

Abstract
Mutations and changes in a protein's environment are well known for their potential to induce misfolding and aggregation, including amyloid formation. Alternatively, such perturbations can trigger new interactions that lead to the polymerization of folded proteins. In contrast to aggregation, this process does not require misfolding and, to highlight this difference, we refer to it as agglomeration. This term encompasses the amorphous assembly of folded proteins as well as the polymerization in one, two, or three dimensions. We stress the remarkable potential of symmetric homo‐oligomers to agglomerate even by single surface point mutations, and we review the double‐edged nature of this potential: how aberrant assemblies resulting from agglomeration can lead to disease, but also how agglomeration can serve in cellular adaptation and be exploited for the rational design of novel biomaterials.
Keywords: agglomeration, protein structures, self-assembly, supramolecular polymerization, symmetry
1. Introduction
As early as 1899, Edmund Wilson reviewed cellular organization as visualized by light microscopy and wrote “[…] the background of all phenomena appears to lie in the invisible organization of a substance which seems to the eye homogeneous.”1 Although it was not known that proteins and RNA form most of the “substance,” it was intuitive that this substance was organized.
Indeed, we know today that living matter is not a mere Brownian soup of molecules. Instead, these molecules assemble with each other to give rise to a dynamic machinery of exquisite complexity. In this Review, we approach the concept of self‐organization by focusing on protein self‐assembly. We distinguish between two main types of assemblies: protein complexes that are finite,2 and potentially infinite assemblies that we term “agglomerates” (see the Glossary). The assembly of proteins into finite complexes has long been viewed as a fundamental level of organization, and today we know thousands of such complexes.3, 4 In contrast, polymeric assemblies that are potentially infinite have been classically viewed as restricted to specific sets of proteins, such as those forming the cytoskeleton.5 However, we will see that numerous reports have been shifting this paradigm in recent years.
In this Review, we first contrast agglomeration and aggregation. Aggregation is sometimes viewed as the only process by which proteins can accidentally form supramolecular polymers in vitro or in vivo. We will see, however, that agglomeration can also occur readily, both in the test tube and during evolution (Section 2). The potential of proteins to agglomerate is largely dependent on the properties of their surfaces and on their internal symmetry. This dependency is the focus of Section 3. In the following two sections, we review instances of naturally occurring protein agglomerates observed in the context of diseases (Section 4) and in the context of normal cellular functions (Section 5). Lastly, we review how agglomeration has been harnessed by chemists and protein engineers in the design of open‐ended protein assemblies (Section 6). Throughout the Review, we place an emphasis on protein symmetry, which is a common theme unifying all these aspects of agglomeration.
2. Agglomeration as a Frequent Process Distinct from Aggregation
2.1. Agglomeration versus Aggregation
The spontaneous assembly and precipitation of proteins is frequently observed when increasing their concentration, changing solvent conditions, or when mutations are introduced.6, 7, 8, 9 It is typical to think of such precipitates as aggregates (see the Glossary), whereby a partially or entirely misfolded form of the protein drives its association into high‐order assemblies.
However, the accidental assembly of proteins does not necessarily involve misfolding. Instead, it may result from associations between folded proteins. Although several terms may be used to describe such a scenario, for example, “Protein X assembles into large structures in which it remains folded” or “Protein X polymerizes while remaining folded” these cannot be conveniently used as verbs to describe the process and do not capture the idea of an accidental occurrence. This led us to use “agglomeration” as a term—which sounds similar to “aggregation” and can be employed succinctly and generically as a verb, for example, “Protein X agglomerates”—to describe a process of infinite, folded‐state assembly. An alternative could be “Protein X adopts a quinary structure,” although this term has been associated to more transient and heterogeneous protein assemblies10 and does not reflect a possible accidental or pathological nature in the way aggregation or agglomeration do.
At a structural level, native contacts are lost in aggregation and are replaced by non‐native contacts, whereas in agglomeration, native contacts preserve the structure of the surface(s) that interact to drive assembly. In both processes, we consider a finite protein unit or protomer11 (see the Glossary) as a starting point and define aggregation or agglomeration as the transition from a finite to an open‐ended, potentially infinite assembly. Such transitions could be triggered by a perturbation such as a mutation7, 12 or a change in the protein's environment.7, 13 We illustrate seven transitions in Figure 1, each involving a different path. These examples show a continuum between pure aggregation, where all native contacts are lost in the assembly, and pure agglomeration, where all native contacts are preserved:
Figure 1.
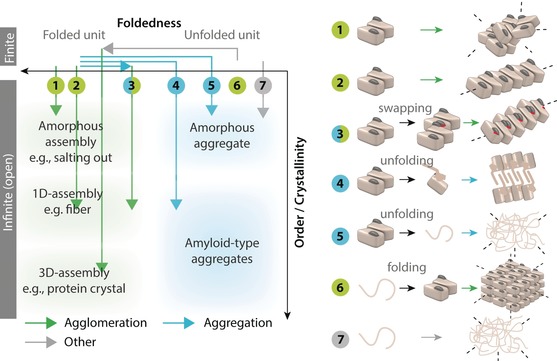
Comparing agglomeration and aggregation. In biology, aggregation is concomitant to a loss of structure, in the form of partial or complete unfolding.7, 8, 9 Thus, the term aggregation is misleading when describing a process where proteins assemble in their folded state. The illustration shows both processes with seven examples of transitions from closed (finite) to open‐ended (infinite) assemblies, each described in the main text (Section 2.1). Each transition is characterized by a pathway with two components: misfolding/folding of the protomers (x‐axis), and orderliness of the assemblies formed (y‐axis). For simplicity, the same dimeric unit is depicted as a starting point in transitions 1–5, but the symmetry of the dimer is only essential for driving transitions 2 and 3.
An increased self‐interaction potential leads to the non‐ordered (amorphous) agglomeration of a homodimer. Such a process can be employed to purify active forms of proteins by “salting them out” using high concentrations of kosmotropic agents such as ammonium sulfate.14, 15
A specific self‐interaction is gained, which drives the ordered assembly of a homodimer into a regular and potentially infinite filament. Such a scenario is seen, for example, in the sickle cell disease16 or in the S134N and apo‐H46R mutants of superoxide dismutase (SOD).17
Part of the structure of the dimer is destabilized, which induces domain swapping with another dimer. The domain swapping creates a new self‐interaction and drives, as in example 2, the formation of an infinite filament. This scenario is halfway between aggregation and agglomeration, since misfolding is required, but filament assembly also depends on the dimer retaining most of its native contacts. Such a scenario is seen in filaments formed by phage T7 endonuclease I, for example.18
Part of the structure of a dimer is destabilized, unfolds, and self‐interacts to form a cross‐beta‐sheet amyloid structure. In contrast to example 3, the unfolded part directly mediates the self‐association and assembly, which does not involve the native contacts. Such filaments are seen with ribonuclease A19 or β‐lactoglobulin filaments, for example.20
The structure or part of the structure of a dimeric unit unfolds, and the residues exposed interact across units to drive the formation of an amorphous aggregate. Here, as well, the native contacts do not mediate the assembly. This is typically seen in thermal denaturation, for example.21
The equilibrium between unfolded and folded states of a polypeptide chain is concentration‐dependent, with the latter being stabilized by homo‐oligomerization. Homomer units form at high peptide concentrations and subsequently assemble into a crystal lattice. Such a behavior has been observed in designed peptides that self‐assemble into a predefined lattice.22
A disordered protein self‐associates and forms an amorphous assembly. Neither agglomeration nor aggregation would be an appropriate term to describe such a transition as no misfolding is necessarily required and yet the polypeptide chains are not folded. Numerous reports of such transitions have been described recently, where intrinsically disordered proteins phase‐separate under specific conditions.23 This transition could be referred to as “phase separation,” although this term is general and applies to other transitions presented Figure 1. Alternatively, “condensation” could be employed.23
The terms “amyloid” and “aggregate” are sometimes used to describe assemblies of folded proteins,24 but this conveys an inaccurate picture. Indeed, making the distinction between assemblies of folded versus misfolded proteins is key because their molecular origin, their formation mechanism, and their cellular consequences are expected to differ:
Aggregation is associated with changes in protein stability,8, 9 whereas agglomeration is dependent on the surface properties of the protein.12, 25
-
Aggregation involves conformational changes, which can dramatically shape the energy landscape of the aggregate growth.26, 27 Activation energy is indeed required to drive partial unfolding,28 and to cross a nucleation barrier in the case of amyloid formation.8 In contrast, the activation energy required for agglomeration drives the disruption of the structured water shell around the protomers.29 Additionally, the growth of an agglomerate can involve a nucleation barrier (e.g. as in crystal formation29) or could be isodesmic,26 and thereby not involve a nucleation barrier. In relation to these differences, aggregates are typically irreversible7—for example, they only resolubilize on addition of denaturants or detergents—whereas agglomeration has often been described as being reversible (Figure 2).12, 13, 30
Figure 2.
 Agglomeration and aggregation show similar macroscopic properties. a) Boiled green fluorescent protein (GFP) unfolds and irreversibly aggregates.30 b) A variant of GFP displaying a net charge of +36 interacts with anionic tRNAs and precipitates. These agglomerates reversibly dissociate upon screening charge–charge interactions with salt.30 c) A homo‐octamer with dihedral symmetry agglomerates through hydrophobic surface interactions. This association is reversible, being induced in the presence of salt and repressed in its absence.12 d) The yeast protein ADE4 (involved in adenine biosynthesis) clusters in response to specific stresses, including adenine depletion.13 This mechanism is reversible upon repleting the growth media. Images reproduced from Ref. 30 [(a) and (b), https://pubs.acs.org/doi/full/10.1021/ja071641y] and Ref. 13 with permission [(d), Copyright 2009, National Academy of Sciences].
Agglomeration and aggregation show similar macroscopic properties. a) Boiled green fluorescent protein (GFP) unfolds and irreversibly aggregates.30 b) A variant of GFP displaying a net charge of +36 interacts with anionic tRNAs and precipitates. These agglomerates reversibly dissociate upon screening charge–charge interactions with salt.30 c) A homo‐octamer with dihedral symmetry agglomerates through hydrophobic surface interactions. This association is reversible, being induced in the presence of salt and repressed in its absence.12 d) The yeast protein ADE4 (involved in adenine biosynthesis) clusters in response to specific stresses, including adenine depletion.13 This mechanism is reversible upon repleting the growth media. Images reproduced from Ref. 30 [(a) and (b), https://pubs.acs.org/doi/full/10.1021/ja071641y] and Ref. 13 with permission [(d), Copyright 2009, National Academy of Sciences]. As to the consequences, misfolding exposes hydrophobic segments that are normally buried and these can recruit chaperones.31, 32 In contrast, agglomeration does not involve the exposure of such segments and, thus, is not expected to recruit chaperones.
Despite these differences at the molecular level, agglomerates and aggregates exhibit similar macroscopic phenotypes: both are visible to the naked eye through turbidity and precipitation in vitro (Figure 2). Similarly, in vivo, agglomerates can cluster into punctate foci12 of similar appearance to those formed by aggregates.33 Such similarities stress further the need for different terminologies to avoid confounding both processes.
2.2. Natural Proteins Are Prone to Self‐Interact
The intrinsic potential of proteins for self‐association leading to agglomeration has been exploited in purification procedures for several decades.15 In these procedures, proteins are fractionated by high concentrations of kosmotropic salts,14 most notably ammonium sulfate. Also called “salting‐out,” this process increases the surface tension of water,34 thus favoring interactions between folded proteins and their precipitation.15
The predisposition of proteins for self‐interaction is perhaps best reflected in X‐ray crystallography experiments, where fortuitous self‐interactions must form a crystal lattice.35 Thus, the success of crystallography underlines that protein surfaces are naturally prone to self‐associate. This idea is confirmed when comparing the chemical composition of protein–protein interfaces (see the Glossary) and solvent‐exposed surface patches. The composition of these two surface types differs, on average, by only two amino acid substitutions36 and highlights that protein surfaces are naturally “sticky.”
To investigate this stickiness potential, Garcia‐Seisdedos et al. examined whether mutations solely designed to increase the surface hydrophobicity of dihedral complexes would often trigger their agglomeration by stacking. All 12 homomers examined underwent agglomeration following the introduction of one to four point mutations,12 thus confirming that protein surfaces evolve on the verge of self‐assembly (Figure 3). Also consistent with this idea is that solely reducing the rotational entropy of a protein by a magnetic field directed its self‐assembly into 2D arrays,37 which shows that the enthalpic component of the association free energy was naturally favorable.
Figure 3.

Increasing surface hydrophobicity triggers supramolecular self‐assembly and is counterbalanced by negative design. a) Top and second row: structure solved by X‐ray crystallography of four homomers with dihedral symmetry. The corresponding PDB codes are indicated at the top. Shown underneath are fluorescence microscopy images of yeast cells expressing the wild‐type homomers fused to a yellow fluorescent protein. The localization is cytoplasmic and homogeneous. The following row shows yeast cells expressing mutant proteins in which surface hydrophobicity was increased by point mutations (shown in red in the structures). The two right‐most mutants formed fibers and the two left‐most formed foci. The last row shows transmission electron microscopy (TEM) images with negative staining for the same mutants, which stack into filaments.12 b) The tip regions of homomers with dihedral symmetry were termed “geometric hot spots” because mutations in those regions triggered agglomeration more frequently than surface mutations away from them.12 Among natural homomers with dihedral symmetry, geometric hot spots were enriched with charged and hydrophilic amino acids relative to the rest of the protein surface.12 Reproduced from Ref. 12 with permission [(a), Copyright 2017, Springer Nature].
In fact, the natural stickiness of protein surface patches can be a burden that needs to be counterbalanced by negative design, whereby specific physicochemical properties evolve to minimize the formation of alternative, unwanted structures.38 For example, from the law of mass‐action, proteins expressed at high levels in cells are more likely to engage in promiscuous interactions than lowly expressed proteins and, accordingly, their surface shows reduced hydrophobicity.25 Negative design can thus be detected on specific proteins but also at sensitive locations on their surface (Figure 3 b) to reduce their potential for mis‐assembly.12, 39
3. Agglomeration in Relation to Protein Quaternary Structure
The symmetry of a protein's quaternary structure has a dramatic impact on its potential to agglomerate. This connection was described formally by Monod and co‐workers.40 The underlying concepts are described in this section. We also refer the reader to a review by Yeates and co‐workers, which describes geometry and symmetry rules underlying finite versus infinite protein assembly.41
3.1. Symmetry of Protein Quaternary Structures
In homo‐oligomers or “homomers,” all protomers are identical in sequence and consequently—as was suggested by Caspar and Klug42—are generally equivalent in terms of their structure and geometry. In other words, they tend to all exist in a chemically identical environment. Only rotation point group symmetries (cyclic, dihedral, and cubic) satisfy this equivalence constraint (Figure 4). These symmetry types are frequent: about 65 % (E. coli) and about 45 % (H. sapiens) of proteins of known structure adopt a point group symmetry (Figure 4). A homomer with cyclic symmetry composed of n protomers (denoted Cn) shows a single n‐fold symmetry axis. Homomers with dihedral symmetry can be viewed as two cyclic complexes stacked “back‐to‐back,” and thus are composed of m=2×n protomers (denoted Dm) related by one n‐fold symmetry axis and n 2‐fold axes (Figure 4). Lastly, the cubic group includes tetrahedral, octahedral, and icosahedral symmetries, which are described further in the literature.42, 43, 44 A hallmark of point group symmetries is that all symmetry axes pass through, and intersect at, the center of mass. It follows that such homomers are only composed of rotational symmetries and never of translational symmetries.
Figure 4.

Point group symmetries of homomers and their frequency among proteins of known structure. The frequency of each symmetry type is shown for proteins from E. coli and H. sapiens, based on a previously published dataset containing homomers from both species.25 The inset illustrates the arrangement of protomers in cyclic, dihedral, and cubic point group symmetries.
3.2. Homotypic and Heterotypic Protein–Protein Interfaces
Protein assembly requires the formation of interfaces with energetically favorable interactions, the physicochemical properties of which have been extensively studied and reviewed.44, 45, 46, 47, 48, 49 In this subsection, we focus on two properties of interfaces that are particularly relevant when rationalizing the formation of open‐ended assemblies: 1) the distinction between homotypic and heterotypic interfaces (see the Glossary), and 2) the potential of protein surfaces to form either type.
Homotypic interfaces involve two identical surfaces interacting with twofold symmetry (Figure 5 a). Thus, any dimer with C 2 symmetry involves a homotypic interface. Heterotypic interfaces are formed when two distinct surface patches are in contact, as happens in hetero‐oligomers and in cyclization events with three or more protomers.2 For example, a cyclic homomer with C 4 symmetry involves four protomers interacting “face‐to‐back” (Figure 5 b).
Figure 5.

New self‐interactions leading to open and closed symmetric assemblies. a) Homotypic interfaces involve two identical surface patches in contact and related by a twofold symmetry axis. b) Heterotypic interfaces involve two distinct surface patches. c) New self‐interactions may drive the formation of open‐ended or closed assemblies depending on 1) the starting symmetry, 2) the type of interface gained (green arrows: homotypic, orange arrows: heterotypic), and 3) the location of the interacting surface patches on the protomer, as described in the main text.
The distinction between homotypic and heterotypic interfaces is fundamental because of the energetic implications. In homotypic interfaces, each amino acid residue is repeated twice by symmetry, so its contribution to the free energy of association is more likely to be extreme on average (i.e. highly favorable or highly unfavorable) when compared to heterotypic interfaces, where each amino acid is present once only.50, 51 Consequently, in a scenario where interfaces are randomly created and where stable ones are selected, homotypic interfaces will be selected more frequently than heterotypic ones,52, 53, 54 which possibly accounts for the high frequency of homotypic interactions in natural homomers.55
3.3. From a Finite Complex to an Agglomerate
A protomer which can interact with itself will form a new assembly, the properties of which depend on several parameters: 1) the starting quaternary structure (see the Glossary), 2) the type of interface gained, and 3) the location of the interface gained on the protomer's structure. As starting quaternary structures, we distinguish monomers, cyclic homomers, and higher order symmetries (dihedral and cubic). For the descriptions below we refer the reader to Figure 5 c:
Upon gaining a new heterotypic interaction, a monomer adopts a closed symmetry (e.g. C 1→C 3). Alternatively, it can form infinite filaments that are straight (e.g. C 1→filament) or contain a helical component (not shown). Upon gaining a homotypic interface, however, a monomer necessarily yields a closed dimer (C 1→C 2).
Starting from a cyclic homomer, further assembly into a finite complex is possible and can involve the creation of both homotypic and/or heterotypic interfaces. The geometry of the new interfaces, however, is decisive in determining the type of the resulting assembly. For example, the gain of a homotypic interface yields a dihedral symmetry if it occurs at the top or bottom parts of the ring (e.g. C 3→D 3), but may also yield an infinite planar assembly if occurring at another location (e.g. C 4→plane).
Lastly, starting from a homomer with dihedral or cubic symmetry, the gain of any new interface—either homotypic or heterotypic—will necessarily result in the formation of an open, infinite assembly (e.g. D 2, D 3, D 4→respective filament, or D 4→plane)
Given these geometric determinants, and considering the energetic advantage of homotypic interfaces over heterotypic ones (Section 3.2), we anticipate that natural open‐ended assemblies will often stem from homomers with dihedral symmetry.
4. Agglomeration and Disease
4.1. Different Modes of Agglomeration Observed with Hemoglobin
The notion that homomers with dihedral symmetry are only one self‐interaction away from forming agglomerates (Section 3.3) suggests that some disease mutations could be linked to agglomeration. Indeed, a well‐known example of agglomeration is that of hemoglobin in the sickle cell disease. A single glutamate‐to‐valine mutation on the hemoglobin surface causes this pathology.16, 56 In the deoxygenated form, mutant hemoglobin tetramers assemble into rigid filaments and deform the shape of the red blood cells. The process is reversible as, upon reoxygenation, the filaments rapidly dissolve, which allows cells to recover their typical shape.16
The filamentous agglomeration of hemoglobin is tied to its C 2 (pseudo‐D 2) symmetry: the mutation E6V at the β subunit surface induces its interaction with a copy of itself, and repeating this interaction through symmetry drives the formation of a filament (Figure 6 a). Interestingly, a lysine mutation at the same residue (E6) can cause another type of agglomeration, where oxyhemoglobin forms intracellular crystals associated with hemolytic anemia in homozygous patients.57, 58
Figure 6.

Open‐ended assemblies related to a disease. a) The mutation E6V triggers the formation of filaments in human deoxyhemoglobin, which causes the sickle cell disease. The Figure shows double strands of hemoglobin S (E6V) tetramers revealed by crystallography.81 b) The mutation P23T on γ‐d‐crystallin monomers induces their amorphous agglomeration, here visualized by EM with negative staining.61, 62 c) The mutation R36S on γ‐d‐crystallin monomers induces their crystallization. Crystals accumulate in the eye lens and cause cataracts. The crystals of γ‐d‐crystallin R36S in phosphate‐buffered saline (PBS) are imaged by inverse light microscopy.63 d) Cytidine triphosphate synthase (CTPS) is an enzyme with D 2 symmetry capable of assembling into filaments. The cryo‐EM reconstruction of a human CTPS is shown.73 e) Lithostatine is a monomeric protein that forms D 2 tetramers after autoproteolysis. The image shows lithostatine protofibrils characterized by AFM.77, 82 f) Zinc promotes insulin to form hexamers, which further assemble into filaments by introducing a covalent modification (purple). Filament formation was dependent on phenol and is visualized here by EM with negative staining.83 Reproduced from Ref. 81 [(a), Copyright 1997, Academic Press. All rights reserved]; Ref. 62 [(b), Copyright 2005, American Chemical Society]; Ref. 63 [(c), Copyright 2000, Oxford University Press]; Ref. 73 [(d), Copyright 2017, Springer Nature]; Ref. 82 [(e), Copyright 2001, John Wiley and Sons], and Ref. 83 [(f), Copyright 2013, American Chemical Society] with permission.
Although the agglomeration of hemoglobin is associated with pathologies in humans, red blood cells in some species of deer naturally exhibit a sickle shape. The sickling mechanism was recently also attributed to a glutamate‐to‐valine change, albeit in a different part of the structure.59 In contrast to the human case, deer hemoglobin also agglomerates in the oxygenated form.
4.2. Other Agglomeration‐Associated Diseases
Agglomeration has been related to pathologies other than the sickle cell. In congenital cataracts, crystallins in the eye lens form large assemblies that scatter light, thus creating opaqueness that leads to blindness. Such assemblies can be triggered by environmental stress, metabolic imbalance, or mutations.60 The P23T mutation on γ‐d‐crystallin, for example, triggers the amorphous assembly of folded crystallins (Figure 6 b), which indicates agglomeration rather than aggregation.61, 62 In another example, Kmoch et al. described a mutation (R36S) that induces γ‐d‐crystallin to assemble into small crystals visible in the eye lens (Figure 6 c).63 The pathological consequences of agglomeration in the sickle cells and cataracts are both linked to physical properties of the agglomerates themselves, which disrupt cell shape or light propagation. In numerous other cases, the impact of agglomerates on cellular functions is less well understood.
Mutations in the enzyme superoxide dismutase (SOD1) are linked to amyotrophic lateral sclerosis (ALS),64 a degenerative disease of the human motor system. Since Rosen et al. first characterized a mutation in SOD1 associated with ALS over two decades ago,64 more than 150 disease‐related mutants have been identified. Nonetheless, the molecular mechanisms underlying the pathology remain unclear. While the loss of dismutase activity has been postulated to be a possible cause,65, 66 the prevalent view is that cytotoxicity is related to misfolding and aggregation of SOD1 mutants.67 It is noteworthy, however, that many of the “aggregates” reported are agglomerate‐like, whereby SOD1 self‐assembles in a largely folded state.17, 68, 69, 70
Similarly, cytidine triphosphate synthase (CTPS, Figure 6 d) and inosine monophosphate dehydrogenase (IMPDH) are two enzymes central to nucleotide biosynthesis that form filamentous agglomerates in various organisms, including humans.71, 72 Their polymerization upregulates their enzymatic activity,73, 74 and the fact that cancer cells display high levels of CTPS and IMPDH activities75, 76 begs the question of an association between their filamentous agglomeration and cancer metabolism and proliferation. Chang et al. recently found that CTPS filaments appear to be substantially enriched in cancerous hepatocytes71 and are associated with high levels of the heat shock protein 90 (HSP90), thus hinting at a possible metabolic adaptation mechanism in hepatocellular carcinoma.
In another example, agglomerates of lithostatine (Figure 6 e) were observed in early onset deposits of Alzheimer's disease.77, 78 More intriguing, 20–40 % of patients treated for chronic inflammation associated with hepatitis C virus infection develop auto‐antibodies recognizing filamentous agglomerates of the protein IMPDH1.79 Interestingly, mutations on the same IMPDH1 enzyme (R224P and D226N) are associated with the autosomal dominant disease retinitis pigmentosa, one of the main causes of visual handicap in developed countries.80 IMPDH1 mutants seem to exhibit the same enzymatic activity as the wild‐type. However, a substantial decrease in protein solubility has been reported, which suggests a possible association between agglomeration and pathology.80
4.3. Agglomeration as a Means To Control Drug Release
The reversible nature of agglomerates has been harnessed as a way to control drug release. Rivera et al. engineered synthetic proteins consisting of insulin fused to tandem repeats of an FKBP12 mutant protein exhibiting a weak dimerization tendency. Dimerization events across multiple polypeptide chains led to amorphous agglomeration by phase separation in a manner described recently,84 and those agglomerates were sequestered in the endoplasmic reticulum. The inhibition of FKBP dimerization with a small molecule would then dissolve the agglomerates and allow insulin release in a controlled fashion.85
Agglomeration of insulin to slow down its release into the blood has been exploited in multiple strategies. In one strategy, three mutations were engineered to induce insulin assembly at neutral pH after injection, whereas insulin would remain soluble at pH 4 in the formulation.86, 87 In an alternative strategy, insulin agglomeration was achieved by covalent modifications of a surface‐exposed lysine residue:88 acylation at lysine 29 with a fatty diacid (hexadecanedioic acid linked through a γ‐l‐glutamate) promoted the stacking of hexamers into filaments termed multihexamers. Inhibition of the stacking by phenol maintained insulin in a low molecular weight form before injection, but diffusion of phenol in the subcutaneous environment allowed insulin to polymerize into a “molecular depot” (Figure 6 f). Subsequently, progressive diffusion of zinc allowed disassembly and slow diffusion of insulin monomers into the blood.83, 89
5. Agglomeration of Natural Proteins
A growing number of studies are identifying proteins that agglomerate in response to environmental stresses90, 91, 92 such as starvation,13, 93, 94, 95 heat shock,96 or DNA damage.97 These assemblies often form in a reversible manner (Figure 2 d),13, 96 thus suggesting a role in cellular adaptation. It is also noteworthy that many of these assemblies form through a process of phase separation, a phenomenon that is stimulating a paradigm shift in the way we view and conceive proteome organization.23, 98 Importantly, the definitions of agglomeration and phase separation converge for some systems (e.g. salting out of proteins99). There is, however, one notable difference: in agglomeration, the identity and structure of protomers are well‐defined, whereas in phase‐separation, protomers can show large conformational and compositional heterogeneity.23, 84
5.1. Symmetry in Natural Filamentous Agglomerates
As we saw in Figure 5 and Section 3, symmetry is intimately tied to the formation of open‐ended assemblies. To highlight this notion, we reviewed filamentous assemblies (Table 1) and identified the quaternary structure found in each assembly. Although monomers represent 35 % and 55 % of proteins of known structure in E. coli and H. sapiens, respectively (Figure 4),43, 55, 100 less than 10 % of the proteins listed in Table 1 are monomeric themselves. In contrast, while only about 15 % of homomers of known structure show a dihedral symmetry,55, 100 more than 60 % of the proteins listed in Table 1 do. This over‐representation of internally symmetric complexes reflects the ease with which homotypic interfaces can evolve (Section 3.2) and that new self‐interactions among dihedral homomers often yield filamentous agglomerates (Section 3.3).
Table 1.
Natural filamentous assemblies.[a]
| Gene | Protein | Variant[b] | Organism | Structure (PDB) | Symmetry | Homologue[b] | Detection method | Ref. | |
|---|---|---|---|---|---|---|---|---|---|
| ACC1 | acetyl‐CoA carboxylase | Wt | S. cerevisiae | 5csk |

|
FM (stationary) | 94 | ||
| ASN1 | asparagine synthetase | Wt | S. cerevisiae | NA |

|
48 % id to 1ct9 (C 2) |
FM (stationary) |
94 | |
| ASN2 | asparagine synthetase | Wt | S. cerevisiae | NA |

|
49 % id to 1ct9 (C 2) |
FM (stationary) |
94 | |
| GDB1 | glycogen debranching enzyme | Wt | S. cerevisiae | NA |

|
71 % id to 5D06 (C 1) |
FM (stationary) |
94 | |
| GDH2 | glutamate dehydrogenase | Wt | S. cerevisiae | NA |

|
37 % id to 1l1f (D 3) |
FM (stationary) |
94 | |
| PFK1 PFK2 |
phosphofructokinase | Wt | S. cerevisiae | 3o8o |

|
FM (stationary) |
94 | ||
| (C 2, pseudo D2) | |||||||||
| TSA1 | thioredoxin peroxidase | Wt | S. cerevisiae | 3sbc |

|
FM (stationary) |
94 | ||
| SUI2 | translation initiation factor | Sui2p | Wt | S. cerevisiae | NA |

|
PDB:6ezo (C 2) |
FM (stationary and log phase) |
95 |
| GCN3 | eIF2B‐α | Wt | S. cerevisiae | NA | 95 | ||||
| GCD7 | eIF2B‐β | Wt | S. cerevisiae | NA | 95 | ||||
| GCD1 | eIF2B‐γ | Wt | S. cerevisiae | NA | note: each “monomer” unit contains all proteins | 94 | |||
| GCD2 | eIF2B‐δ | Wt | S. cerevisiae | NA | 95 | ||||
| GCD6 | eIF2B‐ϵ | Wt | S. cerevisiae | NA | 95 | ||||
| GLN1 | glutamine synthase | Wt | S. cerevisiae | 3Fky |

|
FM (starvation) | 93 | ||
| GLT1 | glutamate synthase | Wt | S. cerevisiae | NA |

|
60 % id to 2vdc (D 3) | FM (stationary) |
95 | |
| PSA1 | GDP‐mannose pyrophosphorylase | Wt | S. cerevisiae | NA | NA | FM (stationary) | 95 | ||
| URA7 | major CTP synthase isozyme | Wt | S. cerevisiae | NA |

|
59 % id to 5u03 (D 2) |
FM (stationary) |
95 | |
| URA8 | minor CTP synthase isozyme | Wt | S. cerevisiae | NA |

|
57 % id to 5u03 (D2) |
FM (stationary) |
95 | |
| PYRG | CTP synthase | Wt | E. coli | 5ktv |

|
FM in vivo, EM in vitro | 120 | ||
| CTPS1 | CTP synthase | Wt | H. sapiens | 2ad5 |

|
IFM in vivo, EM in vitro | 71, 73 | ||
| HBA1 HBA2 |
hemoglobin | E6V | H. sapiens | 1hho |

|
EM in vivo and in vitro | 121, 122 | ||
| (C 2, pseudo D 2) | |||||||||
| GLS | glutaminase C | Wt | H. sapiens | 3voz |

|
EM in vitro | 112 | ||
| HBB | hemoglobin | E22V | O. virginianus | 1hds |

|
EM in vivo | 59, 123 | ||
| (C 2, pseudo D 2) | |||||||||
| SOD1 | superoxide dismutase | H46R | H. sapiens | 4ff9 |

|
crystallography in vitro | 17 | ||
| REG1 | lithostatine | truncation in the N‐term | H. sapiens | 1lit |

|
IHC in vivo and EM and AFM in vitro | 82, 124 | ||
| KIR2DL1 | killer cell Ig‐like receptor | Wt | H. sapiens | NA |

|
SEM in vivo and EM in vitro | 125 | ||
| IMPD2 | inosine monophosphate dehydrogenase | Wt | H. sapiens | 1nf7 |

|
IFM in vivo and EM in vitro | 107 | ||
| PFK1 | phosphofructokinase | Wt | H. sapiens | 4xyj |

|
FM in vivo and EM in vitro | 110 | ||
| GLUD | glutamate dehydrogenase | Wt | B. taurus | 3jcz |

|
EM in vitro | 126 | ||
| GLU1 | β‐glucosidase | Wt | A. sativa | not deposited |

|
EM in vitro | 108 | ||
| ADK1 | adenylate kinase | Wt | Z. mais | 1zak |

|
crystallography in vitro | 103 | ||
[a] The assemblies listed correspond to those formed by proteins that are ordinarily monomeric or form a finite complex. This table does not consider proteins whose sole function involves filament formation, such as actin or tubulin. The gene name and description of each protein identified as forming filaments are given along with its function, the organism, the corresponding structure in the PDB or, if only a homologue is available, the percentage sequence identity shared with the homologue. The symmetry of the protein unit forming the filament (or that of the closest homologue) is shown schematically and the method used to detect it indicated (EM: electron microscopy; AFM: atomic force microscopy; FM: fluorescence microscopy; IFM: immunofluorescence microscopy; IHC: immunohistochemistry) as well as the culture condition in which the filaments were observed. [b] id=identity, NA=not available, Wt=wild‐type.
5.2. Agglomeration and Protein Function
Agglomeration can be induced solely by changes in the environment,101, 102 thus making it a fast and energy‐efficient mechanism for stress response compared to transcription.90, 92 In particular, agglomerates can serve as molecular depots of inactive enzymes that can be disassembled when conditions allow growth93 or a metabolic activity such as photosynthesis to be resumed.103 For example, starvation induces glutamine synthetase in S. cerevisiae to assemble into catalytically inactive filaments.93 Although the molecular mechanisms for the formation of filament and punctate structures upon entry into the stationary phase are largely uncharacterized, it was found that acidification of the cytoplasm can be a trigger in numerous cases,101 and co‐solutes may also play a role.104 The fact that filaments frequently occur upon nutrient depletion is consistent with a “molecular depot” function. Nonetheless, filament assembly does not necessarily lead to catalytic inactivation. For example, CTPS forms filaments that are catalytically active in eukaryotes and inactive in prokaryotes.73, 105, 106 Similarly, IMPDH can assemble into filaments that adopt both active and inactive conformations, shifting from one to the other upon binding to GTP and other substrates.107
A possible burden for the catalytic function of a protein agglomerate is the reduced accessibility of substrates to the active site of the enzyme. However, this handicap can be turned into an asset. In oat β‐glycosidase, the active site of the enzyme is located in a central tunnel formed by a filament, and although filament formation limits substrate accessibility, it also limits its diffusion once it enters into the tunnel, thereby resulting in an increased apparent affinity for its natural substrate. Additionally, the filament increases specificity towards the substrates, as the width of the tunnel acts as a molecular sieve to discriminate the avenacosides from other kinds of β‐glucosides.108
Binding to a substrate can also trigger filament formation of certain proteins. Two examples are acetyl‐CoA carboxylase (ACC)109 and phosphofructokinase (PFK1),110 whose polymerization appears to be promoted by citrate.109, 111 Similarly, the glutaminase inhibitor BPTES induces the dissociation of the glutaminase C filaments and stabilizes the inactive tetrameric form.112
5.3. Agglomeration as a Mechanism for Evolutionary Innovation and its Impact on Fitness
Symmetry has long been harnessed by evolution to generate novel folds, as seen in the TIM barrel and ß‐propeller folds, for example.113, 114 Similarly, in agglomerates, new protein interfaces may create new functionalities such as active sites,115 as seen in natural enzymes.116
More intriguingly, Garcia‐Seisdedos et al. observed that mutations increasing the surface stickiness of homomers frequently resulted in a change of their localization in budding yeast. Whereas all of the wild‐type homomers were expressed in the cytosol, numerous point mutants localized to the nucleus and one formed agglomerates localized at the bud neck.12 These results indicate that proteins can exhibit complex and unexpected behaviors at the cellular level when they agglomerate.
Furthermore, protein agglomerates may create opportunities for the colocalization of other macromolecules and, thereby, seed new functions.117 More straightforwardly, agglomeration can modulate the availability and function of proteins by sequestering them into a confined space. Such a mechanism has been reported for transcription factors containing glutamine‐rich repeats. The expansion of these repeats can induce the transcription factor to self‐assemble, thereby decreasing its activity through sequestration.118 In a similar vein, agglomerates may form phenotypes with deleterious functions that sequester molecular species required for normal cellular function.119
6. Agglomeration for the Design of Biomaterials
Precise control over the structure of materials is a central goal of materials science. Either unstructured127, 128, 129, 130 or fully folded peptides and proteins can be used as building blocks to self‐assemble materials from the bottom‐up. The use of agglomeration for the design of materials provides several benefits. First of all, folded proteins have a well‐defined surface topology that enables precise control over the mode of association. Secondly, the retention of the protein fold can confer catalytic or binding properties to the material. Finally, the reversible nature of agglomeration (Figure 2) reduces the potential for the formation of kinetically trapped intermediates. Additionally, reversibility opens‐up a much bigger opportunity: the design of dynamic materials capable of shifting morphology and of self‐healing. Key to the design of agglomerated state materials are the assembly rules illustrated in Figure 5 c and also described in other reviews.41, 131, 132 Lessons learned from the evolution36, 55, 133, 134, 135, 136, 137, 138, 139, 140 and from the design141, 142, 143, 144, 145 of soluble closed‐symmetry assemblies can be applied to constructing materials by designing outward‐facing interfaces, thus creating assemblies with open symmetries.
6.1. Agglomerates from Coiled‐Coil Protomers
Given the central role of point‐group symmetry in the formation of infinite assemblies, the periodic and symmetric structure of coiled coils makes them ideal building blocks for material design. Moreover, the typical sequence length of the peptide chains (25 to 50 amino acids)146 makes their synthesis attainable by solid‐phase methods. The first example of a synthetic peptide characterized as forming an open‐ended assembly in the folded state was an α‐helical coiled coil that stacked into one‐dimensional fibers.147 Although the sequence of this peptide was not selected for self‐assembly, the observation that self‐assembly was achievable without extensive planning illustrated that agglomeration is a common feature of folded peptides and proteins, as discussed in Section 2.
The serendipitous discovery that coiled coils could readily self‐assemble led to efforts to design folded‐state peptide materials rationally. Woolfson and co‐workers rationally designed coiled‐coil units with protruding uncomplemented sticky ends for unit–unit interactions, which led to their association into filaments.148 This strategy was widely replicated in the design of biocompatible materials.146, 149, 150, 151, 152 The assembly of a helical bundle into filaments by way of surface interactions (as opposed to using “sticky ends”) was achieved later.153 Interestingly, this assembly was designed to be pH‐sensitive and provided a proof‐of‐concept that folded proteins could be designed to self‐assemble into open‐ended filaments by careful consideration of the chemical properties of the surface residues.
In the absence of “sticky ends,” the hydrophobic positions at both extremities of the coiled‐coil bundles provide a natural surface for promoting intermolecular interactions. Conticello et al. exploited this feature to design a heptameric coiled coil prone to self‐assemble by end‐to‐end stacking.154 The large diameter of the 7‐helical oligomer they used created a wide hydrophobic face on the blunt ends of the bundle which, following surface optimization, self‐assembled into infinite fibers. The fibrils were extensively characterized, and their structure was found to be consistent with stacked heptameric coiled coils. This design strategy was subsequently used to illustrate that folded proteins can retain their function in the agglomerated state, with the design of a helical bundle capable of self‐assembling into fibers while retaining the ability to bind curcumin.155 Woolfson and co‐workers expanded on this strategy and generalized it to coiled coils with different oligomerization states, as illustrated in Figure 7 a,b.156 This set of coiled coils was further used as a fusion to a protein cage to create a protein matrix capable of forming in vivo.157
Figure 7.

Engineered agglomerates forming filamentous, planar, amorphous, or crystalline assemblies. a) and b) One‐dimensional assemblies from coiled‐coil peptide subunits by face‐to‐back stacking characterized by TEM with negative staining.156 c) Homomers with dihedral symmetry stack following point mutations solely designed to increase their surface hydrophobicity. Stacks were visualized by TEM with negative staining.12 d) A computationally designed two‐dimensional lattice of homomers with C 6 symmetry interacting by noncovalent interactions, characterized by TEM with negative staining.175 e) An auxetic two‐dimensional lattice of homomers with C 4 symmetry assembled through disulfide bonds. The assembly is seen by TEM with negative staining.176 f) Liquid protein droplet assembled from octahedral ferritin subunits harboring a covalent modification mediating ferritin–ferritin interactions. The droplets were observed by optical microscopy.165 g) Three‐dimensional protein crystal computationally designed from trimeric coiled‐coil subunits and assembled by noncovalent interactions. The image shows an electron density map obtained from crystal X‐ray diffraction experiments.22 Images were reproduced from Ref. 156 [(a,b), Copyright 2013, American Chemical Society], Ref. 12 [(c), Copyright 2017, Springer Nature], Ref. 175 [(d), Copyright 2015, American Association for the Advancement of Science], Ref. 176 [(e), Copyright 2017, Springer Nature], Ref. 165 [(f), Copyright 2009, John Wiley and Sons], and Ref. 22 (g) with permission.
The use of coiled coils as subunits of self‐assembling materials has also been applied to two‐dimensional and three‐dimensional arrays. Saven and co‐workers used a computational approach to identify a sequence variant of a 3‐helix bundle that would self‐interact to form a crystalline lattice with a specific geometry (Figure 7 g).22 This computational approach was also used to design robust arrays of two‐dimensional helical bundles that could tolerate variability in the solution conditions as well as chemical modification of the termini.158, 159
6.2. Agglomerates from Protomers Interacting through Bridging
Notable progress has been achieved through the use of coiled coils as units for self‐assembly. However, the limited structural and functional diversity of coiled‐coil bundles has motivated the use of naturally occurring proteins as alternative building blocks. Indeed, naturally occurring proteins offer a large diversity of shapes and functions, but are difficult to redesign because of their rugged folding landscape and the difficult nature of interface design. The strategies reviewed herein circumvent these difficulties and involve little or no surface redesign. Instead, they are based on bridging pre‐existing symmetric homomers. The bridges can be very diverse, from small molecules to peptides, or can be achieved by genetic fusion of two distinct homomers. The length, branching, and flexibility of the bridging species are key in dictating the resulting structure.
In an early example, Dotan et al. used a lectin with D 2 symmetry as a building block, and a twofold symmetric ligand as a bridge between them. Mixing the two in 1:2 ratio triggered their assembly in a diamond‐like geometry.160 This approach has been formalized and generalized by Yeates and co‐workers,144 who described how 1D, 2D, and 3D materials can be assembled depending on the symmetries of the building blocks and that of the bridge.
6.2.1. Bridging Protomers with Surface Binders
Molecules capable of binding protein surfaces can serve as bridges to create interactions between subunits. For example, multivalent cyclic small molecules that bind to specific types of amino acids can serve as versatile molecular glues to induce polymerization.161, 162 The surfaces of protein targets can be modified with non‐natural amino acids to serve as recognition elements for such small‐molecule polymerizers.163 In a different but related approach, a surface lysine of insulin was covalently modified with a carbohydrate‐binding functional group.164 The presence of a carbohydrate subsequently allowed multiple hexamers of insulin to bridge with each other, thereby leading to their agglomeration.
6.2.2. Assembly into a Low‐Ordered Phase
Highly flexible bridging species will produce only short‐range order and can result in amorphous agglomerates, as in phase separation. Surface‐modified ferritin has been used to create such infinite assemblies displaying short‐range order.165 For example, ferritin was used to create a solvent‐free protein liquid by chemically modifying its surface to create long‐range ionic interactions between the protomers, as illustrated in Figure 7 f.165a
6.2.3. Assembly into One‐Dimensional and Branched Filaments
Directionality and long‐range order can be imposed by rigid linkers. Yeates and co‐workers genetically fused two dimeric proteins by a rigid α‐helix, the orientation of which was designed to promote one‐dimensional propagation.144
Hayashi and co‐workers engineered heme binding proteins covalently bound to heme cofactors.166 The cofactor of one unit bound to the heme binding pocket of another unit, thereby resulting in infinite polymerization. The incorporation of three‐way linkers with heme moieties allowed for branched propagation. In addition, heteromeric structures composed of myoglobin and streptavidin could be constructed through the use of linkers containing both heme and biotin moieties.
Ward and co‐workers also took advantage of the internal D 2 symmetry of streptavidin. They created a linear ligand with biotin groups that dimerized through coordination bonds to iron(II), thus leading to one‐dimensional assembly, the formation of which was dependent on the presence of iron.167 In a different approach, Brunsveld and co‐workers used a ligand capable of stacking into filaments and showed it could act as a scaffold for proteins by way of its appended biotin moieties.168, 169
6.2.4. Assembly into Two‐Dimensional Lattices
The formation of long‐range order in two dimensions will produce periodic planar lattices. However, periodicity over long length scales is difficult to achieve given the potential for the formation of local defects. In that respect, as for one‐dimensional assembly, the rigidity and directionality of the linker are of crucial importance.
Ringler and Schulz combined C 4‐symmetric l‐rhamnulose‐1‐phosphate aldolase (RhuA) with D 2‐symmetric streptavidin to build a two‐dimensional material.170 RhuA was decorated with biotin on its surface to present a binding interface for streptavidin, which served as a bridge to mediate the interaction of RhuA units. The tether to biotin was chosen to be short to maintain the rigidity of the connection and enabled the formation of two‐dimensional networks.
Noble and co‐workers fused multimeric proteins, while considering the compatibility of the internal symmetries in the individual complexes. Complexes that shared a common symmetry operation were selected so that the large‐scale structure of the material could be planned out by specifying the location of each subunit within the 2D lattice.171 By selecting proteins in orientations that would position the point‐of‐fusion termini close to an axis for rotation, the size of the fusion linker could be limited to only two amino acids, thus preventing local defects.
6.2.5. Assembly into Three‐Dimensional Lattices
As indicated in the introduction of this section, the dihedral symmetry of lectin tetramers has been harnessed in the creation of a diamond‐like protein material.160 In this design, the length of the linker between the sugar subunits was carefully selected to ensure that no alternative assembly types would be adopted.
Jiang and co‐workers cemented the idea that the types of materials that can be obtained depend on the flexibility of the bridging molecule.172, 173, 174 In one example, D 2‐symmetric lectin subunits were made to associate by way of a linker composed of a galactose moiety for binding to the lectin as well as a rhodamine B moiety, which dimerized. Changing the length of the linker between the two moieties shifted the dimensionality of the assembly dramatically and produced oligomerization in one, two, or three dimensions.174
6.3. Agglomerates of Protomers Interacting through Designed Interfaces
Recently, advances in computational modeling and an increased understanding of protein interfaces have enabled the design of protein‐based materials by interface design. A protein fiber designed exclusively by mutation was created by Schulz and co‐workers.177 Starting from the cyclic tetramer RhuA, two distinct sets of mutations induced homotypic interactions and stacking into a dihedral octamer, either at the top or the bottom surface of RhuA. When the two sets of mutations were combined (i.e. top and bottom together), this led to an open‐ended assembly by sequential stacking, as illustrated in Figure 5 c (C 4→D 4→filament). Later, Garcia‐Seisdedos et al. generalized this approach and showed that mutations close to the top‐most or bottom‐most surface of large dihedral complexes (D 4 and D 5) and solely designed to increase surface hydrophobicity were often sufficient to trigger stable face‐to‐face stacking into filaments, visible both in vitro and in vivo.12 Similarly, a protein of unknown function named Hcp1 was observed to form C 6 homomers stacking face‐to‐back in the crystal lattice. Mougous and co‐workers stabilized the crystallographic interface using cysteine mutations, which led the protein to form nanotubes in solution.178
Baker and co‐workers have used a computational approach for the design of materials mediated by interactions between globular protein domains.175, 179 For example, from a library of naturally occurring protein complexes with cyclic symmetries, configurations of assemblies within layer space group arrangements with shape complementary of the interface were identified.175a Computational protein design was used to stabilize the selected configurations by mutation. The structure of the lattices, solved using cryo‐EM and electron diffraction, confirmed that the interface geometry could be designed de novo with high precision (Figure 7 d). Remarkably, this approach relaxes the need for an existing binding site and paves the way to the design of 2D materials with any protein and any desired geometry.
6.4. Agglomerates of Protomers Interacting through Designed Metal Coordination and Disulfide Bonds
Tezcan and co‐workers pioneered the design of protein assemblies mediated by metal coordination. They were initially able to build several types of assemblies with closed symmetries by introducing novel metal‐binding sites on the surface of folded proteins.180, 181 The lessons learned from the design of cyclic protein assemblies were then applied to design crystalline materials from natively folded subunits.182, 183 Dynamic features were also incorporated into crystalline materials.176 Cysteine residues introduced at the surface of tetrameric RhuA mediated its assembly into a two‐dimensional lattice upon oxidation (Figure 7 e). Remarkably, these lattices showed dynamic but uniform morphologies, thus indicating that local changes in orientation between RhuA units were able to propagate through the material.
Most recently, Tezcan and co‐workers engineered a system that allowed reversible ferritin agglomeration.184 In this system, a ferritin variant previously designed to crystallize through engineered calcium binding contacts was used. Crystals of this variant were soaked in a solution of poly(acrylate‐acrylamide) precursors, and their polymerization formed a hydrogel matrix within and around the crystals. Expansion of the hydrogel matrix by water absorption followed by dehydration recovered the original structure of the crystal lattice and even improved the resolution of the X‐ray diffraction data. More striking even, the crystal was able to self‐heal fractures occurring during the expansion and contraction cycles.
7. Summary and Outlook
Protein agglomeration connects chemistry and biology through two central features: macromolecular interactions and symmetry. Continued efforts in the characterization of agglomeration will have far‐reaching implications for our understanding of protein function and how it impacts cells, both in healthy and diseased organisms. Additionally, these efforts are paving the way for the design of new types of biomaterials.41, 185, 186 Impressive progress has already been made in the last two decades in terms of agglomerate design, and recent advances in fluorescence microscopy techniques have revealed a multitude of natural protein agglomerates (Table 1). The function of these agglomerates and their potential implication in disease remain poorly characterized, in part because of the difficulty in altering a protein's agglomeration properties in predictable ways. In that respect, advances in protein design could drive a better understanding of agglomerate biology, by allowing the design of mutants constitutively promoting or inhibiting assembly, or by identifying general properties of agglomeration, for example, the fact that agglomerates are highly evolvable from homomers with dihedral symmetry.12
Conversely, assembly rules could be inferred from structural analyses of biological agglomerates, for example, to identify how negative design helps funnel the assembly and promote order over long length scales. Such analyses could also help understand how protein assemblies that are open‐ended can sometimes adopt specific and finite sizes, as in tripeptidyl peptidase II.187 When designing functionalized agglomerates, biological agglomerates may also serve as models to design substrate channels or molecular sieves that increase substrate specificity. In these endeavors, the molecular basis of agglomerates will need to be characterized, which will be facilitated by the development of new cryo‐electron microscopy techniques to characterize protein assemblies in vivo.188
Lastly, agglomerates can easily be dismissed as artifactual aggregation (e.g. when observing precipitation in vitro or punctate structures in vivo). We hope this Review will stimulate awareness of the existence and high likelihood of the agglomeration process in the protein realm, and prompt scientists to actively characterize any potential forms of agglomeration they encounter in test tubes and living cells. Recently, for example, a bacterial protein was found to form cross‐α helical stacks reminiscent of—but different from—the classic amyloid structure involving cross‐β sheets,189 which indicates that new and unexpected types of agglomerates are waiting to be discovered.
Glossary
Protein–protein interface: Contact surface between two proteins.
Homotypic interface: Interface involving two identical protein surfaces related by a twofold symmetry axis.
Heterotypic interface: Interface formed by two distinct protein surfaces.
Protomer: Constituent subunit or groups of subunits within a protein assembly.11 In a homodimer, each polypeptide chain is a protomer. In hemoglobin, each (αβ) pair is a protomer.
Closed assembly or closed symmetry: Protein assembly in which protomers are related by point group rotational symmetry. Such assembly is finite, and all protomers are in equivalent (or quasi‐equivalent) chemical environments.
Open‐ended assembly or open symmetry: Protein assembly that involves translational symmetry between protomers. Assembly by this mode can continue indefinitely, unlike closed assembly. Not all protomers are in equivalent chemical environments. Some protomers exhibit unsatisfied interfaces, potentially leading to open‐ended (infinite) assembly.
Aggregate: Open‐ended assembly of proteins interacting through misfolded regions.
Agglomerate: Open‐ended assembly of protomers interacting through surface regions that retain their native or near‐native fold upon binding. Although the Latin root “glomus” describes a spherical object, we employ agglomerate irrespective of the assembly geometry (amorphous, 1D, 2D, or 3D).
Conflict of interest
The authors declare no conflict of interest.
Biographical Information
Héctor García Seisdedos obtained his PhD in 2012 at the University of Granada, Spain, working on the evolution and design of protein function with Prof. Jose M. Sanchez‐Ruiz. Currently, he is a postdoctoral fellow at the Weizmann Institute of Science in Israel in the group of Dr. Levy, where he is studying how proteins self‐assemble into high order structures from a physical, evolutionary, and cellular perspective.

Biographical Information
José Villegas received his PhD in 2018 from the University of Pennsylvania, USA, working on the computational design of protein–ligand and protein–protein interactions with Prof. Jeffery G. Saven. He is currently a postdoctoral fellow at the Weizmann Institute of Science in the group of Dr. Levy, where he works on elucidating structure–function relationships at protein–protein interfaces.

Biographical Information
Emmanuel Levy received his PhD in 2008 from Cambridge University, UK, working at the MRC Laboratory of Molecular Biology on the classification, evolution, and assembly of protein complexes, with Dr. Sarah Teichmann. He carried out postdoctoral research with Prof. Stephen Michnick at the University of Montreal in Canada, using yeast genetics and high‐throughput methods to characterize protein–protein interactions. In 2012 he became assistant professor at the Weizmann Institute in Israel. His group focuses on understanding biophysical and evolutionary properties of protein–protein interactions, from atomic to cellular scales.

Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank Ulyana Shimanovich, Boris Rybtchinski, Jeffrey Peterson, Yaakov Levy, Justin Kollman, Thomas Høeg‐Jensen, Deborah Fass, and the two anonymous reviewers for their comments on this manuscript, as well as Amnon Horovitz and Yaakov Levy for insightful discussions. H.G.‐S. received support from the Koshland Foundation and a McDonald‐Leapman Grant. J.A.V. is supported by a Fulbright Postdoctoral Fellowship awarded by the United States‐Israel Educational Foundation (USIEF) and a Zuckerman Postdoctoral Scholarship awarded by the Zuckerman STEM Leadership Program. This work was supported by the Israel Science Foundation and the I‐CORE Program of the Planning and Budgeting Committee (grant nos. 1775/12, 2179/14, and 1452/18), by the Marie Curie CIG Program (project no. 711715), by a research grant from the Estelle Funk Foundation, the Estate of Fannie Sherr, the Estate of Albert Delighter, the Merle S. Cahn Foundation, Mrs. Mildred S. Gosden, the Estate of Elizabeth Wachsman, and the Arnold Bortman Family Foundation.
H. Garcia-Seisdedos, J. A. Villegas, E. D. Levy, Angew. Chem. Int. Ed. 2019, 58, 5514.
References
- 1. Wilson E. B., Science 1899, 10, 33–45. [DOI] [PubMed] [Google Scholar]
- 2. Ahnert S. E., Marsh J. A., Hernández H., Robinson C. V., Teichmann S. A., Science 2015, 350, aaa2245. [DOI] [PubMed] [Google Scholar]
- 3. Ruepp A., Brauner B., Dunger-Kaltenbach I., Frishman G., Montrone C., Stransky M., Waegele B., Schmidt T., Doudieu O. N., Stümpflen V., Mewes H. W., Nucleic Acids Res. 2008, 36, D646–D650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pu S., Wong J., Turner B., Cho E., Wodak S. J., Nucleic Acids Res. 2009, 37, 825–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fletcher D. A., Mullins R. D., Nature 2010, 463, 485–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kramer R. M., Shende V. R., Motl N., Pace C. N., Scholtz J. M., Biophys. J. 2012, 102, 1907–1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fink A. L., Fold. Des. 1998, 3, R9–R23. [DOI] [PubMed] [Google Scholar]
- 8. Knowles T. P., Vendruscolo M., Dobson C. M., Nat. Rev. Mol. Cell Biol. 2014, 15, 384–396. [DOI] [PubMed] [Google Scholar]
- 9. Eisenberg D., Jucker M., Cell 2012, 148, 1188–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cohen R. D., Pielak G. J., Protein Sci. 2017, 26, 403–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Monod J., Wyman J., Changeux J. P., J. Mol. Biol. 1965, 12, 88–118. [DOI] [PubMed] [Google Scholar]
- 12. Garcia-Seisdedos H., Empereur-Mot C., Elad N., Levy E. D., Nature 2017, 548, 244–247. [DOI] [PubMed] [Google Scholar]
- 13. Narayanaswamy R., Levy M., Tsechansky M., Stovall G. M., O'Connell J. D., Mirrielees J., Ellington A. D., Marcotte E. M., Proc. Natl. Acad. Sci. USA 2009, 106, 10147–10152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hofmeister F., Naunyn-Schmiedeberg′s Arch. Pharmacol. 1888, 25, 1–30. [Google Scholar]
- 15. Wingfield P., Curr. Protoc. Protein Sci. 2001, A.3F1–A.3F.8. [Google Scholar]
- 16. Eaton W. A., Hofrichter J., Adv. Protein Chem. 1990, 40, 63–279. [DOI] [PubMed] [Google Scholar]
- 17. Elam J. S., Taylor A. B., Strange R., Antonyuk S., Doucette P. A., Rodriguez J. A., Hasnain S. S., Hayward L. J., Valentine J. S., Yeates T. O., Hart P. J., Nat. Struct. Biol. 2003, 10, 461–467. [DOI] [PubMed] [Google Scholar]
- 18. Guo Z., Eisenberg D., Proc. Natl. Acad. Sci. USA 2006, 103, 8042–8047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sambashivan S., Liu Y., Sawaya M. R., Gingery M., Eisenberg D., Nature 2005, 437, 266–269. [DOI] [PubMed] [Google Scholar]
- 20. Hamada D., Tanaka T., Tartaglia G. G., Pawar A., Vendruscolo M., Kawamura M., Tamura A., Tanaka N., Dobson C. M., J. Mol. Biol. 2009, 386, 878–890. [DOI] [PubMed] [Google Scholar]
- 21. Leuenberger P., Ganscha S., Kahraman A., Cappelletti V., Boersema P. J., von Mering C., Claassen M., Picotti P., Science 2017, 355(6327), 10.1126/science.aai7825. [DOI] [PubMed] [Google Scholar]
- 22. Lanci C. J., MacDermaid C. M., Kang S.-G., Acharya R., North B., Yang X., Qiu X. J., DeGrado W. F., Saven J. G., Proc. Natl. Acad. Sci. USA 2012, 109, 7304–7309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Banani S. F., Lee H. O., Hyman A. A., Rosen M. K., Nat. Rev. Mol. Cell Biol. 2017, 18, 285–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Torreira E., Moreno-Del Álamo M., Fuentes-Perez M. E., Fernández C., Martín-Benito J., Moreno-Herrero F., Giraldo R., Llorca O., Structure 2015, 23, 183–189. [DOI] [PubMed] [Google Scholar]
- 25. Levy E. D., De S., Teichmann S. A., Proc. Natl. Acad. Sci. USA 2012, 109, 20461–20466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. De Greef T. F. A., Smulders M. M. J., Wolffs M., Schenning A. P. H. J., Sijbesma R. P., Meijer E. W., Chem. Rev. 2009, 109, 5687–5754. [DOI] [PubMed] [Google Scholar]
- 27. Krieg E., Bastings M. M. C., Besenius P., Rybtchinski B., Chem. Rev. 2016, 116, 2414–2477. [DOI] [PubMed] [Google Scholar]
- 28. Yu H., Dee D. R., Liu X., Brigley A. M., Sosova I., Woodside M. T., Proc. Natl. Acad. Sci. USA 2015, 112, 8308–8313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Vekilov P. G., Soft Matter 2010, 6, 5254–5272. [Google Scholar]
- 30. Lawrence M. S., Phillips K. J., Liu D. R., J. Am. Chem. Soc. 2007, 129, 10110–10112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rüdiger S., Germeroth L., Schneider-Mergener J., Bukau B., EMBO J. 1997, 16, 1501–1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Doyle S. M., Genest O., Wickner S., Nat. Rev. Mol. Cell Biol. 2013, 14, 617–629. [DOI] [PubMed] [Google Scholar]
- 33. Sontag E. M., Vonk W. I. M., Frydman J., Curr. Opin. Cell Biol. 2014, 26, 139–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Arakawa T., Timasheff S. N., Methods Enzymol. 1985, 114, 49–77. [DOI] [PubMed] [Google Scholar]
- 35. Dey S., Levy E. D., in Protein Complex Assembly: Methods and Protocols (Ed.: J. A. Marsh), Springer, New York, 2018, pp. 357–375. [Google Scholar]
- 36. Levy E. D., J. Mol. Biol. 2010, 403, 660–670. [DOI] [PubMed] [Google Scholar]
- 37. Klara S. S., Saboe P. O., Sines I. T., Babaei M., Chiu P.-L., DeZorzi R., Dayal K., Walz T., Kumar M., Mauter M. S., J. Am. Chem. Soc. 2016, 138, 28–31. [DOI] [PubMed] [Google Scholar]
- 38. Doye J. P., Louis A. A., Vendruscolo M., Phys. Biol. 2004, 1, P9–P13. [DOI] [PubMed] [Google Scholar]
- 39. Pechmann S., Levy E. D., Tartaglia G. G., Vendruscolo M., Proc. Natl. Acad. Sci. USA 2009, 106, 10159–10164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Claverie P., Hofnung M., Monod J., C. R. Seances Acad. Sci. 1968, 1616–1618. [Google Scholar]
- 41. Yeates T. O., Liu Y., Laniado J., Curr. Opin. Struct. Biol. 2016, 39, 134–143. [DOI] [PubMed] [Google Scholar]
- 42. Caspar D. L., Klug A., Cold Spring Harbor Symp. Quant. Biol. 1962, 27, 1–24. [DOI] [PubMed] [Google Scholar]
- 43. Goodsell D. S., Olson A. J., Annu. Rev. Biophys. Biomol. Struct. 2000, 29, 105–153. [DOI] [PubMed] [Google Scholar]
- 44. Janin J., Bahadur R. P., Chakrabarti P., Q. Rev. Biophys. 2008, 41, 133–180. [DOI] [PubMed] [Google Scholar]
- 45. Chothia C., Janin J., Nature 1975, 256, 705–708. [DOI] [PubMed] [Google Scholar]
- 46. Wodak S. J., Janin J., Adv. Protein Chem. 2002, 61, 9–73. [DOI] [PubMed] [Google Scholar]
- 47. Deremble C., Lavery R., Curr. Opin. Struct. Biol. 2005, 15, 171–175. [DOI] [PubMed] [Google Scholar]
- 48. Reichmann D., Rahat O., Cohen M., Neuvirth H., Schreiber G., Curr. Opin. Struct. Biol. 2007, 17, 67–76. [DOI] [PubMed] [Google Scholar]
- 49. Keskin O., Gursoy A., Ma B., Nussinov R., Chem. Rev. 2008, 108, 1225–1244. [DOI] [PubMed] [Google Scholar]
- 50. Wales D. J., Chem. Phys. Lett. 1998, 285, 330–336. [Google Scholar]
- 51. Lukatsky D. B., Zeldovich K. B., Shakhnovich E. I., Phys. Rev. Lett. 2006, 97, 178101. [DOI] [PubMed] [Google Scholar]
- 52. Lukatsky D. B., Shakhnovich B. E., Mintseris J., Shakhnovich E. I., J. Mol. Biol. 2007, 365, 1596–1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. André I., Strauss C. E. M., Kaplan D. B., Bradley P., Baker D., Proc. Natl. Acad. Sci. USA 2008, 105, 16148–16152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Schulz G. E., J. Mol. Biol. 2010, 395, 834–843. [DOI] [PubMed] [Google Scholar]
- 55. Levy E. D., Boeri Erba E., Robinson C. V., Teichmann S. A., Nature 2008, 453, 1262–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Pauling L., Itano H. A., Singer S. J., Wells I. C., Science 1949, 110 (2865), 543–548. [DOI] [PubMed] [Google Scholar]
- 57. Charache S., Conley C. L., Waugh D. F., Ugoretz R. J., Spurrell J. R., J. Clin. Invest. 1967, 46, 1795–1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Vekilov P. G., Feeling-Taylor A. R., Petsev D. N., Galkin O., Nagel R. L., Hirsch R. E., Biophys. J. 2002, 83, 1147–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Esin A., Bergendahl L. T., Savolainen V., Marsh J. A., Warnecke T., Nat. Ecol. Evol. 2018, 2, 367–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Graw J., Int. J. Dev. Biol. 2004, 48, 1031–1044. [DOI] [PubMed] [Google Scholar]
- 61. Boatz J. C., Whitley M. J., Li M., Gronenborn A. M., van der Wel P. C. A., Nat. Commun. 2017, 8, 15137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Pande A., Annunziata O., Asherie N., Ogun O., Benedek G. B., Pande J., Biochemistry 2005, 44, 2491–2500. [DOI] [PubMed] [Google Scholar]
- 63. Kmoch S., Brynda J., Asfaw B., Bezouška K., Novák P., Řezáčová P., Ondrová L., Filipec M., Sedláček J., Elleder M., Hum. Mol. Genet. 2000, 9, 1779–1786. [DOI] [PubMed] [Google Scholar]
- 64. Rosen D. R., Siddique T., Patterson D., Figlewicz D. A., Sapp P., Hentati A., Donaldson D., Goto J., O'Regan J. P., Deng H. X., Nature 1993, 362, 59–62. [DOI] [PubMed] [Google Scholar]
- 65. Saccon R. A., Bunton-Stasyshyn R. K. A., Fisher E. M. C., Fratta P., Brain 2013, 136, 2342–2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. McAlary L., Yerbury J. J., Aquilina J. A., Sci. Rep. 2013, 3, 3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Bosco D. A. et al., Nat. Neurosci. 2010, 13, 1396–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Chiti F., Dobson C. M., Nat. Chem. Biol. 2009, 5, 15–22. [DOI] [PubMed] [Google Scholar]
- 69. Pratt A. J. et al., Proc. Natl. Acad. Sci. USA 2014, 111, E4568–E4576. [Google Scholar]
- 70. Broom H. R., Rumfeldt J. A. O., Meiering E. M., Essays Biochem. 2014, 56, 149–165. [DOI] [PubMed] [Google Scholar]
- 71. Chang C.-C., Jeng Y.-M., Peng M., Keppeke G. D., Sung L.-Y., Liu J.-L., Exp. Cell Res. 2017, 361, 292–299. [DOI] [PubMed] [Google Scholar]
- 72. Keppeke G. D., Calise S. J., Chan E. K. L., Andrade L. E. C., J. Genet. Genomics 2015, 42, 287–299. [DOI] [PubMed] [Google Scholar]
- 73. Lynch E. M., Hicks D. R., Shepherd M., Endrizzi J. A., Maker A., Hansen J. M., Barry R. M., Gitai Z., Baldwin E. P., Kollman J. M., Nat. Struct. Mol. Biol. 2017, 24, 507–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Chang C.-C., Lin W.-C., Pai L.-M., Lee H.-S., Wu S.-C., Ding S.-T., Liu J.-L., Sung L.-Y., J. Cell Sci. 2015, 128, 3550–3555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Williams J. C., Kizaki H., Weber G., Morris H. P., Nature 1978, 271, 71–73. [DOI] [PubMed] [Google Scholar]
- 76. Jackson R. C., Weber G., Morris H. P., Nature 1975, 256, 331–333. [DOI] [PubMed] [Google Scholar]
- 77. Laurine E. et al., J. Biol. Chem. 2003, 278, 51770–51778. [DOI] [PubMed] [Google Scholar]
- 78. Ho M.-R., Lou Y.-C., Lin W.-C., Lyu P.-C., Huang W.-N., Chen C., J. Biol. Chem. 2006, 281, 33566–33576. [DOI] [PubMed] [Google Scholar]
- 79. Keppeke G. D., Calise S. J., Chan E. K. L., Andrade L. E. C., World J. Gastroenterol. 2016, 22, 1966–1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Aherne A. et al., Hum. Mol. Genet. 2004, 13, 641–650. [DOI] [PubMed] [Google Scholar]
- 81. Harrington D. J., Adachi K., W. E. Royer Jr , J. Mol. Biol. 1997, 272, 398–407. [DOI] [PubMed] [Google Scholar]
- 82. Grégoire C., Marco S., Thimonier J., Duplan L., Laurine E., Chauvin J., Michel B., Peyrot V., Verdier J., EMBO J. 2001, 20, 3313–3321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Steensgaard D. B., Schluckebier G., Strauss H. M., Norrman M., Thomsen J. K., Friderichsen A. V., Havelund S., Jonassen I., Biochemistry 2013, 52, 295–309. [DOI] [PubMed] [Google Scholar]
- 84. Li P. et al., Nature 2012, 483, 336–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Rivera V. M., Wang X., Wardwell S., Courage N. L., Volchuk A., Keenan T., Holt D. A., Gilman M., Orci L., F. Cerasoli Jr , Rothman J. E., Clarkson T., Science 2000, 287, 826–830. [DOI] [PubMed] [Google Scholar]
- 86. Campbell R. K., White J. R., Levien T., Baker D., Clin. Ther. 2001, 23, 1938–1957; discussion 1923. [DOI] [PubMed] [Google Scholar]
- 87. Coppolino R., Coppolino S., Villari V., J. Pharm. Sci. 2006, 95, 1029–1034. [DOI] [PubMed] [Google Scholar]
- 88. Jonassen I., Havelund S., Ribel U., Plum A., Loftager M., Hoeg-Jensen T., Volund A., Markussen J., Pharm. Res. 2006, 23, 49–55. [DOI] [PubMed] [Google Scholar]
- 89. Jonassen I., Havelund S., Hoeg-Jensen T., Steensgaard D. B., Wahlund P.-O., Ribel U., Pharm. Res. 2012, 29, 2104–2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Minsky A., Shimoni E., Frenkiel-Krispin D., Nat. Rev. Mol. Cell Biol. 2002, 3, 50–60. [DOI] [PubMed] [Google Scholar]
- 91. O'Connell J. D., Zhao A., Ellington A. D., Marcotte E. M., Annu. Rev. Cell Dev. Biol. 2012, 28, 89–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Rabouille C., Alberti S., Curr. Opin. Cell Biol. 2017, 47, 34–42. [DOI] [PubMed] [Google Scholar]
- 93. Petrovska I. et al., Elife 2014, 10.7554/eLife.02409. [DOI] [Google Scholar]
- 94. Shen Q.-J., Kassim H., Huang Y., Li H., Zhang J., Li G., Wang P.-Y., Yan J., Ye F., Liu J.-L., J. Genet. Genomics 2016, 43, 393–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Noree C., Sato B. K., Broyer R. M., Wilhelm J. E., J. Cell Biol. 2010, 190, 541–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Wallace E. W. J. et al., Cell 2015, 162, 1286–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Tkach J. M. et al., Nat. Cell Biol. 2012, 14, 966–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Hyman A. A., Weber C. A., Julicher F., Annu. Rev. Cell Dev. Biol. 2014, 30, 39–58. [DOI] [PubMed] [Google Scholar]
- 99. Dumetz A. C., Chockla A. M., Kaler E. W., Lenhoff A. M., Biophys. J. 2008, 94, 570–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Levy E. D., Teichmann S., Prog. Mol. Biol. Transl. Sci. 2013, 117, 25–51. [DOI] [PubMed] [Google Scholar]
- 101. Munder M. C. et al., Elife 2016, 5, 10.7554/eLife.09347. [DOI] [Google Scholar]
- 102. Sukenik S., Ren P., Gruebele M., Proc. Natl. Acad. Sci. USA 2017, 114, 6776–6781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Wild K., Grafmüller R., Wagner E., Schulz G. E., Eur. J. Biochem. 1997, 250, 326–331. [DOI] [PubMed] [Google Scholar]
- 104. Patel A., Malinovska L., Saha S., Wang J., Alberti S., Krishnan Y., Hyman A. A., Science 2017, 356, 753–756. [DOI] [PubMed] [Google Scholar]
- 105. Barry R. M. et al., Elife 2014, 3, e03638. [Google Scholar]
- 106. Strochlic T. I., Stavrides K. P., Thomas S. V., Nicolas E., O'Reilly A. M., Peterson J. R., EMBO Rep. 2014, 15, 1184–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Anthony S. A., Burrell A. L., Johnson M. C., Duong-Ly K. C., Kuo Y.-M., Simonet J. C., Michener P., Andrews A., Kollman J. M., Peterson J. R., Mol. Biol. Cell 2017, 28, 2589–2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Kim S.-Y., Kim Y.-W., Hegerl R., Cyrklaff M., Kim I.-S., J. Struct. Biol. 2005, 150, 1–10. [DOI] [PubMed] [Google Scholar]
- 109. Beaty N. B., Lane M. D., J. Biol. Chem. 1983, 258, 13051–13055. [PubMed] [Google Scholar]
- 110. Webb B. A., Dosey A. M., Wittmann T., Kollman J. M., Barber D. L., J. Cell Biol. 2017, 216, 2305–2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Kim C.-W., Moon Y.-A., Park S. W., Cheng D., Kwon H. J., Horton J. D., Proc. Natl. Acad. Sci. USA 2010, 107, 9626–9631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Ferreira A. P. S. et al., J. Biol. Chem. 2013, 288, 28009–28020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Eck R. V., Dayhoff M. O., Science 1966, 152, 363–366. [DOI] [PubMed] [Google Scholar]
- 114. Romero Romero M. L., Rabin A., Tawfik D. S., Angew. Chem. Int. Ed. 2016, 55, 15966–15971; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 16198–16203. [Google Scholar]
- 115. Song W. J., Tezcan F. A., Science 2014, 346, 1525–1528. [DOI] [PubMed] [Google Scholar]
- 116. Ali M. H., Imperiali B., Bioorg. Med. Chem. 2005, 13, 5013–5020. [DOI] [PubMed] [Google Scholar]
- 117. Kuriyan J., Eisenberg D., Nature 2007, 450, 983–990. [DOI] [PubMed] [Google Scholar]
- 118. Gemayel R. et al., Mol. Cell 2015, 59, 615–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Bolognesi B., Lorenzo Gotor N., Dhar R., Cirillo D., Baldrighi M., Tartaglia G. G., Lehner B., Cell Rep. 2016, 16, 222–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Ingerson-Mahar M., Briegel A., Werner J. N., Jensen G. J., Gitai Z., Nat. Cell Biol. 2010, 12, 739–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Dykes G. W., Crepeau R. H., Edelstein S. J., J. Mol. Biol. 1979, 130, 451–472. [DOI] [PubMed] [Google Scholar]
- 122. Crepeau R. H., Edelstein S. J., Szalay M., Benesch R. E., Benesch R., Kwong S., Edalji R., Proc. Natl. Acad. Sci. USA 1981, 78, 1406–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Taylor W. J., Simpson C. F., Blood 1974, 43, 907–914. [PubMed] [Google Scholar]
- 124. Duplan L. et al., Neurobiol. Aging 2001, 22, 79–88. [DOI] [PubMed] [Google Scholar]
- 125. Kumar S., Rajagopalan S., Sarkar P., Dorward D. W., Peterson M. E., Liao H.-S., Guillermier C., Steinhauser M. L., Vogel S. S., Long E. O., Mol. Cell 2016, 62, 21–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Munn E. A., Biochim. Biophys. Acta Protein Struct. 1972, 285, 301–313. [DOI] [PubMed] [Google Scholar]
- 127. Petka W. A., Harden J. L., McGrath K. P., Wirtz D., Tirrell D. A., Science 1998, 281, 389–392. [DOI] [PubMed] [Google Scholar]
- 128. Wong Po Foo C. T. S., Lee J. S., Mulyasasmita W., Parisi-Amon A., Heilshorn S. C., Proc. Natl. Acad. Sci. USA 2009, 106, 22067–22072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Sathaye S., Zhang H., Sonmez C., Schneider J. P., MacDermaid C. M., Von Bargen C. D., Saven J. G., Pochan D. J., Biomacromolecules 2014, 15, 3891–3900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Li D., Jones E. M., Sawaya M. R., Furukawa H., Luo F., Ivanova M., Sievers S. A., Wang W., Yaghi O. M., Liu C., Eisenberg D. S., J. Am. Chem. Soc. 2014, 136, 18044–18051. [DOI] [PubMed] [Google Scholar]
- 131. Lai Y.-T., King N. P., Yeates T. O., Trends Cell Biol. 2012, 22, 653–661. [DOI] [PubMed] [Google Scholar]
- 132. Norn C. H., André I., Curr. Opin. Struct. Biol. 2016, 39, 39–45. [DOI] [PubMed] [Google Scholar]
- 133. Perica T., Kondo Y., Tiwari S. P., McLaughlin S. H., Kemplen K. R., Zhang X., Steward A., Reuter N., Clarke J., Teichmann S. A., Science 2014, 346, 1254346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Marsh J. A., Hernández H., Hall Z., Ahnert S. E., Perica T., Robinson C. V., Teichmann S. A., Cell 2013, 153, 461–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Perica T., Chothia C., Teichmann S. A., Proc. Natl. Acad. Sci. USA 2012, 109, 8127–8132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Akiva E., Itzhaki Z., Margalit H., Proc. Natl. Acad. Sci. USA 2008, 105, 13292–13297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Hashimoto K., Panchenko A. R., Proc. Natl. Acad. Sci. USA 2010, 107, 20352–20357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Cohen-Khait R., Dym O., Hamer-Rogotner S., Schreiber G., Structure 2017, 25, 1867–1874. [DOI] [PubMed] [Google Scholar]
- 139. Bershtein S., Mu W., Wu W., Shakhnovich E. I., Proc. Natl. Acad. Sci. USA 2012, 109, 4857–4862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Bennett M. J., Schlunegger M. P., Eisenberg D., Protein Sci. 1995, 4, 2455–2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Fallas J. A. et al., Nat. Chem. 2017, 9, 353–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. King N. P., Sheffler W., Sawaya M. R., Vollmar B. S., Sumida J. P., André I., Gonen T., Yeates T. O., Baker D., Science 2012, 336, 1171–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. King N. P., Bale J. B., Sheffler W., McNamara D. E., Gonen S., Gonen T., Yeates T. O., Baker D., Nature 2014, 510, 103–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Padilla J. E., Colovos C., Yeates T. O., Proc. Natl. Acad. Sci. USA 2001, 98, 2217–2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Pulsipher K. W., Villegas J. A., Roose B. W., Hicks T. L., Yoon J., Saven J. G., Dmochowski I. J., Biochemistry 2017, 56, 3596–3606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Potekhin S. A., Melnik T. N., Popov V., Lanina N. F., Vazina A. A., Rigler P., Verdini A. S., Corradin G., Kajava A. V., Chem. Biol. 2001, 8, 1025–1032. [DOI] [PubMed] [Google Scholar]
- 147. Kojima S., Kuriki Y., Yoshida T., Yazaki K., Miura K.-I., Proc. Jpn. Acad. Ser. B 1997, 73, 7–11. [Google Scholar]
- 148. Pandya M. J., Spooner G. M., Sunde M., Thorpe J. R., Rodger A., Woolfson D. N., Biochemistry 2000, 39, 8728–8734. [DOI] [PubMed] [Google Scholar]
- 149. Zimenkov Y., Conticello V. P., Guo L., Thiyagarajan P., Tetrahedron 2004, 60, 7237–7246. [Google Scholar]
- 150. Wagner D. E., Phillips C. L., Ali W. M., Nybakken G. E., Crawford E. D., Schwab A. D., Smith W. F., Fairman R., Proc. Natl. Acad. Sci. USA 2005, 102, 12656–12661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Zimenkov Y., Dublin S. N., Ni R., Tu R. S., Breedveld V., Apkarian R. P., Conticello V. P., J. Am. Chem. Soc. 2006, 128, 6770–6771. [DOI] [PubMed] [Google Scholar]
- 152. Dublin S. N., Conticello V. P., J. Am. Chem. Soc. 2008, 130, 49–51. [DOI] [PubMed] [Google Scholar]
- 153. Dong H., Paramonov S. E., Hartgerink J. D., J. Am. Chem. Soc. 2008, 130, 13691–13695. [DOI] [PubMed] [Google Scholar]
- 154. Xu C., Liu R., Mehta A. K., Guerrero-Ferreira R. C., Wright E. R., Dunin-Horkawicz S., Morris K., Serpell L. C., Zuo X., Wall J. S., Conticello V. P., J. Am. Chem. Soc. 2013, 135, 15565–15578. [DOI] [PubMed] [Google Scholar]
- 155. Hume J., Sun J., Jacquet R., Renfrew P. D., Martin J. A., Bonneau R., Gilchrist M. L., Montclare J. K., Biomacromolecules 2014, 15, 3503–3510. [DOI] [PubMed] [Google Scholar]
- 156. Burgess N. C., Sharp T. H., Thomas F., Wood C. W., Thomson A. R., Zaccai N. R., Brady R. L., Serpell L. C., Woolfson D. N., J. Am. Chem. Soc. 2015, 137, 10554–10562. [DOI] [PubMed] [Google Scholar]
- 157. Lee M. J., Mantell J., Hodgson L., Alibhai D., Fletcher J. M., Brown I. R., Frank S., Xue W.-F., Verkade P., Woolfson D. N., Warren M. J., Nat. Chem. Biol. 2018, 14, 142–147. [DOI] [PubMed] [Google Scholar]
- 158. Zhang H. V., Polzer F., Haider M. J., Tian Y., Villegas J. A., Kiick K. L., Pochan D. J., Saven J. G., Sci Adv 2016, 2, e1600307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Tian Y., Zhang H. V., Kiick K. L., Saven J. G., Pochan D. J., Org. Biomol. Chem. 2017, 15, 6109–6118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Dotan N., Arad D., Frolow F., Freeman A., Angew. Chem. Int. Ed. 1999, 38, 2363–2366; [PubMed] [Google Scholar]; Angew. Chem. 1999, 111, 2512–2515. [Google Scholar]
- 161. Rennie M. L., Doolan A. M., Raston C. L., Crowley P. B., Angew. Chem. Int. Ed. 2017, 56, 5517–5521; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 5609–5613. [Google Scholar]
- 162. Alex J. M., Rennie M. L., Volpi S., Sansone F., Casnati A., Crowley P. B., Cryst. Growth Des. 2018, 18, 2467–2473. [Google Scholar]
- 163. Guagnini F., Antonik P. M., Rennie M. L., O'Byrne P., Khan A. R., Pinalli R., Dalcanale E., Crowley P. B., Angew. Chem. Int. Ed. 2018, 57, 7126–7130; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 7244–7248. [Google Scholar]
- 164. Hoeg-Jensen T., Havelund S., Nielsen P. K., Markussen J., J. Am. Chem. Soc. 2005, 127, 6158–6159. [DOI] [PubMed] [Google Scholar]
- 165.
- 165a. Perriman A. W., Cölfen H., Hughes R. W., Barrie C. L., Mann S., Angew. Chem. Int. Ed. 2009, 48, 6242–6246; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 6360–6364; [Google Scholar]
- 165b. Bellapadrona G., Sinkar S., Sabanay H, Liljeström V., Kostiainen M., Elbaum M., Biomacromolecules 2015, 16, 2006–2011, https://pubs.acs.org/doi/abs/10.1021/acs.biomac.5b00435. [DOI] [PubMed] [Google Scholar]
- 166. Oohora K., Onoda A., Hayashi T., Chem. Commun. 2012, 48, 11714–11726. [DOI] [PubMed] [Google Scholar]
- 167. Burazerovic S., Gradinaru J., Pierron J., Angew. Chem. Int. Ed. 2007, 46, 5510–5514; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 5606–5610. [Google Scholar]
- 168. Müller M. K., Petkau K., Brunsveld L., Chem. Commun. 2011, 47, 310–312. [DOI] [PubMed] [Google Scholar]
- 169. Petkau-Milroy K., Sonntag M. H., Colditz A., Brunsveld L., Int. J. Mol. Sci. 2013, 14, 21189–21201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Ringler P., Schulz G. E., Science 2003, 302, 106–109. [DOI] [PubMed] [Google Scholar]
- 171. Sinclair J. C., Davies K. M., Venien-Bryan C., Noble M. E., Nat. Nanotechnol. 2011, 6, 558–562. [DOI] [PubMed] [Google Scholar]
- 172. Sakai F., Yang G., Weiss M. S., Liu Y., Chen G., Jiang M., Nat. Commun. 2014, 5, 4634. [DOI] [PubMed] [Google Scholar]
- 173. Yang G. et al., J. Am. Chem. Soc. 2016, 138, 1932–1937. [DOI] [PubMed] [Google Scholar]
- 174. Yang G., Ding H.-M., Kochovski Z., Hu R., Lu Y., Ma Y.-Q., Chen G., Jiang M., Angew. Chem. Int. Ed. 2017, 56, 10691–10695; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 10831–10835. [Google Scholar]
- 175.
- 175a. Gonen S., DiMaio F., Gonen T., Baker D., Science 2015, 348, 1365–1368; [DOI] [PubMed] [Google Scholar]
- 175b. Shen H. et al., Science 2018, 362, 705–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Suzuki Y., Cardone G., Restrepo D., Zavattieri P. D., Baker T. S., Tezcan F. A., Nature 2016, 533, 369–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Grueninger D., Treiber N., Ziegler M. O. P., Koetter J. W. A., Schulze M.-S., Schulz G. E., Science 2008, 319, 206–209. [DOI] [PubMed] [Google Scholar]
- 178. Ballister E. R., Lai A. H., Zuckermann R. N., Cheng Y., Mougous J. D., Proc. Natl. Acad. Sci. USA 2008, 105, 3733–3738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Matthaei J. F., DiMaio F., Richards J. J., Pozzo L. D., Baker D., Baneyx F., Nano Lett. 2015, 15, 5235–5239. [DOI] [PubMed] [Google Scholar]
- 180. Salgado E. N., Ambroggio X. I., Brodin J. D., Lewis R. A., Kuhlman B., Tezcan F. A., Proc. Natl. Acad. Sci. USA 2010, 107, 1827–1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Salgado E. N., Lewis R. A., Faraone-Mennella J., Tezcan F. A., J. Am. Chem. Soc. 2008, 130, 6082–6084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Brodin J. D., Ambroggio X. I., Tang C., Parent K. N., Baker T. S., Tezcan F. A., Nat. Chem. 2012, 4, 375–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Sontz P. A., Bailey J. B., Ahn S., Tezcan F. A., J. Am. Chem. Soc. 2015, 137, 11598–11601. [DOI] [PubMed] [Google Scholar]
- 184. Zhang L., Bailey J. B., Subramanian R. H., Tezcan F. A., Nature 2018, 557, 86–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Okesola B. O., Mata A., Chem. Soc. Rev. 2018, 47, 3721–3736. [DOI] [PubMed] [Google Scholar]
- 186. Luo Q., Dong Z., Hou C., Liu J., Chem. Commun. 2014, 50, 9997–10007. [DOI] [PubMed] [Google Scholar]
- 187. Chuang C. K., Rockel B., Seyit G., Walian P. J., Schönegge A.-M., Peters J., Zwart P. H., Baumeister W., Jap B. K., Nat. Struct. Mol. Biol. 2010, 17, 990–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.F. J. B. Bäuerlein, I. Saha, A. Mishra, M. Kalemanov, A. Martínez-Sánchez, R. Klein, I. Dudanova, M. S. Hipp, F. U. Hartl, W. Baumeister, R. Fernández-Busnadiego, Cell 2017, 171, 179–187.e10. [DOI] [PubMed]
- 189. Tayeb-Fligelman E., Tabachnikov O., Moshe A., Goldshmidt-Tran O., Sawaya M. R., Coquelle N., Colletier J.-P., Landau M., Science 2017, 355, 831–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary