

Abstract
Changes in protein function can lead to changes in the selection acting on specific residues. This can often be detected as evolutionary rate changes at the sites in question. A maximum-likelihood method for detecting evolutionary rate shifts at specific protein positions is presented. The method determines significance values of the rate differences to give a sound statistical foundation for the conclusions drawn from the analyses. A statistical test for detecting slowly evolving sites is also described. The methods are applied to a set of Myc proteins for the identification of both conserved sites and those with changing evolutionary rates. Those positions with conserved and changing rates are related to the structures and functions of their proteins. The results are compared with an earlier Bayesian method, thereby highlighting the advantages of the new likelihood ratio tests.
The explosive growth of available sequence data has necessitated the development of new computerized methods for the functional analysis of proteins. A number of methods have been developed for studying the functions of proteins from their sequences and protein-coding DNAs (1–3). Some of these methods estimate the ratio between nonsynonymous and synonymous rates within protein-coding genes, with ratios >1 and <1 indicating positive versus negative selection, respectively (4). Methods for performing these analyses on a site-specific level also have been developed (5). Along these lines, other methods have focused on amino acid conservation as an indication of protein function (6, 7). This approach is founded on the assumption of functional constraint (i.e., that functionally important residues and sequences are under stronger selective constraints that lower their evolutionary rates).
The concept of amino acid conservation can be taken one step further to yield insights about changes in function over time. This divergence of protein function often is revealed by a rate change in those amino acid residues of the protein that are most directly responsible for its new function (8, 9). To investigate this change in evolution, a likelihood ratio test (LRT) is developed for detecting significant rate shifts at specific sites in proteins.
Such rate changes at a site over evolutionary time trace back to the covarion model of Fitch and Markowitz (10). In this model, the state of a site can change between variable and invariable. Such changes can also occur anywhere in the evolutionary tree relating the sequences under analysis. Furthermore, as the acronym implies (concomitantly variable codons), these rate shifts are tied to sites whose evolution is correlated and is not independent (11). The LRT method assumes that changes occur at a specific point in evolution and that these changes are independent. Here, change is not limited to variable versus invariable, but involves shifts between any two rates. For this reason, we are not dealing with a true covarion model (12, 13). Thus, a site showing a significant rate change will from here on be called a rate shift site, rather than a covarion site.
The reason for focusing on a specific evolutionary point is that gene duplications can create opportunities for functional divergence as one copy of the gene can divergently evolve, whereas the other fulfills the original function (2, 7). Other points in gene evolution where functional change is most likely reflect speciation events that lead to the origins of new major groups [e.g., ciliates versus other eukaryotes in the divergence of their elongation factors (14)].
A slow evolutionary rate at a given site would indicate that this position is functionally important for the protein. Conversely, a high evolutionary rate would indicate that the position is not involved in an important protein function. A significant rate difference between two subfamilies at a given site would thereby mean that the function of this position is probably different in the two groups.
Some work has been done in this area before (2, 8, 15). The approach developed here is unique in that it uses an LRT to determine the significance of the rate differences at specific positions. A test is also developed for deciding whether a given site is evolving slower than the average for the entire protein being analyzed.
Tests for detecting whether two subfamilies have undergone functional divergence have been developed before (15) and will not be the focus of this work. Instead, it is assumed that the subfamilies are known to be functionally divergent, either from biochemical knowledge or previous statistical tests. This work aims to pinpoint the protein positions responsible for this divergence.
The methods are illustrated with a set of Myc proteins and the biochemical significance of these results is discussed. The results are compared with those using the Bayesian method of Gu (15), which calculates the posterior probability of a rate shift. The reasons for the differences between the two approaches are explained.
The Model
The LRT is used as the basis for detecting rate shift sites. The basic idea behind this test, as used in an evolutionary context, was reviewed by Huelsenbeck and Rannala (16).
Position-Specific Rate Shift Test.
To test whether a site from two related groups of sequences is evolving differently, the positions are analyzed individually. An outline of the method is shown in Fig. 1 Left and Center.
Figure 1.

Assume that a gene duplication has resulted in two protein subfamilies. The first consists of sequences Seq1a and Seq1b, whereas the second includes sequences Seq2a, Seq2b, and Seq2c. (Left) H0, where the rates for a site may differ from one protein subfamily to the other. This rate divergence occurs at the root of the tree, where the duplication event occurred. (Center) The situation under H1. The evolutionary rate for a site remains the same throughout the entire tree. If H1 is rejected, rate shift behavior is present at the position under inspection. If H1 is retained, then one can test whether the rate for this site is equal to the average for both proteins. (Right) The testing of this hypothesis (H2). If H2 is rejected, the evolutionary rate for the site is significantly different from the average for all positions.
The test used is as follows. The null hypothesis, H0, states that a given position evolves with different rates in the two sequence subfamilies. The likelihood under this model is calculated by using the method of Felsenstein (17). The rate matrix used is the JTT matrix of Jones et al. (18). The two rates of evolution are varied to obtain the maximum-likelihood (ML) value under this model, L0.
In contrast, hypothesis one (H1) states that the position evolves at the same rate in the two subfamilies. Again, calculations are done according to Felsenstein (17) and with the JTT matrix, but with a single rate used for the two subfamilies. The optimal rate is found, giving the ML value under this model, L1.
Using an LRT statistic, we can evaluate H1. The test statistic can be written as:
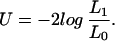 |
Because H1 is a special case of H0 (the hypotheses are nested), the likelihoods will always obey the relationship that L1 ≤ L0. This means that U will never be negative.
There are two degrees of freedom under H0, whereas there is only one under H1. This could indicate that under H1, the distribution of U is approximately χ2 with one degree of freedom, here denoted χ2(1). To investigate how close the distribution of U is to χ2(1), a number of simulations were conducted (Fig. 2). The simulated distributions were quite close to the χ2(1) distribution, so a χ2(1) test can be used with some caution.
Figure 2.

The χ2 distribution with one degree of freedom (smooth curve) compared with a simulation study of U (ragged curve). The simulations consisted of 1,000 samples generated under H1, with rates drawn from a gamma distribution. The calculations are based on the phylogeny and ML conditions used in the Myc protein example. The distribution of U approximately follows that of the χ2(1) statistic, especially in the upper part. Other follow-up simulations indicate that this distribution generally conforms more closely to the χ2(1) curve as the two subfamilies increase, both in terms of their branch lengths and numbers of sequences (Figs. 4–7, which are published as supporting information on the PNAS web site).
Unknown and partially known amino acids are treated as described by Felsenstein (17). This means that unknown amino acids have the effect of pruning the tree to remove the sequences containing them. Gaps are treated like unknown amino acids. This means that all columns in the alignment can be used in the analysis, even though some sequences are unknown or gapped in that region. For moderate numbers of sequences with a gap at a specific position, the test statistic is not influenced much, because this corresponds to using a smaller tree (which would ideally have the same distribution of U).
Advantages and Disadvantages of the Method.
The LRT method has the advantage that it is simple and direct. It answers exactly the question of interest: Does a given position evolve at different rates in different protein subfamilies? The Bayesian method is indirect, because it only uses the JTT matrix to count the expected number of replacements within each subfamily, before comparing these counts (15). The problem is that some replacements are rare (e.g., lysine to cysteine), whereas others are more common (e.g., valine to isoleucine). The JTT rate matrix is fully incorporated in the likelihood calculations presented here to accommodate this fact.
Another advantage of this method, compared with some earlier ones (e.g., ref. 8), is that it acknowledges that the subfamilies are related to each other and are not independent. To illustrate this, consider a given position in the sequences of Fig. 1. Assume that Seq1a and Seq1b have an isoleucine and a leucine, respectively, at this position, whereas Seq2a, Seq2b, and Seq2c have alanines at this site. We know that at least two replacements have occurred. The ML estimations of the individual rates for the two subfamilies give a slow rate to subfamily 2, because there is no direct evidence of a replacement there. Subfamily 1, on the other hand, requires one replacement, and its rate of evolution is estimated to be fast. This means that the model will tend to assume that both of the replacements occurred in subfamily 1. This gives a more significant difference than methods that do not take the relationship between the two subfamilies into account, because they only use the single replacement. Here, then, this hypothetical site would be significant according to our test with the two subfamilies considered together (U = 4.07, P ≈ 0.044), but barely insignificant if the two were analyzed separately (U = 3.17, P ≈ 0.075).
The obvious next question is: Which significance value should be used in these tests? Often a value of P = 0.05 is chosen. The problem here is that multiple tests are being performed. For an alignment of length l, this means that ≈0.05l sites will be significant just by chance when P = 0.05 is used. To correct for this multiple testing, a stricter P value should be chosen, depending on the number of sequences under analysis. For small data sets with relatively few sequences, power is low, so a very strict significance level will yield few results.
Taking all of this into account, we recommend that 0.05l be used to estimate the expected number of sites with P ≤ 0.05 by chance alone. This expectation can then be compared with the observed number of such sites to assess the number of positions with significantly different rates. Those sites with very significant rate changes may stand out among the others. In turn, the entire set of potentially significant sites can be further evaluated for their importance against independent structural and functional data for their proteins (8, 19). A combined approach that uses both perspectives is illustrated below for Myc proteins.
LRT for Conserved Sites.
As outlined in Fig. 1, one can also test whether the rate for a site is different from the average for all protein positions. Such a test is done if H1 is accepted (i.e., both subfamilies have the same rate). The test is done exactly like the rate shift test described above. The test statistic, U, again approximates a χ2(1) distribution according to simulations under this hypothesis (H2) (Fig. 8, which is published as supporting information on the PNAS web site, www.pnas.org). In many cases, the most interesting sites will be those that have a significantly slower rate than the average, because these positions are most likely to be those under the strongest selective constraints and of greatest functional importance.
Analysis of a Set of Myc Sequences
To illustrate the utility of these methods, a set of 38 proteins for c-Myc, N-Myc, and L-Myc (27, seven, and four sequences, respectively) was analyzed for sites with rate shifts and slower rates. These protein sequences included all of those used by Miyamoto and Freire (20), except for those of the intron-less retrogenes, viruses, and nonvertebrates. In addition, these 38 Myc sequences included the five new ones for eutherian mammals reported by Miyamoto et al. (21).
The alignment of the 38 Myc proteins was based on the conserved regions used by Miyamoto and Freire (20). The areas between their conserved regions (including the common boundary between exons 2 and 3) were aligned by using clustal w (22). The final length of the alignment was 583 positions, of which 285 had no gaps. In turn, 440 aligned positions did not have a gap in at least one sequence in both the c-Myc and N-Myc subfamilies. Thus, 440 positions were considered in our analysis of rate changes among sites (see below). All of the position numbers discussed in the following are relative to human c-Myc (23).
The phylogenetic tree used was that of Miyamoto and Freire (20), except that the interordinal relationships of eutherian mammals were fixed according to recent phylogenetic syntheses of both their molecular and morphological data (24–26). The branch lengths of this final phylogeny were optimized by ML using the JTT matrix and gamma distribution for rate heterogeneity among sites (27). The two protein subfamilies compared in our example were c-Myc and N-Myc, whereas L-Myc was used as their outgroup.
The final set of Myc sequences (with accession numbers), multiple sequence alignment, and phylogenetic tree are shown in Table 3, Fig. 9, and Fig. 10, respectively, which are published as supporting information on the PNAS web site.
Results and Interpretation.
The c-myc, N-myc, and L-myc genes encode transcription factors that are important in the regulation of cell proliferation and differentiation (23, 28, 29). The c-myc gene is expressed in many tissues and developmental stages, whereas the expression of both N-myc and L-myc is reduced spatially and temporally. Mutations in these genes have been implicated in many human cancers (30). The proteins of all three genes can be divided into three primary regions: (i) the N-terminal domain (positions 1–144 of human c-Myc); (ii) the central region (positions 145–354); and (iii) the basic helix–loop–helix leucine zipper (bHLHZip) (positions 355–439) (Fig. 3). The N-terminal domain is essential for transcriptional regulation through both transactivation and repression, whereas the bHLHZip is critical for specific DNA binding. The central region includes sites for nonspecific DNA binding, nuclear localization, and additional phosphorylation.
Figure 3.

Summary of results for the 38 Myc proteins, as represented by the c-Myc and N-Myc sequences for human (Homo sapiens), chicken (Gallus gallus), and frog (Xenopus laevis). The full alignment for all 38 Myc sequences is provided in Fig. 9, which is published as supporting information on the PNAS web site. Sites with both blue and red highlighting correspond to those with significant rate differences between the two subfamilies. In these cases, the blue and red distinguish the subfamily with the slower rate from the one with the faster rate, respectively. In turn, sites that are entirely blue or red highlight those with the same rate in the two subfamilies, but with significantly slower or faster rates than the average for all positions, respectively. In all cases, significance refers to the 5% level. Key structural and functional regions of the Myc proteins are labeled above and below the multiple sequence alignment (23, 28, 29). NLS, nuclear localization signal.
Ninety one sites in our evolutionary analyses were defined by rates that were the same in c-Myc and N-Myc, but that were slower than the average for both proteins (Fig. 3). These positions map to different boxes and regions that are of known functional importance to Myc proteins (e.g., Myc boxes 1 and 2 that are critical for the modulation and integration of transcriptional regulation and for transcriptional repression, respectively) (28, 29). Furthermore, these 91 sites with slower rates are not randomly distributed across the three primary regions of Myc proteins (Table 1). This nonrandom pattern identifies the N-terminal domain and bHLHZip as conserved relative to the more variable central region. This greater conservation for the N-terminal domain and bHLHZip is not surprising, given that the primary functions of Myc proteins (transcriptional regulation and specific DNA binding) depend on these two regions.
Table 1.
Distributions of sites with significant rate shifts and equal, but significantly slower rates among the three primary regions of Myc proteins (Fig. 3)
| Myc region | Rate shift sites
|
Equal, but
slow rates
|
||||
|---|---|---|---|---|---|---|
| Significant sites | Other sites | Totals | Significant sites | Other sites | Totals | |
| N-terminal domain | 22 (15.0) | 113 (120.0) | 135 | 36 (26.3) | 77 (86.7) | 113 |
| Central region | 24 (24.7) | 198 (197.3) | 222 | 30 (46.1) | 168 (151.9) | 198 |
| bHLHZip | 3 (9.2) | 80 (73.8) | 83 | 25 (18.6) | 55 (61.4) | 80 |
| Totals | 49 | 391 | 440 | 91 | 300 | 391 |
These summaries are for the 440 positions that could show rate changes between c-Myc and N-Myc. The chi-square test for rate shift sites is significant at the level of 1.5% (X2 = 8.4). The chi-square test for equal, but slow, rates is also significant (X2 = 14.8, P = 0.001%). Expected counts are given in parentheses.
Our LRTs identify 49 sites with significant rate differences at the level of 5% (Table 2). Because the alignment has 440 positions that could show rate shifts, ≈22 such sites are expected by chance alone (440 × 0.05). This indicates that there are ≈27 more sites with significant rate differences than expected. At the 1% level, there are 16 sites with significant rate changes, which again is more than expected by chance (4 or 440 × 0.01). This illustrates the value of using significance levels that are easy to interpret.
Table 2.
The 49 positions with significant rate shifts between the c-Myc and N-Myc subfamilies
| Rank | Position in human c-Myc | Slower subfamily | Significance | Bayesian rank | Rank | Position in human c-Myc | Slower subfamily | Significance | Bayesian rank | Rank | Position in human c-Myc | Slower subfamily | Significance | Bayesian rank |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 94 | c-Myc | 0.000040 | — | 18 | 284 | N-Myc | 0.010 | 4 | 35 | 146 | c-Myc | 0.027 | 11 |
| 2 | 221 | c-Myc | 0.00054 | — | 19 | 230 | c-Myc | 0.012 | 12 | 36 | 285 | N-Myc | 0.028 | 16 |
| 3 | 96 | c-Myc | 0.00062 | — | 20 | 157 | c-Myc | 0.012 | — | 37 | 153 | c-Myc | 0.031 | — |
| 4 | 272 | N-Myc | 0.00081 | — | 21 | 117 | c-Myc | 0.013 | — | 38 | 408 | N-Myc | 0.031 | 15 |
| 5 | 113 | c-Myc | 0.00085 | 2 | 22 | 283 | N-Myc | 0.014 | 6 | 39 | 121 | c-Myc | 0.035 | 13 |
| 6 | 68 | c-Myc | 0.0020 | — | 23 | 111 | c-Myc | 0.014 | 39 | 40 | 150 | c-Myc | 0.035 | 41 |
| 7 | 99 | c-Myc | 0.0025 | 1 | 24 | 277 | N-Myc | 0.015 | 3 | 41 | 83 | c-Myc | 0.036 | — |
| 8 | 114 | c-Myc | 0.0030 | — | 25 | 154 | c-Myc | 0.015 | 80 | 42 | 73 | c-Myc | 0.037 | — |
| 9 | 286 | N-Myc | 0.0040 | — | 26 | 293 | c-Myc | 0.017 | 10 | 43 | 282 | N-Myc | 0.037 | 24 |
| 10 | 66 | c-Myc | 0.0044 | — | 27 | 222 | c-Myc | 0.017 | — | 44 | 340 | c-Myc | 0.040 | 17 |
| 11 | 178 | c-Myc | 0.0071 | — | 28 | 301 | c-Myc | 0.017 | 9 | 45 | 404 | N-Myc | 0.041 | 21 |
| 12 | 273 | N-Myc | 0.0072 | — | 29 | 100 | c-Myc | 0.018 | 19 | 46 | 281 | N-Myc | 0.043 | 8 |
| 13 | 75 | c-Myc | 0.0077 | — | 30 | 122 | c-Myc | 0.022 | 14 | 47 | 414 | N-Myc | 0.048 | 5 |
| 14 | 69 | c-Myc | 0.0082 | — | 31 | 214 | N-Myc | 0.023 | — | 48 | 67 | c-Myc | 0.049 | — |
| 15 | 274 | N-Myc | 0.0084 | 30 | 32 | 109 | c-Myc | 0.024 | 18 | 49 | 93 | c-Myc | 0.050 | — |
| 16 | 116 | c-Myc | 0.0091 | — | 33 | 314 | c-Myc | 0.026 | 47 | |||||
| 17 | 70 | c-Myc | 0.0099 | — | 34 | 115 | c-Myc | 0.027 | — |
These 49 positions are ranked according to their P values. At a significance level of 0.05, approximately 22 significant sites are expected by chance. Thus, approximately 22 of these sites may be random occurrences. Bayesian rank refers to the results from the Bayesian analysis of these Myc sequences (15). As this method cannot accommodate sites with any gaps or unknown positions, several sites in our analysis (marked by dashes) were excluded by the former.
These 49 sites are not randomly distributed across the three primary regions of c-Myc and N-Myc (Table 1). Rather, there are relatively too many and too few sites with significant rate changes in the N-terminal domain versus bHLHZip, respectively (Fig. 3). These results agree with those of Dermitzakis and Clark (31), who showed that the transactivation domains (but not the DNA binding regions) of different transcription factors from the MyoD and Mef2 gene families were characterized by variable rates between duplicate genes. Thus, these results are consistent with their hypothesis that the domains for transcriptional regulation may be more important for the functional differences among transcription factors than their DNA binding regions.
Furthermore, these 49 sites pinpoint more specific boxes and other regions, as of greatest potential importance for the known functional differences between c-Myc and N-Myc. For example, Prendergast (28) hypothesized that positions 107–130 may underlie the functional differences in transactivation and transformation that distinguish c-Myc from N-Myc. Our results identify nine sites with significant rate differences that map to this region (Fig. 3). These nine sites can now serve as specific targets in experiments with site directed mutagenesis for their effects on transactivation and transformation (32).
Comparison to Earlier Work.
The ranking of sites by their significance values differs from that derived from the Bayesian method (15) (Table 2). This is primarily because all replacements are effectively equally weighted in this method. Even though the JTT matrix is used to infer expected numbers of replacements, all replacements are treated equally thereafter. Any method based on comparisons of replacement counts will suffer from this problem. It is not only the number of replacements, but also the nature of those replacements that is important in estimating the significance of an observation.
To illustrate this point, consider position 414 (Fig. 3). It has leucine in all N-Myc sequences, whereas the c-Myc sequences have isoleucine, leucine, threonine, and valine. The latter four amino acids can quickly change between each other, as indicated in the JTT matrix. This means that a relatively slow evolutionary rate can explain the variation at this position in c-Myc. This reason is why this site has a low rank (47 overall and 25 among ungapped sites), compared with the Bayesian method (five among ungapped positions) (Table 2). The latter considers the high number of replacements, but fails to acknowledge that these changes are all very common ones.
Another distinction between our LRT and the Bayesian method (15) is that ours does not assume anything about the distribution of rates across sites. Our distribution-free approach stands in contrast to the latter's reliance on the gamma distribution for the accommodation of rate heterogeneity among sites. Our approach also addresses a different question than the one asked by the Bayesian method. In our approach, the question is: Are the rates for a site the same in two subfamilies (Fig. 1)? In the alternative method, the question is instead: Are the rates for a site independent between two subfamilies? This latter distinction becomes particularly important, when the rates for a site are both fast but different in two subfamilies. Here, our approach is more likely to identify this site as significant, because it tests for rate differences, rather than rate correlations.
The above analysis of rate shift sites by the Bayesian method is based on the fast approximate procedure that is now available in the diverge (version 1.04) computer program (ref. 15; http://xgu1.zool.iastate.edu/doc.html). Recently, Gu (2) presented a full Bayesian alternative for such analyses under the JTT model, thereby correcting for the differences in replacement rates among amino acids. Currently, a finished computer program for general distribution is not available for this alternative, although one is expected soon (X. Gu, personal communication). Furthermore, this alternative still differs from our LRT in its dependence on the gamma distribution to model rate heterogeneity among sites and in its testing of rate correlations, rather than rate differences. It also relies on an indirect procedure for its likelihood calculations of the whole tree, whereby these determinations are made for two extreme lengths of the internal branch that connect its two subtrees. These separate calculations are then linearly combined to obtain the final likelihood of the whole tree. In the Appendix, we present a direct procedure for the calculation of this likelihood.
Power Analysis.
The power of the LRT for rate shift sites was examined with evolutionary simulations using the Myc phylogenetic tree (Fig. 10, which is published as supporting information on the PNAS web site). When the same rates were used at each site between the c-Myc and N-Myc subfamilies, 3.9% of the positions (of 1,000) were significant at the level of 5%. This number should ideally be 5%, but the χ2 distribution of the test statistic is not exact as shown in Fig. 2. When the N-Myc rate at each position was doubled, but halved in c-Myc, for 500 sites, then vice versa for 500 additional sites, the percentage of significant positions of 1,000 increased to 10.4% for this rate ratio of four. When the rate ratio was then increased in this fashion to 16, 34% of the 1,000 sites were now significant. These power analyses indicate that quite high rate ratios are needed to detect rate shift sites between c-Myc and N-Myc. Because of the limited power of the test, it is particularly important to use as many sequences as possible for each subfamily. Furthermore, when using few sequences, evolutionary simulations are recommended, instead of the χ2 approximation, for determining significance levels.
Phylogenetic Errors.
To examine the effects of phylogenetic error on the detection of rate shift sites, the LRTs for the Myc sequences were repeated by using five additional phylogenies. The first two phylogenies were obtained from the neighbor-joining and protein parsimony analyses of the Myc sequences (33), whereas the next two were produced by rerooting the accepted tree at the basal nodes of the c-Myc and N-Myc subfamilies, respectively (20, 24–26). The fifth tree was generated by randomly rearranging the sequences within each subfamily of the accepted phylogeny.
The first two trees were relatively similar topologically to the accepted phylogeny, as they differed from the latter by symmetric differences of 20 and 21, respectively (33). In turn, the two rerooted trees varied from the accepted phylogeny only by their minimized versus maximized basal branches for the c-Myc versus N-Myc subfamilies (and vice versa), respectively. Forty one to 57 sites were significant according to these four alternatives, with 38 to 46 of these positions overlapping with the 49 for the accepted phylogeny (Tables 4 and 5, which are published as supporting information on the PNAS web site). These results indicate that the LRT for rate shift sites is relatively insensitive to rearrangements within the gene tree.
In contrast, the “random” alternative was quite different from the accepted phylogeny, as it varied from the latter by a symmetric difference of 60. One hundred and sixteen sites were significant according to this random alternative, with 41 of these positions overlapping with the 49 for the accepted phylogeny (Tables 4 and 5). These 116 sites document an increase in the frequency of false positives as valid groups are fragmented and additional parallel and back replacements are introduced into one subfamily versus another. This situation becomes most acute when one subfamily is varied for a site, but another is not. In this case, the addition of parallel and back replacements in the first subfamily exaggerates its rate for the site relative to that of the second. Correspondingly, the chance of a significant rate difference between them (i.e., a false positive) becomes exaggerated, too.
Future Directions
A direct statistical test for rate shift sites is presented. It takes the known replacement pattern of amino acids into account through a suitable rate matrix and provides significance values that are easy to interpret. The method is shown to perform well on a protein family that has been studied before for its rate shift positions (15). These comparisons now await further analyses of this protein family with a new ML method (2).
One interesting area of future research is to study the heterogeneity of amino acid frequencies between subfamilies, in addition to their rate shift sites (15). This can be done both on a position-specific level and the whole sequence level. Such investigations would complement the use of rate changes to identify sites of potential functional significance (2).
To compensate for the limited power of the LRT, one can analyze groups of sites rather than individual positions. These groups should be defined a priori according to the structural and functional properties of the protein (e.g., the bHLHZip of Myc). By analyzing positions together, one can increase the power of the test, but at the cost of site specificity. Furthermore, the χ2 method for testing significance becomes questionable in this case, as the small deviations at each site of the group will lead to a large overall departure from this idealized distribution. Thus, when sites are grouped, evolutionary simulations will provide a superior test of significance. Finally, by considering the entire protein as the group, one can test for rate shifts at the whole sequence level in a manner that is analogous to the θ coefficient in the Bayesian method (2, 15).
Availability of Computer Programs
The programs of this study are available at www.daimi.au.dk/∼compbio/rateshift. These programs can analyze both protein and nucleic acid sequences for rate shift sites and conserved positions.
Supplementary Material
Acknowledgments
We thank X. Gu, M. R. Tennant, and an anonymous reviewer for their comments about our research and X. Gu for the use of his program. This research was supported by funds from the Hede Nielsen Family Foundation to B.K. and by the assistance of the Department of Zoology, University of Florida.
Abbreviations
- LRT
likelihood ratio test
- ML
maximum likelihood
- bHLHZip
basic helix–loop–helix leucine zipper
A Bayesian Approach for the Identification of Rate Shift Sites
If the rates among sites are assumed or known to follow some distribution, e.g., a gamma distribution, this information can be used as a prior in a Bayesian analysis of rate shift positions.
The whole tree is designated T, whereas the subtrees for the two subfamilies under investigation are denoted T1 and T2, respectively. Note that T1 and T2 include the branches that connect their most recent common ancestors to the root of the whole tree. Thus, T can be formed directly by joining T1 and T2. The inclusion of these basal branches with their subtrees eliminates the need for separate likelihood calculations, as in the whole tree procedure of the new Bayesian method (2). For a given site, let X then denote the amino acid configuration for all sequences, whereas X1 and X2 represent the configurations in the two respective subfamilies.
We can calculate the probability of the data, P0(X), given that the rates for a site are independent between the two subfamilies. Here, λ1 ⊥ λ2 is used to symbolize that the two rates are independent, with φ referring to their prior distributions. In the equations below, x represents the amino acids at the root of the whole tree:
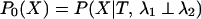 |
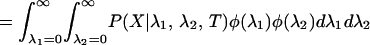 |
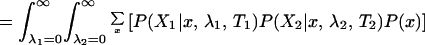 |
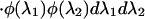 |
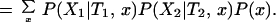 |
Notice that no two-dimensional integration is necessary. The integrals can be computed numerically.
We can also calculate the probability of the data, P1(X), given that the two rates are equal.
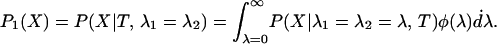 |
The two hypotheses can now be compared by comparing their two probabilities. This can be expressed as the posterior probability that the rates are independent at the site under investigation.
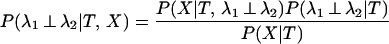 |
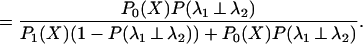 |
A prior probability for the rates being independent, P(λ1 ⊥ λ2), is needed. This probability can be estimated as by Gu (15), who uses θ (the coefficient of functional divergence) as the prior. Using this, we can obtain the probability that the rates at a given site are independent between the two subfamilies.
References
- 1.Bork P, Koonin E V. Nat Genet. 1998;18:313–318. doi: 10.1038/ng0498-313. [DOI] [PubMed] [Google Scholar]
- 2.Gu X. Mol Biol Evol. 2001;18:453–464. doi: 10.1093/oxfordjournals.molbev.a003824. [DOI] [PubMed] [Google Scholar]
- 3.Thornton J M. Science. 2001;292:2095–2097. doi: 10.1126/science.292.5524.2095. [DOI] [PubMed] [Google Scholar]
- 4.Yang Z, Bielawski J P. Trends Ecol Evol. 2000;15:496–503. doi: 10.1016/S0169-5347(00)01994-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Suzuki Y, Gojobori T, Nei M. Bioinformatics. 2001;17:660–661. doi: 10.1093/bioinformatics/17.7.660. [DOI] [PubMed] [Google Scholar]
- 6.Kimura M. The Neutral Theory of Molecular Evolution. Cambridge, U.K.: Cambridge Univ. Press; 1983. [Google Scholar]
- 7.Graur D, Li W-H. Fundamentals of Molecular Evolution. 2nd Ed. Sunderland, MA: Sinauer; 2000. [Google Scholar]
- 8.Gaucher E A, Miyamoto M M, Benner S A. Proc Natl Acad Sci USA. 2001;98:548–552. doi: 10.1073/pnas.98.2.548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang Y, Gu X. Genetics. 2001;158:1311–1320. doi: 10.1093/genetics/158.3.1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fitch W M, Markowitz E. Biochem Genet. 1970;4:579–593. doi: 10.1007/BF00486096. [DOI] [PubMed] [Google Scholar]
- 11.Pollock D D, Taylor W R, Goldman N. J Mol Biol. 1999;287:187–198. doi: 10.1006/jmbi.1998.2601. [DOI] [PubMed] [Google Scholar]
- 12.Tuffley C, Steel M. Math Biosci. 1998;147:63–91. doi: 10.1016/s0025-5564(97)00081-3. [DOI] [PubMed] [Google Scholar]
- 13.Galtier N. Mol Biol Evol. 2001;18:866–873. doi: 10.1093/oxfordjournals.molbev.a003868. [DOI] [PubMed] [Google Scholar]
- 14.Moreira D, Le Guyader H, Philippe H. Mol Biol Evol. 1999;16:234–245. doi: 10.1093/oxfordjournals.molbev.a026105. [DOI] [PubMed] [Google Scholar]
- 15.Gu X. Mol Biol Evol. 1999;16:1664–1674. doi: 10.1093/oxfordjournals.molbev.a026080. [DOI] [PubMed] [Google Scholar]
- 16.Huelsenbeck J P, Rannala B. Science. 1997;276:227–232. doi: 10.1126/science.276.5310.227. [DOI] [PubMed] [Google Scholar]
- 17.Felsenstein J. J Mol Evol. 1981;17:368–376. doi: 10.1007/BF01734359. [DOI] [PubMed] [Google Scholar]
- 18.Jones D T, Taylor W R, Thornton J M. Comput Appl Biosci. 1992;8:275–282. doi: 10.1093/bioinformatics/8.3.275. [DOI] [PubMed] [Google Scholar]
- 19.Landgraf R, Xenarios I, Eisenberg D. J Mol Biol. 2001;307:1487–1502. doi: 10.1006/jmbi.2001.4540. [DOI] [PubMed] [Google Scholar]
- 20.Miyamoto M M, Freire N P. Mol Phylogenet Evol. 2000;16:475–481. doi: 10.1006/mpev.2000.0783. [DOI] [PubMed] [Google Scholar]
- 21.Miyamoto M M, Porter C A, Goodman M. Syst Biol. 2000;49:501–514. doi: 10.1080/10635159950127367. [DOI] [PubMed] [Google Scholar]
- 22.Thompson J D, Higgins D G, Gibson T J. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hesketh R. The Oncogene and Tumor Supressor Gene Factsbook. 2nd Ed. San Diego: Academic; 1997. [Google Scholar]
- 24.Liu F-G R, Miyamoto M M, Freire N P, Ong P Q, Tennant M R, Young T S, Gugel K F. Science. 2001;291:1786–1789. doi: 10.1126/science.1056346. [DOI] [PubMed] [Google Scholar]
- 25.Madsen O, Scally M, Douady C J, Kao D J, DeBry R W, Adkins R, Amrine H M, Stanhope M J, de Jong W W, Springer M S. Nature (London) 2001;409:610–614. doi: 10.1038/35054544. [DOI] [PubMed] [Google Scholar]
- 26.Murphy W J, Eizirik E, Johnson W E, Zhang Y P, Ryder O A, O'Brien S J. Nature (London) 2001;409:614–618. doi: 10.1038/35054550. [DOI] [PubMed] [Google Scholar]
- 27.Yang Z. Comput Appl Biosci. 1997;13:555–556. doi: 10.1093/bioinformatics/13.5.555. [DOI] [PubMed] [Google Scholar]
- 28.Prendergast G C. In: Oncogenes as Transcriptional Regulators: Volume 1, Retroviral Oncogenes. Yaniv M, Ghysdael J, editors. Basel: Birkhäuser; 1997. pp. 1–28. [Google Scholar]
- 29.Facchini L M, Penn L Z. FASEB J. 1998;12:633–651. [PubMed] [Google Scholar]
- 30.Nesbit C E, Tersak J M, Prochownik E V. Oncogene. 1999;18:3004–3016. doi: 10.1038/sj.onc.1202746. [DOI] [PubMed] [Google Scholar]
- 31.Dermitzakis E T, Clark A G. Mol Biol Evol. 2001;18:557–562. doi: 10.1093/oxfordjournals.molbev.a003835. [DOI] [PubMed] [Google Scholar]
- 32.Golding G B, Dean A M. Mol Biol Evol. 1998;15:355–369. doi: 10.1093/oxfordjournals.molbev.a025932. [DOI] [PubMed] [Google Scholar]
- 33.Swofford D L. PAUP*: Phylogenetic Analysis Using Parsimony (*and Other Methods) Sunderland, MA: Sinauer; 1998. , Version 4.0. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.