

Abstract
Human prolyl hydroxylases are involved in the modification of transcription factors, procollagen, and ribosomal proteins, and are current medicinal chemistry targets. To date, there are few reports on inhibitors selective for the different types of prolyl hydroxylases. We report a structurally informed template‐based strategy for the development of inhibitors selective for the human ribosomal prolyl hydroxylase OGFOD1. These inhibitors did not target the other human oxygenases tested, including the structurally similar hypoxia‐inducible transcription factor prolyl hydroxylase, PHD2.
Keywords: barbiturate, epigenetics, histone demethylases, inhibitors, medicinal chemistry, OGFOD1, 2-oxoglutarate oxygenase
Introduction
In humans and other animals, prolyl hydroxylases (PHs) play critical roles in collagen biosynthesis and hypoxia sensing.1, 2, 3 The PHs are FeII and 2‐oxoglutarate (2OG) dependent oxygenases, which normally produce succinate and CO2 as coproducts.4 The procollagen PHs (CP3H and CP4 H) hydroxylate either C3 or C4 of prolyl residues; the latter is essential for maintenance of the collagen triple helix secondary structure (Figure 1 A).5, 6 In humans, the prolyl hydroxylase domain enzymes (PHD1, 2, and 3) act as oxygen sensors in the chronic response to hypoxia by catalyzing oxygen‐limited hydroxylation of prolyl residues in the hypoxia‐inducible factor‐α (HIFα) subunits of HIF transcription factors, leading to HIFα degradation in aerobic conditions (Figure 1 A).1
Figure 1.
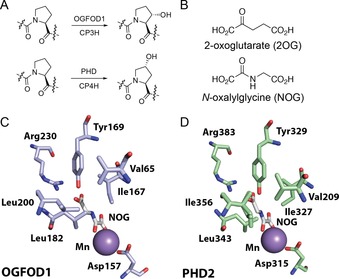
Prolyl hydroxylase reactions and structures: (A) Regio‐ and stereoselectivity of hydroxylations catalyzed by different types of prolyl hydroxylases. Each hydroxylation is coupled to the conversion of 2‐oxoglutarate (2OG) and O2 into succinate and CO2. OGFOD1 acts on a ribosomal protein, the CPHs act on procollagen, and the PHDs act on the hypoxia inducible factor (HIF) transcription factors.5 (B) Structures of 2OG and the 2OG analogue N‐oxalylglycine (NOG). (C, D) Views from the crystallographically observed active sites of OGFOD1 (PDB 4NHX)10 and PHD2 (PDB 5L9R)15 showing the interactions between active site residues, the bound metal [MnII substituting for FeII], and NOG.
The human 2OG oxygenase OGFOD1 has been recently shown to hydroxylate Pro‐62 of the ribosomal protein RPS23.7, 8, 9, 10 Pro‐62RPS23 is situated in the ribosomal decoding site, which is responsible for ensuring the fidelity of mRNA codon recognition by tRNA and release factor proteins during protein synthesis.11, 12 While the role of this hydroxylation in human and animal cells is not yet understood, the Saccharomyces cerevisiae OGFOD1 homologue Tpa1p is proposed to catalyze dihydroxylation of the corresponding prolyl residue, and to regulate translational accuracy in an mRNA sequence context‐dependent manner.8, 13, 14
Of the ∼60–70 human 2OG oxygenases, some are current medicinal chemistry targets, including enzymes involved in chromatin modification and lipid metabolism.5, 16 Inhibition of the procollagen hydroxylases is under consideration as a target to limit the overproduction of collagen associated with certain cancers and fibrotic diseases.17 The PHDs are presently being targeted for the treatment of hypoxia‐related diseases, with inhibitors in late‐stage clinical trials for anaemia.18 If OGFOD1 is indeed involved in mRNA codon recognition, as suggested based on studies of yeast homologues,8, 13 small‐molecule‐mediated inhibition of ribosomal hydroxylation could prove useful for the treatment of diseases such as muscular dystrophy that are caused by premature stop‐codons through nonsense suppression.19 However, due to the uncertainty regarding the specific roles of OGFOD1 and OGFOD1‐catalysed hydroxylation in animals, it is unclear how exactly its inhibition might manifest. Thus, such OGFOD1 inhibitors are also of interest as chemical probes to decipher the biological role of RPS23 hydroxylation, as well as those of other recently reported ribosome‐associated hydroxylations.20, 21, 22, 23
As the 2OG oxygenases are involved in diverse biological processes, developing inhibitors selective for particular oxygenases is an important therapeutic consideration. The available biophysical evidence, principally from crystallography, implies that key features in the active sites of the different types of human PHs are substantially, but not completely, conserved.4 Therefore, there is the potential that inhibitors targeting OGFOD1 could also interfere with hypoxia sensing and collagen biosynthesis through inhibition of other PHs (or vice versa). To date, no detailed evidence for inhibitors selective for the different types of human PHs have been reported. Here, we establish the viability of a structure‐guided template‐based approach for the development of selective OGFOD1 inhibitors which do not target the other human oxygenases tested, including the human hypoxia‐sensing enzyme PHD2.
Results
To assess the viability of developing inhibitors selective for particular PHs, with a focus on OGFOD1, we first compared crystal structures of OGFOD1 and PHD2.10, 24, 25 Although there are differences in the OGFOD1 and PHD2 active sites, the binding modes of 2OG [and the 2OG analogue N‐oxalylglycine (NOG)] appear largely conserved (Figure 1 B–D). The 2‐oxoacid group of 2OG (or NOG) binds to the metal in a bidentate manner, while the 2OG C‐5 carboxylate is positioned to interact with conserved tyrosine and arginine residues (Tyr169 and Arg230 in OGFOD1, Tyr329 and Arg383 in PHD2; Figure 1 C, D). These comparisons, along with those shown for other human oxygenases,4 suggest that inadvertent inhibition of the PHDs may represent a challenge in developing selective OGFOD1 inhibitors, and vice versa.
Given the conserved elements of the OGFOD1 and PHD2 active sites,10 and evidence for induced fit and conformational movements in prolyl hydroxylase catalysis,15 we contemplated a structurally informed template‐derivatization approach for developing OGFOD1 inhibitors.26 We first considered the 4‐hydroxyphenylpyruvate dioxygenase (HPPD) inhibitor nitisinone 1 (which is clinically used for the treatment of tyrosinaemia)27 and related plant growth inhibitors sulcotrione 2, mesotrione 3, prohexadione‐calcium 4, and trinexapac‐ethyl 5 (Figure 2 A).28 These compounds are related to the “tricarbonyl” chemotype found in the PHD inhibitor GSK1278 863 6, which is currently in clinical trials for the treatment of anaemia.18 In addition to GSK1278863 6, we tested PHD inhibitors FG2216 7, FG4592 8, and IOX2 9.29, 30
Figure 2.

Oxygenase inhibitors and their binding modes: (A) Structures of inhibitors of 4‐hydroxyphenylpyruvate dioxygenase (HPPD) and PHD2. Like OGFOD1 and PHD2, HPPD is an FeII‐dependent 2‐oxoacid oxygenase, but from a different structural family. (B) Comparison of the common chelation motifs of sulcotrione 2, GSK1278863 6, and FG2216 7. The structures of sulcotrione 2 and GSK1278863 6 were modeled onto the crystallographically observed structure of PHD2 with bound FG2216 7 (PDB 3HQU).25 A larger version is shown in Figure S5 in the Supporting Information. (C) In silico binding model of GSK1278863 6 with OGFOD1 (PDB 4NHX)10 and PHD2 (PDB 5L9R).15 The tricarbonyl is expected to interact with the bound metal, while the N‐cyclohexyl groups likely occupy the substrate binding position.
These inhibitors were screened for binding to OGFOD1 and PHD2 by differential scanning fluorimetry (DSF) and for inhibition using matrix‐assisted laser desorption ionization time‐of‐flight mass spectrometry (MALDI‐TOF MS) (Figures S1–S4, Supporting Information).31, 32 As expected based on previous studies,33 PHD inhibitors FG2216 7, FG4592 8, and IOX2 9 inhibited OGFOD1 (Figure S1). Notably, triketone‐based HPPD and plant growth inhibitors 1–5 (Figure 2 A), related to GSK1278863 6, displayed moderate inhibition of OGFOD1, while not inhibiting the activity of PHD2 within our limits of detection (Figure S2). Additionally, these inhibitors increased the apparent thermal stability of OGFOD1, as observed by DSF (Figures S3, S4). The inhibition of OGFOD1 by prohexadione‐calcium 4 and trinexapac‐ethyl 5 (Figure S2) suggests that further investigation of these agrochemicals is warranted, given their potential interactions with other human 2OG oxygenases.
GSK1278863 6 and sulcotrione 2 were modeled into the crystallographically observed OGFOD1 and PHD2 active sites, with consideration for potential metal‐chelating properties, salt bridge interactions, and crystallographic studies of PHD2 with inhibitors (such as FG2216 7; PDB 3HQU) (Figure 2 B and Figure S5).25 These analyses imply that both 6 and 2 will engage in FeII chelation via an enolate form of their 1,3‐diketone motif (Figure S5). Similarly to the C‐5 carboxylate of 2OG/NOG,10 and the carboxylate of FG2216 7,25 the GSK1278863 6 carboxylate is predicted to interact with Tyr169 and Arg230 of OGFOD1. The OGFOD1:6 model suggests that the inhibitor ring systems bind in two approximately perpendicular planes, one comprising the diketone ring and glycinamide side chain, and the other formed from the cyclohexyl rings (Figure 2 C). The model also implies that the cyclohexyl rings engage differently with the OGFOD1 and PHD2 active sites (Figure 2 C). By contrast, compounds based on an isoquinoline chemotype (e.g., FG2216 7) are predicted to bind in a more co‐planar manner25 and were considered less likely to readily lead to OGFOD1‐selective inhibitors.
While many reported PHD2 inhibitors possess a glycinamide side chain, the triketone plant growth/HPPD inhibitors do not (Figure 2 A). Modeling results suggest that the sulcotrione 2 methyl sulfonyl group and the nitro group of mesotrione 3 and nitisinone 1 may mimic the 2OG/glycinamide side‐chain binding at their active sites (Figure 2 B). It is possible that the enzyme active site may accommodate the side chains of nitisinone 1, sulcotrione 2, and mesotrione 3, which are bulkier than the glycinamide side chain of FG2216 7; however, in the absence of structural information, we cannot preclude the possibility that these inhibitors with bulky side chains interact with the enzyme in an alternate orientation.
Both the barbiturate‐based PHD inhibitors (e.g., 6) and the HPPD/plant growth inhibitors (e.g., 1–5) have a tri‐carbonyl motif. However, triketones 1–5 are likely more conformationally flexible than the barbiturates. Notably, the DSF results suggest that the triketones stabilize OGFOD1 more than PHD2 (Figure S3), whereas the glycinamide‐containing PHD2 inhibitors (e.g., 6–9) stabilize PHD2 more than OGFOD1 (Figure S4). We thus explored whether modification of the barbiturate/cyclohexane‐1,3‐dione‐based ring scaffolds could be exploited in the development of selective OGFOD1 inhibitors.
Di‐carbonyl compounds (e.g., diethyl malonate 10 and 2‐acetylcyclohexanone 11), triketones (e.g., triacetylmethane 12 and substituted 1,3‐cyclohexanediones 13–15), and structurally related compounds (e.g., 2′,6′‐dihydroxyacetophenone 16 and barbituric acid 17) were screened for OGFOD1 inhibition (Figure 3). The preliminary results suggested that the degree of OGFOD1 inhibition may in part relate to the propensity for inhibitor enolization. Thus, whereas the diketones tested (i.e., 10, 11) were poor inhibitors, the triketones (e.g., 12–15) were more potent (Figure 3 B). However, the phenolic triketone “mimic” 2′,6′‐dihydroxyacetophenone 16 was a relatively poor OGFOD1 inhibitor. The most potent template identified for OGFOD1 inhibition was manifested in the C‐5 substituted barbiturate derivatives (18–20); such compounds are readily enolized, which is likely beneficial for metal chelation. N‐Methylation of the barbiturate ring (e.g., 20) improved potency, as did the introduction of an acyl substituent on C‐5 of the barbiturate core (e.g., 18 compared to 17). Replacement of the C‐2 oxygen with sulfur (thiobarbituric acid 19) did not substantially increase the potency relative to the oxygen analogue (i.e., 18), while replacement of the oxygen at C‐4 with a nitrogen (e.g., 21) abolished any inhibitory activity (Figure 3 B). We therefore focused on obtaining selective OGFOD1 inhibitors by modifying the C‐5 position of a 1,3‐dimethyl barbiturate core. Notably, compounds 22 and 23, which combine a barbiturate core with a glycinamide ethyl ester side chain, did not manifest clear selectivity for OGFOD1 over PHD2 (Figure 4; Figure S6).
Figure 3.

Fragment‐based screening approach for OGFOD1 inhibition: (A) Structures of diketones, triketones, and structurally related compounds used for fragment‐based screening. (B) Inhibitory effect of the fragments on the hydroxylation activity of OGFOD1 (1 μm). The plotted data represent the mean percentage inhibition for the experiment performed in triplicate, whereas the error bars indicate the standard deviation.
Figure 4.
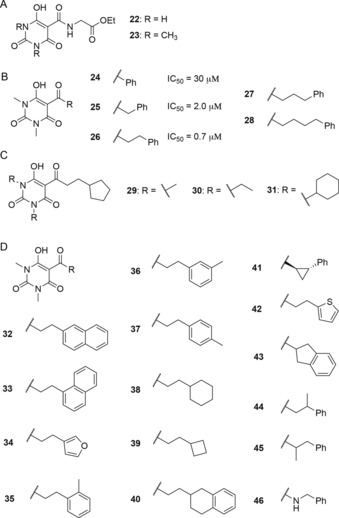
Optimization studies of barbiturate inhibitors: (A) Structures of the barbiturate glycinamide ethyl esters tested against OGFOD1 and PHD2. (B) Panel of phenyl‐substituted N,N′‐dimethylbarbiturates, demonstrating the impact of increasing acyl chain length on OGFOD1 inhibition. (C) Panel of barbiturates synthesized with different N‐alkyl substituents. (D) Panel of C‐5 substituted N,N′‐dimethylbarbiturates based on lead compound CCT3 (26).
Therefore, 24 was prepared, in which structural features of 22 are “merged” with HPPD/plant growth inhibitors 1–3, by introducing a C‐5 aromatic acyl substituent in place of a glycinamide. To investigate the optimal spacing between the barbiturate and the aromatic side chain, the initial screen encompassed a 1,3‐dimethyl barbiturate core bearing acetyl 20, benzoyl 24, phenylacetyl 25, and hydrocinnamoyl 26 (or CCT3) substituents. The resulting aromatic compounds potently inhibited OGFOD1, with an increase in potency being observed upon extending the carbon chain length from 24 (IC50=30 μm) to 25 (IC50=2 μm), and from 25 to 26 (CCT3) (IC50=0.7 μm; assays performed using 1 μm OGFOD1; Figure S7). Extending the side chain further (e.g., 27, 28) did not provide a substantial increase in potency. Importantly, this series displayed no observable PHD2 inhibition at concentrations up to 100 μm (Figure S8). These results suggest that the C‐5 substituent is important for obtaining selectivity between OGFOD1 and PHD2.
The importance of the N‐alkyl substituents for inhibition was examined with barbiturates bearing C‐5 cyclopentyl substituents (Figure 4 C). Analogues bearing N,N′‐dimethyl (29), N,N′‐diethyl (30), and N,N′‐dicyclohexyl (31) groups were prepared (Figure 4 C); analogues 30 and 31 were less potent than 29, suggesting that substituents larger than methyl groups may not be favorable for OGFOD1 inhibition (Figure S9). Note, however, that GSK1278863 6 potently inhibits OGFOD1 despite the presence of N,N′‐dicyclohexyl groups (Figure 2 A).33
Modeling suggests that the aryl side chains of 25 and CCT3 likely do not fit in the OGFOD1 2OG binding site due to steric constraints; instead, the aryl ring may bind in the substrate binding site, potentially contributing to the selectivity of these inhibitors. Varying the C‐5 side chain with different mono‐ and bicyclic aromatic and saturated substituents (32–45) did not have a substantial impact on potency (Figure 4 D, Figure S10). Similarly, changing the nature of the C‐5 link from a ketone to an amide (i.e., 46), or extending the carbon chain length beyond two carbons between the carbonyl and the substituent had little effect (Figure 4D).
On the basis of these SAR studies, in particular those examining the impact of the C‐5 and barbiturate N‐alkyl substituents, the sub‐micromolar potency inhibitors CCT3 (26) and CCT4 (42) were selected for further characterization (Figure 4 and Figure S11). These two inhibitors were screened for inhibitory activity against a panel of human 2OG oxygenases (Figure 5 A). Of the human enzymes screened, including the HIF prolyl hydroxylase PHD2, the ribosomal oxygenases MINA53 and NO66 (which hydroxylate histidinyl residues in ribosomal proteins)20 and the asparaginyl hydroxylase factor‐inhibiting HIF (FIH), CCT3 and CCT4 only potently inhibited OGFOD1 (with IC50 values of 0.73 and 0.69 μm, respectively; Figures S7, S11). Additionally, these compounds showed poor, or no, inhibition of more distantly related 2OG oxygenases, such as the histone demethylases KDM4A, JARID1B, and JARID1C (Figure 5 A). CCT3 and CCT4 did potently inhibit the structurally close yeast OGFOD1 homologues Tpa1p and Ofd1 (which hydroxylate RPS23 prolyl residues; Figure S12), consistent with the high levels of similarity between the homologous active sites.10 It is also notable that the sensitivity of the current hydroxylation assay is a limitation for ranking the activities of the potent inhibitors, as the IC50 values obtained are close to the OGFOD1 concentration used (i.e., the minimum IC50 value that can be measured is 0.5 μm); thus, these inhibitors may be more potent than represented by the currently reported IC50 values.
Figure 5.
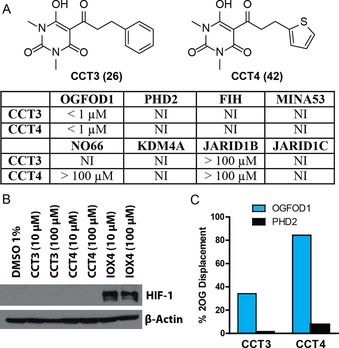
Selectivity of lead compounds against 2OG oxygenases: (A) Comparison of inhibition data for compounds CCT3 and CCT4 against OGFOD1 and other human 2OG oxygenases. NI: no inhibition observed. (B) Western blot (antibody specific for HIFα) showing the lack of impact of CCT3 and CCT4 on the activity of PHD enzymes in HeLa cells.33 In comparison, the known PHD inhibitor IOX4 inhibits the hydroxylation of HIFα, preventing proteasomal degradation.34 (C) Extent of displacement of 2OG from the active site of OGFOD1 (blue) and PHD2 (black) by CCT3 and CCT4 as observed by CPMG‐edited 1H‐NMR. See Figure S15 (Supporting Information) for additional details.
To validate the in vitro inhibition results, direct binding interactions between inhibitors CCT3 and CCT4 and OGFOD1 and PHD2 were assessed by NMR analysis. While both inhibitors were observed to strongly bind to OGFOD1, as observed using 1H Carr‐Purcell‐Meiboom‐Gill (CPMG) analyses (Figure S13), only weak binding to PHD2 was observed by water‐Ligand Observe Gradient Spectroscopy (wLOGSY) experiments (Figure S14). Competition experiments between the inhibitors and enzyme‐bound 2OG were conducted by monitoring the recovery of the enzyme‐free 2OG methylene peak at 2.35 ppm using CPMG‐edited 1H‐NMR upon addition of the inhibitor.35 The results indicate that CCT3 and CCT4 are capable of displacing bound 2OG from the active site of OGFOD1, but not from that of PHD2 (Figure 5 C, S15).
We examined the potential inhibition of PHD enzymes by CCT3 and CCT4 using the HeLa human cell line.36 Compared to the known PHD inhibitor IOX4,34 CCT3 and CCT4 did not stabilize HIFα (Figure 5 B). PHD‐catalyzed hydroxylation targets HIFα for proteasomal degradation, indicating that these compounds do not inhibit the PHDs in cells. Based on a MDR1‐MDCK assay (performed by Cyprotex, UK; see the Supporting Information), CCT3 and CCT4 demonstrate good cell permeability properties, and are predicted to be permeable to the blood brain barrier. Liver microsome stability studies indicate only low levels of clearance of CCT3 and CCT4 (Cyprotex, UK).
Discussion
Our results demonstrate the viability of a template‐based approach for the development of selective 2OG oxygenase/prolyl hydroxylase inhibitors capable of differentiating between closely related active sites, such as those of the human prolyl hydroxylases OGFOD1 and PHD2. Furthermore, the optimized inhibitors also did not inhibit the other 2OG oxygenases tested, including other ribosomal oxygenases, as well as histone demethylases. The results suggest that specific inhibitor ‘templates’ may be preferred for certain oxygenases or oxygenase subfamilies, as supported by work implying differential selectivity between PH and JmjC histone demethylase inhibitors.37 This preference for particular templates may even extend to enzymes with closely related active sites. Appropriately modified barbiturate‐based inhibitors may selectively inhibit OGFOD1 because of their ability to support substituents, which extend towards active‐site residues present in OGFOD1 but not in PHD2. By contrast, the glycinamide side chain present in many PHD2 inhibitors (including several compounds in clinical trials, e.g., 6 and 7) is clearly not required for potent OGFOD1 inhibition. It should also be noted that potent PHD2 inhibitors without a glycinamide side chain are known.33, 34 In future work, it will be of interest to further explore the selectivity of the compounds reported here. In this regard, studies with the procollagen C‐4 (and C‐3) prolyl hydroxylases are of particular interest, especially as the procollagen C‐4 PHs are potential therapeutic targets.38
There is considerable academic and pharmaceutical interest in developing chemical probe compounds to investigate the biological functions of 2OG oxygenases.39 Our results suggest that the development of leads based on known pharmaceutical and agrochemical ‘templates’ (some of which can penetrate the blood–brain barrier) with well‐studied physicochemical properties, such as barbiturates, will be a productive strategy. The combination of the tricarbonyl barbiturate template of the PHD2 inhibitor GSK1278863 6 with the side chains of agrochemical oxygenase inhibitors (e.g., prohexadione‐calcium), followed by subsequent optimization, yielded potent and selective OGFOD1 inhibitors. Future work can now be focused on applying these OGFOD1 inhibitors to investigate the biological roles of OGFOD1, and applying a similar inhibitor development strategy to other ribosomal oxygenases. Based on what is observed in these functional studies, further optimization of these inhibitors may be warranted (e.g., if penetration of the blood–brain barrier is desirable).
It is important to note that many PHD2 inhibitors reported in the literature, including those screened in this work, and those currently in clinical trials, also inhibit OGFOD1.33 Indeed, they may also inhibit other human prolyl hydroxylases and 2OG oxygenases, including those for which assays are currently not available.10, 40 From a clinical perspective, it is also important to note that the barbiturate‐related ′triketone′ HPPD inhibitor nitisinone, which is used in the treatment of type I tyrosinaemia,27 is an OGFOD1 inhibitor, something that might be taken into consideration if nitisinone successors with improved properties are pursued. In the present work, we have demonstrated that it is possible to attain selectivity between different 2OG oxygenases, with lead compounds that inhibit OGFOD1, but not PHD2. Such “biochemical selectivity” is not necessarily an issue with clinical applications as the desired pharmacological effect/safety profile may be achieved by controlling metabolism and tissue distribution. However, we propose that, at least for chronic applications, biochemical selectivity could and should be optimized during the development of 2OG oxygenase inhibitors. We also hope that inhibitors selective for particular 2OG oxygenases may help enable their individual biological roles to be deciphered.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank the Wellcome Trust, Cancer Research UK, and the Biotechnology and Biological Sciences Research Council (BBSRC) for supporting our research on 2OG‐dependent oxygenases. C.L. thanks the Leverhulme Trust for an Early Career Fellowship.
C. C. Thinnes, C. T. Lohans, M. I. Abboud, T.-L. Yeh, A. Tumber, R. P. Nowak, M. Attwood, M. E. Cockman, U. Oppermann, C. Loenarz, C. J. Schofield, Chem. Eur. J. 2019, 25, 2019.
References
- 1. Schofield C. J., Ratcliffe P. J., Nat. Rev. Mol. Cell Biol. 2004, 5, 343–354. [DOI] [PubMed] [Google Scholar]
- 2. Wilkins S. E., Abboud M. I., Hancock R. L., Schofield C. J., ChemMedChem 2016, 11, 773–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Webb J. D., Coleman M. L., Pugh C. W., Cell. Mol. Life Sci. 2009, 66, 3539–3554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Aik W. S., Chowdhury R., Clifton I. J., Hopkinson R. J., Leissing T., McDonough M. A., Nowak R., Schofield C. J., Walport L. J., 2-Oxoglutarate-Dependent Oxygenases, Royal Society of Chemistry, Cambridge, 2015, p. 59–94. [Google Scholar]
- 5. Loenarz C., Schofield C. J., Nat. Chem. Biol. 2008, 4, 152–156. [DOI] [PubMed] [Google Scholar]
- 6. Gorres K. L., Raines R. T., Crit. Rev. Biochem. Mol. Biol. 2010, 45, 106–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Singleton R. S., Liu-Yi P., Formenti F., Ge W., Sekirnik R., Fischer R., Adam J., Pollard P. J., Wolf A., Thalhammer A., Loenarz C., Flashman E., Yamamoto A., Coleman M. L., Kessler B. M., Wappner P., Schofield C. J., Ratcliffe P. J., Cockman M. E., Proc. Natl. Acad. Sci. USA 2014, 111, 4031–4036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Loenarz C., Sekirnik R., Thalhammer A., Ge W., Spivakovsky E., Mackeen M. M., McDonough M. A., Cockman M. E., Kessler B. M., Ratcliffe P. J., Wolf A., Schofield C. J., Proc. Natl. Acad. Sci. USA 2014, 111, 4019–4024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Katz M. J., Acevedo J. M., Loenarz C., Galagovsky D., Liu-Yi P., Pérez-Pepe M., Thalhammer A., Sekirnik R., Ge W., Melani M., Thomas M. G., Simonetta S., Boccaccio G. L., Schofield C. J., Cockman M. E., Ratcliffe P. J., Wappner P., Proc. Natl. Acad. Sci. USA 2014, 111, 4025–4030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Horita S., Scotti J. S., Thinnes C., Mottaghi-Taromsari Y. S., Thalhammer A., Ge W., Aik W., Loenarz C., Schofield C. J., McDonough M. A., Structure 2015, 23, 639–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Schmeing T. M., Ramakrishnan V., Nature 2009, 461, 1234–1242. [DOI] [PubMed] [Google Scholar]
- 12. Gilbert W. V., Trends Biochem. Sci. 2011, 36, 127–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nesterchuk M. V., Sergiev P. V., Dontsova O. A., Acta Naturae 2011, 3, 22–33. [PMC free article] [PubMed] [Google Scholar]
- 14. Alksne L. E., Anthony R. A., Liebman S. W., Warner J. R., Proc. Natl. Acad. Sci. USA 1993, 90, 9538–9541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chowdhury R., Leung I. K., Tian Y. M., Abboud M. I., Ge W., Domene C., Cantrelle F. X., Landrieu I., Hardy A. P., Pugh C. W., Ratcliffe P. J., Claridge T. D., Schofield C. J., Nat. Commun. 2016, 7, 12673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rose N. R., McDonough M. A., King O. N., Kawamura A., Schofield C. J., Chem. Soc. Rev. 2011, 40, 4364–4397. [DOI] [PubMed] [Google Scholar]
- 17. Vasta J. D., Andersen K. A., Deck K. M., Nizzi C. P., Eisenstein R. S., Raines R. T., ACS Chem. Biol. 2016, 11, 193–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chan M. C., Holt-Martyn J. P., Schofield C. J., Ratcliffe P. J., Mol. Aspects Med. 2016, 47–48, 54–75. [DOI] [PubMed] [Google Scholar]
- 19. Keeling K. M., Xue X., Gunn G., Bedwell D. M., Annu. Rev. Genomics Hum. Genet. 2014, 15, 371–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ge W., Wolf A., Feng T., Ho C. H., Sekirnik R., Zayer A., Granatino N., Cockman M. E., Loenarz C., Loik N. D., Hardy A. P., Claridge T. D., Hamed R. B., Chowdhury R., Gong L., Robinson C. V., Trudgian D. C., Jiang M., Mackeen M. M., McCullagh J. S., Gordiyenko Y., Thalhammer A., Yamamoto A., Yang M., Liu-Yi P., Zhang Z., Schmidt-Zachmann M., Kessler B. M., Ratcliffe P. J., Preston G. M., Coleman M. L., Schofield C. J., Nat. Chem. Biol. 2012, 8, 960–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chowdhury R., Sekirnik R., Brissett N. C., Krojer T., Ho C. H., Ng S. S., Clifton I. J., Ge W., Kershaw N. J., Fox G. C., Muniz J. R. C., Vollmar M., Phillips C., Pilka E. S., Kavanagh K. L., von Delft F., Oppermann U., McDonough M. A., Doherty A. J., Schofield C. J., Nature 2014, 510, 422–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Markolovic S., Zhuang Q., Wilkins S. E., Eaton C. D., Abboud M. I., Katz M. J., McNeil H. E., Leśniak R. K., Hall C., Struwe W. B., Konietzny R., Davis S., Yang M., Ge W., Benesch J. L. P., Kessler B. M., Ratcliffe P. J., Cockman M. E., Fischer R., Wappner P., Chowdhury R., Coleman M. L., Schofield C. J., Nat. Chem. Biol. 2018, 14, 688–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Feng T., Yamamoto A., Wilkins S. E., Sokolova E., Yates L. A., Münzel M., Singh P., Hopkinson R. J., Fischer R., Cockman M. E., Shelley J., Trudgian D. C., Schödel J., McCullagh J. S., Ge W., Kessler B. M., Gilbert R. J., Frolova L. Y., Alkalaeva E., Ratcliffe P. J., Schofield C. J., Coleman M. L., Mol. Cell 2014, 53, 645–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. McDonough M. A., Loenarz C., Chowdhury R., Clifton I. J., Schofield C. J., Curr. Opin. Struct. Biol. 2010, 20, 659–672. [DOI] [PubMed] [Google Scholar]
- 25. Chowdhury R., McDonough M. A., Mecinović J., Loenarz C., Flashman E., Hewitson K. S., Domene C., Schofield C. J., Structure 2009, 17, 981–989. [DOI] [PubMed] [Google Scholar]
- 26. Thinnes C. C., Tumber A., Yapp C., Scozzafava G., Yeh T., Chan M. C., Tran T. A., Hsu K., Tarhonskaya H., Walport L. J., Wilkins S. E., Martinez E. D., Müller S., Pugh C. W., Ratcliffe P. J., Brennan P. E., Kawamura A., Schofield C. J., Chem. Commun. 2015, 51, 15458–15461. [DOI] [PubMed] [Google Scholar]
- 27. McKiernan P. J., Drugs 2006, 66, 743–750. [DOI] [PubMed] [Google Scholar]
- 28. Moran G. R., Arch. Biochem. Biophys. 2005, 433, 117–128. [DOI] [PubMed] [Google Scholar]
- 29. Koury M. J., Haase V. H., Nat. Rev. Nephrol. 2015, 11, 394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chowdhury R., Candela-Lena J. I., Chan M. C., Greenald D. J., Yeoh K. K., Tian Y. M., McDonough M. A., Tumber A., Rose N. R., Conejo-Garcia A., Demetriades M., Mathavan S., Kawamura A., Lee M. K., van Eeden F., Pugh C. W., Ratcliffe P. J., Schofield C. J., ACS Chem. Biol. 2013, 8, 1488–1496. [DOI] [PubMed] [Google Scholar]
- 31. Chowdhury R., Yeoh K. K., Tian Y. M., Hillringhaus L., Bagg E. A., Rose N. R., Leung I. K., Li X. S., Woon E. C., Yang M., McDonough M. A., King O. N., Clifton I. J., Klose R. J., Claridge T. D., Ratcliffe P. J., Schofield C. J., Kawamura A., EMBO Rep. 2011, 12, 463–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Niesen F. H., Berglund H., Vedadi M., Nat. Protoc. 2007, 2, 2212–2221. [DOI] [PubMed] [Google Scholar]
- 33. Yeh T.-L., Leissing T. M., Abboud M. I., Thinnes C. C., Atasoylu O., Holt-Martyn J. P., Zhang D., Tumber A., Lippl K., Lohans C. T., Leung I. K. H., Morcrette H., Clifton I. J., Claridge T. D. W., Kawamura A., Flashman E., Lu X., Ratcliffe P. J., Chowdhury R., Pugh C. W., Schofield C. J., Chem. Sci. 2017, 8, 7651–7668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chan M. C., Atasoylu O., Hodson E., Tumber A., Leung I. K., Chowdhury R., Gómez-Pérez V., Demetriades M., Rydzik A. M., Holt-Martyn J., Tian Y. M., Bishop T., Claridge T. D., Kawamura A., Pugh C. W., Ratcliffe P. J., Schofield C. J., PLoS One 2015, 10, e0132004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Abboud M. I., McAllister T. E., Leung I. K. H., Chowdhury R., Jorgensen C., Domene C., Mecinović J., Lippl K., Hancock R. L., Hopkinson R. J., Kawamura A., Claridge T. D. W., Schofield C. J., Chem. Commun 2018, 54, 3130–3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tian Y. M., Yeoh K. K., Lee M. K., Eriksson T., Kessler B. M., Kramer H. B., Edelmann M. J., Willam C., Pugh C. W., Schofield C. J., Ratcliffe P. J., J. Biol. Chem. 2011, 286, 13041–13051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Thinnes C. C., England K. S., Kawamura A., Chowdhury R., Schofield C. J., Hopkinson R. J., Biochim. Biophys. Acta Gene Regul. Mech. 2014, 1839, 1416–1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Vasta J. D., Raines R. T., J Med Chem. 2018, 61, 10403–10411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Arrowsmith C. H., Audia J. E., Austin C., Baell J., Bennett J., Blagg J., Bountra C., Brennan P. E., Brown P. J., Bunnage M. E., Buser-Doepner C., Campbell R. M., Carter A. J., Cohen P., Copeland R. A., Cravatt B., Dahlin J. L., Dhanak D., Edwards A. M., Frederiksen M., Frye S. V., Gray N., Grimshaw C. E., Hepworth D., Howe T., Huber K. V., Jin J., Knapp S., Kotz J. D., Kruger R. G., Lowe D., Mader M. M., Marsden B., Mueller-Fahrnow A., Müller S., O'Hagan R. C., Overington J. P., Owen D. R., Rosenberg S. H., Roth B., Roth B., Ross R., Schapira M., Schreiber S. L., Shoichet B., Sundström M., Superti-Furga G., Taunton J., Toledo-Sherman L., Walpole C., Walters M. A., Willson T. M., Workman P., Young R. N., Zuercher W. J., Nat. Chem. Biol. 2015, 11, 536–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. McMurray F., Demetriades M., Aik W., Merkestein M., Kramer H., Andrew D. S., Scudamore C. L., Hough T. A., Wells S., Ashcroft F. M., McDonough M. A., Schofield C. J., Cox R. D., PLoS One 2015, 10, e0121829. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary