

Abstract
Background
Chronic infection with Burkholderia cepacia complex species remains a significant problem for clinicians treating people with cystic fibrosis. Colonisation with Burkholderia cepacia complex species is linked to a more rapid decline in lung function and increases morbidity and mortality. There remain no objective guidelines for strategies to eradicate Burkholderia cepacia complex in cystic fibrosis lung disease, as these are inherently resistant to the majority of antibiotics and there has been very little research in this area. This review aims to examine the current treatment options for people with cystic fibrosis with acute infection with Burkholderia cepacia complex and to identify an evidence‐based strategy that is both safe and effective. This is an updated version of the review.
Objectives
To identify whether treatment of Burkholderia cepacia complex infections can achieve eradication, or if treatment can prevent or delay the onset of chronic infection. To establish whether following eradication, clinical outcomes are improved and if there are any adverse effects.
Search methods
We searched the Cochrane Cystic Fibrosis Trials Register, compiled from electronic database searches and handsearching of journals and conference abstract books. We also searched the reference lists of relevant articles and reviews.
Last search: 12 March 2019.
We also searched electronic clinical trials registers for the USA and Europe.
Date of last search: 12 March 2019.
Selection criteria
Randomised or quasi‐randomised studies in people with cystic fibrosis of antibiotics or alternative therapeutic agents used alone or in combination, using any method of delivery and any treatment duration, to eradicate Burkholderia cepacia complex infections compared to another antibiotic, placebo or no treatment.
Data collection and analysis
Two authors independently assessed for inclusion in the review the eligibility of 52 studies (79 references) identified by the search of the Group's Trial Register and the other electronic searches.
Main results
No studies looking at the eradication of Burkholderia cepacia complex species were identified.
Authors' conclusions
The authors have concluded that there was an extreme lack of evidence in this area of treatment management for people with cystic fibrosis. Without further comprehensive studies, it is difficult to draw conclusions about a safe and effective management strategy for Burkholderia cepacia complex eradication in cystic fibrosis. Thus, while the review could not offer clinicians evidence of an effective eradication protocol for Burkholderia cepacia complex, it has highlighted an urgent need for exploration and research in this area, specifically the need for well‐designed multi‐centre randomised controlled studies of a variety of (novel) antibiotic agents.
Plain language summary
Treatments to cure long‐term infections with Burkholderia cepacia in people with cystic fibrosis
Review question
We reviewed the evidence for antibiotic treatment to cure early infection with Burkholderia cepacia complex in people with cystic fibrosis and prevent it becoming permanent.
Background
Cystic fibrosis is an inherited disease and people who have this disease produce large amounts of thick mucus which is difficult to clear. This mucus blocks up their lungs and digestive systems. People with cystic fibrosis suffer from lots of chest infections, which cause scarring of their airways. Eventually, they develop infections that can't be cured with antibiotics, so their lungs always contain lots of bugs, this is described as being chronically infected. One of these bugs, Burkholderia cepacia, causes a lot of problems for people with cystic fibrosis because it is very difficult to treat and makes their lung disease deteriorate faster than it otherwise would. This is an updated version of the review.
Search date
We last searched for any evidence on 12 March 2019.
Study characteristics
We looked for studies of treatments which could eliminate Burkholderia cepacia from the lungs of people with cystic fibrosis. We did not find any relevant studies. This review highlights an urgent need for more research into new ways of treating long‐term Burkholderia cepacia infection in people with cystic fibrosis.
Background
Description of the condition
Cystic fibrosis (CF) is the most common autosomal recessive condition affecting people of Northern European descent (Farrell 2018), with an incidence of approximately 1 in 2500 (Ratjen 2003). A multisystem disease, CF primarily affects the lungs, pancreas and gastrointestinal (GI) tract, but in the majority of cases it is progressive lung disease that causes premature death. Frequent early bacterial infections progress to colonisation and chronic infection, resulting in an exaggerated neutrophil inflammatory response that leads to extensive lung damage (bronchiectasis), which over many years progresses to respiratory failure and death.
A mutation in an ion transporter, cystic fibrosis transmembrane conductance regulator (CFTR), results in abnormal sodium chloride exchange on the epithelia of people with CF. In the respiratory tract, it is likely that bacterial colonisation occurs because of reduced chloride secretion and increased sodium reabsorption. In the airway epithelium this leads to reduced water content of secretions as well as reduced depth of periciliary fluid, which in turn leads to the trapping of inhaled bacteria and slower clearance (Saiman 2004). There is also a growing body of evidence to suggest that the CF neutrophil displays delayed apoptosis (programmed cell death), resulting in prolonged inflammation and release of pro‐inflammatory cytokines, which contributes to airway damage (Moriceau 2009; Moriceau 2010).
Over the course of their lives people with CF are vulnerable to bacterial infections caused by many different species, but over the last few decades a specific subgroup of pathogens affecting the CF population has been characterised. The most troubling of these are Staphylococcus aureus, Pseudomonas aeruginosa and Burkholderia cepacia complex pathogens (LiPuma 2010). The B cepacia complex (BCC) is the collective name for a group of at least 21 closely related bacteria that have been isolated from both human infections and the natural environment (Drevinek 2010; Loveridge 2017). Although not the most commonly isolated pathogen in people with CF, affecting approximately only 2% to 4% of such people in the USA (Drevinek 2010), it is amongst the most virulent; and chronic infection has been independently associated with increased morbidity and mortality in those affected (Corey 1996). In the UK, it was reported as being responsible for 1.4% and 5.1 %of lung infections in children and adults in 2017 (CF Trust 2017). The species are grouped into 'genomovars', a term which describes different strains of B cepacia according to their genetic content, each of which displays independent modes of transmission and clinical effects. There are currently 21 genomovars, and while all species within the complex have been isolated from humans, two species ‐ B cenocepacia (genomovar III) and B multivorans (genomovar II), cause between 85% and 97% of BCC infections in people with CF (Drevinek 2010). A subset of species have been shown to be transmissible from one person to another and can cause epidemics, and as a result it is important for people with CF to be segregated according to presence or absence of BCC species (LiPuma 2010; St Denis 2007). Despite the fact that many people culture genotypically distinct strains of BCC and thus it is likely that they are contracted from independent environmental sources, the majority of cases are a result of transmission from other people with CF (LiPuma 2010; Mahenthiralingam 2001; Millar‐Jones 1998).
In some people, infection is transient, while others remain stable for long periods despite chronic infection (CF Trust 2004); in rare cases people with CF succumb to a rapidly progressive pneumonia known as 'cepacia syndrome' (CF Trust 2009). For most people, however, chronic BCC infection develops and causes an unpredictable decline in lung function ranging from relatively mild to rapidly deteriorating, leading to a significant increase in both time spent in hospital and mortality (CF Trust 2004; Zlosnik 2011). Furthermore, presence of BCC infection before transplant results in a significant excess in post‐transplant mortality, generally caused by overwhelming BCC sepsis (CF Trust 2004). The reasons for this variable outcome are not yet fully understood, but it has been suggested that the ability of some BCC species to produce copious exomucopolysaccharides (mucoid species) is linked to a milder course than those non‐mucoid species, which appear to produce a more rapid and severe decline in lung function (Zlosnik 2011). Chronic infection is usually with a single species, but super‐ or co‐infection with additional species can occur, and in addition there is in vitro evidence that commonly used antibiotics, e.g. ciprofloxacin, can cause mucoid strains to become non‐mucoid (LiPuma 2010; Zlosnik 2011). In summary, there is a great deal of good quality evidence from well‐designed studies that shows a significant clinical deterioration as a result of BCC infections.
This review will aim to identify intervention strategies to eradicate BCC infection or prevent or delay chronicity of that infection in people with CF. There is as yet no vaccine for any of the BCC species, which is linked in part to their variability, but there have been early animal studies that suggest vaccine therapy may be possible in the future (Sokol 2000). Until such time as an effective preventive measure is developed, it is of the utmost importance to develop strategies for treating infection once it has occurred. Species of BCC, particularly B cenocepacia, are intrinsically resistant to aminoglycosides, most beta lactams and polymyxins and are also capable of developing in vivo resistance to almost any antimicrobial agent (Drevinek 2010), with some UK centres reporting pan‐resistance in over 80% of isolates (Moore 2001). Resistance can be observed in all strains (Nzula 2002), but environmentally‐ rather than clinically‐acquired infections are usually more amenable to therapy (CF Trust 2009); it has been suggested that resistance is highest in Burkholderia dolosa (Vermis 2003).
Description of the intervention
The increasing longevity and reduced morbidity in people with CF observed in recent years is, in major part, due to the aggressive use of appropriate antibiotics to prevent, eradicate, or control respiratory infections. Flucloxacillin prophylaxis from diagnosis until three years of age is effective in reducing the incidence of S aureus infection (Smyth 2003), though such an approach has been unsuccessful with P aeruginosa (Tramper‐Stranders 2010). However, early identification and eradication of P aeruginosa can successfully prevent or delay chronic infection (Douglas 2009; Gibson 2003; Gibson 2007; Langton Hewer 2009; Ratjen 2001; Ratjen 2006; Taccetti 2005); and once chronic infection is established, regular nebulised antibiotics or intermittent courses of intravenous antibiotics, or both, help in maintaining lung function and reducing sputum bacterial load (CF Trust 2009). It is possible that early aggressive treatment of certain BCC species with appropriate antibiotics may delay or prevent chronic infection.
How the intervention might work
It has been shown that chronicity develops in 94% of B cenocepacia infections and 50% of B multivorans infections despite therapy, which as previously described, represent the vast majority of BCC infections, although in some people this may be transient (Ball 2010; Etherington 2003). Strategies that have been implemented to help prevent or delay infection include segregation according to infection or strain status and emphasis on hygiene of shared equipment, which appears to be effective (CF Trust 2004).
Despite the difficulties in determining an effective therapeutic regimen for BCC, some progress has been made in identifying strategies to eradicate other troublesome CF pathogens, such as P aeruginosa, (Langton Hewer 2009), and it is to be hoped that the principles of this therapy may be applicable to cases of BCC infection.
Why it is important to do this review
The outcome of BCC infection in people with CF is variable, but generally results in an increase in morbidity and mortality related to a decline in lung function. The epidemiology of these infections seems to be changing and treatment of these infections is challenging because of the inherent antibiotic resistance. There is no real consensus on the best way to treat these infections and as yet no standard treatment regimens for eradication. Successful eradication would hopefully reduce decline in lung function, and therefore morbidity and mortality, and improve quality of life. It is important to systematically analyse the evidence available in order to try and find a strategy that may help alleviate the burden of disease currently caused by BCC infections in people with CF. This is an updated version of the review (Regan 2012; Regan 2014; Regan 2016).
Objectives
To identify whether early, aggressive therapy of BCC infections is able to achieve eradication after initial acquisition or prevent or delay onset of chronic infection and whether this improves clinical outcome measures such as lung function, nutritional status, clinical scores and mortality.
To determine whether these therapies are linked to any adverse effects or cause an increase the isolates of other species in the lower respiratory tract.
To assess any evidence of superiority between different therapies or therapeutic regimens with respect to cost‐effectiveness or clinical outcome.
Methods
Criteria for considering studies for this review
Types of studies
Randomised or quasi‐randomised controlled studies.
Types of participants
Any person with a clinical diagnosis of CF that has been confirmed by sweat testing or genetic analysis, or both, who acquires a new infection or a re‐infection with BCC. People of all ages and disease severity will be included.
Types of interventions
Any antibiotic or antibiotic adjuvant therapy used alone or in combination to eradicate BCC infection. Treatments may be compared to an alternative antimicrobial agent, a placebo or no treatment, (excluding the participant's usual therapeutic regimen). The mode of delivery of the intervention may be inhaled, oral or intravenous and there is no limit to the duration of therapy or dosage used.
Types of outcome measures
Primary outcomes
Eradication (i.e. no BCC positive cultures from bronchoalveolar lavage (BAL), sputum or oropharyngeal aspirate for a period of 12 months over a minimum of six samples)
Length of time remaining infection free (post‐eradication therapy)
Secondary outcomes
-
Spirometric lung function, expressed as a per cent predicted based on age, sex and height
forced expiratory volume at one second (FEV1)
forced vital capacity (FVC)
-
Growth and nutritional status
body mass index (BMI) z score
weight z score
height (in children only) z score
Mortality
Quality of life (QoL) assessment (measured using all instruments, validated or not, e.g. the Cystic Fibrosis Questionnaire‐Revised version (CFQ‐R) (Quittner 2009) and the Cystic Fibrosis Quality of Life Questionnaire (CFQoL) (Gee 2000))
-
Adverse events of the eradication treatment used (as classified by the review authors), including microbiological sequelae (i.e. does eradication of B cepacia result in alternative colonisation?) and drug interactions or hypersensitivity reactions:
mild: resulted in no change to treatment, e.g. mild sensitivity reactions
moderate: resulted in a change in treatment, e.g. renal or auditory impairment
severe: required hospital admission or is life‐threatening, e.g. anaphylaxis
Search methods for identification of studies
Studies are eligible for inclusion in the review irrespective of publication status (e.g. abstract or online trial report) or language.
Electronic searches
We identified relevant studies from the Group's CF Trials Register using the terms: Burkholderia cepacia OR (mixed infections AND (eradication OR unknown)).
The Cystic Fibrosis Trials Register is compiled from electronic searches of the Cochrane Central Register of Controlled Trials (CENTRAL) (updated each new issue of The Cochrane Library), weekly searches of MEDLINE, a search of Embase to 1995 and the prospective handsearching of two journals ‐ Pediatric Pulmonology and the Journal of Cystic Fibrosis. Unpublished work is identified by searching the abstract books of three major cystic fibrosis conferences: the International Cystic Fibrosis Conference; the European Cystic Fibrosis Conference and the North American Cystic Fibrosis Conference. For full details of all searching activities for the register, please see the relevant sections of the Cochrane Cystic Fibrosis and Genetic Disorders Group website..
Date of the last search: 12 March 2019.
We searched a number of online trials registries as detailed in the appendices (Appendix 1).
Date of the last search: 12 March 2019.
Searching other resources
In addition to our search of the Group's CF Trials Register, we would also have handsearched the reference list of any study that we included. If, in future updates of the review, we identify any relevant papers in the reference lists of included studies, we will contact the authors for further information. To the best of our knowledge, there are no novel anti‐BCC antimicrobial agents on that market at the current time, so we did not approach any manufacturers of antibiotics.
Data collection and analysis
Selection of studies
The two authors (KR, JB) independently applied the selection criteria to determine the studies to be included in the review. There was no disagreement between the authors. Where it was unclear which pathogens participants were colonised with, the authors contacted the relevant authors to find out if any participants had BCC species.
Data extraction and management
Both authors planned to extract data independently from any included studies using standard data acquisition forms. If there had been any disagreements on the risk of bias or suitability of a study, they planned to reach a consensus by discussion. The authors planned to extract information on the mode of delivery of drug treatment. If data become available in future, they will initially combine all data in the analysis and later undertake a subgroup analysis by mode of delivery (see below).
With cases of P aeruginosa, infection is defined as chronic when more than 50% of months, when samples had been taken, were P aeruginosa‐culture positive and intermittent when 50% or less of months, when samples had been taken, were P aeruginosa‐culture positive, People with CF are considered free of infection if no growth of P aeruginosa has been identified during the previous 12 months if they were previously P aeruginosa‐culture positive or if P aeruginosa has never been cultured from sputum or a cough swab (Lee 2003). In accordance with this, the authors classified chronic infection as BCC cultured from more than 50% samples over 12 months; initial infection as those with a first isolation of BCC; and recent infection as those with less than 12 months history of BCC isolation. In future versions of this review, in those studies where this information is not included, the review authors will contact the study authors to seek this additional information.
In cases where participants are infected with multiple species, the review authors still planned to utilise data on eradication of B. cepacia as it remains clinically relevant since people with CF are often colonised with multiple species.
If data become available for future versions of this review, the authors plan to group data relating to the outcomes measured according to the time elapsed from baseline. Standard clinical practice is to take a sample at the end of any eradication therapy and thereafter at any routine clinical encounter and upon exacerbation. Thus the review authors will group data according to samples taken at: post‐eradication therapy; up to three months; up to six months; up to nine months; up to one year; and over one year. In those cases where eradication is defined differently than above, the authors will discuss the alternative definition and decide whether it is comparable to their own or whether they should exclude the study from statistical analysis.
Assessment of risk of bias in included studies
Both authors planned to independently determine the risk of bias using the Cochrane Collaboration's tool for assessing risk of bias as detailed in Chapter 8 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). The authors aimed to examine the following from reported data for each included study: randomisation process; method of allocation concealment; degree of blinding; completeness of outcome data and whether intention‐to‐treat analyses were possible; and selective reporting.
If any studies are included in future updates of the review, where the authors view the description and methods of randomisation and allocation concealment to be adequate, they will consider this to be a low risk of bias and where methods are inadequate, a high risk of bias. If the methods are not sufficiently described, they will consider this to constitute an unclear risk of bias. The greater the degree of blinding in the intervention, the lower the risk of bias that the authors will attribute to the study, (e.g. a double‐blinded study has a lower risk of bias than a single‐blinded study). Where study investigators properly account for withdrawals from studies and these are of similar quantity across groups, the authors will judge the study to be at low risk of bias. However, where the authors deem that investigators inadequately justify withdrawals or their number are unevenly distributed between groups, the review authors will consider the study to be at high risk of bias. If the authors identify any evidence of selective outcome reporting as described in 'Assessment of reporting biases' below, they will consider the study to be at high risk of bias, if they find no evidence of selective reporting , they will consider the study to be at a low risk of bias.
In future, where there is disagreement over any aspect of the risk of bias for a given study, the authors will reach a consensus by discussion.
Measures of treatment effect
The absence of data available within the scope of this review has prevented the application of the methodology described below to the current version, but should any data become available in the future, we intend to use this protocol to analyse data.
The wide range of outcome measures the authors hope to assess in this review will produce data of distinct types and they will therefore assess these by different measures. They plan to collect data on all participants regardless of compliance or later decisions by study authors about suitability for inclusion in the results.
For binary (dichotomous) data, they will identify the number of participants with each outcome event by allocated treatment group, and will use these data to calculate the odds ratio (OR) and 95% confidence intervals (CIs). For continuous data, the authors will separate outcome data (means and standard deviations (SDs)) according to allocated treatment group and calculate the mean difference (MD) and 95% CIs for each group. If outcomes are measured using different units of measurement, they will calculate the standardised mean difference (SMD). For time‐to‐event data, they plan to calculate the hazard ratio (HR) and 95% CIs according to the outcomes for each of the groups in the study.
Unit of analysis issues
The authors' inclusion criteria for studies in the review do not permit the use of cross‐over studies or cluster‐randomised studies. For a highly variable and chronic condition like CF, a cross‐over study design is not appropriate as baseline values at the start of the second treatment arm are likely to be significantly different from those at the start of the first. Cluster‐randomised studies present a unit of analysis issue, unless data are analysed only at the level of the group rather than an individual level. Furthermore, individuals in the same cluster tend to be more similar to one another than to individuals in other clusters, which represents a risk of bias. When analysing the data, the unit of analysis will be the individual and not the number of episodes of a given event (e.g. infection or adverse reaction).
Dealing with missing data
In future versions of this review, in those studies where data on outcome measures are not available for all participants enrolled in the studies, the authors will perform an available‐case analysis using the available data. Where all randomised participants are accounted for they will perform an intention‐to‐treat (ITT) analysis.
Assessment of heterogeneity
In the event that data are available on this subject in future versions of this review, where sufficient studies are available, the authors will assess statistical heterogeneity using the I² statistic (Higgins 2003). They will consider I² values of under 25% to be of low heterogeneity; those between 26% and 50% to be moderate heterogeneity; those between 51% and 75% to be substantial heterogeneity; and those over 75% to be considerable heterogeneity. Their analysis will use 95% CIs, which should be sufficient for the number of participants they are likely to be considering, and they will perform a Chi² test. The Chi² analysis will allow the authors to determine whether any differences in results between studies are a result of chance. They will use the test to compare the statistical heterogeneity of studies in accordance with the Cochrane Handbook for Systematic Reviews of Interventions, and will also use it in the visual assessment of forest plots (Deeks 2011).
Assessment of reporting biases
The authors plan to assess reporting bias by comparing the published outcome measures with those outcomes mentioned in the description of the methods within the published papers. In the event that important outcome measures have not been accounted for, they will contact the authors for information about both the missing data and the original study protocol. In accordance with guidance in the Cochrane Handbook for Systematic Reviews of Interventions (Sterne 2011), they will use the funnel plot tool to assess the publication bias of each study to be included.
Data synthesis
When data are available, the authors will use a fixed‐effect model to analyse the data from the included studies where possible. However, if they detect at least substantial statistical heterogeneity using the I² statistic (over 50%), they will apply a random‐effects model.
Subgroup analysis and investigation of heterogeneity
Where sufficient evidence is available (minimum 10 studies for meta‐analysis ‐ not achieved with this version of the review) the authors will investigate the effects of:
dosage;
duration of treatment, e.g. up to 14 days, up to one month, up to three months, up to six months, up to 12 months;
antibiotic therapies used alone or when delivered in combination with adjuvant therapies;
mode of delivery, e.g. inhaled and oral agents versus intravenous delivery.
We will achieve this by categorising participants into the related subgroups and performing meta‐analyses on each of these subgroups.
Sensitivity analysis
The authors will test the robustness of their results using sensitivity analyses relating to:
fixed‐effect versus random‐effects analysis;
multicentre versus single‐centre studies.
Summary of findings tables
We will construct a summary of findings table for each comparison included in the review using the GRADEpro software. We will consider the following outcomes:
eradication of BCC;
length of time remaining infection free (post eradication therapy);
FEV1 (change from baseline);
BMI;
mortality;
QoL; and
adverse events.
Using the GRADE approach, described in Chapter 12 of the Cochrane Handbook of Systematic Review for Interventions (Schünemann 2011), we will classify the body of evidence as high, moderate, low or very low. Where we judge the evidence not to be high quality, we will describe the rationale for this judgement in footnotes to the table.
Results
Description of studies
Results of the search
Searches of the CFGD Group's CF Trials Register identified 50 studies (77 references) and two studies (one reference each) were identified by an additional electronic search (NCT00298922; Uluer 2013). None of these met the inclusion criteria for the review (see Excluded studies).
This process is graphically represented in a study flow diagram (Figure 1).
1.
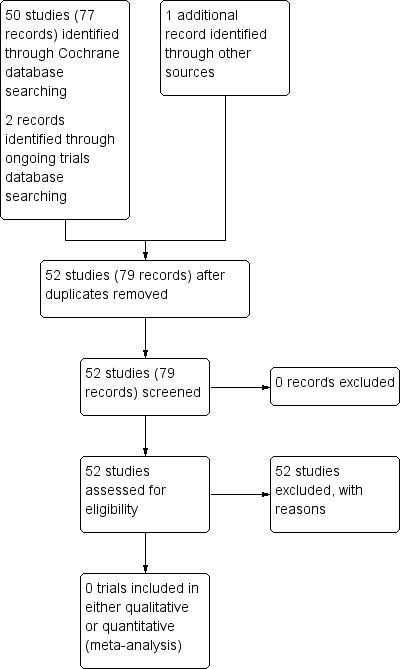
Study flow diagram.
Included studies
No studies were eligible for inclusion in this review.
Excluded studies
A total of 52 studies were excluded from the review for a number of reasons and full details are available in the tables 'Characteristics of excluded studies'. Where it was unclear which pathogens participants were colonised with, we contacted the relevant authors to find out if any participants had BCC species. Three studies specifically included participants infected with BCC: two of these were excluded as although they looked at eradication of BCC species, they were of cross‐over design (Ledson 2002; Rye 2015); while a further study was excluded as it recruited participants with chronic BCC infection where eradication is not possible (Tullis 2014). Six studies were excluded as they looked at treatment for P aeruginosa (Adeboyeku 2001; Carswell 1987; Huang 1982; Kapranov 1995; Knight 1979; Loening‐Bauke 1979). In a further 10 studies no pathogen was specified (Chua 1990; Conway 1996; Cooper 1985; Heininger 1993; Nathanson 1985; Postnikov 2001a; Romano 1991; Romano 1992; Salh 1992; Stutman 1987). One study was focused on reducing early pulmonary infections in CF, not including BCC (Singh 2013), and a further study had no participants infected with BCC (Frederiksen 2006). The most common reason for exclusion was that studies examined pharmacokinetics or safety issues of antibiotic therapy; 19 studies were excluded for this reason (Davis 1987; Degg 1996; Dodd 1997; Geller 2004; Goldfarb 1986; Griffith 2008; Gulliver 2003; Hodges 2014; Huls 2000; Kruger 2001; Labiris 2004; Pai 2006; Postnikov 2000; Postnikov 2001; Prayle 2016, Roberts 1993; Rosenfeld 2006; Smith 1997; Wood 1996). Two studies examined the role of zinc supplementation in reducing frequency of pulmonary exacerbations in CF (Khorasani 2009; Sharma 2016). Five studies were excluded as they specifically looked at the effects of treatment in relation to the mode of delivery: three studies looked at the effects of home intravenous antibiotic treatment (Amelina 2000; Hjelte 1988; Ramstrom 2000); one study looked at the efficacy of delivery of a nebulised antibiotic (Keller 2010); and one study looked at the effect of pancreatic enzymes on the absorption of oral antibiotics (Vitti 1975). One study was excluded as it had an open‐label design and did not have a control group (Uluer 2013). One study considered compliance to a treatment regimen (Dodd 1998) and one study looked at the development of a scoring system for the efficacy of antibiotic regimens (Huang 1979). The remaining study was not able to enrol the intended sample size and initial attempts to get it published were unsuccessful and hence the investigators did not persist (NCT00298922).
Risk of bias in included studies
No studies were included in the review.
Effects of interventions
No studies were included in the review.
Discussion
Summary of main results
For this review, we could not find any study that met the inclusion criteria of any antibiotic or antibiotic adjuvant therapy used alone or in combination to eradicate BCC infection which was compared to an alternative antimicrobial agent, a placebo or no treatment, (excluding the participant's usual therapeutic regimen).
Overall completeness and applicability of evidence
From a selection of studies, the Ledson study was the only one that was a randomised controlled trial of eradication therapy for BCC in CF (Ledson 2002). However, it was of cross‐over design, comparing nebulised taurolidine (an antibiotic to which BCC has displayed in vitro sensitivity) to 0.9% saline as a placebo, and hence excluded from the review.
As we were unable to identify any studies for inclusion in this review, we were not able to address our aim of establishing options for BCC eradication in people who are infected with BCC and no data were available on attempts to delay or prevent chronic infection as no such studies were identified. The lack of suitable studies also meant that we were unable to compare treatment options and assess any differences in efficacy or adverse events.
Quality of the evidence
There was no evidence identified for this review.
Potential biases in the review process
While every possible effort has been made by the authors to limit bias in the review process, there remains the possibility of sources of bias. While comprehensive searches have been performed, it is possible that relevant studies were missed or that studies have been undertaken and results of these have not been published.
Agreements and disagreements with other studies or reviews
The Ledson study is in many ways representative of the albeit small pool of evidence available on eradication of BCC in CF. In this cross‐over study, the eradication of BCC was not achieved in any participant, and there was no improvement in either FEV1 or FVC. It is likely that, as with P aeruginosa, eradication is most likely to be successful if attempted at first detection, rather than once chronic infection has become established, and thus these results cannot reliably be extrapolated to people with a first isolation of BCC in their sputum, when they may be more susceptible to eradication strategies. Despite the lack of efficacy in this study, the lack of adverse events indicates that nebulised taurolidine may be a viable treatment on a case‐to‐case basis where specific colonising organisms are known to be sensitive. The specificity of BCC infection is such that there are almost no data on BCC from other conditions, since it is rarely encountered outside the context of CF and chronic granulomatous disease (a very rare disorder of the immune system). The evidence which is available, most of which is in the form of individual case reports or small, non‐randomised or non‐blinded studies, rarely reports successful eradication of BCC from the CF airways. This is due to both the difficulties of accessing a severely diseased airway and also the inherent resistance of BCC species themselves. Other evidence on eradication has not been systematically reviewed to the best of our knowledge due to the nature of the reports.
Consensus documents acknowledge the lack of high quality evidence in this area, but do make recommendations for the treatment of BCC (CF Trust 2009) which are summarised as follows:
antimicrobial therapy should be directed by in vitro sensitivities where available;
combination therapy should be used for treatment of BCC exacerbations and "cepacia syndrome";
the routine use of synergy testing to guide therapy of BCC cannot be recommended at this time;
the use of eradication therapy for all new growths of BCC should be considered.
Authors' conclusions
Implications for practice.
No studies were identified which met the reviews's inclusion criteria and hence there is insufficient evidence from the literature to determine an effective strategy for the eradication of Burkholderia cepacia complex (BCC) species from adults with cystic fibrosis (CF).
Implications for research.
This review highlights a clear lack of reliable evidence on which to base management decisions for people with CF, either chronically or newly infected with BCC species, despite a wealth of knowledge on the adverse long‐term effects of BCC infection. There is a need for well‐designed, randomised, multi‐centre studies of a variety of eradication strategies, based on in vitro studies of sensitivity or individual case reports of successful eradication, that will allow the examination of the effects of different agents on BCC infection and provide sufficient participants to ensure the studies have adequate power. In order for progress to be made more rapidly in this field, it may be appropriate for studies to address a number of alternative regimens in order for successful therapies to be identified sooner. Such studies should be specifically designed to address the issue of eradicating BCC species from people with CF while minimising adverse effects and preventing harmful changes in airway colonisation. Of particular interest would be randomised studies examining aggressive management at first isolation of BCC, as it is now standard practice to delay or prevent onset of chronicity of Pseudomonas aeruginosa infection in CF in most centres. Such strategies may be the only realistic hope of eradication during a potential window of susceptibility before infection is established and in the absence of novel antimicrobial agents to which BCC is sensitive. Outcome measurements for these studies should focus on clinically relevant parameters including improvements in forced expiratory volume in one second (FEV1) and forced vital capacity (FVC), body mass index (BMI) and nutritional status as well as the primary measure of reduction in bacterial colonisation assessed by airway secretions. These outcome measures offer good clinical evidence as to the general health of a person with CF, and can thus indicate the relevance of any alterations in colonisation. Furthermore, evidence from people with both CF and non‐CF bronchiectasis suggests that while, in the short term, eradication of chronic airway pathogens is difficult, longer‐term regimens (up to 12 months) can often prove effective. This is something that should be considered when designing studies, and it may be that effects on colony counts are not seen until a number of months after the initiation of therapy.
What's new
| Date | Event | Description |
|---|---|---|
| 18 March 2019 | New search has been performed | A search of the Cystic Fibrosis and Genetic Disorders Review Group's Cystic Fibrosis Trials Register identified seven new references for potential inclusion in the review. All seven were additional references to the already excluded studies (Griffith 2008; Prayle 2016; Rye 2015; Tullis 2014). Two potential new references were identified from searches of online trial registries and were also excluded (NCT00298922; Uluer 2013). |
| 18 March 2019 | New citation required but conclusions have not changed | No new studies or data were included in this updated review, hence our conclusions remain the same. |
History
Protocol first published: Issue 5, 2012 Review first published: Issue 10, 2014
| Date | Event | Description |
|---|---|---|
| 1 November 2016 | New search has been performed | A search of the Cochrane Cystic Fibrosis and Genetic Disorders Group's Cystic Fibrosis Trials Register identified nine references to seven studies potentially eligible for inclusion in this review. These were assessed by the authors and unfortunately none were eligible for inclusion. Two studies previously listed as awaiting classification have now been excluded (NCT00298922; Uluer 2013). |
| 1 November 2016 | New citation required but conclusions have not changed | No new data have been included in this updated review, hence there are no changes to the conclusions. |
Acknowledgements
The review authors would like to thank Nikki Jahnke for her invaluable support during the process of writing this review and advising the authors on all aspects of it during its completion.
Appendices
Appendix 1. Additional electronic searches
| Database | Search terms | Date of latest search |
| European Union Clinical Trials Register (www.clinicaltrialsregister.eu) |
"cystic fibrosis" AND "Burkholderia cepacia" | 12 March 2019 |
| ClinicalTrials.gov (clinicaltrials.gov) |
"cystic fibrosis" AND "Burkholderia cepacia" | 12 March 2019 |
| WHO ICTRP (apps.who.int/trialsearch/) |
"cystic fibrosis" AND "Burkholderia cepacia" | 12 March 2019 |
| UK Clinical Trials Gateway (www.ukctg.nihr.ac.uk/). |
"cystic fibrosis" AND "Burkholderia cepacia" | 12 March 2019 |
Characteristics of studies
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Adeboyeku 2001 | Study of safety and tolerability of an eradication protocol for P. aeruginosa. |
| Amelina 2000 | Study of effects of home IVs for exacerbations of CF bronchiectasis on lung function and QoL in CF. |
| Carswell 1987 | Study of eradication protocols for P. aeruginosa colonisation. |
| Chua 1990 | Assessment of bronchial responsiveness to nebulised antibiotics, pathogen not specified. |
| Conway 1996 | Assessment of different combinations of IV antibiotics in the treatment of exacerbations of CF bronchiectasis, pathogen not specified. |
| Cooper 1985 | Assessment of different combinations of IV antibiotics in the treatment of exacerbations of CF bronchiectasis, pathogen not specified. |
| Davis 1987 | Study of the pharmacokinetics of ciprofloxacin in people with CF, pathogen not specified. |
| Degg 1996 | Evaluation of the ototoxic effects of gentamicin on people with CF, pathogen not specified. |
| Dodd 1997 | Study of the correlation between tonicity and adverse effects of nebulised colistin in CF, pathogen not specified. |
| Dodd 1998 | Discussion of the discrepancy between patient‐reported and objectively assessed non‐compliance in clinical studies, neither drug or pathogen specified. |
| Frederiksen 2006 | No participants infected with BCC in study. |
| Geller 2004 | Pharmacokinetics, safety and efficacy of nebulised tobramycin in CF, pathogen not specified. |
| Goldfarb 1986 | Pharmacokinetics of single dose oral ciprofloxacin in people with CF, pathogen not specified. |
| Griffith 2008 | Pharmacokinetics of single dose nebulised levofloxacin in people with CF, pathogen not specified. |
| Gulliver 2003 | Study of tolerance of nebulised tobramycin in people with CF, pathogen not specified. |
| Heininger 1993 | Comparison of different dose regimens of aminoglycosides in exacerbations of CF bronchiectasis, pathogen not specified. |
| Hjelte 1988 | Study of QoL for people with CF with home versus inpatient IV antibiotics for exacerbations. |
| Hodges 2014 | Pharmacokinetic study. |
| Huang 1979 | Study of a novel scoring system for assessing the efficacy of IV antibiotic regimens in exacerbations of CF bronchiectasis. |
| Huang 1982 | Strategy for eradication of P. aeruginosa. |
| Huls 2000 | Study of the effects of lung function on serum concentrations of antibiotics, pathogen not specified. |
| Kapranov 1995 | Strategy for eradication of P. aeruginosa. |
| Keller 2010 | Investigation of the efficacy of delivery of a novel therapeutic preparation of nebulised tobramycin, pathogen not specified. |
| Khorasani 2009 | Study examined the role of zinc supplementation in reducing frequency of pulmonary exacerbations in CF. |
| Knight 1979 | Eradication strategy for P. aeruginosa. |
| Kruger 2001 | Report of ototoxicity of IV tobramycin in people with CF, pathogen not specified. |
| Labiris 2004 | Investigation of whether inhalation of preservatives from IV tobramycin preparations causes airway inflammation in CF, pathogen not specified. |
| Ledson 2002 | Cross‐over study of B. cepacia therapy, study design not eligible. |
| Loening‐Bauke 1979 | Comparison of long‐term options for eradication of P. aeruginosa. |
| Nathanson 1985 | Study of efficacy of nebulised gentamicin in CF, pathogen not specified. |
| NCT00298922 | Due to delays in starting related to getting placebo, the investigators were not able to enrol the intended sample size. The study was negative but was underpowered. Initial attempts to get it published were unsuccessful and hence the investigators did not persist. |
| Pai 2006 | Pharmacokinetics of levofloxacin in adults with CF, pathogen not specified. |
| Postnikov 2000 | Study of tolerance and efficacy of pefloxacin as a prophylaxis and treatment of severe infections in children with CF and aplastic anaemia, pathogen not specified. |
| Postnikov 2001 | Study of safety of fluoroquinolones in children, pathogen not specified. |
| Postnikov 2001a | Comparison of growth rates of children with CF either treated or not treated with ciprofloxacin, pathogen not specified. |
| Prayle 2016 | Pharmacokinetic study. |
| Ramstrom 2000 | Study of the effects of altered methods of preparation of home IV antibiotic therapies in CF. |
| Roberts 1993 | Study of interactions of tobramycin and ticarcillin in CF, pathogen not specified. |
| Romano 1991 | Non‐blinded study of ofloxacin for people with CF requiring long‐term antibiotics with 'sensitive' sputum culture (pathogens unknown). |
| Romano 1992 | Non‐blinded study of ofloxacin for people with CF requiring long‐term antibiotics with 'sensitive' sputum culture (pathogens unknown). |
| Rosenfeld 2006 | Study of accumulation of nebulised tobramycin in respiratory secretions, pathogen not specified. |
| Rye 2015 | Cross‐over study of B. cepacia therapy, study design not eligible. |
| Salh 1992 | Study of antibiotic regimens for treatment of CF bronchiectasis exacerbations, pathogen not specified. |
| Sharma 2016 | Study examined the role of zinc supplementation in reducing frequency of pulmonary exacerbations in CF. |
| Singh 2013 | Study focused on reducing early pulmonary infections in CF, not including BCC. |
| Smith 1997 | Study of the uses of salivary ciprofloxacin concentrations in people with CF, pathogen not specified. |
| Stutman 1987 | Comparison of different dose regimens of oral ciprofloxacin in chronically infected people with CF, pathogen not specified. |
| Tullis 2014 | RCT of treatment for people with chronic B. cepacia infection. |
| Uluer 2013 | Open‐label design with no control group; treatment offered to all people with CF with Burkholderia dolosa. |
| Vitti 1975 | Study of effectiveness of pancreatic enzyme supplements on absorption of oral antibiotics in CF. |
| Wood 1996 | Study of minimisation of aminoglycoside toxicity in CF. |
B. cepacia: Burkholderia cepacia CF: cystic fibrosis IV: intravenous P aeruginosa: Pseudomonas aeruginosa QoL: quality of life RCT: randomised controlled trial
Contributions of authors
| Roles and responsibilities | |
| TASK | WHO WILL UNDERTAKE THE TASK? |
| Protocol stage: draft the protocol | Kate Regan |
| Review stage: select which studies to include (2 + 1 arbiter) | Kate Regan & Jayesh Bhatt |
| Review stage: extract data from studies (2 people) | Kate Regan & Jayesh Bhatt |
| Review stage: enter data into RevMan | Kate Regan |
| Review stage: carry out the analysis | Kate Regan |
| Review stage: interpret the analysis | Kate Regan & Jayesh Bhatt |
| Review stage: draft the final review | Kate Regan & Jayesh Bhatt |
| Update stage: update the review | Kate Regan & Jayesh Bhatt |
Sources of support
Internal sources
No sources of support supplied
External sources
-
National Institute for Health Research, UK.
This systematic review was supported by the National Institute for Health Research, via Cochrane Infrastructure funding to the Cochrane Cystic Fibrosis and Genetic Disorders Group.
Declarations of interest
Kate Regan declares no conflict of interest.
Jayesh Bhatt has received lecture fees from Vertex Pharmaceuticals Inc. for a talk on pulmonary exacerbations in cystic fibrosis; Vertext do not produce any drugs relevant to this review.
New search for studies and content updated (no change to conclusions)
References
References to studies excluded from this review
Adeboyeku 2001 {published data only}
- Adeboyeku DU, Agent P, Jackson V, Hodson M. A double blind randomised study to compare the safety and tolerance of differing concentrations of nebulised colistin administered using HaloLite in cystic fibrosis (CF) patients. Pediatric Pulmonology 2001;32 Suppl 22:288. [CFGD Register: PI165; MEDLINE: ] [Google Scholar]
Amelina 2000 {published data only}
- Amelina E, Senkevich N, Cherniak A, Cherniaev A, Chuchalin A. Home intravenous therapy in adult cystic fibrosis patients. The impact on lung function and quality of life. European Respiratory Journal 2000;16 Suppl 31:123s. [CFGD Register: PI181; MEDLINE: ] [Google Scholar]
Carswell 1987 {published data only}
- Carswell F, Ward C, Cook DA, Speller DC. A controlled trial of nebulized aminoglycoside and oral flucloxacillin versus placebo in the outpatient management of children with cystic fibrosis. British Journal of Diseases of the Chest 1987;81(4):356‐60. [CFGD Register: PI54] [DOI] [PubMed] [Google Scholar]
Chua 1990 {published data only}
- Chua H, Collis G, Souef P. Bronchial response of children with cystic fibrosis to nebulised antibiotics [abstract]. Australian and New Zealand Journal of Medicine 1990;20:537. [CFGD Register: PI66b] [Google Scholar]
- Chua HL, Collis GG, Souef PN. Bronchial response to nebulized antibiotics in children with cystic fibrosis. European Respiratory Journal 1990;3(10):1114‐6. [CFGD Register: PI66a] [PubMed] [Google Scholar]
Conway 1996 {published data only}
- Conway SP. Ceftazidime 3G BD is as effective as ceftazidime 2G TDS in the treatment of respiratory exacerbations in cystic fibrosis. Israel Journal of Medical Sciences 1996;32 Suppl:S256. [CFGD Register: PI78; MEDLINE: ] [Google Scholar]
Cooper 1985 {published data only}
- Cooper DM, Harris M, Mitchell I. Comparison of intravenous and inhalation antibiotic therapy in acute pulmonary deterioration in cystic fibrosis. American Review of Respiratory Disease 1985;131:A242. [CFGD Register: PI129] [Google Scholar]
Davis 1987 {published data only}
- Davis RL, Koup JR, Williams Warren J, Weber A, Heggen L, Stempel D, et al. Pharmacokinetics of ciprofloxacin in cystic fibrosis. Antimicrobial Agents and Chemotherapy 1987;31(6):915‐9. [CFGD Register: PI50] [DOI] [PMC free article] [PubMed] [Google Scholar]
Degg 1996 {published data only}
- Degg C, Mulheran M. The effect of frequent exposure to gentamicin on distortion product OAEs in patients with cystic fibrosis. British Journal of Audiology 1996;30(2):99‐100. [CFGD Register: PI167] [Google Scholar]
Dodd 1997 {published data only}
- Dodd M, Maddison J, Abbott J, Webb AK. The effect of the tonicity of nebulised colistin on lung function in adults with cystic fibrosis [abstract]. Proceedings of the 18th European Cystic Fibrosis Conference; 1993 May 21‐26; Madrid, Spain. 1993:121. [CFGD Register: PI100a; MEDLINE: ]
- Dodd ME, Abbott J, Maddison J, Moorcroft AJ, Webb AK. Effect of tonicity of nebulised colistin on chest tightness and pulmonary function in adults with cystic fibrosis. Thorax 1997;52(7):656‐8. [CFGD Register: PI100b; MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodd ME, Maddison J, Abbot J, Webb AR. The effect of the tonicity of nebulised colistin on chest tightness and lung function in adults with cystic fibrosis. European Respiratory Journal 1993;6 Suppl 17:515s. [CFGD Register: PI100c; MEDLINE: ] [Google Scholar]
Dodd 1998 {published data only}
- Dodd ME, Haworth CS, Moorcroft AJ, Miles J, Webb AK. Is medicine evidence‐based when there is discrepancy between patient reported and objective measures of compliance in clinical trials? [abstract]. Pediatric Pulmonology 1998;26 Suppl 17:389‐90. [CFGD Register: PI237; MEDLINE: ] [Google Scholar]
Frederiksen 2006 {published data only}
- Frederiksen B, Koch C, Hoiby N, Pressler T, Hansen A. Effect of aerosolised rhDnase (Pulmozyme®) on pulmonary infections in CF: an open randomised study [abstract]. Pediatric Pulmonology 2000;Suppl 20:246. [CENTRAL: 792917; CFGD Register: BD97a; CRS: 5500100000003555] [Google Scholar]
- Frederiksen B, Pressler T, Hansen A, Koch C, Hoiby N. Effect of aerosolized rhDNase (Pulmozyme) on pulmonary colonization in patients with cystic fibrosis. Acta Paediatrica 2006;95(9):1070‐4. [CENTRAL: 571885; CFGD Register: BD97b; CRS: 5500100000002869; EMBASE: 2006449673; PUBMED: 16938752] [DOI] [PubMed] [Google Scholar]
Geller 2004 {published data only}
- Geller DE, Rodriguez CA, Howenstine M, Murphy T, Voter K, Nickerson B, et al. The effects of doubling concentration of tobramycin solution for inhalation on pharmacokinetics (PK), safety and delivery time in patients with cystic fibrosis (CF) [abstract]. American Journal of American Journal of Respiratory and Critical Care Medicine 2004;169(7):A391. [CFGD Register: PI183a; MEDLINE: ] [Google Scholar]
- Rosenfeld M, Geller DE, Rodriguez CA, Howenstine M, Konstan M, Ordonez C, et al. Serum pharmacokinetics of two preparations of tobramycin solution for inhalation in young cystic fibrosis patients [abstract]. American Journal of Respiratory and Critical Care Medicine 2004;169(7):A386. [CFGD Register: PI183b; MEDLINE: ] [Google Scholar]
Goldfarb 1986 {published data only}
- Goldfarb J, Wormser GP, Inchiosa MA Jr, Guideri G, Diaz M, Gandhi R, et al. Single‐dose pharmacokinetics of oral ciprofloxacin in patients with cystic fibrosis. Journal of Clinical Pharmacology 1986;26(3):222‐6. [CFGD Register: PI44] [DOI] [PubMed] [Google Scholar]
Griffith 2008 {published data only}
- Geller DE, Flume P, Schwab R, Fornos P, Conrad DJ, Morgan E, et al. A phase 1 safety, tolerability and pharmacokinetic (PK) study of MP‐376 (levofloxacin solution for inhalation) in stable cystic fibrosis (CF) patients. Pediatric Pulmonology 2008;43 Suppl 31:315. [Abstract no.: 321; CFGD Register: PI210b; MEDLINE: ] [Google Scholar]
- Griffith DC, Hansen C, Pressler T, Balchen T, Jensen TJ, Geller DE, et al. Single‐dose pharmacokinetics of aerosol MP‐376 (levofloxacin solution for inhalation) in cystic fibrosis patients: PK‐PD implications. Journal of Cystic Fibrosis 2008;7 Suppl 2:S26. [CFGD Register: PI210a; MEDLINE: ] [Google Scholar]
- Kearns GL, Rubino CM, Griffith DC, Geller DE, Forrest A, Bhavnani SM, et al. Levofloxacin pharmacokinetics (PK) after administration of MP‐376 (Levofloxacin inhalation solution; Aeroquin) in children with cystic fibrosis. Journal of Cystic Fibrosis 2011;10 Suppl 1:S23. [Abstract no.: 88; CFGD Register: PI210d] [Google Scholar]
- Stockmann C, Hillyard B, Ampofo K, Spigarelli MG, Sherwin CM. Levofloxacin inhalation solution for the treatment of chronic Pseudomonas aeruginosa infection among patients with cystic fibrosis. Expert Review of Respiratory Medicine 2015;9(1):13‐22. [CFGD Register: PI210c] [DOI] [PubMed] [Google Scholar]
Gulliver 2003 {published data only}
- Gulliver T, Wilson S, Williams G, Harris M, Cooper D. Nebulized tobramycin (intravenous solution) is tolerated without inducing cough and wheeze in cystic fibrosis patients [abstract]. Proceedings of the Thoracic Society of Australia & New Zealand Annual Scientific Meeting; 2003 April 4‐9; Adelaide, Australia. 2003:Abstract no: P139. [CFGD Register: PI184; MEDLINE: ]
Heininger 1993 {published data only}
- Heininger U, Bowing B, Stehr K, Solbach W. Aminoglycosides in patients with mucoviscidosis and pulmonary exacerbation. Comparison of once or three times daily administration [Aminoglykoside bei Patienten mit Mukoviszidose und pulmonaler Exazerbation: Vergleich von Einmal‐ und Dreimalgabe.]. Klinische Padiatrie 1993;205(1):18‐22. [CFGD Register: PI74] [DOI] [PubMed] [Google Scholar]
Hjelte 1988 {published data only}
- Hjelte L, Widen B, Malmborg AS, Freyschuss U, Strandvik B. Intravenous administration of antibiotics at home in patients with cystic fibrosis improves quality of life [Intravenos antibiotikabehandling i hemmet vid cystisk fibros ger okad livskvalitet]. Lakartidningen 1988;85(18):1614‐7. [CFGD Register: PI206; MEDLINE: ] [PubMed] [Google Scholar]
Hodges 2014 {published data only}
- Hodges L, MacGregor G, Stevens H, Dessen A, Myrset A. An open label, randomised, two‐way crossover scintigraphic study to investigate lung deposition of radiolabelled alginate oligosaccharide delivered as a dry powder and as a nebulised solution in cystic fibrosis patients. Pediatric Pulmonology 2014;49 Suppl 38:305. [Abstract no.: 251; CENTRAL: 1012384; CFGD Register: BD215; CRS: 5500131000000154] [Google Scholar]
Huang 1979 {published data only}
- Huang N, Palmer J, Schidlow D, Hsuan F, Hsu C, Goldberg M, et al. Evaluation of antibiotic therapy in patients with cystic fibrosis. Chest 1979;76(3):354‐5. [CFGD Register: PI113; MEDLINE: ] [Google Scholar]
Huang 1982 {published data only}
- Huang NN, Palmer J, Braverman S, Keith HH, Schidlow D. Therapeutic efficacy of ticarcillin and carbenicillin in patients with cystic fibrosis: a double blind study [abstract]. Proceedings of 23rd Annual Meeting Cystic Fibrosis Club Abstracts; 1982 May 14; Washington D.C.. 1982:124. [CFGD Register: PI114; MEDLINE: ]
Huls 2000 {published data only}
- App EM, Huls G, Bittner‐Dersch P, Stolz S, Lindemann H, Matthys H. Impared lung function influences the serum concentration of inhaled drugs in cystic fibrosis. Pediatric Pulmonology 2000;30 Suppl 20:279‐80. [CFGD Register: PI156b; MEDLINE: ] [Google Scholar]
- Huls G, App EM, Bittner‐Dersch P, Stolz S, Lindemann H. Impaired lung function influences the serum concentration of inhaled drugs in cystic fibrosis. Proceedings of 13th International Cystic Fibrosis Congress; 2000 June 4‐8; Stockholm, Sweden. 2000:177. [CFGD Register: PI156a; MEDLINE: ]
Kapranov 1995 {published data only}
- Kapranov NI, Belousov YB, Kashyrskaya NY, Smirnova EY. Quinoline therapy in children with cystic fibrosis. Proceedings of 20th European Cystic Fibrosis Conference; 1995 June 18‐21; Brussels, Belgium. 1995:P19. [CFGD Register: PI104; MEDLINE: ]
Keller 2010 {published data only}
- Keller M, Coates AL, Griese M, Denk O, Schierholz J, Knoch M. In‐vivo data support equivalent therapeutic efficacy of a new tobramycin inhalation solution (150mg/1.5ml) administered by the eFlow® electronic nebuliser compared to TOBI® in the PARI LC PLUS®. Journal of Cystic Fibrosis 2010;9 Suppl 1:S22. [Abstract no.: 84; CFGD Register: PI241; MEDLINE: ] [Google Scholar]
Khorasani 2009 {published data only}
- Khorasani EN, Mansouri F. Effect of zinc supplementation on respiratory infections in children with cystic fibrosis. European Respiratory Society Annual Congress; 2009; Sept 12‐16; Vienna, Austria. 2009:722s. [Abstract no.: P4032; CENTRAL: 793188; CFGD Register: GN255; CRS: 5500050000000367]
Knight 1979 {published data only}
- Knight RK, Batten JC, Mearns M. A double blind trial of cephalexin in cystic fibrosis patients with pseudomonas in the sputum. Proceedings of 9th Meeting European Working Group for Cystic Fibrosis; 1979 June 12‐13; Noordwijkerhout, The Netherlands. 1979:52. [CFGD Register: PI124; MEDLINE: ]
Kruger 2001 {published data only}
- Junge S, Kruger K, Schonweiler R, Ptok M, Ballmann M. Once daily dosage of intravenous tobramycin ‐ increased risk for cochlea damage in children with cystic fibrosis (CF)?. Pediatric Pulmonology 2001;32 Suppl 22:291. [CFGD Register: PI160b] [Google Scholar]
- Kruger K, Junge S, Schonweiler R, Ptok M, Ballmann M. Once daily dosage of intravenous tobramycin in patients with cystic fibrosis ‐ increased risk for cochlea damage?. Proceedings of 24th European Cystic Fibrosis Conference; 2001 June 6‐9; Vienna, Austria. 2001:P191. [CFGD Register: PI160a; MEDLINE: ]
Labiris 2004 {published data only}
- Labiris R, Freitag A, Pratt B, Efthimiadis A, Hargreave F, Dolovich M. Does inhalation of preservatives from IV tobramycin preparations (TOB) cause airway inflammation?. American Journal of Respiratory and Critical Care Medicine 2004;169(7):A307. [CFGD Register: PI182; MEDLINE: ] [Google Scholar]
Ledson 2002 {published data only}
- Ledson MJ, Gallagher MJ, Cowperthwaite C, Robinson M, Convery RP, Walshaw MJ. A randomised double blind placebo controlled crossover trial of nebulised taurolidine in adult CF patients colonised with B Cepacia. Proceedings of 22nd European Cystic Fibrosis Conference; 1998 June 13‐19; Berlin, Germany. 1998:120. [CFGD Register: PI128a; MEDLINE: ]
- Ledson MJ, Gallagher MJ, Robinson M, Cowperthwaite C, Williets T, Hart CA, et al. A randomized double‐blinded placebo‐controlled crossover trial of nebulized taurolidine in adult cystic fibrosis patients infected with Burkholderia cepacia. Journal of Aerosol Medicine 2002;15(1):51‐7. [CFGD Register: PI128b] [DOI] [PubMed] [Google Scholar]
Loening‐Bauke 1979 {published data only}
- Loening‐Baucke VA, Mischler E, Myers MG. A placebo‐controlled trial of cephalexin therapy in the ambulatory management of patients with cystic fibrosis. Journal of Pediatrics 1979;95(4):630‐7. [CFGD Register: PI19b] [DOI] [PubMed] [Google Scholar]
- Loening‐Baucke VA, Mischler EH, Myers MG. Cephalexin in cystic fibrosis: a placebo‐controlled study. Pediatric Research 1978;12(4 Pt 2):495. [CFGD Register: PI19c; MEDLINE: ] [Google Scholar]
- Loening‐Bauke V, Mischler EH, Myers MG. Cephalexin compared to placebo in the management of patients with cystic fibrosis. Proceedings of 19th Cystic Fibrosis Club Abstracts; 1978. 1978:69. [CFGD Register: PI19a; MEDLINE: ]
Nathanson 1985 {published data only}
- Nathanson I, Cropp GJA, Li P, Neter E. Effectiveness of aerosolized gentamicin in cystic fibrosis (CF). Cystic Fibrosis Club Abstracts; 1985. 1985:145. [CFGD Register: PI130; MEDLINE: ]
NCT00298922 {published data only (unpublished sought but not used)}
- NCT00298922. Azithromycin in patients with CF, infected with Burkholderia Cepacia Complex. http://clinicaltrials.gov/show/NCT00298922 (accessed 28 November 2013).
Pai 2006 {published data only}
- Pai MP, Allen SE, Amsden GW. Altered steady state pharmacokinetics of levofloxacin in adult cystic fibrosis patients receiving calcium carbonate. Journal of Cystic Fibrosis 2006;5(3):153‐7. [CFGD Register: PI199; MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Postnikov 2000 {published data only}
- Postnikov SS, Semykin SIu, Kapranov NI, Perederko LV, Polikarpova SV, Khamidullina KF. Evaluation of tolerance and efficacy of pefloxacin in the treatment and prevention of severe infections in children with mucoviscidosis and aplastic anemia [Otsenka effektivnosti i perenosimosti pefloksatsina pri lechenii i profilaktike tiazhelykh infektsii u detei s mukovistsidozom i aplasticheskoi anemiei.]. Antibiotiki i Khimioterapiia 2000;45(8):25‐30. [CFGD Register: PI171] [PubMed] [Google Scholar]
Postnikov 2001 {published data only}
- Postnikov SS, Semykin SJ, Najimov VP. Safety of fluoroquinolones in children [abstract]. Proceedings of 24th European Cystic Fibrosis Conference; 2001 June 6‐9; Vienna, Austria. 2001:P213. [CFGD Register: PI162; MEDLINE: ]
Postnikov 2001a {published data only}
- Postnikov SS, Semiakin SI, Nazhimov VP, Kapranov NI. Comparative yearly growth rate of children with mucoviscidosis treated and not treated with ciprofloxacin:clinicomorphological comparisons [Cravnitel'naiia godovaia skorost' rosta u detei s mukovistsidozom, poluchavshikh i nepolychavshikh tsiprofloksatsin:klinicomorfologicheskie copostavleniia.]. Antibiotiki I Khimioterapiia 2001;46(10):11‐3. [CFGD Register: PI169] [PubMed] [Google Scholar]
Prayle 2016 {published data only}
- Prayle A, Jain K, Watson A, Smyth AR. Are morning doses of intravenous tobramycin less nephrotoxic than evening? Evidence from urinary biomarkers in the critic study. Pediatric Pulmonology 2013;48 Suppl 36:299. [Abstract no.: 261; CENTRAL: 980338; CFGD Register: CO55; CRS: 5500125000000420] [Google Scholar]
- Prayle AP, Jain K, Touw DJ, Koch BCP, Knox AJ, Watson A, et al. The pharmacokinetics and toxicity of morning vs. evening tobramycin dosing for pulmonary exacerbations of cystic fibrosis: a randomised comparison. Journal of Cystic Fibrosis 2016;15(4):510‐7. [CFGD Register: CO55b] [DOI] [PMC free article] [PubMed] [Google Scholar]
Ramstrom 2000 {published data only}
- Ramstrom H, Erwander I, Mared L, Kornfalt R, Seiving B. Pharmaceutical intervention in the care of cystic fibrosis patients. Journal of Clinical Pharmacy and Therapeutics 2000;25(6):427‐34. [CFGD Register: PI159] [DOI] [PubMed] [Google Scholar]
Roberts 1993 {published data only}
- Roberts GW, Nation RL, Jarvinen AO. Measurement of serum tobramycin in the presence of ticarcillin or piperacillin. Australian Journal of Hospital Pharmacy 1992;22(2):152‐4. [CFGD Register: PI132b] [Google Scholar]
- Roberts GW, Nation RL, Jarvinen AO, Martin AJ. An in vivo assessment of the tobramycin/ticarcillin interaction in cystic fibrosis patients. British Journal of Clinical Pharmacology 1993;36:372‐5. [CFGFD Register: PI132a] [DOI] [PMC free article] [PubMed] [Google Scholar]
Romano 1991 {published data only}
- Romano L, Minicucci L, Spallone E, Girosi D, Campelli A, Fabbri A, et al. Role of home therapy with ofloxacin in patients with cystic fibrosis (CF) [Ruolo della terapia domiciliare con ofloxacin in pazienti con fibrosi cistica (FC)]. Giornale Italiano de Chemioterapia 1991;38(1‐3):181‐3. [CFGD Register: PI68] [PubMed] [Google Scholar]
Romano 1992 {published data only}
- Romano L, Girosi D, Spallone E, Parisi F, Minicucci L, Romano C. The use of ofloxacin in cystic fibrosis patients [Uso dell'ofloxacin nei pazienti con fibrosi cistica.]. Minerva Pediatrica 1992;44(3):79‐86. [CFGD Register: AB59] [PubMed] [Google Scholar]
Rosenfeld 2006 {published data only}
- Rosenfeld M, Emerson J, Uh D, Anderson G, Genatossio A, McNamara S, et al. Does tobramycin accumulate in respiratory secretions with repeated aerosol administration: a pilot study. Pediatric Pulmonology 2006;41 Suppl 29:327. [CFGD Register: PI203; MEDLINE: ] [Google Scholar]
Rye 2015 {published data only}
- Rye PD, Mahenthiralingam E, Weiser R, Marchesi JR, Opitz P, Trentmann M, et al. Microbiota analysis of samples from a randomized double‐blind, placebo‐controlled cross‐over study of inhaled alginate oligosaccharide (OLIGOG) in cystic fibrosis individuals using aztreonam due to chronic colonization with burkholderia SPP. Pediatric Pulmonology 2015;50 Suppl 41:328. [Abstract no.: 363; CENTRAL: 1092173; CFGD Register: BD223a; CRS: 5500135000001369] [Google Scholar]
- Weiser R, Mahenthiralingham E, Marchesi J, Opitz P, Trentmann M, Rye PD, et al. Standardised cultivation‐independent methods to understand the effect of OligoG on Burkholderia and microbial diversity in the CF lung during a clinical trial. Journal of Cystic Fibrosis 2016;15 Suppl 1:S60. [Abstract no.: 35; CENTRAL: 1157480; CFGD Register: BD223b; CRS: 5500135000001560] [Google Scholar]
- Weiser R, Marchesi J, Opitz P, Trentmann M, Rye PD, Onsoyen E, et al. Microbiota analysis of samples from a randomized double‐blind, placebo‐controlled cross‐over study of inhaled alginate oligosaccharide (oligog) in cystic fibrosis individuals using aztreonam due to chronic colonization with Burkholderia SPP. Pediatric Pulmonology 2016;51 Suppl 45:332. [CFGD Register: BD223c] [Google Scholar]
- Weiser R, Smith A, Marchesi J, Rye PD, Dessen A, Hilde Myrset A, et al. Application of microbiota analysis to phase IIb trials of inhaled alginate oligosaccharide (Oligo G) in cystic fibrosis patients. Journal of Cystic Fibrosis 2017;16 Suppl 1:S79. [CFGD Register: BD223d] [Google Scholar]
Salh 1992 {published data only}
- Salh B, Bilton D, Dodd M, Abbot J, Webb K. A comparison of aztreonam and ceftazidime in the treatment of respiratory infections in adults with cystic fibrosis. Scandinavian Journal of Infectious Diseases 1992;24(2):215‐8. [CFGD Register: PI72] [DOI] [PubMed] [Google Scholar]
Sharma 2016 {published data only}
- Sharma G, Lodha R, Shastri S, Saini S, Kapil A, Singla M, et al. Zinc supplementation for one year among children with cystic fibrosis does not decrease pulmonary infection. Respiratory Care 2016;61(1):78‐84. [CFGD Register: GN256; CRS: 5500135000001517; PUBMED: 26443019] [DOI] [PubMed] [Google Scholar]
Singh 2013 {published data only}
- Singh SB, Shelton AU, Kotek K, Starner TD. A clinically‐embedded trial to evaluate the efficacy of interventions for pre‐pseudomonal pathogens [abstract]. Pediatric Pulmonology 2013;48 Suppl 36:335, Abstract no: 358. [CENTRAL: 999884; CFGD Register: PI274 ; CRS: 5500127000000006] [Google Scholar]
Smith 1997 {published data only}
- Smith A, Weber A, Pandher R, Williams‐Warren J, Cohen ML, Ramsey B. Utilization of salivary concentrations of ciprofloxacin in subjects with cystic fibrosis. Infection 1997;25(2):106‐8. [CFGD Register: PI145] [DOI] [PubMed] [Google Scholar]
Stutman 1987 {published data only}
- Shalit I, Stutman HR, Marks MI, Chartrand SA, Hilman BC. Randomized study of two dosage regimens of ciprofloxacin for treating chronic bronchopulmonary infection in patients with cystic fibrosis. American Journal of Medicine 1987;82 Suppl 4A:189‐95. [CFGD Register: PI48b] [PubMed] [Google Scholar]
- Stutman HR, Shalit I, Marks MI, Greenwood R, Chartrand SA, Hilman BC. Pharmacokinetics of two dosage regimens of ciprofloxacin during a two‐week therapeutic trial in patients with cystic fibrosis. American Journal of Medicine 1987;82 Suppl 4A:142‐5. [CFGD Register: PI48a] [PubMed] [Google Scholar]
Tullis 2014 {published data only}
- Balfour‐Lynn IM. At last, Burkholderia spp. is one of the inclusion criteria‐‐a negative (but published) randomised controlled trial. Journal of Cystic Fibrosis 2014;13(3):241‐2. [CFGD Register: PI259i] [DOI] [PubMed] [Google Scholar]
- Burns J, LiPuma JJ, Retsch‐Bogart G, Bresnik M, Henig N, McKevitt M, et al. No antibiotic cross‐resistance after 1 year of continuous aztreonam for inhalation solution (AZLI) in cystic fibrosis (CF) patients (pts) with chronic Burkholderia (BURK) infection. Journal of Cystic Fibrosis 2012;11 Suppl 1:S71. [Abstract no.: 58; CFGD Register: PI259d; ] [Google Scholar]
- Burns JL, LiPuma J, McKevitt M, Lewis S, Retsch‐Bogart GZ, Bresnik M, et al. The effect of Burkholderia colony morphology in cystic fibrosis (CF) patients with chronic Burkholderia species in a randomized trial of aztreonam for inhalation solution (AZLI). Pediatric Pulmonology 2012;47 Suppl 35:333. [Abstract no.: 307; CFGD Register: PI259g; ] [Google Scholar]
- Tullis DE, Burns JL, Retsch‐Bogart GZ, Bresnik M, Henig NR, Lewis SA, et al. Inhaled aztreonam for chronic Burkholderia infection in cystic fibrosis: a placebo‐controlled trial. Journal of Cystic Fibrosis 2014;13(3):296‐305. [CFGD Register: PI259h] [DOI] [PubMed] [Google Scholar]
- Tullis E, Burns J, Retsch‐Bogart G, Bresnik M, Henig N, Lewis S, et al. Aztreonam for inhalation solution (AZLI) in cystic fibrosis (CF) patients with chronic Burkholderia species (BURK) infection: final results from a randomized, placebo‐controlled trial. Journal of Cystic Fibrosis 2012;11 Suppl 1:S11. [Abstract no.: WS5.4; CFGD Register: PI259e; ] [Google Scholar]
- Tullis E, Burns JL, Retsch‐Bogart G, Bresnik M, Henig NR, Lewis S, et al. Aztreonam for inhalation solution (AZLI) in cystic fibrosis (CF) patients with chronic burkholderia species (Burk) infection: initial results from a randomized, placebo‐controlled trial. Pediatric Pulmonology 2011;46 Suppl 34:296. [Abstract no.: 234; CFGD Register: PI259b; ] [Google Scholar]
- Tullis E, Burns JL, Retsch‐Bogart G, Bresnik M, Lewis S, Montgomery AB, et al. Aztreonam 75mg powder and solvent for nebuliser solution (AZLI) in cystic fibrosis (CF) patients with chronic Burkholderia species (Burk) infection: baseline demographics and microbiology from randomized , placebo‐controlled trial. Journal of Cystic Fibrosis 2011;10 Suppl 1:S22. [Abstract no.: 85; CFGD Register: PI259c; ] [Google Scholar]
- Tullis E, Burns JL, Retsch‐Bogart GZ, Bresnik M, Lewis S, LiPuma J. Lung function in cystic fibrosis (CF) patients with chronic Burholderia (Burk) species infection over the course of a prospective, randomized trial of aztreonam for inhalation solution (AZLI). Pediatric Pulmonology 2012;47 Suppl 35:334. [Abstract no.: 310; CFGD Register: PI259f; ] [Google Scholar]
- Tullis E, LiPuma JJ, Retsch‐Bogart G, Bresnik M, Henig N, McKevitt M, et al. Effects of continous aztreonam for inhalation solution (AZLI) use on pathogens and antibiotic susceptibility in cystic fibrosis (CF) patients with chronic Burkholderia species infection. Pediatric Pulmonology 2011;46 Suppl 34:305. [Abstract no.: 257; CFGD Register: PI259a; ] [Google Scholar]
Uluer 2013 {published data only}
- Uluer AZ, Waltz DA, Kalish LA, Adams S, Gerard C, Ericson DA. Inhaled amiloride and tobramycin solutions fail to eradicate Burkholderia dolosa in patients with cystic fibrosis. Journal of Cystic Fibrosis 2013;12(1):54‐9. [DOI] [PubMed] [Google Scholar]
Vitti 1975 {published data only}
- Vitti TG, Berg TJ, Pagtakhan RD. The effect of pancreatic enzyme supplement on the intestinal absorption of ampicillin and cloxacillin in children with cystic fibrosis [abstract]. 16th Cystic Fibrosis Club Abstracts; 1975. 1975:56. [CFGD Register: PI81; MEDLINE: ]
Wood 1996 {published data only}
- Wood PJ, Ioannides Demos LL, Li SC, Williams TJ, Hickey B, Spicer WJ, et al. Minimisation of aminoglycoside toxicity in patients with cystic fibrosis. Thorax 1996;51(4):369‐73. [CFGD Register: PI109; MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Additional references
Ball 2010
- Ball R, Brownlee KG, Duff AJ, Denton M, Conway SP, Lee TW. Can Burkholderia cepacia complex be eradicated with nebulised amiloride and TOBI?. Journal of Cystic Fibrosis 2010;9(1):73‐4. [DOI] [PubMed] [Google Scholar]
CF Trust 2004
- Littlewood J, Dodd M, Elborn S, Geddes D, Govan J, Hart CA, et al. The Burkholderia cepacia complex. Suggestions for prevention and infection control ‐ 2nd Edition. Report of the UK Cystic Fibrosis Trust Infection Control Group September 2004. [ISBN: 0‐9540536‐9‐9]
CF Trust 2009
- UK CF Trust Antibiotic Working Group. Antibiotic treatment for cystic fibrosis ‐ 3rd Edition. Report of the UK Cystic Fibrosis Trust Antibiotic Working Group May 2009. [ISBN: 0‐9548511‐3‐7]
CF Trust 2017
- UK CF Trust. Annual Data Report 2017. UK CF Registry 2018.
Corey 1996
- Corey M, Farewell V. Determinants of Mortality from Cystic Fibrosis in Canada, 1970‐1989. American Journal of Epidemiology 1996;143(10):1007‐17. [DOI] [PubMed] [Google Scholar]
Deeks 2011
- Deeks J, Higgins J, Altman D. Chapter 9 Analysing data and undertaking meta‐analysis. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Douglas 2009
- Douglas TA, Brennan S, Gard S, Berry L, Gangell C, Stick SM, et al. Acquisition and eradication of P. aeruginosa in young children with cystic fibrosis. European Respiratory Journal 2009;33(2):305‐11. [DOI] [PubMed] [Google Scholar]
Drevinek 2010
- Drevinek P, Mahenthiralingam E. Burkholderia cenocepacia in cystic fibrosis: epidemiology and molecular mechanisms of virulence. Clinical Microbiology and Infection 2010;16(7):821‐30. [DOI] [PubMed] [Google Scholar]
Etherington 2003
- Etherington C, Peckham DG, Conway SP, Denton M. Burkholderia cepacia complex infection in adult patients with cystic fibrosis ‐ is early eradication possible?. Journal of Cystic Fibrosis 2003;2(4):220‐1. [DOI] [PubMed] [Google Scholar]
Farrell 2018
- Farrell P, Férec C, Macek M, Frischer T, Renner S, Riss K, et al. Estimating the age of p.(Phe508del) with family studies of geographically distinct European populations and the early spread of cystic fibrosis. European Journal of Human Genetics 2018;26(12):1832‐9. [DOI: 10.1038/s41431-018-0234-z] [DOI] [PMC free article] [PubMed] [Google Scholar]
Gee 2000
- Gee L, Abbott J, Conway S, Etherington C, Webb A. Development of a disease specific health related quality of life measure for adults and adolescents with cystic fibrosis. Thorax 2000;55(11):946‐54. [DOI] [PMC free article] [PubMed] [Google Scholar]
Gibson 2003
- Gibson RL, Emerson J, McNamara S, Burns JL, Rosenfeld M, Yunker A, et al. Significant microbiological effect of inhaled tobramycin in young children with cystic fibrosis. American Journal of Critical Care Medicine 2003;167(6):841‐9. [DOI] [PubMed] [Google Scholar]
Gibson 2007
- Gibson RL, Emerson J, Mayer‐Hamblett N, Burns JL, McNamara S, Accurso FJ, et al. Duration of treatment effect after tobramycin solution for inhalation in young children with cystic fibrosis. Pediatric Pulmonology 2007;42(7):610‐23. [DOI] [PubMed] [Google Scholar]
Higgins 2003
- Higgins JPT, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta‐analyses. BMJ 2003;327(7414):557‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JPT, Altman DG (editors). Chapter 8: Assessing risk of bias in included studies. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Langton Hewer 2009
- Langton Hewer SC, Smyth AR. Antibiotic strategies for eradicating Pseudomonas aeruginosa in people with cystic fibrosis. Cochrane Database of Systematic Reviews 2009, Issue 4. [DOI: 10.1002/14651858.CD004197.pub3] [DOI] [PubMed] [Google Scholar]
Lee 2003
- Lee TWR, Brownlee KG, Conway SP, Denton M, Littlewood JM. Evaluation of a new definition for chronic Pseudomonas aeruginosa infection in cystic fibrosis patients. Journal of Cystic Fibrosis 2003;2(1):29‐34. [DOI] [PubMed] [Google Scholar]
LiPuma 2010
- LiPuma JJ. The changing microbial epidemiology in cystic fibrosis. Clinical Microbiology Reviews 2010;23(2):299‐323. [DOI] [PMC free article] [PubMed] [Google Scholar]
Loveridge 2017
- Loveridge EJ, Jones C, Bull MJ, Moody SC, Kahl MW, Khan Z, et al. Reclassification of the specialized metabolite producer Pseudomonas mesoacidophila ATCC 31433 as a member of the Burkholderia cepacia complex. Journal of Bacteriology 2017;199(13):e00125‐17. [PUBMED: 28439036] [DOI] [PMC free article] [PubMed] [Google Scholar]
Mahenthiralingam 2001
- Mahenthiralingam E, Vandamme P, Campbell ME, Henry DA, Gravelle AM, Wong LTK, et al. Infection with Burkholderia cepacia complex genomovars in patients with cystic fibrosis: virulent transmissible strains of genomovar III can replace Burkholderia multivorans. Clinical Infectious Diseases 2001;33(9):1469‐75. [DOI] [PubMed] [Google Scholar]
Millar‐Jones 1998
- Millar‐Jones L, Ryley HC, Paull A, Goodchild MC. Transmission and prevalence of Burkholderia cepacia in welsh cystic fibrosis patients. Respiratory Medicine 1998;92(2):178‐83. [DOI] [PubMed] [Google Scholar]
Moore 2001
- Moore JE, Crowe M, Shaw A, McCaughan J, Redmond AO. Antibiotic resistance in Burkholderia cepacia at two regional cystic fibrosis centres in Northern Ireland: is there a need for synergy testing?. Journal of Clinical Microbiology 2001;48(2):319‐21. [DOI] [PubMed] [Google Scholar]
Moriceau 2009
- Moriceau S, Kantari C, Mocek J, Davezac N, Gabillet J, Guerrera IC, et al. Coronin‐1 is associated with neutrophil survival and is cleaved during apoptosis: potential implication in neutrophils from cystic fibrosis patients. Journal of Immunology 2009;182(11):7254‐63. [DOI] [PubMed] [Google Scholar]
Moriceau 2010
- Moriceau S, Lenoir G, Witko‐Sarsat V. In cystic fibrosis homozygotes and heterozygotes, neutrophil apoptosis is delayed and modulated by diamide or roscovitine: evidence for an innate neutrophil disturbance. Journal of Innate Immunity 2010;2(3):260‐6. [DOI] [PubMed] [Google Scholar]
Nzula 2002
- Nzula S, Vandamme P, Govan JR. Influence of taxonomic status on the in vitro antimicrobial susceptibility of the Burkholderia cepacia complex. Journal of Antimicrobial Chemotherapy 2002;50(2):265‐9. [DOI] [PubMed] [Google Scholar]
Quittner 2009
- Quittner AL, Modi AC, Wainwright C, Otto K, Kirihara J, Montgomery AB. Determination of the minimal clinically important difference scores for the Cystic Fibrosis Questionnaire‐Revised respiratory symptom scale in two populations of patients with cystic fibrosis and chronic Pseudomonas aeruginosa airway infection. Chest 2009;135(6):1610‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Ratjen 2001
- Ratjen F, Doring G, Nikolaizik WH. Effect of inhaled tobramycin on early Pseudomonas aeruginosa colonisation in patients with cystic fibrosis. Lancet 2001;358(9286):983‐4. [DOI] [PubMed] [Google Scholar]
Ratjen 2003
- Ratjen F, Doring G. Cystic Fibrosis. Lancet 2003;361(9358):681‐9. [DOI] [PubMed] [Google Scholar]
Ratjen 2006
- Ratjen F. Treatment of early Pseudomonas aeruginosa infection in patients with cystic fibrosis. Current Opinion in Pulmonary Medicine 2006;12(6):428‐32. [DOI] [PubMed] [Google Scholar]
Saiman 2004
- Saiman L. Microbiology of early CF lung disease. Paediatric Respiratory Review 2004;5 Suppl A:S367‐9. [DOI] [PubMed] [Google Scholar]
Smyth 2003
- Smyth A, Walters S. Prophylactic anti‐staphylococcal antibiotics for cystic fibrosis. Cochrane Database of Systematic Reviews 2003, Issue 3. [DOI: 10.1002/14651858.CD001912] [DOI] [PubMed] [Google Scholar]
Sokol 2000
- Sokol PA, Kooi C, Hodges RS, Cachia P, Woods DE. Immunization with a Pseudomonas aeruginosa elastase peptide reduces severity of experimental lung infections due to P. aeruginosa or Burkholderia cepacia. Journal of Infectious Diseases 2000;181(5):1682‐92. [DOI] [PubMed] [Google Scholar]
St Denis 2007
- Denis M, Ramotar K, Vandemheen K, Tullis E, Ferris W, Chan F, et al. Infection with Burkholderia cepacia complex bacteria and pulmonary exacerbations of cystic fibrosis. Chest 2007;131(4):1188‐96. [DOI] [PubMed] [Google Scholar]
Sterne 2011
- Sterne J, Egger M, Moher D. Chapter 10: Addressing reporting biases. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1 [updated March 2011]. The CochraneCollaboration, 2011. Available from www.cochrane‐handbook.org.
Taccetti 2005
- Taccetti G, Campana S, Festini F, Mascherini M, Döring G. Early eradication therapy against Pseudomonas aeruginosa in cystic fibrosis. European Respiratory Journal 2005;26(3):458‐61. [DOI] [PubMed] [Google Scholar]
Tramper‐Stranders 2010
- Tramper‐Stranders GA, Wolfs TF, Haren Noman S, Aalderen WM, Nagelkerke AF, Nuijsink M, et al. Controlled trial of cycled antibiotic prophylaxis to prevent initial Pseudomonas aeruginosa infection in children with cystic fibrosis. Thorax 2010;65(10):915‐20. [DOI] [PubMed] [Google Scholar]
Vermis 2003
- Vermis K, Vandamme PA, Nelis HJ. Burkholderia cepacia complex genomovars: utilization of carbon sources, susceptibility to antimicrobial agents and growth on selective media. Journal of Applied Microbiology 2003;95(6):1191‐9. [DOI] [PubMed] [Google Scholar]
Zlosnik 2011
- Zlosnik JEA, Costa PS, Brant R, Mori PYB, Hird TJ, Fraenkel MC, et al. Mucoid and nonmucoid Burkholderia cepacia complex bacteria in cystic fibrosis infections. American Journal of Respiratory and Critical Care Medicine 2011;183(1):67‐72. [DOI] [PubMed] [Google Scholar]
References to other published versions of this review
Regan 2012
- Regan K, Bhatt J. Eradication therapy for Burkholderia cepacia complex in people with cystic fibrosis. Cochrane Database of Systematic Reviews 2012, Issue 5. [DOI: 10.1002/14651858.CD009876] [DOI] [PMC free article] [PubMed] [Google Scholar]
Regan 2014
- Regan KH, Bhatt J. Eradication therapy for Burkholderia cepacia complex in people with cystic fibrosis. Cochrane Database of Systematic Reviews 2014, Issue 10. [DOI: 10.1002/14651858.CD009876.pub2] [DOI] [PubMed] [Google Scholar]
Regan 2016
- Regan KH, Bhatt J. Eradication therapy for Burkholderia cepacia complex in people with cystic fibrosis. Cochrane Database of Systematic Reviews 2016, Issue 11. [DOI: 10.1002/14651858.CD009876.pub3] [DOI] [PubMed] [Google Scholar]