

Abstract
Background
Quantitative genetic studies suggest the existence of variation at the genome level that affects the ability of cattle to resist to parasitic diseases. The objective of the current study was to identify regions of the bovine genome that are associated with resistance to endo-parasites.
Methods
Individual cattle records were available for Fasciola hepatica-damaged liver from 18 abattoirs. Deregressed estimated breeding values (EBV) for F. hepatica-damaged liver were generated for genotyped animals with a record for F. hepatica-damaged liver and for genotyped sires with a least one progeny record for F. hepatica-damaged liver; 3702 animals were available. In addition, individual cow records for antibody response to F. hepatica on 6388 genotyped dairy cows, antibody response to Ostertagia ostertagi on 8334 genotyped dairy cows and antibody response to Neospora caninum on 4597 genotyped dairy cows were adjusted for non-genetic effects. Genotypes were imputed to whole-sequence; after edits, 14,190,141 single nucleotide polymorphisms (SNPs) and 16,603,644 SNPs were available for cattle with deregressed EBV for F. hepatica-damaged liver and cows with an antibody response to a parasitic disease, respectively. Association analyses were undertaken using linear regression on one SNP at a time, in which a genomic relationship matrix accounted for the relationships between animals.
Results
Genomic regions for F. hepatica-damaged liver were located on Bos taurus autosomes (BTA) 1, 8, 11, 16, 17 and 18; each region included at least one SNP with a p value lower than 10−6. Five SNPs were identified as significant (q value < 0.05) for antibody response to N. caninum and were located on BTA21 or 25. For antibody response to F. hepatica and O. ostertagi, six and nine quantitative trait loci (QTL) regions that included at least one SNP with a p value lower than 10−6 were identified, respectively. Gene set enrichment analysis revealed a significant association between functional annotations related to the olfactory system and QTL that were suggestively associated with endo-parasite phenotypes.
Conclusions
A number of novel genomic regions were suggestively associated with endo-parasite phenotypes across the bovine genome and two genomic regions on BTA21 and 25 were associated with antibody response to N. caninum.
Electronic supplementary material
The online version of this article (10.1186/s12711-019-0457-7) contains supplementary material, which is available to authorized users.
Background
Parasitic diseases are highly prevalent worldwide in cattle production systems [1] and this prevalence is forecasted to increase further as a repercussion of the anticipated change in climate [2]. In Ireland, between 75.4% [3] and 83% [4] of the Irish dairy herds, show evidence of exposure to Fasciola hepatica. In addition, parasitic diseases are associated with significant economic losses in both dairy and beef cattle [5–7]. Details on endo-parasite diseases in cattle are in Sekiya et al. [1]. Using pedigree-based relationships between individuals, Twomey et al. [8] documented the presence of genetic variation for two phenotypes due to F. hepatica in cattle and proposed that selection and breeding animals resistant to F. hepatica would complement current control strategies for F. hepatica (i.e., anthelmintic treatments; [9]). Genetic variability has also been documented for other endo-parasites in cattle such as Ostertagia ostertagi [10, 11] and Neospora caninum [10, 12]. Since genetic gain is a function of selection accuracy [13], the accuracy of traditional genetic evaluations that exploit only pedigree relationships are hindered by the lack of available phenotypic data; this is especially true for low heritability traits. Although the study of Twomey et al. [8] was limited by the low accuracy of the genetic evaluations, they reported a 6% unit difference in the prevalence of F. hepatica-damaged liver between cows with an estimated breeding value (EBV) for F. hepatica in the top 10% versus the bottom 10%; the mean EBV reliability of those animals was 0.18. It is hypothesised that the inclusion of genomic information within these evaluations will improve the reliability of the EBV for F. hepatica and thus improve the prediction of whether or not an animal will have F. hepatica-damaged liver.
Few studies have attempted to locate genomic regions associated with endo-parasitic diseases in cattle. Using a genotyping panel of only 153 microsatellite markers, Coppieters et al. [11] documented that Bos taurus autosomes (BTA) 9 and 19 harboured quantitative trait loci (QTL) associated with nematode burden in 768 Dutch dairy cows that were selected to be in the top 10% and bottom 10% of their respective sire family (n = 12) for faecal egg count (FEC). Using 305 Aberdeen Angus calves genotyped for 190 microsatellite markers only, Kim et al. [14] reported that regions on BTA8 and 12 were associated with FEC. To the best of our knowledge, Kim et al. [15] is the only available study that used dense genome-wide SNP genotypes (31,165 SNPs after edits) to map genomic regions associated with endo-parasitic diseases in cattle; they reported that 12 regions in the bovine genome contributed to significant genetic variation in FEC.
The objective of our study was to identify single nucleotide polymorphisms (SNPs) associated with three endo-parasitic diseases in cattle using imputed whole-genome sequence data in a large multi-breed population. Results from this study will improve our understanding of the underlying genetic mechanisms of resistance in cattle to parasitic diseases. This could contribute to enhance the development of further control strategies for parasitic diseases, such as vaccine development, and to improve diagnostics of parasitic diseases. In addition, our results provide information that would be useful in future studies that attempt to increase the accuracy of genetic evaluations, through the exploration of targeted genomic regions.
Methods
The phenotypic data analysed in this study originated from two sources: (1) national abattoir cattle data containing records for F. hepatica-damaged liver, and (2) serological testing of individual cows in 68 dairy herds for endo-parasites.
Phenotypic data for F. hepatica-damaged liver
Individual animal records for F. hepatica-damaged liver were available from dairy and beef cattle that were slaughtered in 18 Irish abattoirs between February 2012 and February 2018. Cattle livers were diagnosed as either “liver exhibits F. hepatica damage and live F. hepatica observed in the liver” or “liver exhibits F. hepatica damage without the identifiable presence of live F. hepatica”. Animals were considered to have no F. hepatica-damaged liver if none of the above diagnostic elements were detected and were slaughtered in the same abattoir and at the same date than an animal diagnosed with a F. hepatica-damaged liver. Twomey et al. [8] described in detail the processes to generate F. hepatica-damaged liver phenotypes, and also the procedures and criteria imposed to maximise the likelihood that only animals exposed to the parasite were included in the analysis based on F. hepatica-damaged liver information available from their herd-mates. In summary, only animals with no herd movement after 90 days of age were retained. Animals were considered as exposed if they had resided in a herd 100 days prior to the slaughter of a herd-mate with live F. hepatica present in the liver. If the slaughtered herd-mate had a F. hepatica-damaged liver with no observable live F. hepatica, an additional criterion was imposed, i.e. as well as having resided in the herd 100 days prior to the slaughter of the slaughtered herd-mate, animals also had to be born within 100 days of the birth of the slaughtered herd-mate, to be considered as exposed.
For the purposes of our study, liver damage caused by F. hepatica was dichotomized; animals were considered either infected (i.e., observation of F. hepatica damage in the liver) or not infected with F. hepatica (i.e., liver did not exhibit F. hepatica damage). The final dataset consisted of 187,584 animals with a F. hepatica-damaged liver record, either positive or negative.
Breeding values and associated reliabilities for the binary trait of F. hepatica-damaged liver were estimated with an animal linear mixed model in the MIX99 genetic evaluation software suite [16]. The model and variance components specified in the genetic evaluations were those previously documented for the genetic evaluation of F. hepatica-damaged liver [8]. Fixed effects in the model were parity/age group, stage of lactation, heterosis and recombination coefficients, animal gender, herd-level contemporary group, and date-by-abattoir of slaughter. The random effects were the direct additive genetic effects, where with representing the additive variance and representing the numerator relationships matrix, and a residual effect, where with representing the residual variance and representing an identity matrix. The pedigree of each animal was traced back to the founder population, which was allocated to 11 genetic groups based on breed (i.e., Holstein, Friesian, Belgian Blue, Hereford, Aberdeen Angus, Simmental Limousin, Charolais, several other French breeds not listed, and other breeds not listed and unknown); the pedigree consisted of 865,173 animals. The genetic and the residual variances were equal to 0.001 and 0.115, respectively, as estimated by Twomey et al. [8] in a large subset of animals used in the present study. Only EBV for animals that had both a genotype and an EBV reliability higher than 0.01 were considered for further analyses. Moreover, 16,196 animals were retained if they had a record themselves for F. hepatica-damaged liver (n = 10,837) or were sires that had at least one progeny record for F. hepatica-damaged liver (n = 5359). The effective record contribution (ERC) for each animal was estimated and used as the weighting factor for the deregression of EBV. Deregression was undertaken using the Secant method in the MiX99 software [17]. Deregressed EBV for animals (i.e., a combination of animals with a record themselves and sires with at least one progeny record) with an ERC lower than 1 were not considered further; finally, 3702 animals remained for the analyses.
Phenotypic data for antibody response to endo-parasites
Blood samples were collected in autumn 2016 from 10,879 dairy cows as part of a cross-sectional seroprevalence study of 68 Irish dairy herds. All blood samples were tested separately for the presence of antibodies to F. hepatica using the Svanovir F. hepatica-Ab ELISA kit, to O. ostertagi using the Svanovir O. ostertagi-Ab ELISA kit, and to N. caninum using the Svanovir Neospora-Ab ELISA kit (Boehringer Ingelheim Svanova, Uppsala, Sweden). ELISA tests for all blood samples were carried out by the same commercial laboratory (FarmLab Diagnostics, Co. Roscommon, Ireland). Antibody responses to F. hepatica and O. ostertagi were expressed as optical density ratio (ODR), and antibody response to N. caninum was expressed as percent positivity (PP). Data edits were as described by Twomey et al. [10]. In summary, the positively skewed ODR for F. hepatica was transformed using the natural logarithm to approximate a normally distributed variable, whereas a reciprocal transformation was used to normalize the positively skewed PP for N. caninum. Records were removed if, at blood sampling, the cow resided in a herd different to that in which it had been present at 90 days of age. Only animals with a record for antibody response to F. hepatica were retained if they resided in a herd that had at least five cows positive for antibody response to F. hepatica (i.e., an ODR ≥ 0.4 was previously shown to lead to production losses [18] and, thus, this cut-off was regarded as the positive threshold for antibody response to F. hepatica in the current study) and a within-herd prevalence higher than 5% with a positive antibody response to F. hepatica on the day of the blood test. Since the majority of grazing cattle are exposed to O. ostertagi [19, 20] and since cows in our study were known to graze grass for most of the year, all cows were considered as exposed to O. ostertagi. For antibody response to N. caninum, we retained only the records as positive for cows that resided in a herd with a within-herd cow prevalence of more than 1% (i.e., ELISA test manufacturer stated that a PP ≥ 20 indicates a positive result for N. caninum, and was thus treated as such in our study) for antibody response to N. caninum. In addition, the likelihood that progeny from a cow infected with N. caninum will also be infected with N. caninum [19] is high, thus, we discarded progeny from dams that had a positive result for N. caninum (86 cows were discarded). The final dataset consisted of 6892 records for antibody response to F. hepatica, 9260 records for antibody response to O. ostertagi and 5289 records for antibody response to N. caninum.
Each observed animal phenotype for the continuous traits for antibody response to F. hepatica, O. ostertagi and N. caninum was adjusted for non-genetic effects. Genetic and non-genetic effects for antibody response to each of the three parasites were estimated using the MIX99 software suite [16]. The statistical model was as previously described by Twomey et al. [10]. Fixed effects for antibody response to each of the three parasites were parity, age relative to parity median, stage of lactation, heterosis and recombination coefficients, and contemporary group; random effects were the direct additive genetic effects, where with representing the additive variance and representing the numerator relationships matrix, and a residual effect, where with representing the residual variance and representing an identity matrix. The pedigree of each animal was traced back to the founder population, which was allocated to 11 genetic groups based on breed (i.e., Holstein, Friesian, Belgian Blue, Hereford, Aberdeen Angus, Simmental Limousin, Charolais, several other French breeds not listed, and other breeds not listed and unknown). Variance components were as previously described by Twomey et al. [10] for the current dataset; the fitted genetic variance was 0.095 for antibody response to F. hepatica, 0.003 for antibody response to O. ostertagi and 0.001 for antibody response to N. caninum, while the residual variance was 0.734 for F. hepatica, 0.034 for O. ostertagi and 0.010 for N. caninum, which resulted in heritability values of 0.12, 0.08 and 0.09, respectively. For each cow, phenotypes for the three parasites were adjusted for fixed effects (estimated with the linear mixed model) from the cow’s respective phenotype. Only animals that had both an adjusted phenotype and an available genotype were retained for further analysis; 6388 animals remained for F. hepatica, 8334 animals remained for O. ostertagi and 4597 animals remained for N. caninum.
Genotypes
Genotypes were available from six different genotyping panels, and the number of animals genotyped per panel is in Additional file 1: Table S1. In the dataset of deregressed EBV for F. hepatica-damaged liver, 87% of the animals were genotyped using either the Illumina bovine High-Density BeadChip (HD; n = 777,962 SNPs; 20% of animals) or the International Dairy and Beef version 1 (IDBv1; n = 17,137 SNPs; 10% of animals), version 2 (IDBv2; n = 18,004 SNPs; 32% of animals) or version 3 (IDBv3; n = 53,450 SNPs; 25% of animals) genotyping panels. In the dataset of adjusted phenotypes for antibody response to the three different phenotypes, most of the animals were genotyped on the IDBv1 (2% of animals), IDBv2 (76% of animals) or IDBv3 (14% of animals) genotyping panel. All animals had a call rate higher than 90% and only autosomal SNPs, SNPs with a reported chromosomal position, and SNPs with a call rate higher than 90% were retained for each panel.
All animals were imputed to HD using a two-step approach with the FImpute2 software [21]; first, the IDB genotyped animals were imputed to the Bovine SNP50 density (50 k) and subsequently all resulting genotypes (including the Bovine SNP50 genotypes) were imputed to HD using a multi-breed reference population of 5504 HD genotyped animals. Second, imputation to whole-genome sequence (WGS) was undertaken using a multi-breed reference population of 2333 Bos taurus animals from Run6.0 of the 1000 Bulls Genomes Project. All variants in the reference population were called using SAMtools, and genotype calls were improved using the Beagle software to provide a consensus SNP density across all animals. Details of the alignment to the UMD 3.1 Bos taurus assembly, variant calling and quality controls that were completed within the multi-breed sequence reference population are as described by Daetwyler et al. [22]. In total, 41.39 million SNPs in Run6.0 were identified across the genome and the average coverage (standard deviation) was 12.85 (6.94)X. Imputation of the HD genotypes to WGS was then completed by first phasing all HD genotypes using Eagle ([23]; version 2.3.2) and subsequently imputing to WGS using minimac3 [24].
Regions with poor WGS imputation accuracy, perhaps due to local mis-assemblies or mis-orientated contigs, were identified by using a larger dataset of 147,309 verified parent-progeny relationships. Mendelian errors, defined as the proportion of opposing homozygotes in a parent-progeny pair, were estimated for each relationship and the subsequent Mendelian error rate per SNP was calculated. To accurately identify genomic regions with poor imputation, the R package GenWin [25] which fits a β-spline to the data to find likely inflection points, was used to determine the breakpoints of genomic regions with high Mendelian errors. Windows were analysed using an initial window size of 5 kb and Genwin pooled windows for which the SNP Mendelian error rates were similar. The average SNP Mendelian error rate per window was estimated and all variants within windows in which the mean SNP Mendelian error rate was higher than 0.02 were removed (i.e. 687,137 SNPs were removed).
Further quality control edits were also imposed on the imputed sequence genotypes, including the removal of SNPs with a minor allele frequency (MAF) lower than 0.002 and of SNPs that deviated from Hardy–Weinberg equation (P < 6 × 10−6). After these edits, 14,190,141 SNPs remained for animals with a deregressed EBV for F. hepatica-damaged liver, whereas 16,603,644 SNPs remained for cows with an antibody response to a parasitic disease.
The final dataset of animals with both a deregressed EBV for F. hepatica-damaged and an imputed sequence genotype contained 3702 animals. The mean breed composition of the animals in the dataset of F. hepatica-damaged liver was 38% Holstein–Friesian, 17% Limousin, 11% Charolais, 11% Aberdeen Angus and the remaining 23% was another breed (Fig. 1). The final dataset of animals with both an adjusted phenotype for antibody response and an imputed sequence genotype contained 6388 animals for F. hepatica, 8334 animals for O. ostertagi and 4597 animals for N. caninum. In the dataset of antibody response to the three parasitic diseases, the mean breed composition of the cows was 84% Holstein–Friesian, 9% Jersey with the remaining 7% from another breed.
Fig. 1.
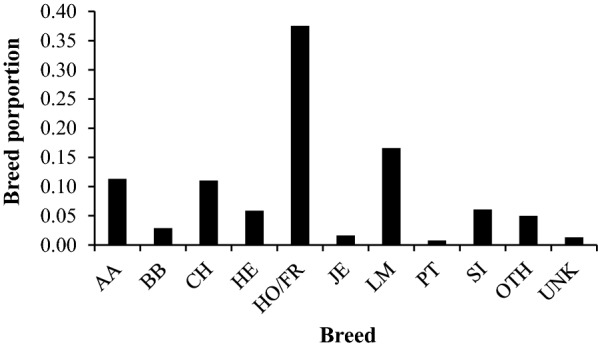
The mean breed composition of animals (n = 3702) in the dataset for F. hepatica-damaged liver for Aberdeen Angus (AA), Belgian Blue (BB), Charolais (CH), Hereford (HE), Holstein–Friesian (HO/FR), Jersey (JE), Limousin (LM), Parthenaise (PT), Simmental (SI), all other known breeds (OTH) and unknown breed (UNK)
Association analyses
A genomic relationship matrix was calculated separately for each of the traits using Method I of VanRaden [26] as:
where is an incidence matrix for the HD genotypes, centred by allele frequencies, and and are the dominant and recessive allele, respectively.
For all the traits in this study, SNP effects were estimated using linear mixed models in the WOMBAT software [27]. For each of the dependent variables, animal was included as a random effect with relationships between animals accounted for via the genomic relationship matrix. When the dependent variable was a deregressed EBV for F. hepatica-damaged liver, the phenotype was weighted based on the information available. The weight was calculated as [28]:
where is the weighting factor of the ith deregressed EBV, is the heritability estimate for F. hepatica-damaged liver, is the reliability of the ith deregressed EBV, and is the proportion of genetic variance not accounted for by the SNPs and was set at 0.9. Purfield et al. [29] tested various values of (i.e., 0.1, 0.2, 0.8 and 0.9) and reported that all had a minimal impact on the results. The proportions of Holstein–Friesian, Limousin, Charolais, Simmental, Aberdeen Angus, Belgian Blue, Parthenaise, Jersey and other breeds were fitted as covariates in the model for F. hepatica-damaged liver; a constraint was imposed by setting the effect of the Hereford breed to zero to avoid linear dependencies. The genomic inflation was estimated for each association analysis and all estimates were less than 1.1, i.e. 1.004 for F. hepatica-damaged liver, 1.0002 for antibody response to F. hepatica, 1.0133 for antibody response to O. osteragi and 1.0320 for antibody response to N. caninum. Multiple-testing correction was applied to each analysis by transforming the p values into their corresponding q values assuming a false discovery rate of 5% [30]. Therefore, SNPs with a q value lower than 0.05 were regarded as significant for the purpose of this study. Suggestive SNPs were defined as SNPs with a p value lower than 10−5.
Quantitative trait loci (QTL) regions
Using PLINK [31], QTL regions were defined around each SNP of interest, based on the flanking LD pattern. For each significant or suggestive SNP, its squared correlation () with all the other SNPs within 5 Mb upstream and 5 Mb downstream was calculated. Using a threshold of 0.5 for , the start of the QTL region was defined as the position of the SNP that was furthest upstream from the significant or suggestive SNP with an higher than 0.5; the end of the QTL region was defined as the position of the SNP that was furthest downstream with an with the suggestive or significant SNP higher than 0.5. For QTL regions that overlapped, QTL regions were combined into a single QTL region and maximum and minimum positions were set as start and end boundaries.
Annotation and gene set enrichment analysis
Gene search using Ensembl (http://ensembl.org) on the UMD 3.1 genome build was carried out by focusing on the defined QTL regions. In addition, protein-coding genes, which were identified either as overlapping with or located within QTL regions, were subject to enrichment analysis on The Database for Annotation, Visualization and Integrated Discovery (DAVID) v. 6.8. Functional annotations (Gene Ontology (GO) Biological Process, GO Cellular Component, GO Molecular Function and Kyoto Encyclopedia of Genes and Genomes (KEGG) and Reactome Pathways) were assigned to genes using the functional annotation tool. P values were calculated by EASE (an adoption of the Fisher Exact test to measure the gene-enrichment in annotation terms) to identify functional annotations that were associated with traits of interest.
Results
The mean effective record contribution (ERC) was equal to 4.53 (ranging from 1.01 to 277.69) in the dataset of F. hepatica-damaged liver (Fig. 2).
Fig. 2.
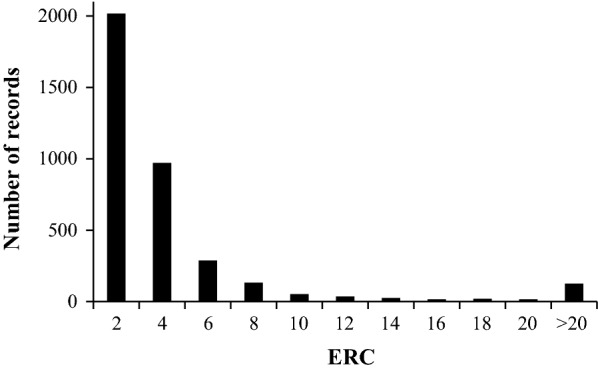
Distribution of effective record contributions (ERC) of animals in the dataset of deregressed F. hepatica-damaged liver EBV
F. hepatica-damaged liver
The associations between each SNP and F. hepatica-damaged liver are shown on the Manhattan plot in Fig. 3. We found no SNP with a q value lower than 0.05 for F. hepatica-damaged liver. Details of the most strongly associated SNPs (with a p value < 10−6) in each QTL region for F. hepatica-damaged liver are in Table 1. Six SNPs, located on BTA1, 8, 11, 16, 17 and 18, had a p value lower than 10−6 (Table 1); five of these six SNPs were classified as intronic variants in one of each of the FTO, AGPAT3, ENSBTAG00000017404, YWHAQ, or MLLT3 genes. The sixth SNP for F. hepatica-damaged liver was classified as an intergenic variant on BTA1. SNP rs384464701 on BTA18 was the most strongly associated with F. hepatica-damaged liver (p value = 1.92 × 10−8) and was classified as an intronic variant located in the FTO gene. The frequency of the favourable allele for each of the six SNPs with a p value lower than 10−6 ranged from 0.002 to 0.998 (Table 1). In total, 121 suggestive SNPs were identified (p value < 10−5) and were located across 23 chromosomes.
Fig. 3.

Manhattan plot showing − log10(p values) of the association between the effect of each single nucleotide polymorphism (n = 16,603,644 SNPs) and the deregressed estimated breeding value for F. hepatica-damaged liver. The blue line is the threshold for suggestive SNPs
Table 1.
Details of the most strongly associated SNPs (with a p value < 10−6) in each QTL region with deregressed EBV for F. hepatica-damaged liver
| BTA | SNP name | Pos (bp) | p value | Gene | Favourable allele (freq) | Effecta | Annotation | Start of QTL | End of QTL | Other genes in the QTL region |
|---|---|---|---|---|---|---|---|---|---|---|
| 18 | rs384464701 | 22,279,521 | 1.92 × 10−8 | FTO | A (0.988) | 0.09 | Intron | 22,279,521 | 22,279,521 | |
| 1 | – | 146,726,679 | 3.76 × 10−8 | AGPAT3 | G (0.998) | 0.18 | Intron | 146,465,269 | 14,6811,109 | PDXK, CSTB, RRP1, TRAPPC10 |
| 17 | rs110361447 | 71,554,867 | 1.26 × 10−7 | SEC14L2 | A (0.470) | 0.02 | Intron | 71,554,867 | 71,554,867 | |
| 16 | rs800135980 | 14,007,063 | 5.27 × 10−7 | – | A (0.002) | 0.19 | Intergenic | 14,007,063 | 14,007,063 | |
| 11 | rs384697649 | 87,844,503 | 7.79 × 10−7 | YWHAQ | T (0.996) | 0.13 | Intron | 87,844,503 | 87,844,503 | |
| 8 | rs109859381 | 24,008,154 | 8.79 × 10−7 | MLLT3 | T (0.888) | 0.02 | Intron | 24,008,154 | 24,008,154 |
BTA: Bos taurus chromosome number, SNP: name of the single nucleotide polymorphism, pos: position of base pair on the chromosome, p value: p value of the SNP, gene: gene name in which the SNP is located, favourable allele (freq): allele with favourable effect (frequency of the favourable allele), effect: effect of the favourable allele, annotation: annotation of the SNP, start of QTL: base pair on the chromosome at the start of the QTL, end of QTL: base pair on the chromosome at the end of the QTL, other genes in the QTL region: protein-coding genes located in the QTL region that the SNP is not located in
aBinary trait (0/1)
Forty-eight QTL regions were suggestively associated with F. hepatica-damaged liver (see Additional file 2: Table S2). On BTA7, 21 SNPs were suggestively associated with F. hepatica-damaged liver in a single QTL region that spanned from 65.23 to 67.30 Mb and this region included three protein-coding genes (NMUR2, GRIA1 and FAM114A2); the SNP with the strongest association in this region was rs475893299 (p value = 2.23 × 10−6). In the QTL region between 93.61 and 93.79 Mb on BTA11, 18 SNPs were suggestively associated with F. hepatica-damaged liver; all the genes located in this region encoded olfactory receptors (OR1J2, OR1N1, ENSBTAG00000047728, ENSBTAG00000047112, ENSBTAG00000046536, ENSBTAG00000045527, ENSBTAG00000045545, ENSBTAG00000020660, ENSBTAG00000038551, OR1Q1, and OR1B1); in addition, the only QTL region associated with antibody response to F. hepatica that overlapped with a QTL associated with F. hepatica-damaged liver was on BTA11 between 93.74 and 93.89 Mb.
Antibody response to endo-parasites
The associations between each SNP and antibody response to F. hepatica, O. ostertagi, and N. caninum are shown on the Manhattan plots in Fig. 4. Details of the most strongly associated SNPs (with a p value < 10−6) in each QTL region for antibody response to F. hepatica, O. ostertagi, and N. caninum are in Tables 2, 3 and 4, respectively.
Fig. 4.

Manhattan plot showing − log10(p values) of the association between the effect of each single nucleotide polymorphism and antibody response to a F. hepatica, b O. ostertagi, c N. caninum. The blue line is the threshold for suggestive SNPs
Table 2.
Details of the most strongly associated SNPs (with a p value < 10−6) in each QTL region with antibody response to F. hepatica
| BTA | SNP name | Pos (bp) | p value | Gene | Favourable allele (freq) | Effecta | Annotation | Start of QTL | End of QTL | Other genes in the QTL region |
|---|---|---|---|---|---|---|---|---|---|---|
| 14 | rs801151638 | 82,704,197 | 2.02 × 10−8 | – | A (0.988) | 0.42 | Intergenic | 80,104,349 | 83,212,505 | RALYL, SNX16, CHMP4C |
| 20 | rs382942594 | 21,005,570 | 1.77 × 10−7 | – | T (0.063) | 0.19 | Intergenic | 20,574,362 | 22,008,166 | RAB3C, ENSBTAG00000005229, GAPT, PLK2, ENSBTAG00000026505 |
| 6 | rs439064316 | 44,643,846 | 1.96 × 10−7 | – | C (0.008) | 0.46 | Intergenic | 44,643,846 | 44,643,846 | – |
| 15 | rs134727358 | 67,661,877 | 5.12 × 10−7 | PRR5L | G (0.003) | 0.66 | Upstream | 67,661,877 | 67,661,877 | – |
| 15 | rs379765497 | 23,526,098 | 5.44 × 10−7 | – | A (0.008) | 0.44 | Intergenic | 23,515,275 | 2,3581,425 | – |
| 18 | rs110443247 | 4,974,911 | 6.04 × 10−7 | – | C (0.375) | 0.08 | Intergenic | 4,974,911 | 5,158,840 | NUDT7, VAT1L |
BTA: Bos taurus chromosome number, SNP: name of the single nucleotide polymorphism, pos: position of base pair on the chromosome, p value: p value of the SNP, gene: gene name in which the SNP is located, favourable allele (freq): allele with favourable effect (frequency of the favourable allele), effect: effect of the favourable allele, annotation: annotation of the SNP, start of QTL: base pair on the chromosome at the start of the QTL, end of QTL: base pair on the chromosome at the end of the QTL, other genes in the QTL region: protein-coding genes located in the QTL region that the SNP is not located in
aBinary trait (0/1)
Table 3.
Details of the most strongly associated SNPs (with a p value < 10−6) in each QTL region with antibody response to O. osteratgi
| BTA | SNP name | Pos (bp) | p value | Gene | Favourable allele (freq) | Effecta | Annotation | Start of QTL | End of QTL | Other genes in the QTL region |
|---|---|---|---|---|---|---|---|---|---|---|
| 23 | rs455561091 | 33,302,807 | 3.61 × 10−7 | – | A (0.004) | 0.13 | Intergenic | 33,029,305 | 34,481,525 | GPLD1, MRS2, DCDC2, NRSN1, PRP4, PRP8, PRP6 |
| 21 | rs475710014 | 65,072,681 | 4.26 × 10−7 | – | A (0.054) | 0.03 | Intergenic | 65,072,681 | 65,072,681 | – |
| 14 | rs523857350 | 35,375,134 | 4.62 × 10−7 | – | A (0.005) | 0.11 | Intergenic | 35,375,134 | 35,740,556 | SULF1, ENSBTAG00000037399, SLCO5A1 |
| 4 | rs29026148 | 62,981,078 | 4.85 × 10−7 | – | A (0.650) | 0.02 | Intergenic | 62,981,078 | 62,993,200 | – |
| 19 | – | 7,584,712 | 5.43 × 10−7 | – | A (0.198) | 0.02 | Intergenic | 7,584,712 | 7,584,712 | – |
| 14 | rs439873982 | 7,893,299 | 7.67 × 10−7 | – | T (0.008) | 0.08 | Intergenic | 7,866,148 | 7,899,389 | – |
| 3 | – | 1,746,600 | 7.74 × 10−7 | GPA33 | T (0.004) | 0.12 | Intron | 945,572 | 6,388,604 | MPZL1, RCSD1, CREG1, CD247, POU2F1, DUSP27, MAEL, ILDR2, TADA1, POGK, ENSBTAG00000022504, ENSBTAG00000022954, FAM78B, UCK2, TMCO1, ALDH9A1, MGST3, LRRC52, RXRG, LMX1A, PBX1, ENSBTAG00000045987, ENSBTAG00000034449, NUF2, RGS5, RGS4 |
| 12 | rs379990763 | 9,621,581 | 8.25 × 10−7 | – | A (0.010) | 0.08 | Intergenic | 9,402,063 | 9,801,565 | – |
| 13 | rs209722101 | 70,057,265 | 9.50 × 10−7 | – | G (0.002) | 0.15 | Intergenic | 70,057,265 | 70,057,265 | – |
BTA: Bos taurus chromosome number, SNP: name of the single nucleotide polymorphism, pos: position of base pair on the chromosome, p value: p value of the SNP, gene: gene name in which the SNP is located, favourable allele (freq): allele with favourable effect (frequency of the favourable allele), effect: effect of the favourable allele, annotation: annotation of the SNP, start of QTL: base pair on the chromosome at the start of the QTL, end of QTL: base pair on the chromosome at the end of the QTL, other genes in the QTL region: protein-coding genes located in the QTL region that the SNP is not located in
aBinary trait (0/1)
Table 4.
Details of the most strongly associated SNPs (with a p value < 10−6) in each QTL with antibody response to N. caninum
| BTA | SNP name | Pos (bp) | p value | Gene | Favourable allele (freq) | Effecta | Annotation | Start of QTL | End of QTL | Number of genes in the QTL region |
|---|---|---|---|---|---|---|---|---|---|---|
| 25 | rs133449464 | 13,507,716 | 2.56 × 10−10 | PARN | T (0.005) | 0.09 | Intron | 13,037,560 | 14,102,058 | 9 |
| 21 | – | 55,835,765 | 2.26 × 10−9 | PPIP5K1 | A (0.003) | 0.11 | Intron | 55,069,089 | 56,273,280 | 29 |
| 1 | rs466868917 | 46,464,860 | 4.08 × 10−8 | NXPE3 | C (0.007) | 0.08 | Intron | 45,131,004 | 49,146,658 | 18 |
| 1 | rs381312356 | 111,186,754 | 1.24 × 10−7 | VEPH1 | C (0.039) | 0.03 | Intron | 106,928,970 | 113,261,468 | 32 |
| 9 | rs714447117 | 15,859,873 | 3.19 × 10−7 | ENSBTAG00000021175 | C (0.004) | 0.07 | Downstream | 15,859,873 | 15,859,873 | – |
| 3 | – | 10,934,190 | 3.89 × 10−7 | – | C (0.037) | 0.03 | Intergenic | 9,428,852 | 15,031,457 | 126 |
| 5 | rs208471794 | 70,717,913 | 7.34 × 10−7 | – | G (0.929) | 0.02 | Intergenic | 70,717,913 | 70,717,913 | – |
| 21 | rs478339448 | 9,874,248 | 7.83 × 10−7 | – | C (0.006) | 0.07 | Intergenic | 9,874,248 | 10,045,196 | – |
BTA: Bos taurus chromosome number, SNP: name of the single nucleotide polymorphism, pos: position of base pair on the chromosome, p value: p value of the SNP, gene: gene name in which the SNP is located, favourable allele (freq): allele with favourable effect (frequency of the favourable allele), effect: effect of the favourable allele, annotation: annotation of the SNP, start of QTL: base pair on the chromosome at the start of the QTL, end of QTL: base pair on the chromosome at the end of the QTL, other genes in the QTL region: protein-coding genes located in the QTL region that the SNP is not located in
aBinary trait (0/1)
F. hepatica
We did not detect any SNP that was significantly associated (q value < 0.05) with antibody response to F. hepatica. Fifty-seven SNPs had a p value lower than 10−6 and were located on BTA6 (1 SNP), BTA14 (1 SNP), BTA15 (43 SNPs), BTA18 (1 SNP) and BTA20 (11 SNPs). The most highly associated SNP was rs801151638 (p value = 2.02 × 10−8), which was classified as an intergenic variant on BTA14 (Table 2). The frequency of the favourable allele ranged from 0.003 to 0.988 for the 57 SNPs with a p value lower than 10−6. In total, 226 SNPs were detected as suggestively associated with antibody response to F. hepatica (p value < 10−5) and 37 QTL regions were identified for antibody response to F. hepatica; the largest QTL was 7,593,750 bp long (between 25.26 and 32.86 Mb) on BTA19; (see Additional file 3: Table S3). Some suggestive SNPs were not in linkage disequilibrium (LD; r2 < 0.5) with flanking SNPs, thus nine of the 37 detected QTL regions harboured only one SNP. Fifty SNPs were suggestively associated with antibody response to F. hepatica in the QTL region between 80.81 and 81.30 Mb on BTA16, which contained 4268 tested SNPs (average LD between 50 suggestive SNPs was 0.99); SNP rs434571591 had the strongest association with antibody response to F. hepatica (p value < 3.28 × 10−6). Five protein-coding genes were located between 80.8 and 81.30 Mb on BTA16, namely NR5A2, ENSBTAG00000047428, KIF14, DDX59, and CAMSAP2. In the QTL region between 23.52 and 23.58 Mb on BTA15, 43 of the 709 tested SNPs were detected as suggestively associated with antibody response to F. hepatica; no gene was identified in, or overlapped with, this QTL region.
O. ostertagi
We found no SNP significantly associated with antibody response to O. ostertagi (q value < 0.05), but nine SNPs were detected for antibody response to O. ostertagi with a p value lower than 10−6 (Table 3). Of these nine SNPs, seven had a frequency of the favourable allele lower than 0.1, and one SNP was classified as an intronic variant in the GPA33 gene and the remaining eight as intergenic variants. Forty-two QTL were identified as suggestively associated with antibody response to O. ostertagi (p value < 10−5; see Additional file 4: Table S4). In total, 134 SNPs were identified as suggestively associated with antibody response to O. ostertagi. The QTL region between 94.02 and 98.01 Mb on BTA12 harboured 21 SNPs suggestively associated with antibody response to O. ostertagi; no gene was located in this region.
N. caninium
Five SNPs with a q value lower than 0.05 for antibody response to N. caninium were located on BTA21 (2 SNPs) and 25 (3 SNPs), respectively. In the QTL region between 13.04 and 14.10 Mb on BTA25, one of the three SNPs was classified as an intronic variant in the PARN gene and the other two as downstream variants on the micro-RNA gene, bta-mir-365-1. SNP rs133449464, that was classified as an intronic variant in the PARN gene, was the most strongly associated (p value = 2.56 × 10−10) with antibody response to N. caninium (Table 4). The frequency of the favourable allele at SNP rs133449464 was only 0.005 and none of the cows included in the dataset was homozygous for the favourable allele. The two SNPs on BTA21 with a q value lower than 0.05 were located within the QTL between 55.07 and 56.27 Mb and classified as intronic variants in the genes PPIP5K1 and PDIA3; one of these two SNPs was also classified as a downstream variant in the gene CATSPER2.
In total, 248 suggestive and significant SNPs were associated with antibody response to N caninium; only five of these 248 SNPs had a MAF higher than 0.1. Forty-one QTL were identified as associated with antibody response to N. caninium, of which eight contained only one SNP, since the suggestive SNP was not in LD (r2 > 0.5) with any other SNP in the flanking region of 5 Mb; the largest of the 41 QTL regions was 6,332,498 bp long (between 106.93 and 113.26 Mb) on BTA1 (see Additional file 5: Table S5). Almost half of the suggestive SNPs associated with antibody response to N. caninium were located in only three QTL regions, between 45.13 and 49.15 Mb on BTA1 (24 SNPs), between 106.93 and 113.26 Mb on BTA1 (66 SNPs) and between 57.83 and 58.95 Mb on BTA14 (30 SNPs).
Gene set enrichment
Of the 102 protein-coding genes, which either overlapped with, or were within QTL regions of suggestive SNPs for F. hepatica-damaged liver, DAVID identified 97 for the gene set enrichment analysis. The most strongly associated pathways and gene ontology (GO) sets with F. hepatica-damaged liver are in Additional file 6: Table S6. Three pathways and 11 GO sets were identified with a p value lower than 0.05. The pathway R-BTA-381753 had the strongest association with F. hepatica-damaged liver and included 11 genes from the gene set. The GO set with the strongest association (p value = 9.50 × 10−3) with F. hepatica-damaged liver was the biological process of ion transmembrane transport, which included four genes from the gene set.
Gene sets submitted to DAVID consisted of 324 genes for antibody response to F. hepatica, 333 genes for antibody response to O. ostertagi, and 429 genes for antibody response to N. caninum. In total, DAVID recognised 307, 289, and 373 genes for antibody response to F. hepatica, O. ostertagi and N. caninum, respectively. The top five ranked pathways for antibody response to F. hepatica, O. ostertagi, and N. caninum are in Additional file 7: Table S7 and the five GO sets that were most highly associated with antibody response to F. hepatica, O. ostertagi, and N. caninum are in Additional file 8: Table S8.
Five pathways and 28 GO sets were significantly (p value < 0.05) associated with antibody response to F. hepatica. The top ranked pathway and GO set for antibody response to F. hepatica was R-BTA-2142712 (which included three genes from the gene set; see Additional file 7: Table S7) and lipoxygenase pathway (included six genes from the gene set; see Additional file 8: Table S8), respectively. For antibody response to O. ostertagi, we found 14 pathways and 28 GO sets with a p value lower than 0.05. A strong association was identified between the pathway olfactory transduction (p value = 2 × 10−15) and antibody response to O. ostertagi; 60 genes were included in this pathway and were also identified in QTL regions associated with antibody response to O. ostertagi (see Additional file 7: Table S7). The GO biological process of G-protein coupled receptor signalling pathway was strongly associated with antibody response to O. ostertagi (p value = 5.40 × 10−24; see Additional file 8: Table S8).
In the gene set enrichment analysis of antibody response to N. caninum, 19 pathways and 23 GO sets were identified as significant (p value < 0.05). The alpha-linolenic acid metabolism pathway was associated with antibody response to N. caninum with a p value lower than 2.70 × 10−8 (see Additional file 7: Table S7). The GO metabolic function, phospholipase A2 activity, had the strongest association with antibody response to N. caninum (p value = 4.90 × 10−7; see Additional file 8: Table S8).
Discussion
The globally widespread prevalence of endo-parasites in dairy and beef cattle is of major concern [1], since parasitic infection is associated with reduced overall animal performance and compromised animal health and welfare [32, 33]. Although there are many shortcomings of using anthelmintic treatments, a single annual anthelmintic treatment is still the mainstay control strategy for F. hepatica and O. ostertagi in cattle [9, 34]. For the prevention of N. caninum infection, herd biosecurity is the major control strategy adopted in herds [35]. Since several studies have shown that F. hepatica, O. ostertagi, and N. caninum display genetic diversity, genetic selection for resistant cattle could be a complementary approach to current control strategies [8, 10, 11]. In addition, the existence of large genetic variation for endo-parasites in cattle suggests the presence of variability at the genome level, which governs inter-animal variability in susceptibility to endo-parasites. Information at the genome-level, pertaining to the ability of cattle to resist to parasitic infection, could help develop novel pharmaceuticals and diagnostic tests and implement genome-enabled selection. Consequently, knowledge of the biology that underlies such resistance to parasitic infection could help reduce the prevalence of endo-parasites in cattle, and at the same time reduce the reliance on anthelmintic treatments. Therefore, the objective of our study was to locate genomic regions that contribute to the ability of cattle to resist infection to F. hepatica, O. ostertagi and N. caninum.
Benefit of using whole-sequence data
It is important to note that our analyses were done on imputed sequence data, which may contain errors [36], and thus to alleviate this issue, rare variants with a MAF lower than 0.2%, SNPs that deviated from Hardy–Weinberg equilibrium (in a larger population to more conclusively identify genotyping errors and abnormalities), and SNPs in regions with a high Mendelian error rate were removed, as described in the Methods section.
To evaluate the benefit of using (imputed) whole-genome sequence in association analyses in cattle, we carried out an additional analysis in which only 50 k and HD SNPs were used (see Additional file 9: Figure S1). When the associations that we detected were limited to only SNPs on either the 50 k or HD panels, no SNP was identified with a p value lower than 10−6 for any of the parasite phenotypes investigated. In addition, only 4 and 2% of the QTL detected for antibody response to F. hepatica and O. ostertagi, respectively, and no QTL for F. hepatica-damaged liver and antibody response to N. caninum would have been identified if only 50 k SNPs were used instead of the imputed whole-genome sequence data. Furthermore, if we had used only the HD SNPs instead of the imputed whole-sequence data, only 4% of QTL reported here would have been detected for F. hepatica-damaged liver and only 11, 5 and 0% of QTL would have been detected for antibody response to F. hepatica, O. ostertagi, and N. caninum, respectively. Therefore, the majority of the causal SNPs or SNPs in LD with causal SNPs for parasitic diseases in cattle are absent on the 50 k and HD panels, which highlights the benefit of using whole-sequence data to identify more precisely the regions of the genome associated with specific traits. It should also be noted, that our study included multiple breeds, which may not be a representative dataset of other studies.
Comparison to previous genomic studies on parasitic diseases
A limited number of previous studies have attempted to locate genomic regions associated with endo-parasitic diseases in cattle, but none used antibody response or liver damage phenotypes to identify regions of the bovine genome associated with parasitic diseases. In their analysis of 768 Dutch dairy cows using 153 microsatellite markers, Coppieters et al. [11] documented that FEC was associated with the QTL between 22.96 and 25.23 Mb on BTA19 and concluded that the gene located in this region, ITGAE, was involved in the immune response to parasitic diseases. Although, in our study, the QTL between 22.96 and 25.23 Mb on BTA19 did not include any suggestive SNPs, a nearby region, located between 25.26 and 32.86 Mb, contained six suggestive SNPs for antibody response to F. hepatica (p values ranging from 6.59 × 10−6 to 8.09 × 10−6). Thus, one or more QTL regions on BTA19 may be associated with resistance to parasitic diseases.
In a study on 584 Angus cattle, Kim et al. [15] reported that 12 regions on the bovine genome contributed to significant genetic variation in FEC; to our knowledge, this is the only previous study that used dense genome-wide SNP genotypes (31,165 SNPs after edits) to map genomic regions associated with endo-parasitic diseases in cattle. In our study, the QTL between 65.23 and 67.30 Mb on BTA7 contained the most suggestive SNPs (21 SNPs) associated with F. hepatica-damaged liver, and this region overlaps with one of the QTL (64.19 to 66.89 Mb) identified by Kim et al. [15]. One of the genes that we detected in this region, i.e. GRIA1, was previously reported to be associated with ovulation rate in 639 Japanese Black cattle [37]. However, Cushman et al. [38] reported that GRIA1 had no influence on ovulation rate in a study of 139 crossbred beef cows. In humans, GRIA1 has been linked to the neurological disease schizophrenia [39, 40], asparaginase hypersensitivity [41], and migraines [42]. In our study, we found that GRIA1 was involved in two pathways, which were over-represented for F. hepatica-damaged liver and are linked to the neurology of the animal, amyotrophic lateral sclerosis, and neuroactive ligand-receptor interaction. Although ectopic F. hepatica infection has been reported to cause neurological diseases in humans [43, 44], there are no clinical reports of neurological diseases in cattle associated with lesions in the central nervous system.
Kim et al. [15] also documented a QTL region, located between 48.67 and 66.21 Mb on BTA6, which was suggestively associated with FEC in 584 Aberdeen Angus cattle. In our study, we detected a QTL region (containing 18 suggestive SNPs) on BTA6 associated with antibody response to F. hepatica within the region previously reported by Kim et al. [15]. We found four protein-coding genes (UBE2 K, N4BP2, PDS5A, and RHOH) and one RNA gene (SnRNA) that were located within this region on BTA6. The RHOH gene likely plays a role in the determination of the antibody response to F. hepatica, since it is involved in the activation of mast cells, which are known to increase in numbers in response to F. hepatica infection [45]. There was no overlap between the associations identified by Kim et al. [15] with FEC and those identified here for antibody response to either O. ostertagi or N. caninum.
In a study based on only four sheep that were experimentally infected with F. hepatica, Alvarez Rojas et al. [46] identified the lipoxygenase pathway with an over-representation of genes that were both up-regulated and down-regulated in peripheral blood mononuclear cells, 2 weeks after infection. These results are supported by our findings on the association of the lipoxygenase pathway and other pathways linked to arachidonic acid with antibody response to F. hepatica. This was expected since arachidonic acid, which is a polyunsaturated omega-6 fatty acid, is documented as suppressing the Th1 immune response in mice [47, 48] that decreases during F. hepatica infection [49]. Furthermore, studies comparing cows supplemented or not with a range of polyunsaturated fatty acids showed that those that were supplemented had higher levels of T-helper cells, T-cytotoxic cells, cells that expressed IL-2 receptors, and CD62 adhesion molecules [50], as well as a higher mean lymphocyte proliferation and higher titers of anti-OVA IgG [51].
In our study, we also expected that there would be very little overlap between QTL regions associated to the two phenotypes for F. hepatica, since Twomey et al. [8] found only a weak genetic relationship between F. hepatica-damaged liver and antibody response to F. hepatica (0.37; SE = 0.28).
Regions strongly associated with N. caninum
To date, the significant regions associated with antibody response to N. caninum on BTA21 and 25 had not been reported to be associated with endo-parasitic diseases in cattle. The favourable alleles for the most significant SNP on BTA21 and 25 had a frequency of only 0.003 and 0.005, respectively, which suggests that, if this frequency increased, more genetic gain is potentially possible. However, when we analysed exclusively the 3038 purebred Holstein–Friesians in our study, the allele frequency of the most significant SNP on BTA21 and 25 dropped to 0.002 and 0.001, respectively and these SNPs were not associated with antibody response to N. caninum (p value > 0.06). Nevertheless, the most significant SNP on BTA25 in the multi-breed analysis was an intronic variant located in the PARN gene, which encodes an enzyme (PARN) that has a role in early foetal development [52]. In cows, the gene PARN has been linked to embryo loss, since the level of PARN mRNA was lower in persistent follicles (‘ageing’ follicles associated with embryo loss) than in growing follicles [53]. N. caninum infection in females can result in abortion or embryo loss [35] and in reduced reproductive performance [10], thus the association that we observed between PARN and antibody response to N. caninum is supported by the association between PARN and embryo loss.
Identification of possible novel candidate genes
We identified that SNP rs384464701, an intronic variant in the FTO gene on BTA18, was associated with F. hepatica-damaged liver but the frequency of its favourable allele (0.988) was very high, thus, the potential gains for phenotypic improvement in resistance are very limited; in fact, the allele is fixed in the two British breeds of the 301 purebred Aberdeen Angus and 164 purebred Herefords included in our study. Analysis of the 1248 purebred Holstein–Friesians (i.e., ≥ 87.5% Holstein–Friesian) in our study revealed that this SNP rs384464701 had a p value of 3.91 × 10−4 and the frequency of its favourable allele was 0.003. FTO is known to have a strong association with human obesity [54]. Bravard et al. [55] suggested that FTO has a role in the metabolic regulation of the liver. In cattle, variants of the FTO gene have previously been associated with both growth and carcass traits [56, 57] as well as with fat and protein yield in milk [58]; taken together, these results suggest that FTO may be involved in energy homeostasis. However, Twomey et al. [10] reported that F. hepatica-damage liver had little or no genetic association with milk production and carcass merit in cattle.
Regarding the antibody response to O. ostertagi, we detected a SNP at 1,746,600 bp located on BTA3 in the GPA33 gene that was associated with a p value of 7.74 × 10−7; there were 17 other suggestive SNPs identified in this region between 0.95 and 6.39 Mb on BTA3. Although, the frequency of the favourable allele (< 0.004) was low for all these suggestive SNPs, GPA33 is a likely candidate gene associated with antibody response to O. ostertagi. Indeed, in a study on mice, Williams et al. [59] reported that GPA33 was important for the intestinal barrier function, and thus it may have a role in the defence of gastrointestinal nematodes such as O. ostertagi.
Avoidance of parasitic diseases
Research on animal resistance to disease focuses mainly on the immune response [60, 61]. Nevertheless, the coevolution of animals and parasites has led to the development of animal behavioural strategies, including an avoidance strategy, to minimize parasite infection [62]. One such avoidance strategy is avoiding faecal areas caused by an odour cue, since the majority of parasites will be located in areas near (or within) faecal material [62]. In a study on 16 Aberdeen Angus steers, Smith et al. [63] observed that cattle avoided grazing areas contaminated by faeces, with the exception of rabbit faecal material. In a study on 20 sheep, Hutchings et al. [64] reported that ewes classified as genetically resistant to parasitic diseases displayed a different grazing behaviour compared to ewes classified as genetically susceptible to parasitic diseases; ewes that were considered as genetically resistant did not graze areas contaminated by faeces. As suggested by Twomey et al. [8], avoidance strategies may contribute to the genetic differences observed among cattle for different parasite phenotypes. Although, avoidance techniques by livestock may depend on visual clues, odour clues are most likely also involved.
Our findings suggest that cattle use odour cues as part of an avoidance strategy for the three endo-parasites studied here, since at least one pathway and GO set involved in the olfactory system were significant (p value < 0.05) for each of the parasite phenotypes. In addition, the only region on the bovine genome, that contained QTL that were suggestively associated to F. hepatica-damaged liver and antibody response to F. hepatica, included a large number of olfactory receptor genes. This suggests that a proportion of the genetic variation observed for parasitic diseases may be linked to the ability of cattle to avoid infective larvae. Nevertheless, since genes in these suggestive regions were used in the gene set enrichment analysis, further research is needed to support the concept that cattle avoid infection through odour cues.
Conclusions
We identified a large number of QTL regions that were associated with the endo-parasite phenotypes investigated in our study. We observed strong associations between genomic regions on BTA21 and 25 with antibody response to N. caninum; the associated favourable alleles had a low frequency in the studied population, which suggests that large genetic gain could be achieved.
Additional files
Additional file 1: Table S1. Number of animals with an Illumina Bovine High-Density BeadChip (HD), Illumina Bovine50 beadchip (50 k) and Low-Density BeadChip (LD) and with an International Dairy and Beef version 1 (V1), version 2 (V2) and version 3 (V3) genotype in the current study for deregressed EBV for F. hepatica-damaged liver, as well as for the adjusted phenotype of antibody response to F. hepatica, O. ostertagi and N. caninum.
Additional file 2: Table S2. Chromosome number, start of quantitative trait locus (QTL) region, end of QTL region and number of single nucleotide polymorphisms (SNPs) with a p value < 1 × 10−5 for each QTL region identified as suggestively associated with F. hepatica-damaged liver.
Additional file 3: Table S3. Chromosome number, start of quantitative trait locus (QTL) region, end of QTL region and number of single nucleotide polymorphisms (SNPs) with a p value < 1 × 10−5 for each QTL region identified as suggestively associated with antibody response to F. hepatica.
Additional file 4: Table S4. Chromosome number, start of quantitative trait locus (QTL) region, end of QTL region and number of single nucleotide polymorphisms (SNPs) with a p value < 1 × 10−5 for each QTL region identified as suggestively associated with antibody response to O. ostertagi.
Additional file 5: Table S5. Chromosome number, start of quantitative trait locus (QTL) region, end of QTL region and number of single nucleotide polymorphisms (SNPs) with a p value < 1 × 10−5 for each QTL region identified as suggestively associated with antibody response to N. caninium.
Additional file 6: Table S6. Name, type, p value and genes for the top 5 ranked pathways and gene ontology (GO) sets for F. hepatica-damaged liver based on the EASE p value (an adoption of the Fisher Exact test to measure the gene-enrichment in annotation terms).
Additional file 7: Table S7. Name, source, p value and number of genes for the top 5 ranked pathways for antibody response to F. hepatica, O. ostertagi and N. caninum based on the EASE p value (an adoption of the Fisher Exact test to measure the gene-enrichment in annotation terms).
Additional file 8: Table S8. Name, type, p value and genes for the top 5 ranked gene ontology (GO) sets for antibody response to F. hepatica, O. ostertagi and N. caninum based on the EASE p value (an adoption of the Fisher Exact test to measure the gene-enrichment in annotation terms).
Additional file 9: Figure S1. Manhattan plot showing -log10(p values) of association between each SNP effect and F. hepatica-damaged liver for (a) 50 k data and (b) HD data. The blue line is the threshold for suggestive SNPs.
Authors’ contributions
AJT designed the study, performed the statistical analyses, and drafted the manuscript. DCP performed the imputation, and together with DPB, participated in the design of the study. DCP and DPB helped to draft the manuscript. All authors read and approved the final manuscript.
Acknowledgements
We gratefully acknowledge the 1000 Bull Genomes Consortium for providing accessibility to whole-genome sequence data, which were used in this study.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
All data generated during this study are included in this published article.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Funding
Funding from the Irish Department of Agriculture, Food and the Marine STIMULUS research grants HEALTHYGENES and FLUKELESS, funding from the Science Foundation Ireland principal investigator award grant (14/IA/2576) and funding from a joint research grant from Science Foundation Ireland and the Department of Agriculture, Food and Marine on behalf of the Government of Ireland under the Grant 16/RC/3835 (VistaMilk) are greatly appreciated.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Alan J. Twomey, Email: alan.twomey@teagasc.ie
Donagh P. Berry, Email: donagh.berry@teagasc.ie
Ross D. Evans, Email: revans@icbf.ie
Michael L. Doherty, Email: michael.doherty@ucd.ie
David A. Graham, Email: dgraham@animalhealthireland.ie
Deirdre C. Purfield, Email: deirdre.purfield@teagasc.ie
References
- 1.Sekiya M, Zintl A, Doherty ML. Bulk milk ELISA and the diagnosis of parasite infections in dairy herds: a review. Ir Vet J. 2013;66:14. doi: 10.1186/2046-0481-66-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.van Dijk J, Sargison ND, Kenyon F, Skuce PJ. Climate change and infectious disease: helminthological challenges to farmed ruminants in temperate regions. Animal. 2010;4:377–392. doi: 10.1017/S1751731109990991. [DOI] [PubMed] [Google Scholar]
- 3.Bloemhoff Y, Forbes A, Danaher M, Good B, Morgan E, Mulcahy G, et al. Determining the prevalence and seasonality of Fasciola hepatica in pasture-based dairy herds in Ireland using a bulk tank milk ELISA. Ir Vet J. 2015;68:16. doi: 10.1186/s13620-015-0042-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Selemetas N, de Waal T. Detection of major climatic and environmental predictors of liver fluke exposure risk in Ireland using spatial cluster analysis. Vet Parasitol. 2015;209:242–253. doi: 10.1016/j.vetpar.2015.02.029. [DOI] [PubMed] [Google Scholar]
- 5.Charlier J, Claerebout E, Duchateau L, Vercruysse J. A survey to determine relationships between bulk tank milk antibodies against Ostertagia ostertagi and milk production parameters. Vet Parasitol. 2005;129:67–75. doi: 10.1016/j.vetpar.2004.11.024. [DOI] [PubMed] [Google Scholar]
- 6.Schweizer G, Braun U, Deplazes P, Torgerson PR. Estimating the financial losses due to bovine fasciolosis in Switzerland. Vet Rec. 2005;157:188–193. doi: 10.1136/vr.157.7.188. [DOI] [PubMed] [Google Scholar]
- 7.Reichel MP, Alejandra Ayanegui-Alcérreca M, Gondim LFP, Ellis JT. What is the global economic impact of Neospora caninum in cattle—the billion dollar question. Int J Parasitol. 2013;43:133–142. doi: 10.1016/j.ijpara.2012.10.022. [DOI] [PubMed] [Google Scholar]
- 8.Twomey AJ, Sayers RG, Carroll RI, Byrne N, Brien EO, Doherty ML, et al. Genetic parameters for both a liver damage phenotype caused by Fasciola hepatica and antibody response to Fasciola hepatica phenotype in dairy and beef cattle. J Anim Sci. 2016;94:4109–4119. doi: 10.2527/jas.2016-0621. [DOI] [PubMed] [Google Scholar]
- 9.Bloemhoff Y, Danaher M, Andrew F, Morgan E, Mulcahy G, Power C, et al. Parasite control practices on pasture-based dairy farms in the Republic of Ireland. Vet Parasitol. 2014;204:352–363. doi: 10.1016/j.vetpar.2014.05.029. [DOI] [PubMed] [Google Scholar]
- 10.Twomey AJ, Carroll RI, Doherty ML, Byrne N, Graham DA, Sayers RG, et al. Genetic correlations between endo-parasite phenotypes and economically important traits in dairy and beef cattle. J Anim Sci. 2018;96:407–421. doi: 10.1093/jas/sky008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Coppieters W, Mes THM, Druet T, Farnir F, Tamma N, Schrooten C, et al. Mapping QTL influencing gastrointestinal nematode burden in Dutch Holstein–Friesian dairy cattle. BMC Genomics. 2009;10:96. doi: 10.1186/1471-2164-10-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pan Y, Jansen GB, Duffield TF, Hietala S, Kleton D, Lin CY, et al. Genetic susceptibility to Neospora canium infection in Holstein cattle in Ontario. J Dairy Sci. 2004;87:3967–3975. doi: 10.3168/jds.S0022-0302(04)73537-7. [DOI] [PubMed] [Google Scholar]
- 13.Rendel J, Robertson A. Estimation of genetic gain in milk yield by selection in a closed herd of dairy cattle. J Genet. 1950;50:1–8. doi: 10.1007/BF02986789. [DOI] [PubMed] [Google Scholar]
- 14.Kim ES, Sonstegard TS, da Silva MVG, Gasbarre LC, Van Tassell CP. Identification of quantitative trait loci affecting gastrointestinal parasite resistance in an experimental Angus population. Anim Genet. 2014;45:117–121. doi: 10.1111/age.12101. [DOI] [PubMed] [Google Scholar]
- 15.Kim ES, Sonstegard TS, da Silva MVG, Gasbarre LC, Van Tassell CP. Genome-wide scan of gastrointestinal nematode resistance in closed Angus population selected for minimized influence of MHC. PLoS One. 2015;10:e0119380. doi: 10.1371/journal.pone.0119380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Strandén I, Lidauer M. Solving large mixed linear models using preconditioned conjugate gradient iteration. J Dairy Sci. 1999;82:2779–2787. doi: 10.3168/jds.S0022-0302(99)75535-9. [DOI] [PubMed] [Google Scholar]
- 17.Strandén I, Mäntysaari EA. A recipe for multiple trait deregression. Interbull Bull. 2010;42:21–24. [Google Scholar]
- 18.Charlier J, Hostens M, Jacobs J, Ranst B, Duchateau L, Vercruysse J. Integrating fasciolosis control in the dry cow management: the effect of closantel treatment on milk production. PLoS One. 2012;7:e43216. doi: 10.1371/journal.pone.0043216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bloemhoff Y, Forbes A, Good B, Morgan E, Mulcahy G, Strube C, et al. Prevalence and seasonality of bulk milk antibodies against Dictyocaulus viviparus and Ostertagia ostertagi in Irish pasture-based dairy herds. Vet Parasitol. 2015;209:108–116. doi: 10.1016/j.vetpar.2015.01.021. [DOI] [PubMed] [Google Scholar]
- 20.Forbes AB, Vercruysse J, Charlier J. A survey of the exposure to Ostertagi ostertagi in dairy cow herds in Europe through the measurement of antibodies in milk samples from the bulk tank. Vet Parasitol. 2008;157:100–107. doi: 10.1016/j.vetpar.2008.06.023. [DOI] [PubMed] [Google Scholar]
- 21.Sargolzaei M, Schenkel FS, Jansen GB, Schaeffer LR. Extent of linkage disequilibrium in Holstein cattle in North America. J Dairy Sci. 2008;9:2106–2117. doi: 10.3168/jds.2007-0553. [DOI] [PubMed] [Google Scholar]
- 22.Daetwyler HD, Capitan A, Pausch H, Stothard P, van Binsbergen R, Brøndum RF, et al. Whole-genome sequencing of 234 bulls facilitates mapping of monogenic and complex traits in cattle. Nat Genet. 2014;46:858–865. doi: 10.1038/ng.3034. [DOI] [PubMed] [Google Scholar]
- 23.Loh PR, Danecek P, Palamara PF, Fuchsberger C, Reshef YA, Finucane HK, et al. Reference-based phasing using the Haplotype Reference Consortium panel. Nat Genet. 2016;48:1443–1448. doi: 10.1038/ng.3679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Das S, Forer L, Schönherr S, Sidore C, Locke AE, Kwong A, et al. Next-generation genotype imputation service and methods. Nat Genet. 2016;48:1284–1287. doi: 10.1038/ng.3656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Beissinger TM, Rosa GJ, Kaeppler SM, Gianola D, De Leon N. Defining window-boundaries for genomic analyses using smoothing spline techniques. Genet Sel Evol. 2015;47:30. doi: 10.1186/s12711-015-0105-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.VanRaden PM. Efficient methods to compute genomic predictions. J Dairy Sci. 2008;91:4414–4423. doi: 10.3168/jds.2007-0980. [DOI] [PubMed] [Google Scholar]
- 27.Meyer K, Tier B. “SNP Snappy”: a strategy for fast genome-wide association studies fitting a full mixed model. Genetics. 2012;190:275–277. doi: 10.1534/genetics.111.134841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Garrick DJ, Taylor JF, Fernando RL. Deregressing estimated breeding values and weighting information for genomic regression analyses. Genet Sel Evol. 2009;41:55. doi: 10.1186/1297-9686-41-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Purfield DC, Bradley DG, Evans RD, Kearney FJ, Berry DP. Genome-wide association study for calving performance using high-density genotypes in dairy and beef cattle. Genet Sel Evol. 2015;47:47. doi: 10.1186/s12711-015-0126-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Storey JD, Base AJ, Dabney A, Robinson D. qvalue: Q-value estimation for false discovery rate control. R package version 2.10.0. 2015. http://github.com/jdstorey/qvalue. Accessed 13 Feb 2019.
- 31.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mezo M, González-Warleta M, Castro-Hermida JA, Muiño L, Ubeira FM. Association between anti-F. hepatica antibody levels in milk and production losses in dairy cows. Vet Parasitol. 2011;180:237–242. doi: 10.1016/j.vetpar.2011.03.009. [DOI] [PubMed] [Google Scholar]
- 33.Charlier J, De Cat A, Forbes A, Vercruysse J. Measurement of antibodies to gastrointestinal nematodes and liver fluke in meat juice of beef cattle and associations with carcass parameters. Vet Parasitol. 2009;166:235–240. doi: 10.1016/j.vetpar.2009.09.040. [DOI] [PubMed] [Google Scholar]
- 34.Selemetas N, Phelan P, O’Kiely P, de Waal T. The effects of farm management practices on liver fluke prevalence and the current internal parasite control measures employed on Irish dairy farms. Vet Parasitol. 2015;207:228–240. doi: 10.1016/j.vetpar.2014.12.010. [DOI] [PubMed] [Google Scholar]
- 35.Dubey JP, Schares G, Ortega-Mora LM. Epidemiology and control of neosporosis and Neospora caninum. Clin Microbiol Rev. 2007;20:323–367. doi: 10.1128/CMR.00031-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brøndum RF, Ma P, Lund MS, Su G. Genotype imputation within and across Nordic cattle breeds. J Dairy Sci. 2012;95:6795–6800. doi: 10.3168/jds.2012-5585. [DOI] [PubMed] [Google Scholar]
- 37.Sugimoto M, Sasaki S, Watanabe T, Nishimura S, Ideta A, Yamazaki M, et al. Ionotropic glutamate receptor AMPA 1 is associated with ovulation rate. PLoS One. 2010;5:e13817. doi: 10.1371/journal.pone.0013817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cushman RA, Miles JR, Rempel LA, McDaneld TG, Kuehn LA, Chitko-McKown CG, et al. Identification of an ionotropic glutamate receptor AMPA1/GRIA1 polymorphism in crossbred beef cows differing in fertility. J Anim Sci. 2013;91:2640–2646. doi: 10.2527/jas.2012-5950. [DOI] [PubMed] [Google Scholar]
- 39.Choi KH, Zepp ME, Higgs BW, Weickert CS, Webster MJ. Expression profiles of schizophrenia susceptibility genes during human prefrontal cortical development. J Psychiatry Neurosci. 2009;34:450–458. [PMC free article] [PubMed] [Google Scholar]
- 40.Magri C, Gardella R, Barlati SD, Podavini D, Iatropoulos P, Bonomi S, et al. Glutamate AMPA receptor subunit 1 gene (GRIA1) and DSM-IV-TR schizophrenia: a pilot case-control association study in an Italian sample. Am J Med Genet B Neuropsychiatr Genet. 2006;141B:287–293. doi: 10.1002/ajmg.b.30294. [DOI] [PubMed] [Google Scholar]
- 41.Chen SH, Pei D, Yang W, Cheng C, Jeha S, Cox NJ, et al. Genetic variations in GRIA1 on chromosome 5q33 related to asparaginase hypersensitivity. Clin Pharmacol Ther. 2010;88:191–196. doi: 10.1038/clpt.2010.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Formicola D, Aloia A, Sampaolo S, Farina O, Diodato D, Griffiths LR, et al. Common variants in the regulative regions of GRIA1 and GRIA3 receptor genes are associated with migraine susceptibility. BMC Med Genet. 2010;11:103. doi: 10.1186/1471-2350-11-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mas-Coma S, Agramunt VH, Valero MA. Neurological and ocular fascioliasis in humans. Adv Parasitol. 2014;84:27–149. doi: 10.1016/B978-0-12-800099-1.00002-8. [DOI] [PubMed] [Google Scholar]
- 44.Mas-Coma S, Valero MA, Bargues MD. Fasciola, lymnaeids and human fascioliasis, with a global overview on disease transmission, epidemiology, evolutionary genetics, molecular epidemiology and control. Adv Parasitol. 2009;69:41–146. doi: 10.1016/S0065-308X(09)69002-3. [DOI] [PubMed] [Google Scholar]
- 45.Vukman KV, Adams PN, Dowling D, Metz M, Maurer M, O’Neill SM. The effects of Fasciola hepatica tegumental antigens on mast cell function. Int J Parasitol. 2013;43:531–539. doi: 10.1016/j.ijpara.2013.01.011. [DOI] [PubMed] [Google Scholar]
- 46.Alvarez Rojas CA, Scheerlinck JP, Ansell BRE, Hall RS, Gasser RB, Jex AR. Time-course study of the transcriptome of peripheral blood mononuclear cells (PBMCs) from sheep infected with Fasciola hepatica. PLoS One. 2016;11:e0159194. doi: 10.1371/journal.pone.0159194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wallace FA, Miles EA, Evans C, Stock TE, Yaqoob P, Calder PC. Dietary fatty acids influence the production of Th1- but not Th2-type cytokines. J Leukoc Biol. 2001;69:449–457. [PubMed] [Google Scholar]
- 48.Monk JM, Turk HF, Fan YY, Callaway E, Weeks B, Yang P, et al. Antagonizing arachidonic acid-derived eicosanoids reduces inflammatory Th17 and Th1 cell-mediated inflammation and colitis severity. Mediat Inflamm. 2014;2014:917149. doi: 10.1155/2014/917149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.O’Neill SM, Brady MT, Callanan JJ, Mulcahy G, Joyce P, Mills KH, et al. Fasciola hepatica infection downregulates Th1 responses in mice. Parasite Immunol. 2000;22:147–155. doi: 10.1046/j.1365-3024.2000.00290.x. [DOI] [PubMed] [Google Scholar]
- 50.Gandra JR, Barletta RV, Mingoti RD, Verdurico LC, Freitas JE, Jr, Oliveira LJ, et al. Effects of whole flaxseed, raw soybeans, and calcium salts of fatty acids on measures of cellular immune function of transition dairy cows. J Dairy Sci. 2016;99:4590–4606. doi: 10.3168/jds.2015-9974. [DOI] [PubMed] [Google Scholar]
- 51.Caroprese M, Marzano A, Entrican G, Wattegedera S, Albenzio M, Sevi A. Immune response of cows fed polyunsaturated fatty acids under high ambient temperatures. J Dairy Sci. 2009;92:2796–2803. doi: 10.3168/jds.2008-1809. [DOI] [PubMed] [Google Scholar]
- 52.Godwin AR, Kojima S, Green CB, Wilusz J. Kiss your tail goodbye: the role of PARN, Nocturnin, and Angel deadenylases in mRNA biology. Biochim Biophys Acta. 2013;1829:571–579. doi: 10.1016/j.bbagrm.2012.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lingenfelter BM, Dailey RA, Inskeep EK, Vernon MW, Poole DH, Rhinehart JD, et al. Changes of maternal transcripts in oocytes from persistent follicles in cattle. Mol Reprod Dev. 2007;74:265–272. doi: 10.1002/mrd.20568. [DOI] [PubMed] [Google Scholar]
- 54.Fawcett KA, Barroso I. The genetics of obesity: FTO leads the way. Trends Genet. 2010;26:266–274. doi: 10.1016/j.tig.2010.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bravard A, Vial G, Chauvin MA, Rouillé Y, Bailleul B, Vidal H, et al. FTO contributes to hepatic metabolism regulation through regulation of leptin action and STAT3 signalling in liver. Cell Commun Signal. 2014;12:4. doi: 10.1186/1478-811X-12-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jevsinek Skok D, Kunej T, Kovac M, Malovrh S, Potocnik K, Petric N, et al. FTO gene variants are associated with growth and carcass traits in cattle. Anim Genet. 2016;47:219–222. doi: 10.1111/age.12403. [DOI] [PubMed] [Google Scholar]
- 57.Rempel LA, Casas E, Shackelford SD, Wheeler TL. Relationship of polymorphisms within metabolic genes and carcass traits in crossbred beef cattle. J Anim Sci. 2012;90:1311–1316. doi: 10.2527/jas.2011-4302. [DOI] [PubMed] [Google Scholar]
- 58.Zielke LG, Bortfeldt RH, Reissmann M, Tetens J, Thaller G, Brockmann GA. Impact of variation at the FTO locus on milk fat yield in Holstein dairy cattle. PLoS One. 2013;8:e63406. doi: 10.1371/journal.pone.0063406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Williams BB, Tebbutt NC, Buchert M, Putoczki TL, Doggett K, Bao S, et al. Glycoprotein A33 deficiency: a new mouse model of impaired intestinal epithelial barrier function and inflammatory disease. Dis Model Mech. 2015;8:805–815. doi: 10.1242/dmm.019935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Råberg L, Graham AL, Read AF. Decomposing health: tolerance and resistance to parasites in animals. Philos Trans R Soc Lond B Biol Sci. 2009;364:37–49. doi: 10.1098/rstb.2008.0184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.McManus C, do Prado Paim T, de Melo CB, Brasil BS, Paiva SR. Selection methods for resistance to and tolerance of helminths in livestock. Parasite. 2014;21:56. doi: 10.1051/parasite/2014055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hart BL. Behavioral adaptations to pathogens and parasites: five strategies. Neurosci Biobehav Rev. 1990;14:273–294. doi: 10.1016/S0149-7634(05)80038-7. [DOI] [PubMed] [Google Scholar]
- 63.Smith LA, White PCL, Marion G, Hutchings MR. Livestock grazing behavior and inter- versus intraspecific disease risk via the faecal–oral route. Behav Ecol. 2009;20:426–432. doi: 10.1093/beheco/arn143. [DOI] [Google Scholar]
- 64.Hutchings MR, Knowler KJ, McAnulty R, McEwan JC. Genetically resistant sheep avoid parasites to a greater extent than do susceptible sheep. Proc Biol Sci. 2007;274:1839–1844. doi: 10.1098/rspb.2007.0398. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Table S1. Number of animals with an Illumina Bovine High-Density BeadChip (HD), Illumina Bovine50 beadchip (50 k) and Low-Density BeadChip (LD) and with an International Dairy and Beef version 1 (V1), version 2 (V2) and version 3 (V3) genotype in the current study for deregressed EBV for F. hepatica-damaged liver, as well as for the adjusted phenotype of antibody response to F. hepatica, O. ostertagi and N. caninum.
Additional file 2: Table S2. Chromosome number, start of quantitative trait locus (QTL) region, end of QTL region and number of single nucleotide polymorphisms (SNPs) with a p value < 1 × 10−5 for each QTL region identified as suggestively associated with F. hepatica-damaged liver.
Additional file 3: Table S3. Chromosome number, start of quantitative trait locus (QTL) region, end of QTL region and number of single nucleotide polymorphisms (SNPs) with a p value < 1 × 10−5 for each QTL region identified as suggestively associated with antibody response to F. hepatica.
Additional file 4: Table S4. Chromosome number, start of quantitative trait locus (QTL) region, end of QTL region and number of single nucleotide polymorphisms (SNPs) with a p value < 1 × 10−5 for each QTL region identified as suggestively associated with antibody response to O. ostertagi.
Additional file 5: Table S5. Chromosome number, start of quantitative trait locus (QTL) region, end of QTL region and number of single nucleotide polymorphisms (SNPs) with a p value < 1 × 10−5 for each QTL region identified as suggestively associated with antibody response to N. caninium.
Additional file 6: Table S6. Name, type, p value and genes for the top 5 ranked pathways and gene ontology (GO) sets for F. hepatica-damaged liver based on the EASE p value (an adoption of the Fisher Exact test to measure the gene-enrichment in annotation terms).
Additional file 7: Table S7. Name, source, p value and number of genes for the top 5 ranked pathways for antibody response to F. hepatica, O. ostertagi and N. caninum based on the EASE p value (an adoption of the Fisher Exact test to measure the gene-enrichment in annotation terms).
Additional file 8: Table S8. Name, type, p value and genes for the top 5 ranked gene ontology (GO) sets for antibody response to F. hepatica, O. ostertagi and N. caninum based on the EASE p value (an adoption of the Fisher Exact test to measure the gene-enrichment in annotation terms).
Additional file 9: Figure S1. Manhattan plot showing -log10(p values) of association between each SNP effect and F. hepatica-damaged liver for (a) 50 k data and (b) HD data. The blue line is the threshold for suggestive SNPs.
Data Availability Statement
All data generated during this study are included in this published article.