the reaction intermediates formed in iEDDA cycloaddition can lead to the formation of new types of fluorophores. The influence of various substituents on their photophysical properties and the generality of the approach with use of various trans‐cyclooctene derivatives were studied. Model bioimaging experiments demonstrate the application potential of fluorogenic iEDDA cycloaddition.
Keywords: bioorthogonal chemistry, click chemistry, cycloaddition, heterocycles, tetrazines
Bioorthogonal reactions are a set of chemical transformations that enable labeling and study of biomolecules under native biological conditions.1 The properties of these reactions include, among other beneficial factors, high degrees of selectivity, fast kinetics, formation of stable covalent products, and compatibility with strict biological conditions. A number of reactions featuring these attributes are currently available and they have been developed into valuable tools in biology, chemical biology, materials sciences, and biomedical research.2
One especially useful property of bioorthogonal reactions is fluorogenicity. This attribute is based on the production of fluorescent products from nonfluorescent starting materials, thus providing a convenient means of detection through the production of a fluorescent signal.3 There are several ways in which this can be achieved. One is based on quenching of fluorescence through intramolecular energy transfer in bioorthogonal fluorophore conjugates. The subsequent reaction provides products as different chemical species, and this leads to restoration of the fluorescence of the attached fluorophore. One prominent example of a class of reagents of this type is that of 1,2,4,5‐tetrazines.4
Another, comparably rare, example of a fluorogenic reaction is based on the formation of a fluorophore as a result of reaction between two non‐fluorescent bioorthogonal reagents. An example belonging to this class of fluorogenic reactions is the formation of fluorescent pyrazolines through reaction between nitrile imines and various dipolarophiles.5
We recently discovered that pyridinium 1,2,4‐triazines containing a push–pull substitution pattern also form fluorescent products in reaction with strained trans‐cyclooctenes (TCOs).6 In addition, we and others have reported that reactions between 1,2,4,5‐tetrazines and particular dienophiles also lead to fluorescent products without the need for attachment of an extra fluorophore moiety.7 Despite successful application of this chemistry for bioimaging, we found that only the axially substituted hydroxy trans‐cyclooctene and azabicyclononene8 dienophiles afforded fluorescent dihydropyridazine products. This limited scope of dienophiles suitable for fluorogenic labeling hampers broader utility of the methodology. An alternative and more general approach is hence desirable. In continuation of our work in this direction we show here that by using 1,2,4,5‐tetrazines containing specific electron‐donating substituents it is possible to extend the fluorogenic tetrazine cycloaddition to other dienophiles. We have characterized the photophysical properties of the reaction products and in this sense evaluated the effects of various substituents on the tetrazine core, as well as the influence of the TCO structure. In addition, we show that the inherent fluorogenic nature of the chemistry is also operative under biological conditions and can be applied for bioimaging.
The first reaction step of inverse‐electron‐demand Diels–Alder (iEDDA) cycloaddition between a 1,2,4,5‐tetrazine and a dienophile involves the formation of a tetraazabicyclic system, which, after a retro‐Diels–Alder reaction and extrusion of molecular nitrogen, is transformed into a 4,5‐dihydropyridazine (Figure 1 A). This can further isomerize to the corresponding 1,4‐dihydropyridazine through, for example, addition and elimination of a water molecule or alternatively through intramolecular interaction with an appropriately placed heteroatom substituent on the TCO.7b, 9 The initially formed 4,5‐dihydropyridazine heterocyclic core is a unique π‐conjugated system formed upon reaction between tetrazines and all TCO derivatives. Therefore, we thought that it offers an exclusive opportunity to be used for potential formation of fluorophores. Indeed, during experimentation with various 1,2,4,5‐tetrazines we found that tetrazine derivative 1 a, bearing a p‐dimethylamino substituent, forms a fluorescent product when combined with equatorially hydroxy‐substituted TCO 2 a (eqTCO). As we have previously reported,7b the same derivative leads to fluorescent products, with different photophysical properties, upon reaction with the corresponding axially hydroxy‐substituted TCO 2 b (axTCO, Figure 1 B and C). We ascribed the formation of the two different fluorescent species to the production of different tautomers of the dihydropyridazine core (Figures S1 and S2 in the Supporting Information). The structure of the central dihydropyridazine heterocycle thus influences the photophysical properties of the products, enabling control over the outcome of the reaction by simply changing the configuration of the starting TCO. This interesting behavior prompted us to explore the phenomenon in more detail.
Figure 1.
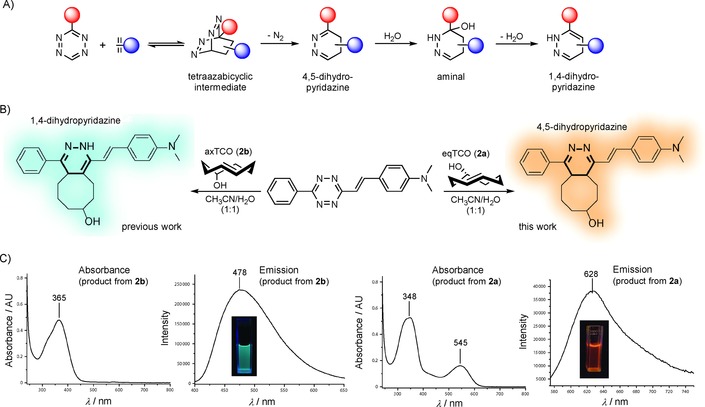
A) General mechanism of iEDDA cycloaddition between a 1,2,4,5‐tetrazine and an alkene dienophile. B) Scheme showing formation of two different products in the reactions between tetrazine 1 a and different TCO isomers. The dihydropyridazine core is highlighted in bold. For clarity only one regioisomer of each product is shown. C) Absorption and emission spectra of products formed in the reactions between 1 a and 2 a or 2 b.
We first synthesized a series of tetrazine derivatives by the Heck cross‐coupling methodology developed by Devaraj and co‐workers4b and measured the photophysical properties of the products formed after reaction with eqTCO (Table 1).
Table 1.
Photophysical properties of 4,5‐dihydropyridazine products.
| |||||
|---|---|---|---|---|---|
| λ abs/λ em [a] | Stokes | Φ fl [b] | Intensity | ||
| R1 | R2 | [nm] | shift | [%] | increase |
| [nm] | |||||
|
|
545/628 | 83 | 0.55 | 13‐fold |
|
|
295/– | – | – | – |
|
|
546/626 | 80 | 0.52 | 18‐fold |
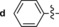
|
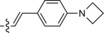
|
546/626 | 80 | 0.45 | 10‐fold |
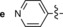
|
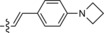
|
566/643 | 77 | 0.21 | threefold |
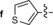
|
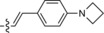
|
549/626 | 77 | 0.47 | 12‐fold |
[a] Absorption and emission maxima were measured in CH3CN/H2O 1:1. [b] Relative fluorescence quantum yields were determined by using Nile Red in methanol as standard.
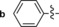
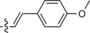
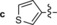
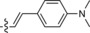
The derivative formed from the tetrazine 1 b, bearing a methoxy substituent, was found to be non‐fluorescent. This indicates that the weaker electron‐donating ability of the methoxy group is not sufficient to promote formation of the fluorophore. On the other hand, the presence of a phenyl or thiophene moiety at position 6 in the tetrazine structure, in combination with a dimethylamino substituent (tetrazines 1 a and 1 c), led to the formation of fluorophores with higher fluorescence quantum yields. The enhancement of the fluorescence signals after the iEDDA click cycloaddition reached 13‐ and 18‐fold for the phenyl‐ and the thiophene‐substituted tetrazines, respectively (Figures 2 and S3–S8, Tables 1 and S2).
Figure 2.
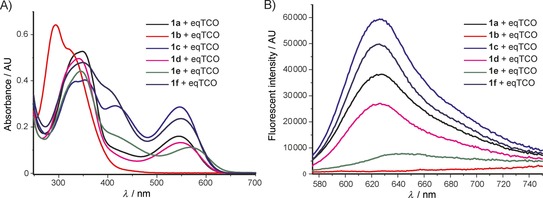
Summary of A) absorption, and B) emission spectra of the products formed in the reactions between tetrazines 1 a–f and eqTCO.
A recent literature example prompted us to explore the possibility of introducing an azetidine moiety as an alternative electron‐donating substituent; this had been shown to improve the photophysical properties of some xanthene dyes.10 We also varied the substituent on the other side of the tetrazine and introduced phenyl, thiophene, and pyridyl groups at position 6. Surprisingly the iEDDA cycloaddition between the pyridyl‐substituted tetrazine 1 e and eqTCO provided only modestly increased fluorescence (Table 1). Possibly, the presence of an electron‐withdrawing substituent (pyridyl) at this position is not beneficial for fluorescence generation and/or other quenching mechanisms are responsible for the observed lower fluorescence. By comparing the absorption and emission maxima of all derivatives it can be concluded that none of these substituents influence the absorption and emission maxima to any great extent. Although the fluorescence quantum yields of the fluorophores formed in the reactions are rather low, their photophysical properties enabled successful use in bioimaging application, as we show below.
An important aspect of bioconjugation reactions is the reactivity of the reagents, which are usually used at low‐micromolar concentrations. Accordingly, we determined the second‐order rate constants of iEDDA cycloaddition between tetrazines 1 a–f and the eqTCO 2 a (Table S1). All of the tetrazines reacted with this dienophile, displaying second‐order rate constants of approximately 7 m −1 s−1 in a 1:1 mixture of CH3CN and H2O at room temperature. For purposes of comparison, we also determined the second‐order rate constants of cycloaddition between the same tetrazines and the axTCO 2 b. Our data are in good agreement with previous observations in which the equatorial isomer was found to react four to five times more slowly than the corresponding axial isomer (Table S1).11
As mentioned above, the 4,5‐dihydropyridazine system is an intermediate in the iEDDA reaction. Depending on the TCO structure and the environment (e.g., presence of water), it can further isomerize to the corresponding 1,4‐dihydropyridazine (Figure 1 A). This means that the fluorescence of the initially formed 4,5‐dihydropyridazine should decrease over time as the tautomerization proceeds. This decrease in fluorescence is an important factor for potential application in, for example, bioimaging. Consequently, we measured the decay in the fluorescent signal over time for tetrazine derivatives 1 c and 1 f (Figures 3, S9, and S10).
Figure 3.
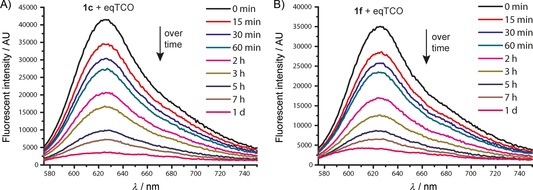
The decay in fluorescence signals over time for the click products formed from eqTCO 2 a and tetrazines 1 c and 1 f.
Our data show that the fluorescence of the click products persists over hours with a half‐life of about two hours, almost completely disappearing within one day. Concomitant appearance of the fluorescence of the newly formed 1,4‐dihydropyridazine tautomer was detected after this time period (Figures S9 and S10). In addition, formation of a small amount of the fully oxidized pyridazine product was observed by HPLC‐MS analysis (Figure S1). This product can also partially contribute to the observed decay in the fluorescent signal. Although this enables only temporary labeling, the timescale is sufficient for bioimaging applications (see below).
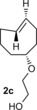
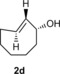
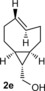
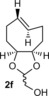
To study the scope of the fluorogenic reaction we tested a series of TCOs12—2 c to 2 f—for their propensity to form fluorescent products with tetrazines 1 c and 1 f (Figures 4, S11, and S12, Tables 2, S3, and S4). We found that all TCOs tested gave rise to the formation of fluorophores with similar photophysical properties to those observed with the eqTCO 2 a. The only exception was the dioxolane‐fused TCO 2 f, which provided a less pronounced fluorescence enhancement of only sixfold. The absorption and emission maxima of all derivatives were similar, being centered around 550 nm and 630 nm, respectively. These data demonstrate that the formation of 4,5‐dihydropyridazines in cycloadditions between tetrazines and TCOs can be considered a more general approach for the development of fluorogenic reactions based on this type of chemistry.
Figure 4.
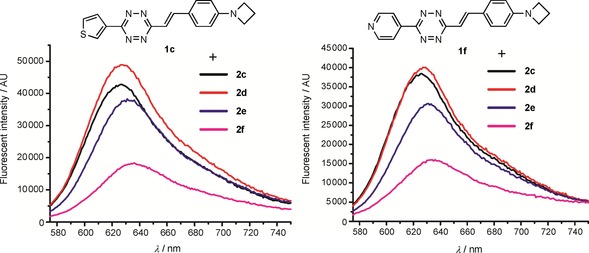
Fluorescence enhancement of the reaction between tetrazines 1 c/1 f and different TCOs.
Table 2.
Absorption and emission maxima [nm] and fluorescence enhancement of the products formed in reaction of 1 c/1 f with different TCOs.
| Tetrazine |
|
|
|
|
|---|---|---|---|---|
| 1 c | 547/627 | 550/627 | 547/631 | 558/638 |
| Fl. intensity increase | 13‐fold | 14‐fold | 11‐fold | sixfold |
| 1 f | 550/627 | 550/627 | 550/631 | 551/631 |
| Fl. intensity increase | ninefold | ninefold | eightfold | fourfold |
We next performed a series of experiments to test whether we could apply these fluorogenic reactions for bioimaging. We first prepared triphenylphosphonium‐functionalized TCO (TPP‐TCO). The TPP moiety is known to target various cargoes to mitochondria13 and thus enables organelle‐specific intracellular labeling. We incubated live HeLa cells with 5 μm TPP‐TCO for 15 min, then washed the cells to remove any excess of the probe, and finally added 5 μm tetrazine 1 c to initiate the fluorogenic reaction. We observed the formation of a marked fluorescence signal inside the cells after only 5 min incubation, thus indicating good cell permeability of probe 1 c and confirming successful reaction. Co‐staining with commercially available mitochondrion‐specific MitoTracker Deep Red dye further confirmed specific labeling of this organelle (Figures 5 A and S13). The fluorescent signal was still detectable even after 2 h, in good agreement with our previous time‐lapse fluorescence‐decay experiments.
Figure 5.
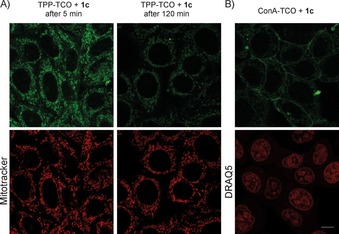
Confocal microscope images from fluorogenic live cell labeling. A) Live HeLa cells were incubated with the mitochondrion‐selective TPP‐TCO probe followed by incubation with 1 c. Mitochondrial labeling was confirmed by co‐staining with Mitotracker Deep Red. B) Labeling of glycoconjugates with ConA‐TCO and 1 c. Excitation for the click product: λ=561 nm (intensity 50 %). Emission was collected in the λ=568–620 nm window. MitoTracker Deep Red and DRAQ5 were excited with a λ=633 nm laser, intensity 20 % and 25 %, respectively. Emission was collected in the λ=643–703 nm window. Scale bar: 10 μm.
To examine the potential of the fluorogenic reaction for bioimaging further, we also conjugated the TCO moiety to concanavalin A (ConA‐TCO). Concanavalin A is a lectin with high specificity for α‐d‐mannose‐ and α‐d‐glucose‐containing glycoconjugates.14 After incubation of live HeLa cells with ConA‐TCO and addition of tetrazine 1 c, specific cell membrane labeling was observed by confocal fluorescence microscopy, whereas cells treated only with ConA‐TCO were not fluorescent (Figures 5 B and S14). These experiments demonstrate that the fluorogenic nature of the cycloaddition is well preserved under biological conditions and can be used for intracellular labeling as well as for fluorescent labeling of cell membrane compartments.
In conclusion, we have shown that the 4,5‐dihydropyridazine reaction intermediate is a unique structural motif that can be utilized for the formation of fluorescent products in iEDDA cycloaddition between 1,2,4,5‐tetrazines and trans‐cyclooctenes. Tetrazines containing electron‐donating dimethylamino or azetidine groups form fluorescent products upon treatment with various TCOs. The fluorogenic nature of the cycloaddition enables application for bioimaging, as we have shown by fluorogenic labeling of intracellular compartments and cell membrane glycoconjugates.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was supported by the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (grant agreement no. 677465). We acknowledge the Light Microscopy Facility at the Institute of Molecular Genetics AS CR supported by the Czech‐BioImaging large RI project (LM2015062 funded by MEYS CR) and by European Regional Development Fund‐Project “National infrastructure for biological and medical imaging” (no. CZ.02.1.01/0.0/0.0/16_013/0001775) for their support with obtaining scientific data presented in this paper. We thank Michael Downey for critical reading of the manuscript.
S. J. Siegl, J. Galeta, R. Dzijak, A. Vázquez, M. Del Río-Villanueva, M. Dračínský, M. Vrabel, ChemBioChem 2019, 20, 886.
References
- 1.
- 1a. Baskin J. M., Bertozzi C. R., QSAR Comb. Sci. 2007, 26, 1211–1219; [Google Scholar]
- 1b. Debets M. F., van Hest J. C. M., Rutjes F. P. J. T., Org. Biomol. Chem. 2013, 11, 6439–6455; [DOI] [PubMed] [Google Scholar]
- 1c. King M., Wagner A., Bioconjugate Chem. 2014, 25, 825–839; [DOI] [PubMed] [Google Scholar]
- 1d. Lim R. K. V., Lin Q., Chem. Commun. 2010, 46, 1589–1600; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1e. Sletten E. M., Bertozzi C. R., Angew. Chem. Int. Ed. 2009, 48, 6974–6998; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 7108–7133. [Google Scholar]
- 2.
- 2a. Kluger R., J. Am. Chem. Soc. 2010, 132, 6611–6612; [Google Scholar]
- 2b. Nwe K., Brechbiel M. W., Cancer Biother. Radiopharm. 2009, 24, 289–302; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2c. Spicer C. D., Davis B. G., Nat. Commun. 2014, 5, 4740; [DOI] [PubMed] [Google Scholar]
- 2d. Spicer C. D., Pashuck E. T., Stevens M. M., Chem. Rev. 2018, 118, 7702–7743; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2e. Xi W., Scott T. F., Kloxin C. J., Bowman C. N., Adv. Funct. Mater. 2014, 24, 2572–2590; [Google Scholar]
- 2f. Topics in Current Chemistry, Vol. 374: Cycloadditions in Bioorthogonal Chemistry (Eds.: M. Vrabel, T. Carell), Springer, Switzerland, 2016. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Le Droumaguet C., Wang C., Wang Q., Chem. Soc. Rev. 2010, 39, 1233–1239; [DOI] [PubMed] [Google Scholar]
- 3b. Nadler A., Schultz C., Angew. Chem. Int. Ed. 2013, 52, 2408–2410; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 2466–2469. [Google Scholar]
- 4.
- 4a. Devaraj N. K., Hilderbrand S., Upadhyay R., Mazitschek R., Weissleder R., Angew. Chem. Int. Ed. 2010, 49, 2869–2872; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 2931–2934; [Google Scholar]
- 4b. Wu H. X., Yang J., Seckute J., Devaraj N. K., Angew. Chem. Int. Ed. 2014, 53, 5805–5809; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 5915–5919; [Google Scholar]
- 4c. Wieczorek A., Werther P., Euchner J., Wombacher R., Chem. Sci. 2017, 8, 1506–1510; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4d. Kozma E., Girona G. E., Paci G., Lemke E. A., Kele P., Chem. Commun. 2017, 53, 6696–6699; [DOI] [PubMed] [Google Scholar]
- 4e. Kozma E., Demeter O., Kele P., ChemBioChem 2017, 18, 486–501; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4f. Knorr G., Kozma E., Schaart J. M., Nemeth K., Torok G., Kele P., Bioconjugate Chem. 2018, 29, 1312–1318. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Yu Z., Ohulchanskyy T. Y., An P., Prasad P. N., Lin Q., J. Am. Chem. Soc. 2013, 135, 16766–16769; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5b. Kaya E., Vrabel M., Deiml C., Prill S., Fluxa V. S., Carell T., Angew. Chem. Int. Ed. 2012, 51, 4466–4469; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 4542–4545; [Google Scholar]
- 5c. Song W., Wang Y., Qu J., Madden M. M., Lin Q., Angew. Chem. Int. Ed. 2008, 47, 2832–2835; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 2874–2877; [Google Scholar]
- 5d. Song W., Wang Y., Qu J., Lin Q., J. Am. Chem. Soc. 2008, 130, 9654–9655. [DOI] [PubMed] [Google Scholar]
- 6. Siegl S. J., Dzijak R., Vázquez A., Pohl R., Vrabel M., Chem. Sci. 2017, 8, 3593–3598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.
- 7a. Shang X., Song X., Faller C., Lai R., Li H., Cerny R., Niu W., Guo J., Chem. Sci. 2017, 8, 1141–1145; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7b. Vázquez A., Dzijak R., Dracinsky M., Rampmaier R., Siegl S. J., Vrabel M., Angew. Chem. Int. Ed. 2017, 56, 1334–1337; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 1354–1357. [Google Scholar]
- 8. Siegl S. J., Vázquez A., Dzijak R., Dračínský M., Galeta J., Rampmaier R., Klepetářová B., Vrabel M., Chem. Eur. J. 2018, 24, 2426–2432. [DOI] [PubMed] [Google Scholar]
- 9. Blackman M. L., Royzen M., Fox J. M., J. Am. Chem. Soc. 2008, 130, 13518–13519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.
- 10a. Grimm J. B., Muthusamy A. K., Liang Y. J., Brown T. A., Lemon W. C., Patel R., Lu R. W., Macklin J. J., Keller P. J., Ji N., Lavis L. D., Nat. Methods 2017, 14, 987–994; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10b. Grimm J. B., English B. P., Chen J. J., Slaughter J. P., Zhang Z. J., Revyakin A., Patel R., Macklin J. J., Normanno D., Singer R. H., Lionnet T., Lavis L. D., Nat. Methods 2015, 12, 244–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rossin R., van den Bosch S. M., ten Hoeve W., Carvelli M., Versteegen R. M., Lub J., Robillard M. S., Bioconjugate Chem. 2013, 24, 1210–1217. [DOI] [PubMed] [Google Scholar]
- 12.
- 12a. Taylor M. T., Blackman M. L., Dmitrenko O., Fox J. M., J. Am. Chem. Soc. 2011, 133, 9646–9649; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12b. Darko A., Wallace S., Dmitrenko O., Machovina M. M., Mehl R. A., Chin J. W., Fox J. M., Chem. Sci. 2014, 5, 3770–3776; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12c. Versteegen R. M., Rossin R., ten Hoeve W., Janssen H. M., Robillard M. S., Angew. Chem. Int. Ed. 2013, 52, 14112–14116; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 14362–14366. [Google Scholar]
- 13. Zielonka J., Joseph J., Sikora A., Hardy M., Ouari O., Vasquez-Vivar J., Cheng G., Lopez M., Kalyanaraman B., Chem. Rev. 2017, 117, 10043–10120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Goldstein I. J., Poretz R. D. in The Lectins: Properties, Functions, and Applications in Biology and Medicine., Vol. 32 (Eds.: I. E. Liener, N. Sharon, I. J. Goldstein), Academic Press, Orlando, 1986, pp. 33–247. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary