

Abstract
Intrinsically disordered proteins (IDPs) do not have rigid 3D structures, showing changes in their folding depending on the environment or ligands. Intrinsically disordered proteins are widely spread in eukaryotic genomes, and these proteins participate in many cell regulatory metabolism processes. Some IDPs, when aberrantly folded, can be the cause of some diseases such as Alzheimer′s, Parkinson′s, and prionic, among others. In these diseases, there are modifications in parts of the protein or in its entirety. A common conformational variation of these IDPs is misfolding and aggregation, forming, for instance, neurotoxic amyloid plaques. In this review, we discuss some IDPs that are involved in neurodegenerative diseases (such as beta amyloid, alpha synuclein, tau, and the “IDP-like” PrP), cancer (p53, c-Myc), and diabetes (amylin), focusing on the structural changes of these IDPs that are linked to such pathologies. We also present the IDP modulation mechanisms that can be explored in new strategies for drug design. Lastly, we show some candidate drugs that can be used in the future for the treatment of diseases caused by misfolded IDPs, considering that cancer therapy has more advanced research in comparison to other diseases, while also discussing recent and future developments in this area of research. Therefore, we aim to provide support to the study of IDPs and their modulation mechanisms as promising approaches to combat such severe diseases.
Keywords: intrinsically disordered proteins (IDPs), neurodegenerative diseases, aggregation, drugs, drug discovery
1. Introduction
The protein structure–function paradigm was established in the 20th century. The key point of this paradigm is that an ordered (rigid) and unique 3D structure of a protein is an obligatory prerequisite for protein function [1,2]. Nevertheless, recent studies have provided broad and convincing evidences that some proteins do not adopt only one structure, but still are fully functional [3]. The different possible protein conformations are structured (folded), molten globular, pre-molten globular, and unstructured (unfolded) [4].
Since the beginning of the 2000s, a new class of unstructured proteins started to be studied more due to the improvement of techniques to elucidate protein structure. Crystal-structure analysis using X-ray diffraction cannot provide information on unstructured states, with only the absence of electron density in some regions being observed. However, the nuclear magnetic resonance (NMR) technique allowed for the better characterization of these disordered proteins, confirming the flexibility of protein segments that are missing in crystallography experiments [5]. They are defined mainly as intrinsically disordered proteins (IDPs), in spite of some authors defining these proteins as natively denatured [6], natively unfolded [7], intrinsically unstructured [8], and natively disordered proteins [9], among other definitions. We will use the IDP definition to refer to these proteins. The disorder could be also present in some regions of proteins; these regions are named intrinsically disordered regions (IDRs). Intrinsically disordered proteins /IDRs have no single, well-defined equilibrium structure and exist as heterogeneous ensembles of conformers [10].
There are significant differences between the amino acid sequences of IDPs/IDRs in comparison with structured globular proteins and/or domains. These differences are related to amino acid composition, sequence complexity, hydrophobicity, aromaticity, charge, flexibility, type and rate of amino acid substitutions over evolutionary time [11]. Some features of IDPs are the low content of hydrophobic residues and the high load of charged residues [12]. Intrinsically disordered proteins/IDPRs present large hydrodynamic volumes, low content of ordered secondary structure, and high structural heterogeneity. These proteins are very flexible. However, some of them show transitions from the disordered to the ordered state in the presence of natural ligands [10]. The ability of IDPs to return to the highly flexible conformations after performing their biological function, and their predisposition to acquire different conformations according to the environment, are unique properties of IDPs [13] (Figure 1).
Figure 1.

Example of intrinsically disordered proteins (IDP) conformational plasticity. Shown are ordered and disordered extremes in the conformational ensemble described for the photoactive yellow protein from Halorhodospira halophile (PDB IDs 3PHY and 2KX6).
It was demonstrated that these IDPs are highly prevalent in many genomes, including humans’, and are important in several cellular processes, such as regulation of transcription and translation, cell cycle control, and signaling [3]. It is important to highlight that they are much more common in eukaryotes, in comparison to Eubacteria and Archaea, reflecting the greater importance of disorder-associated signaling and regulation for eukaryotic cells [13]. Intrinsically disordered proteins are present in major disease pathways, such as cancer, amyloidosis, diabetes, cardiovascular, and neurodegenerative diseases. Changes in the environment and/or mutation(s) of IDPs would be expected to affect their normal function, leading to misidentification and missignaling. Consequently, it can result in misfolding and aggregation, which are known to be associated with the pathogenesis of numerous diseases. Some IDPs, such as α-synuclein, tau protein, p53, and BRCA1 are important in neurodegenerative diseases and cancer, being attractive targets for drugs modulating protein–protein interactions. Based on these IDPs and other examples, novel strategies for drug discovery have been developed [11,13]. The ability to modulate the interactions of these proteins offers tremendous opportunities of investigation in chemical biology and molecular therapeutics. Several recent small molecules, such as potential drugs, have been shown to act by blocking protein–protein interactions based on intrinsic disorder of one of the partners [14].
In this review, we will focus on IDPs involved in some neurodegenerative diseases, such as α-synuclein, amyloid β-peptide, and tau protein, while also commenting on cancer associated IDPs, such as p53 and c-Myc, and diabetes-related amylin. In addition, we will summarize the strategies to modulate IDPs action in some diseases and the promising drugs in this field, which are currently more developed for non-neurodegenerative disorders, prompting the need of focusing strategies on IDP-centered drug development for them.
2. Intrinsically Disordered Proteins in Some Diseases
Inside the cell, protein folding is promoted by chaperone machinery that allows the protein to adopt a folded, biologically active form [15]. However, IDPs remain partially or totally unfolded and could cause many neurodegenerative disorders due to some changes in their folding [10]. Neurodegenerative diseases are disorders characterized by progressive loss of neurons associated with deposition of proteins showing altered physicochemical properties in the brain and in peripheral organs. These proteins show misbehavior and disarrangement, affecting negatively their processing, functioning, and/or folding [16,17]. In some of these disorders, there is a conversion of the functional state of specific proteins into an aggregate state that can accumulate as fibrils, causing loss of native function, and consequent gain of a toxic function. The toxicity of these fibrils is caused by disrupting intracellular transport, overwhelming protein degradation pathways, and/or disturbing vital cell functions [16,18]. Misfolding and aggregation of IDPs/IDPRs are especially common in neurodegeneration [16,19,20].
If these misfolded proteins accumulate as deposits of aggregates, they can originate many neurodegenerative diseases such as Alzheimer’s, Parkinson’s, Huntington’s, and prionic diseases, among others [21]. Proteins that accumulate as amyloid fibrils are called amyloidogenic proteins. In order to facilitate the understanding, they can be divided in two groups: 1) proteins that present a well-defined structure with only part of the molecule being disordered, as in the case of prion protein; 2) IDPs like amyloid-β (Aβ), tau and α-synuclein, that show changes in the entire protein [22]. In addition to neurodegenerative diseases, IDPs are also involved in diabetes and different types of cancer. Here, we briefly summarize and cover the general characteristics of some IDPs that can accumulate as fibril aggregates rich in β-structure, and their association with some neurodegenerative diseases, as well as features of cancer- and diabetes-related IDPs.
2.1. α-Synuclein and Parkinson’s Disease
Synucleinopathies refer to a group of neurodegenerative diseases, namely Parkinson’s disease (PD), dementia with Lewy bodies, and multiple system atrophy, characterized histologically by the presence of inclusions (Lewy bodies and Lewy neurites) composed of aggregated α-synuclein in the central nervous system (CNS) [23,24]. Aggregates containing α-synuclein can be found also in microglia and astrocytes, and in neurons of the peripheral nervous system associated with rarer autonomic diseases. This protein, encoded by the SNCA gene located on chromosome 4 region q21, is predominantly expressed in the brain, where it concentrates in nerve terminals. Three isoforms of synuclein, α, β, and γ, are known, but only the α isoform is found in Lewy bodies and neurites. α-synuclein is a single chain with 140 amino acids, and displays 61% identity compared to β-synuclein (134 amino acids), and its sequence contains seven imperfect repeats of eleven amino acids, each with a -KTKEGV- conserved core, separated by nine amino acid residues [25]. Weinreb and co-workers [7] reported, in 1996, the intrinsically disordered nature of α-synuclein in solution, and the protein was found to maintain its disordered state in physiological cell conditions [26]. Upon reversible binding to negatively charged phospholipids, α-synuclein oligomerizes and undergoes structural changes to assume a highly dynamic α-helical conformation while still maintaining partially disordered stretches [27,28].
The physiological role of α-synuclein is still elusive. Mice lacking all three synucleins developed only mild neurodegenerative pathology [29,30]. Lipid-bound α-synuclein accumulates in the plasma membrane of synaptic terminals and synaptic vesicles suggesting a role in neurotransmitter release [31]. The protein has been shown to possess some chaperone activity, interacting with components of the SNARE complex [32] and promoting dilatation of the exocytotic fusion pore [33]. In synucleinopathies, misfolding of lipid-bound α-synuclein occurs leading to β-sheet rich amyloid fibrils, found as the main component of Lewy bodies and Lewy neurites [20,34]. The core of fibrillated protein comprises about 70 amino acids of its repeat region, organized in parallel, in-register β-sheets in a Greek key topology [35].
In contrast to its normal, physiological form, pathological aggregated α-synuclein is extensively phosphorylated at S129 and S87. Other posttranslational modifications present in pathological α-synuclein include nitration, oxidation (for which oxidized by-products of dopamine might contribute) and truncation. Not all of these modifications contribute to accelerate the fibrillation process, since nitration and oxidation decrease fibril formation and stabilize oligomers and protofibrils of α-synuclein. On the other hand, truncated α-synuclein, typically at its C-terminal, shows increased propensity towards fibrillation. In some of the familial forms of PD, point mutations in α-synuclein (E46K, A30P, A53T) alter its propensity to fibrillate [36]. However, about 90% of Parkinson’s disease cases are idiopathic [23]. The identification of which α-synuclein species are indeed toxic is yet incomplete and is an intense field of debate. There is a growing perception that soluble oligomeric forms of α-synuclein are the most relevant in terms of toxicity, suggesting that Lewy inclusions might represent a protective response, and that interventions to favor the fibrillation process could be of therapeutic value.
Aggregation of α-synuclein apparently starts in the synapses and the aggregates propagate to nearby neurons through a prion-like mechanism [37,38]. Initial brain structures accumulating intracellular α-synuclein inclusions are the olfactory bulb, glossopharyngeal, and vagal nerves; then the Lewy pathology spreads to other regions of the brain reaching the amygdala and substantia nigra, where it causes death of dopaminergic neurons consequently leading to the motor symptoms characteristic of PD. In the more advanced cases, Lewy bodies and neurites are found in the neocortex, accounting for the cognitive impairment associated to the disease [23,39,40].
2.2. Amyloid β-Peptide, Tau Protein, and Alzheimer′s Disease
Alzheimer′s disease (AD) is one of the most prevalent neurodegenerative diseases that affects the learning and memory processes beyond the reduction of the brain area, degenerationand death of neurons [41,42]. Diagnosis of AD requires the identification of senile plaques composed by fibril β-amyloid peptides and tangles of tau protein aggregates [41,43]. Amyloid-β (Aβ) peptide is a well-known IDP with several oligomeric forms [44]. Amyloid-β aggregates are formed mainly by peptides containing 39 to 43 amino acids yielded by proteolytic cleavage of amyloid precursor protein (APP) [45,46,47]. Its aggregated form is significantly linked to Alzheimer′s disease, and the generation of Aβ and plaque pathology is linked to the presence of mutations or transport defects related to this protein [48,49].
The amyloid precursor protein (APP) is a transmembrane glycoprotein (type I) that is suggested to be involved in the development of the neurosystem, acting as a cell adhesion molecule [50]. The gene that encodes APP is located in human chromosome 21 [51,52] and this gene yields different isoforms by alternative splicing. Nevertheless, the function of APP is still not understood [43]. The APP proteolytic processing occurs via α, β, and γ-secretase [53]. This process can happen via two pathways: the non-amyloidogenic and the amyloidogenic route (producing toxic Aβ1–40/42) [54]. Aβ peptides occur in two major lengths, Aβ1–40 and Aβ1–42 amino acids, both present in senile plaques [11,46]. Some studies showed that Aβ1–42 accumulate as an early event in neuronal dysfunction, acting as seeding in the formation of amyloid plaques [55,56].
The two alloforms, Aβ1–40 and Aβ1–42, have identical sequences with the exception of two residues in the C-terminus of Aβ1–42, causing major differences in conformational behavior, with Aβ1–42 being much more folded than Aβ1–40 [57]. The amyloid plaques could also be associated to other molecules and metal ions, playing an important role in their assembly and toxicity [58,59]. If some mutations occur in the substrate (APP) or in the γ-secretase regulator proteins (prenisilin-1 and prenisilin-2) it may cause an alteration of APP processing, increasing the levels of Aβ1–42 or Aβ1–43 peptides formed [60,61]. These mutations are known to be involved in development of early onset AD [62,63,64,65].
In order to support the idea that Aβ peptides possess an important role in AD, Simmons and co-workers [66] demonstrated that aggregation of Aβ increased the neurotoxic effect in rat embryonic neuronal cells. Kirkitadze and co-workers [67] studied the Aβ1–40 and Aβ1–42 oligomerization and assembly into fibrils, showing that the early features of fibril assembly were the increase of intermediates containing α-helix and then their decrease by the assembly of fibrils. Yan and Wang [68] showed that Aβ1–42 possesses more tendencies to aggregate in comparison with Aβ1–40, and that their C-terminal domain is more rigid.
A structural model for amyloid Aβ1-40 using solid state NMR (ssNMR) spectroscopy was proposed. It was found that the first 10 residues are disordered, a β-strand conformation forming β-sheet structure was found between residues 12–24 and 30–40 [69]. After that, other studies were performed with different forms of preparation of the fibrils, with the binding of Cu2+, with mutant forms of the peptide, among others [70,71,72,73].
Recently, the peptide Aβ1–42 was studied also using ssNMR and in one of the studies it displayed triple parallel β-sheet segments, which is formed by three β-sheets encompassing residues 12–18 (β1), 24–33 (β2), and 36–40 (β3) [74]. Another NMR study of Aβ1–42, demonstrated that the fibril core is formed by a dimeric form of the peptide, containing four β-strands in an S-shaped amyloid fold [75]. Wälti and coworkers [76] found similar results: the fibril in dimeric form, forming a double-horseshoe. The different results of these groups were probably due to the differences in the preparation of the fibrils, such as pH, peptide concentration, agitation and ionic strength, as well as the source of the peptide (recombinant or synthetic) [42]. For a detailed review about the structural features of the two peptides, see Reference [42]. Besides the importance of Aβ1–40 and Aβ1–42, some studies demonstrated that the presence of minor isoforms of Aβ peptides could be involved in aggregation and/or or neurotoxicity [49,77,78], although their effect in AD is not fully understood.
Tau is a microtubule-associated protein initially identified as a protein involved in microtubule (MT) assembly and stabilization [79] and in the axonal transport of proteins [80]. Nowadays, the list of physiological functions of tau has expanded to include diverse roles such as protection against DNA damage and cell signaling [81]. Recent data revealed that tau physiologically interacts with various proteins and subcellular structures, and upon release from neurons, it may even act on other cells, widening the spectrum of its repercussions in health and in diseased states [82].
The single gene encoding the tau protein is present in one copy in the human genome, located in chromosome 17q21 [83,84]. Alternative splicing of this gene can yield six different isoforms of tau with polypeptide chains varying from 352 to 441 amino acids [85,86], all containing either three or four tandem repeats of 31 or 32 amino acid residues, the so-called microtubule binding repeats [81,82]. Tau is composed of 25 to 30% of charged amino acids and contains many proline residues, rendering it full intrinsically disordered. Tau undergoes many types of posttranslational modifications such as phosphorylation, glycosylation, methylation, acetylation, ubiquitinilation, SUMOylation (interaction with Small Ubiquitin-like Modifiers), nitration, among others, which are thought to finely regulate the involvement of the protein in its various biological functions. As a result of “abnormal” phosphorylation, glycosylation, oxidation, truncation or other posttranslational modification [6,82], tau becomes prone to aggregation and forms intracellular deposits, a feature of several neurodegenerative diseases collectively known as “tauopathies”. The abnormal tau adopts many transient local foldings among which β-structures of hydrophobic regions, characteristic of neurofibrillary tangles, and paired helical filaments of its microtubule binding domains [87,88] (for a review, see Reference 81]). Tau aggregates can mediate the spreading of the neuropathology to neighboring cells through its paired helical filaments, emerging as a possible target for taoupathy therapies [89]. Oxidation status of tau cysteine residues plays an important role in aggregation. While the formation of intermolecular disulfide bridges aggregates the protein, intramolecular cystine bonds prevent aggregation [90]. Truncation and/or proteolysis of tau yielding lower molecular mass forms of the protein, either in the intracellular or extracellular compartments, were also reported to lead to conformational changes that culminate in toxic, aggregated fibrillar tau [91,92].
In the most common tauopathy, Alzheimer’s disease, and in some forms of frontotemporal dementia, the sites of neurodegeneration correlate with deposits of an aberrant hyperphosphorylated tau. All six isoforms of hyperphosphorylated tau are found in tauopathies, resulting in loss of the protein’s ability to bind to microtubules and causing disturbance of axonal transport [82]. Phosphorylation of tau may occur in more than 85 putative sites, and distinct kinases and phosphatases are involved in controlling the protein’s phosphate content. On the other hand, the glycosylation and/or acetylation status of tau determines its phosphorylation pattern [93].
As a consequence of its disordered nature, tau interacts with a diverse array of partners inside the cell, among which are proteins, small molecules, nucleic acids, and metal ions, with many of these interactions modifying tau’s structural properties and biological functions. At least 33 distinct protein partners bind to tau’s different domains or motifs, as reviewed in Reference [81]. The multifunctionality of tau resulting from the combination of the wide range of its binding partners and a plethora of posttranslational modifications guarantees its place among true moonlighting proteins [94]. One of such interactions is with the β-amyloid peptide, in a manner that the neurotoxicity of both partners is thought to be reinforced [95,96].
2.3. Prion Protein in Prion Diseases
The term prion was introduced to describe a small proteinaceous agent that was causing neurodegenerative disease in humans and other animals [97]. It was identified as an abnormal form of the prion protein [98,99]. The prion protein (PrP), encoded by the Prnp gene, is a glycoprotein, natively found in cells and that could be involved in the maintenance of myelin in neurons among other functions [100,101,102]. Structural studies of PrP using NMR demonstrated that the N-terminal portion of the recombinant murine PrP is unstructured and flexible, and that the C-terminal portion is globular, containing 3 α-helices and a short anti-parallel β-sheet [103]. A similar structure was found for murine PrP [103], hamster PrP [104], human PrP [105] and bovine Prp [106]. Prions are not considered IDPs per se due to their mixed structural features. Some authors argue in favor of prion-specific classification [107], while others consider them to be IDP-like or IDR-containing proteins [11,108].
In prion diseases, PrP changes are predominantly from an α-helical conformation (PrPC) into a β-sheet-rich structure acquiring a PrPSc form that is misfolded, aggregated and that causes transmissible and fatal neurodegenerative diseases [18,109]. Three kinds of prion diseases have been reported: sporadic, infectious, and hereditary forms, including human disorders like Creutzfeldt–Jakob disease (CJD), Gerstmann–Sträussler–Scheinker disease (GSS), familial atypical dementia, Kuru, and veterinary disorders such as scrapie in sheep, goats, mouse, etc. [110,111]. The basic neurocytological characteristics of these diseases are a progressive vacuolation of neurons and gray matter changing to a spongiform aspect with extensive neuronal loss [112].
Interspecies transmission of prions has been postulated [113], although some interspecific barrier for transmission of PrPSc prions has been established [114]. One factor involved in this barrier could be the difference between the donor and host amino acids sequence [115,116,117]. A recent study brings new insights on prion replication during species transition [118].
The structural modifications involved in prion propagation and infectivity is the transition of α-helices of PrPc into aggregated β-sheet of PrPSC [109,119]. The presence of PrPSC abnormal form seems to stimulate and serves as template for transition of PrPC into the infectious conformation [120]. Makarava and colleagues [121] reported that prion disease could be induced in wild-type animals by injection of recombinant PrPC fibrils. In order to understand how this transition occurs, Stahl and co-workers performed a study using mass spectrometry and Edman sequencing. They demonstrated that the primary structures of PrPSc were the same as the one predicted for the PrPC gene, suggesting that the difference between them is not in RNA modification nor splicing events. In the same study, no covalent modifications were identified in this transition [122]. In another study, using Fourier-transform infrared (FTIR) spectroscopy and circular dichroism (CD), it was shown that PrPC contains around 42% of α-helices in its structure and only 3% of β-sheet content. On the other hand, the modified isoform PrPSc contain a higher content of β-sheet (43%) and a lower content of α-helices (30%) [109].
The PrPSc aggregates present resistance to proteolytic degradation at the C-terminal region, differently from the PrPc normal form [123]. Saverioni and co-workers [124] demonstrated that human PrPSc isolates showed strain-specific differences in their resistance to proteolytic digestion, something that could be linked to aggregate stability. Such aggregates can have heterogeneous sizes [124,125].
When PrPc obtains a β-sheet-rich conformation and misfolded form, it has a tendency to accumulate as amyloid fibers, a useful characteristic for detection and diagnostic of diseases [126,127]. In spite of that, the formation of amyloid plaques is not an obligatory event in prion infectivity [128]. Thinking in a therapeutic target for prion diseases, one approach would be blocking the conversion of PrPC into PrPSc [129].
2.4. p53, c-Myc, and Cancer
Several human diseases, such as cancer, diabetes, and autoimmune disorders, have been found to be associated with deregulation of transcription factors [130]. Carcinogenesis is a multi-step process, resulting in uncontrolled cell growth. Mutations in DNA that lead to cancer disturb these orderly processes by disrupting their regulation. This disruption results in uncontrolled cell division leading to cancer development [131]. Deregulation of multiple transcription factors has been reported in cancer progression. Extensively studied transcription factors that have shown a major role in progression of different types of cancer are p53 and c-Myc, two intrinsically disordered proteins [132,133].
Fifty percent of all human cancer present mutations in TP53, and on many other cancers, the function of the p53 protein is compromised. Thus, p53 is a very important target in cancer therapy [134]. Mutations in p53 are found in several types of cancer such as colon, lung, esophagus, breast, liver, brain, reticuloendothelial, and hemopoietic tissues [135]. Additionally, many p53 mutants, instead of losing functions, acquire oncogenic properties, enabling them to promote invasion, metastasis, proliferation, and cell survival [136].
p53 is a key transcription factor involved in the regulation of cell proliferation, apoptosis, DNA repair, angiogenesis, and senescence. It acts as an important defense protein against cancer onset and evolution and is negatively regulated by interaction with the oncoprotein MDM2 (murine double minute 2). In human cancers, the TP53 gene is frequently mutated or deleted, or the wild-type p53 function is inhibited by high levels of MDM2, leading to the downregulation of tumor suppressive p53 pathways [137,138,139]. When DNA damage occurs, p53 is activated to promote the elimination or repair of the damaged cells. p53 is phosphorylated by DNA damage response (DDR) kinase, leading to cell cycle arrest, senescence, or apoptosis. In addition, p53 stimulates DNA repair by activating genes encoding components of the DNA repair machinery [140].
Human p53 is a homotetramer of 393 amino acids composed of an intrinsically disordered N-terminal transactivation domain (TAD), followed by a conserved proline-rich domain, a central and structured DNA-binding domain, and an intrinsically disordered C-terminal encoding its nuclear localization signals and oligomerization domain required for transcriptional activity [138,141,142,143,144,145,146,147]. Natively unfolded regions account for about 40% of the full-length protein and the disordered regions are extensively used to mediate and modulate interactions with other proteins. Disorder is crucial for p53 function, since its numerous posttranslational modifications are majorly found within the disordered regions [11,146,148]. The full TAD of p53 consists of the N-terminal containing 73 residues and with a net charge of −17, due to its richness in acidic amino acid residues, such as aspartic acid and glutamic acid [143]. The C-terminus, on the other hand, is rich in basic amino acids (mainly lysines) and binds DNA non-specifically [146].
Transactivation domain is a promiscuous binding site for several interacting proteins, including negative regulators as MDM2 and MDM4 [146,149,150,151]. Transactivation domain is an IDR that undergoes coupled folding and binding when interacting with partner proteins like the E3 ligase, RPA70 (the 70 kDa subunit of replication protein A) and MDM2. p53 forms an amphipathic helix when it binds to the MDM2 in a hydrophobic cleft in its N-terminal domain [137,138,149,152,153,154,155]. The p53–MDM2 interaction blocks the binding of p53 to several transcription factors. In addition, MDM2 tags p53 for ubiquitination and consequent degradation by the proteasome and the p53–MDM2 complex tends to be exported from the nucleus, preventing p53 to act as a “cellular gatekeeper” [138,144,156].
The proto-oncogene c-MYC encodes a transcription factor that is implicated in various cellular processes such as cell growth, proliferation, loss of differentiation and apoptosis [157]. Elevated or deregulated expression of c-MYC has been detected in various human cancers and is frequently associated with aggressive and poorly differentiated tumors. Some of these cancers include breast, colon, cervical, small-cell lung carcinomas, osteosarcomas, glioblastomas, melanoma, and myeloid leukemia [158,159,160]. c-Myc is a very important protein for understanding and developing therapeutics against cancers and cancer stem cells [161].
c-Myc is an IDP and becomes transcriptionally functional when it forms an heterodimer with its obligate partner Max to assume a coiled-coil structure that recognizes the E-box (enhancer-box)-sequence 5′-CACGTG-3′. The c-Myc N-terminus, its TAD, can activate transcription in mammalian cells when fused to a heterologous DNA-binding domain. The C-terminus of this protein contains a basic-helix-loop-helix-leucine zipper (b-HLH-LZ) domain, and it promotes its interaction with Max, that has the same (b-HLH-LZ) domain, and the sequence-specific DNA binding mentioned above [162,163,164,165]. Nuclear magnetic resonance studies of c-Myc disordered region have attributed to it the protein functional plasticity and multiprotein complex formation capacity [166]. Computational and experimental investigations show that c-Myc extensively employs its disorder regions to perform diverse interactions with other partners [161].
It is important to highlight that Max protein is critical for c-Myc’s transcriptional activities, both gene activation and repression [162,167]. Considering c-Myc as a target for cancer therapy, one approach to c-Myc inhibition has been to disrupt the formation of this dimeric complex [132]. However, the disruption of c-Myc-Max dimerization is not easy, since both proteins are IDPs and protein–protein interaction involving large flat surface areas are difficult to target with small molecules, such as drugs [168,169].
2.5. Amylin and Diabetes
Diabetes (Type II) is a multifactorial disease characterized by dysfunction of insulin action (insulin resistance) and failure of insulin secretion by pancreatic β-cells [11,170]. One hallmark feature of this disease is the accumulation of amyloid fibrils into pancreatic islets (islets of Langerhans). These amyloid deposits are majority composed by islet amyloid polypeptides (IAPP), also called amylin. Islet amyloid polypeptides are IDPs composed of 37 amino acid residues, co-secreted with insulin by the same pancreatic cells, and its gene is located on chromosome 12 in humans [171,172,173].
The process of aggregation of IAPP seems to be initiated by interaction of one IAPP monomer to another, progressively leading to the formation of aggregates [174,175]. Analysis of human IAPP using circular dichroism spectroscopy demonstrated that the fibril formation was accompanied by a conformational change of random coil to β-sheet/α-helical structure [176]. These transient conformations were further confirmed by other studies [177,178,179].
Cytotoxicity of IAPP accumulated as amyloid deposits could be associated with loss of pancreatic β-cells functions and cells apoptosis [180,181]. Recent reviews of computational studies provided mechanistic insights of IAPP structure as monomers and oligomers and their interaction with lipid bilayers in order to understand the IAPP cytotoxicity mediated by membranes [175,182].
3. Strategies for IDP Modulation
Intrinsically disordered proteins can rapidly populate different conformations in solution, usually not assuming a well-defined three-dimensional structure in their native state, as a result of their signature low-sequence complexity and low proportion of bulky hydrophobic amino acids (instead, charged and hydrophilic residues are common) that lead to a flexible, dynamically disordered behavior and larger interaction surface areas than analogous folded regions in globular proteins [183,184,185,186]. Considering that IDPs participate in numerous key processes in cell metabolism, it is expected that their activity would be regulated by multiple mechanisms at transcriptional, post-transcriptional, and translational levels, which makes active IDPs accessible in shorter periods compared to structured proteins [187,188]. Some aspects of IDP modulation (mainly for IDPs involved in cell signaling) are covered in this section, arbitrarily grouped in mechanisms that engage in direct structural changes on IDPs in order to achieve stabilization (coupled folding and binding, post-translational modifications), and mechanisms that control IDPs abundance in the cell (mRNA decay, IDP proteasomal degradation, nanny model for stabilization).
3.1. Regulation of IDP Activity through Structural Changes
Intrinsically disordered proteins acting in intracellular pathways contain conserved motifs for interaction with nucleic acids and other proteins, and frequently form low-affinity complexes advantageous to processes like signal transduction [184]. The recognition elements (being often called “SLiMs”—short linear motifs ranging from 3 to 10 amino acids [189,190,191]) determine a pivotal feature associated with disordered proteins, that is their binding promiscuity, which is carried out through “one-to-many” and “many-to-one” mechanisms [192]. Interestingly, many IDPs are able to adopt ordered structures when interacting with certain targets, characterizing the coupled folding and binding phenomenon. Also, structural polymorphisms can emerge from the IDP conformational landscape in cases where the same disordered protein can assume different defined structures as it binds to different targets. Some of the recognition elements are also targeted for post-translational modifications by regulatory enzymes, enabling disordered-to-ordered transitions in IDPs. However, induced folding in IDPs is not mandatory for activity, as many regions that remain disordered upon partner binding are important to function, constituting “fuzzy” complexes [193,194].
3.1.1. Coupled Folding and Binding
The folding induction by partner interaction is possibly one of the most reported characteristics of IDPs, despite not being a phenomenon absolutely widespread across this class of proteins (considering the fuzzy complexes), nor a completely understood process. There are numerous examples of disordered-to-ordered transitions in proteins implicated in the regulation of gene expression, like transcription factors that assume folded motifs when interacting with DNA. One particular example is the leucine zipper protein GCN4, which presents a basic region that is unstructured in the absence of DNA but becomes a stable helical structure when interacting to its cognate AP-1 site [195]. The transition begins with transient nascent helical forms, observed in the unbound state, that interact with DNA and lead to dramatic structural changes, explained by a reduction of the entropic cost of DNA binding due to restriction of the conformational space accessible to the basic region [8,196]. Thus, the induced folding is usually explained by loss of conformational entropy (from the unbound IDP state) upon target binding and compensatory favorable contributions from reduction in exposed hydrophobic surfaces and enhanced electrostatic interactions [196]. Exhaustive kinetic studies are necessary for the investigation on what order the events of binding and folding occur, and segregate them in schemes of induced fit (IF, when the IDP binds to a partner and then folds) or conformational selection (CS, when the partner only binds to IDPs in a certain conformation) [197]. However, trying to define the coupled folding and binding in two categories may offer a rather simplistic explanation for the IDPs behavior, since the mechanisms can overlap depending upon the system conditions [197,198].
3.1.2. Post-Translational Modifications
Because of their accessibility to modifying enzymes, IDPs are frequent targets for post-translational modifications (PTMs), which expand their functional versatility [185,199]. The occurrence of PTMs causes structural changes on IDPs by affecting their energy landscapes, due to modifications of the physicochemical properties of the primary sequence [185]. Post-translational modifications engage in the addition of chemical functional groups (usually small radicals such as phosphoryl, alkyl, acyl or glycosyl) or involve the direct modification of residues through reactions of oxidation, deimidation, and deamidation. Intrinsically disordered proteins suffering PTMs may have their electrostatic, steric, and hydrophobic properties modified, possibly inducing transformations on the structure due to enhancement/inhibition of contacts of motifs within the IDP chain or with binding partners [185,200,201,202]. Phosphorylation is one of the most prevalent PTM and constitutes a major regulatory mechanism in various cellular processes involving signal transduction. Replacing a neutral hydroxyl group with a tetrahedral phosphoryl results in new possibilities for intra- and intermolecular electrostatic interactions (e.g., salt bridges and hydrogen bonds). Many other types of PTMs work in a similar fashion (but modulating different chemical properties), providing new forms of interaction that can ultimately cause alterations in IDPs activity, including disordered to ordered transitions.
3.2. Regulation of IDPs Abundance
Intrinsically disordered proteins levels are carefully monitored in the cell, and changes in their abundance are associated with disease, mainly due to defective signal transduction (linked to the occurrence of some cancers [11,203]) and non-specific interactions that generate fibrillar aggregates (present in many neurodegenerative disorders [199,204]). Tight regulation can be achieved in different levels, controlling the half-lives of mRNAs encoding IDPs and the abundance of IDPs themselves. Obviously, there are outliers for these global trends and certain IDPs are present in cells in large amounts or for long periods of time [205]—usually not the IDPs involved in dynamic processes such as cell signaling. Examples include the fibrous muscle protein titin [206,207] and the curious case of tardigrade-specific IDPs, that are constitutively expressed and upregulated in some tardigrade species and are essential for desiccation tolerance [208].
3.2.1. IDP-Encoding mRNAs
A robust study monitoring gene expression in Saccharomyces cerevisiae (with similar trends detected for human genes [205]) demonstrated that mRNAs encoding sequences categorized as “highly unstructured” have lower half-lives than mRNAs encoding more structured proteins, having a comparable number of transcription factors regulating them [206]. One of the reasons for the increased mRNA decay is hypothesized to be the short poly(A) tails observed in IDP-encoding mRNAs, that foment RNA degradation pathways. Moreover, other factors related to transcript instability, like the binding of RNA-binding PUF proteins (that usually facilitate deadenylation and subsequent RNA clearance [209,210]) were found to be increased in mRNA coding for disordered proteins [206].
3.2.2. Proteasomal Degradation
Intrinsically disordered proteins undergo proteasomal degradation through two different (but not mutually exclusive) pathways, ubiquitin-dependent (UD) and ubiquitin-independent (UI) [211]. The UD pathway relies on the addition of ubiquitin to the substrate to be degraded, in a process regulated by a series of enzymes and is mediated collectively by the 26S proteasome [212]. It was found that IDPs present a high content of predicted ubiquitination sites [185,205], and, in an analysis of ubiquitinated proteins, there seems to be a correlation between confirmed degradation sites and regions of disorder [213]. Alternatively, the UI pathway is mainly orchestrated by the core of the 20S proteasome, being a default process for degradation of free disordered proteins. Some evidences support the hypothesis that the flexible and extended structures of IDPs (as well as disordered terminal segments in folded proteins) facilitates the interaction with the proteasome, considering that bulky particles have reduced cleavage rates [214,215].
3.2.3. Stabilization through “Nanny” Proteins
As IDPs are usually prone to degradation, there is a need for protein stabilization in some contexts. The ubiquitous enzyme NQO1 functions as a “gatekeeper” of the 20S proteasomes, binding and regulating the degradation of some IDPs, in a mechanism consuming NADH [211,216]. An analogous mechanism characterizes the “nanny” model for IDP protection from cleavage, where there is sequestration of ID segments by interactions with other proteins, resulting in evasion from 20S proteasomal digestion. The binding of the nanny is transient, beginning at the initial stage of IDPs’ life cycle, when they are newly synthesized (assuming they are more sensible to digestion in this stage) [154]. Despite the fact that nanny proteins also bind to nascent polypeptide chains, they are not considered chaperones because they do not induce a fixed three-dimensional organization on targets, only assisting on IDP conservation without permanently affecting their disordered structure.
3.3. Modulation of IDPs by Chaperones and Co-Chaperones
Aggregates produced in neurodegenerative diseases have been shown to respond to changes in levels of molecular chaperones, suggesting the possibility of therapeutic intervention and a role for chaperones in disease pathogenesis [217]. The heat shock protein Hsp90 promotes neurodegenerative disorders indirectly [218]. Tau protein accumulation is regulated by a (Hsp90) chaperone system. This chaperone is able to bind Tau, causing a conformational change that allows tau’s phosphorylation by glycogen synthase kinase (GSK3β), leading to tau aggregation [219]. The inhibition of this chaperone results in the reduction of tau phosphorylation levels, due to reduction of GSK3β levels [220]. Another approach was performed, the use of a co-chaperone of Hsp90, ATPase homolog 1 (Aha1), this protein is an activator of Hsp90. This approach promoted the increase of the production of aggregated tau in vitro and in mouse model of neurodegenerative disease. Moreover, inhibition of Aha1 reduced tau accumulation in cultured cells. Thus, Aha1 is an interesting target to the treatment of Alzheimer’s disease [221].
The Hsp70 is a protein stabilizer, has a cellular protection against neurodegeneration of the central nervous system [218]. Members of the Hsp70 family, such as Hsp70 and Hsc70, bind to misfolded proteins and somehow send them to the lysosome–autophagy pathway or ubiquitin–proteasome system for degradation [222,223]. Folding and degradation of proteins are linked through co-chaperones, such as C-terminus of HSP70-interacting protein (CHIP) and HSJ1 (DNAJB2) [224] which regulate the decisions determining whether misfolded proteins are refolded or degraded. The CHIP is associated with α-synuclein inclusions and act as a co-chaperone, altering its aggregation and enhancing the degradation of the misfolded α-synuclein [225].
Another important chaperone is cyclophilin 40 (CyP40) that is a cis/trans peptidyl-prolyl isomerase (PPIase) and is involved in regulation and orientation of proline residues [226,227]. Tau protein is rich in proline residues and its residues, usually found in β-turns, are involved in tau aggregation propensity [228]. Based on this information, Baker and coworkers [229] demonstrated that CyP40 possess the ability to dissolve amyloids fibrils in vitro. Nuclear magnetic resonance experiments showed that CyP40 acts specifically on proline rich residues performing the disaggregation of tau fibrils and oligomers. This cyclophilin could also interact with others aggregated proteins containing proline, like α-synuclein [229].
4. Known Drugs Acting on IDPs
Despite IDPs abundance in eukaryotes, currently there are no FDA-approved drugs specifically targeting these proteins, only experimental and speculative ones (i.e. drugs that have been evaluated by the United States Food and Drug Administration agency and had their marketing sanctioned). Some experimental drug examples are prevalent in the literature, such as those targeting p53-MDM2, c-Myc-Max, and EWS-Fli1 complexes, while some others are less discussed [230,231,232,233]. In this section we provide an overview of the pharmacological modulation of IDPs, neurodegenerative and otherwise.
Intrinsically disordered proteins are normally considered aggregation-avoidant, due to their high proportion of charged residues (as opposed to patches of folding-inducing, hydrophobic residues). Such “non-folding” plasticity is proposed to be advantageous for proteins with multiple partners [234,235]. However, some of the IDPs, as described earlier, are found in conformational diseases and amyloid formation (e.g., in Alzheimer’s disease, Parkinson’s disease). The suppression of fibril formation, thus, is of therapeutic interest. Drug candidates in this front include molecular tweezers (Table 1), which are ligands designed to bind lysine and arginine specifically, perturbing aggregation [236,237,238]. These positively-charged residues are prone to interact with negatively-charged regions in the fibril-forming monomers [237]. The SEN1576 compound, a 5-aryloxypyrimidine inhibitor of synaptotoxic Aβ aggregation (Table 1) was shown to be safe and orally bioavailable with good brain penetration [239].
Table 1.
Known drugs acting on IDPs (selected examples).
| Compound Name * | Targets | Compound Structure |
|---|---|---|
| CLR01 (Molecular tweezers) | Lysine and arginine residues in amyloid proteins |
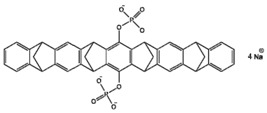
|
| ELN484228 | α-Synuclein |
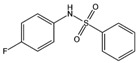
|
| SEN1576 | Amyloid β |
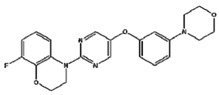
|
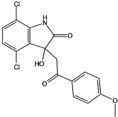
| NQTrp | PHF6 (Tau protein) |
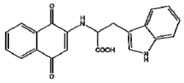
|
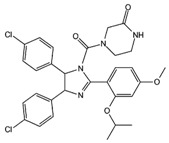
| Nutlin-3 | p53-MDM2 complex |
|
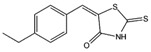
| 10058-F4 | c-Myc-Max complex |
|
| 10074-G5 | c-Myc-Max complex |
|
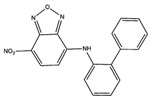
| YK-4–279 | EWS-Fli1 |
|
| Trodusquemine | PTP1B |
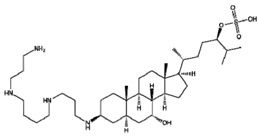
|
* Unless otherwise specified, these names are directly taken from their lead-compound coding and have no meaning on their own. EWS: Ewing Sarcoma; MDM2: oncoprotein (murine double minute 2); Myc: Myelocytomatosis- transcription factor homolog; NQTrp: naphthoquinone-tryptophan hybrid; PHF6: Tau-derived peptide (amino acid sequence VQIVYK); PTP1B: protein-tyrosine phosphatase 1B.
Fragments of amyloid fibrils also served as templates for non-natural amino acid inhibitors of amyloid fibril formation (d-TLKIVW) [240], while the ELN484228 (Table 1) compound was shown to be protective in cell models for vesicular dysfunction via α-Synuclein [235]. Alterations of the neuroleptic agent chlorpromazine allowed for enhanced 20S proteasome activation, inducing degradation of IDPs, such as tau and α-synuclein, but not of structured proteins [241]. These chlorpromazine-derived molecules, despite showing noteworthy potential (even as tools to study the proteasome 20S gate regulation), may interfere with other, physiological, non-pathologic disordered proteins to a still unstudied extent. A naphthoquinone-tryptophan hybrid (NQTrp) (Table 1) was shown to be effective in model systems for tau aggregation [242].
Regarding tumor-associated IDPs, the most commonly mutated gene in human cancers, the tumor suppressor protein p53, is key to cell cycle signaling. It is regulated by binding to various partners, including MDM2 and Taz2 [243,244,245]. Despite being highly disordered, p53 is not itself the target for the currently screened drug candidates aiming the p53-MDM2 complex. Instead, these ligands aim to occupy the p53-binding site in MDM2. The inhibitors nutlins (Table 1) (cis-imidazoline analogs currently in phase-I clinical trials), were shown to be potent against multiple cancerous cell lines, including breast cancer, colorectal cancer, lung cancer, osteosarcoma, prostate cancer, and renal cancer [246,247,248,249].
The c-Myc-Max complex involves the IDP transcription factor c-Myc that is activated by binding to Max, being expressed constitutively in various cancer cells [250]. Inhibitor candidates target the disordered c-Myc in this case, including peptidomimetic inhibitors [249,250], the small molecules 10058-F4, 10074-G5 (Table 1), and some others [250,251,252,253,254,255,256,257]. The oncogenic fusion protein EWS-Fli1 is an IDP exclusively present in Ewing’s sarcoma [257]. As with c-Myc-Max inhibitors, the small molecule inhibitor YK-4–279 (Table 1) targets the disordered EWS-Fli1 protein directly [257,258].
The AF9-AF4 dimer is found in acute leukemias and is composed by two disordered fusion proteins. The AF9 protein is responsible for turning hematopoietic cells oncogenic [259,260]. The AF4-derived peptide of amino acid sequence PFWT was shown to inhibit AF9 when used in combination with established chemotherapeutic agents [261,262], while some non-peptidic inhibitor candidates have been identified by high-throughput screening [263]. The protein-tyrosine phosphatase 1B (PTP1B), a reticular non-transmembrane enzyme, has been validated as therapeutic target for diabetes, obesity, and breast cancer, due to its role as negative regulator of insulin and leptin signaling [264]. Protein-tyrosine phosphatase 1B has an elongated disordered carboxy-terminus, to which trodusquemine (Table 1) (MSI-1436, a natural product) binds [265]. This aminosterol acts allosterically, stabilizing an inactive form of the enzyme by binding to a non-catalytic disordered site [265].
Inhibitors of α-synuclein aggregation are considered a promising approach. From in vitro studies, a few lead molecules were identified, such as EGCG (epigallocatechin gallate) [266]. Iron is known to induce the aggregation of α-synuclein. Deferiprone is an iron chelator used in thalassemic patients. Two clinical trials have shown a decrease in iron content in the substantia nigra of some PD patients, while a trend for improved motor scores was seen for all enrolled patients [267]. Although copper can induce aggregation of α-synuclein in vitro, levels of copper in the substantia nigra of PD patients are up to 50% lower than that of age-matched controls. The Cu2+ complex of diacetylbis-(4-methylthiosemicarbazone), called Cu2+(atsm), showed neuroprotective action in different animal models of PD [268], prompting for a phase I trial. From in vivo studies, promising results were obtained for KYP-2047, an inhibitor of prolyl oligopeptidase, an enzyme shown to interact with α-synuclein [269,270,271]; the diphenyl-pyrazole compound anle138b, was shown to cross the blood-brain barrier of mice and to reduce aggregation of α-synuclein [269]. The NPT200-11 compound prevented the formation of oligomers of α-synuclein and improved neuropathological symptoms in transgenic mice [272] and it has already been subjected to a phase I clinical trial. Another promising compound, NPT088, is a fusion protein of a general amyloid interaction motif derived from a bacteriophage [273] and a fragment of human immunoglobin, developed by Proclara Biociences, entered a phase I clinical trial for AD [274]. Phase II clinical trial of NPT088 is expected to include PD patients. Intrabodies, a single chain variable fragment of immunoglobulin expressed intracellularly, have been developed to target oligomeric and fibrillary α-synuclein, conferred neuroprotection, apparently by shifting the dynamics of the aggregation process [275,276,277]. Addition of a proteasome-addressing sequence to intrabodies targeted pathological forms of α-synuclein to degradation, NbSyn87PEST, directed towards the C-terminal region, and VH14PEST, directed against the NAC hydrophobic interaction domain, effectively degraded α-synuclein in cultured cells [278]. In rats overexpressing wild-type α-synuclein, these proteasome-targeted intrabodies (or nanobodies) decreased the levels of pathological aggregates, increased striatal dopamine levels and improved motor function [279]. Research in this promising field moves to find ways to deliver these compounds in adequate levels in specific areas of the brain, probably by using viral vectors.
Immunotherapies against α-synuclein, based on the evidence of an extracellular pathological protein during spreading of PD to different brain structures, show promising results in animal models [280]. Besides opsonization of the pathological protein for clearance, it is likely that antibodies could block further oligomerization of α-synuclein. Both passive (humanized monoclonal antibodies) and active (vaccine) immunization are being pursued. Pharmaceutical companies have joined the efforts and early clinical trials have been concluded or are under way. A brief description of the more advanced planned immunotherapies follows. A phase I trial was conducted by Roche for PRX002, a monoclonal antibody against the C-terminus of α-synuclein. It was well tolerated and reduced by 96% the levels of serum α-synuclein [281,282]. Affitope PD01A, a synthetic α-synuclein-mimicking peptide developed by Affiris for active immunization, had the first pilot study in 21 PD patients concluded in May 2018. It elicited a specific antibody response and showed good safety and tolerability profiles in a long-term (4 years) outpatient setting. Results of Affitope PD03A phase I clinical trial indicated no severe off-target effects, and a dose-dependent production of antibodies that cross-reacted with the intended α-synuclein epitope. Results from animal studies demonstrated that the antibodies raised against these antigens crossed the blood-brain barrier, decreasing the levels of aggregated α-synuclein, thereby improving motor function [283,284].
Regarding tauopathies, various therapeutic approaches have been tested, aiming to inhibit aggregation of tau, either directly or by preventing its interaction with some partners, and removal of toxic conformers and fibrillated tau [80]. However, the enormous effort put on finding ways to revert or delay the neurodegeneration symptoms associated to fibrillar tau, or to prevent the onset of tauopathies, has been so far unsuccessful, partly due to the intrinsically disordered nature of tau, which hampers drug design based on structural approaches. Small molecules that inhibit tau aggregation in vitro are considered promising leads to anti-tauopathy drugs [285] and the number of new tau inhibitory molecules grows steadily [286]. Nevertheless, there are unanswered questions regarding their effectiveness in vivo and the potential non-specific effects on normal tau physiology that could impact heavily on the CNS. The most studied small inhibitory molecules belong to distinct chemical groups, such as phenotiazines, cyanines, rhodanines, and arylmethines [287,288]. Peptides derived from neuroprotective proteins like NAP (amino acid sequence NAPVSIPQ) and d-SAL (all D-amino acid sequence SALLRSIPA) [289,290], enantiomeric peptides [291], and RNA/DNA aptamers [292] are also attractive components of future anti-tauopathy therapies. Some natural molecules present in cellular medium, such vitamin B12 [293] and 8-nitro-CGMP [294], are known to inhibit tau aggregation through oxidation of its cysteine residues. Drugs that bind tau, inducing formation of intramolecular disulfide bonds, such as methylene blue [295] or cinnamon derivatives [296], are potential frames for developing specific tau aggregation inhibitors. Taking into account that accumulation of hyperphosphorylated tau is a hallmark of AD and other neurodegenerative disorders, inhibitors of kinases, particularly of glycogen sintase kinase 3β and of Fyn, a member the Src-family of non-receptor tyrosine kinases, have drawn much attention for their anti-tauopathy potential [80]. Another strategy focuses on dual inhibitors that would interfere on tau aggregability and simultaneously block its interaction with protein partners, particularly kinases [297,298]. Other attempts to develop an anti-tauopathy drug have focused on inhibiting tau interaction with proteases like beta-secretases [299], caspases [300], and calpain [301], and chaperones such as Hsp90 [302], among others.
Tau-targeted immunotherapy began in 2013 [303], and since then a dozen of different types of immunological strategies were subject of clinical trials, including two active immunizations (vaccines) and humanized monoclonal antibodies directed towards distinct tau epitopes aiming passive immunization (reviewed in References [304,305]). These clinical trials are still at early phases and only limited data on the outcomes have been disclosed so far [306,307]. To achieve a successful immunotherapy to treat tauopathies, antibodies should be capable of neutralizing at least one of the many diseased isoforms of tau, either intracellularly or in the extracellular space, and interrupt the processes that lead to tau fibrillation and the neuron-to-neuron spreading. Ideally, the antibodies do not bind to normal tau and are able to cross the blood–brain barrier. In the case of a tau vaccine, the senescence of the immune system of the elderly has to be considered [304]. AADvac1 was conceived as the first vaccine against AD, using as immunogen a tau peptide previously identified to be essential for its pathological aggregation. Active immunization with this peptide elicits antibodies against a stretch of tau’s primary sequence (amino acid residues 294–305) and to conformational epitopes as well, targeting mainly extracellular tau, reducing its oligomerization. Tested in different animal models of AD, AADvac1 raised a protective humoral immune response with antibodies that discriminated between normal and pathological tau, reduced the level of neurofibrillary pathology in rat brains and lowered the content of disease-specific hyperphosphorylated tau [308]. Phase I clinical trial of AADvac1, conducted in 2013–2015 in patients aged 50–85 years with mild-to-moderate AD immunized weekly for 12 weeks, revealed a favorable safety profile and 29 out of 30 patients given AADvac1 developed an IgG response [309]. After 72 weeks, and booster doses of AADvac1, patients who had developed higher IgG titers showed lower hippocampal atrophy and cognitive decline rates and only mild adverse side effects [310]. A second active immunotherapy against tau has the compound ACI-35 as the immunogen, a peptide containing tau’s phospho-epitope pS396/pS404, in a liposome-based formulation able to elicit antibodies against abnormal hyperphosphorylated tau in P301L tau-mice [311].
Attempts of passive immunotherapy utilize humanized monoclonal antibodies, mostly of IgG1 or IgG4 isotypes, which are directed towards stretches of tau’s primary sequence known to be involved in the oligomerization of the protein, or to extracellular seeding-capable forms of truncated tau [304]. Phase I clinical trial of ABBV-8E12, one of such humanized monoclonal antibodies [312], revealed a satisfactory safety profile in 30 patients with the progressive supranuclear palsy tauopathy, receiving single doses (2.5 to 50 mg/kg) of the antibody, with no signs of immunogenicity against it [313]. Phase I trials of other two anti-tau antibodies, C2N-8E12 and BMS-986168, were also conducted [307].
Something that is noteworthy, and that demonstrates the close interplay between amyloid β peptide and tau in causing neurodegenerative diseases, therapeutic interventions aimed at one pathology can ameliorate symptoms of the other. This is the case of immunization of triple transgenic AD-like mice with a full-length DNA of amyloid β1–42 peptide, which showed a 40% reduction in the brain content of the amyloid β1–42 concomitant with a 25–50% decrease of total tau and different phosphorylated tau isoforms [314]. Conversely, passive immunization with antibodies against tau’s fragments 6–18 and 184–195 protected triple transgenic AD-like mice by reducing amyloid precursor protein in the CA1 region of hypothalamus and in amyloid plaques [315,316].
5. Status and Challenges in Drug Development for IDPs
The limited number of drugs targeting IDPs currently available (see previous section) may look disappointing, considering the physiological relevance of these proteins. One should be careful, though, to not exaggerate the current lack of IDP-specific drugs as being a reflection of disorder as a limitation for drug development.
The rational drug design strategy has been used successfully since the 1980s [317,318,319]. It depends on knowledge of the three-dimensional structure of the target protein, based on which ligands (usually inhibitors) are planned with aid of computational tools [320]. By definition, IDPs do not have a single, major conformation, occurring in dynamic conformational ensembles [10], and there is difficulty in using such traditional techniques to design IDP ligands [321]. Hence, most cases of IDP drug development were carried out by experimental screening and not by rational design [322]. Still, detection of IDP “hits” (potential initial drug candidates) through high throughput screening of compounds has been challenging [321] and computational methods achieved some success in predicting good candidates [323].
For IDPs with recognizable/determined metastable structures in their conformational ensembles, such structures could be used for rational drug design. However, IDPs are expected to be promiscuous, acting as hubs for multiple cellular processes [324]. This scenario, described as “protein clouds” [325] has its complexity further increased, with IDP ligands being described as “ligand clouds around protein clouds” [326]. Such roles in protein–protein interactions (PPIs) make IDPs especially interesting as drug targets, but the development of molecules targeting PPIs has been in itself, challenging [327,328,329,330].
A large difference is expected between the entropic loss and the enthalpic gain upon binding of a small ligand to an IDP, but some of them were shown to be capable of forming adaptable, specific interfaces for small molecule binding [231,235,321]. Intrinsically disordered proteins are difficult targets, since their interactions with small molecules are weaker and more transient, and the entropic loss is greater, in comparison to structured proteins [331]. The fragment-based drug design approach allows fragments to sample large amounts of chemical space, reducing the number of compounds for screening, with different fragments that bind at different regions of IDPs being able to be linked together via an appropriate linker [331]. Such fragments usually require hydrogen bonds to achieve detectable binding, generating an enthalpic gain that compensates for the entropic loss upon binding of the small molecules, lowering the free energy of the protein upon binding [331].
Still, the over-representation of IDPs in disorders, as summarized by the D2 concept (for “disorder in disorders”) [11] and the D3 concept (for “disorder in degenerative disorders”) [16], points to these proteins as promising therapeutic targets. Attempts to detect potentially druggable cavities in IDPs have identified at least 14 targets that could be subjected to rational drug design [233]. A more general estimate is that 9% of detected cavities may be druggable in IDPs, in comparison to 5% in ordered proteins [233]. These observations are especially interesting, considering that current drugs target around 500 proteins, less than 10% of the estimated potential target list, with very strict classes (such as enzymes and G-protein coupled receptors) accounting for more than 70% of them [230,331,332].
There have been major advances in the detection and prediction of IDP features in protein sequences [293], something that will surely help in the identification of these special drug targets. Major breakthroughs are also being achieved by the combination of experimental methods (especially NMR and fluorescence techniques) with computational modeling and molecular dynamics simulation of IDPs [294,322]. The latter is one of the few methods that allow for the description of IDPs in their conformational ensemble, instead of a single (or just a few) conformations [132,322,324,333,334,335,336,337].
6. Conclusion and Perspectives
The pharmacological strategies developed so far (and reviewed here) that target IDPs can be separated as binding directly to IDPs and hampering their aggregation by keeping them in the interaction incompetent conformation; interacting with the IDP and promoting the stabilization of non-toxic/ non-amyloidogenic oligometric species; and interacting with the amyloidogenic protein and greatly accelerating its aggregation to minimize the period of toxic oligomer formation [338]. Still, as we described here, there are many pathways acting on IDP control, and these are still unexplored targets for pharmaceutical interference. As the binding mechanisms of IDPs are being better described from a physical chemical standpoint [339,340], it is becoming clear that for candidate molecules to act on IDPs they must deviate from traditional prediction rules for drug-likeness [319,320]. One standout feature of IDP ligands is that they are larger and more three-dimensional than traditional drugs [341]. Adding another layer of complexity to this scenario, some proteins are shown to be conditionally unfolded [342], being disordered only under specific conditions.
Despite being abundant in eukaryotes, in which IDPs have evolutionarily conserved interaction partners [343], the occurrence of disorder in proteins from other organisms is being described. It includes the description of IDPs in Trypanosomatid parasites [344] and in some paramyxoviruses, including measles, Nipah and Hendra viruses [345]. These proteins constitute prospective targets for drug design endeavors, as was also observed for multiple disordered targets in prostate cancer [346]. Furthermore, recent evaluations indicate that IDP-targeted drug development may not be irreconcilable with structure-based drug design [347].
The development of drugs specifically tailored for IDPs is still in its infancy. As with the whole pipeline for drug discovery, there has been continuous progress in this area, and as we proposed in this work, there are many untapped pathways and unexplored targets regarding these proteins. As the biophysical techniques advance to catch up with the diversity of disordered behaviors in proteins, one can expect major developments in this front. Taking the limited but solid cases of success in IDP-specific drug design, we may face a future in which target disorder may be taken as the rule and not the exception.
Abbreviations
| AD | Alzheimer’s Disease |
| APP | Amyloid Precursor Protein |
| CJD | Creutzfeldt–Jakob Disease |
| CNS | Central Nervous System |
| CS | Conformational Selection |
| GSS | Gerstmann–Sträussler–Scheinker Disease |
| IAPP | Islet Amyloid Polypeptide (Amylin) |
| IDP | Intrinsically Disordered Protein |
| IDR | Intrinsically Disordered Region |
| IF | Induced Fit |
| MT | Microtubules |
| NMR | Nuclear Magnetic Resonance |
| PDB | RCSB Protein Databank |
| PrP | Prion Protein |
| PrPSc | Prion Protein, Alternate Conformation |
| PTM | Post-Translational Modification |
| SLiMs | Short Linear Motifs |
| ssNMR | Solid-State Nuclear Magnetic Resonance |
| TAD | Transactivation Domain (of p53) |
| UD | Ubiquitin-Dependent |
| UI | Ubiquitin-Independent |
Author Contributions
Conceptualization, C.R.C. and R.L.-B.; Writing—Original Draft Preparation, A.H.S.M., F.C.L., E.O.J., C.R.C., and R.L.-B.; Writing—Review & Editing, C.R.C. and R.L.-B.; Funding Acquisition, C.R.C.
Funding
The authors of this work have been funded by grants from Coordenação de Aperfeiçoamento de Pessoal de Nível Superior-CAPES and Conselho Nacional de Desenvolvimento Científico e Tecnológico - CNPq.
Conflicts of Interest
The authors declare no conflict of interest.
References
- 1.Dunker A.K., Lawson J.D., Brown C.J., Williams R.M., Romero P., Oh J.S., Oldfield C.J., Campen A.M., Ratliff C.M., Hipps K.W., et al. Intrinsically disordered protein. J. Mol. Graph. Model. 2001;19:26–59. doi: 10.1016/S1093-3263(00)00138-8. [DOI] [PubMed] [Google Scholar]
- 2.Uversky V.N. Natively unfolded proteins: A point where biology waits for physics. Protein Sci. 2002;11:739–756. doi: 10.1110/ps.4210102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wallin S. Intrinsically disordered proteins: Structural and functional dynamics. Res. Rep. Biol. 2017;8:7–16. doi: 10.2147/RRB.S57282. [DOI] [Google Scholar]
- 4.Uversky V.N. Protein folding revisited. A polypeptide chain at the folding—Misfolding—Nonfolding cross-roads: Which way to go? Cell. Mol. Life Sci. 2003;60:1852–1871. doi: 10.1007/s00018-003-3096-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dyson H.J., Wright P.E. Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol. 2005;6:197–208. doi: 10.1038/nrm1589. [DOI] [PubMed] [Google Scholar]
- 6.Schweers O., Schönbrunn-Hanebeck E., Marx A., Mandelkow E. Structural studies of tau protein and Alzheimer paired helical filaments show no evidence for beta-structure. J. Biol. Chem. 1994;269:24290–24297. [PubMed] [Google Scholar]
- 7.Weinreb P.H., Zhen W., Poon A.W., Conway K.A., Lansbury P.T. NACP, a protein implicated in Alzheimer’s disease and learning, is natively unfolded. Biochemistry. 1996;35:13709–13715. doi: 10.1021/bi961799n. [DOI] [PubMed] [Google Scholar]
- 8.Wright P.E., Dyson H.J. Intrinsically unstructured proteins: Re-assessing the protein structure-function paradigm. J. Mol. Biol. 1999;293:321–331. doi: 10.1006/jmbi.1999.3110. [DOI] [PubMed] [Google Scholar]
- 9.Daughdrill G.W., Pielak G.J., Uversky V.N., Cortese M.S., Dunker A.K. Natively disordered proteins. In: Buchner J., Kiefhaber T., editors. Protein Fold Handbook. Wiley VCH; Weinheim, Germany: 2005. pp. 275–357. [Google Scholar]
- 10.Uversky V.N. A decade and a half of protein intrinsic disorder: Biology still waits for physics. Protein Sci. 2013;22:693–724. doi: 10.1002/pro.2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Uversky V.N., Oldfield C.J., Dunker A.K. Intrinsically Disordered Proteins in Human Diseases: Introducing the D2 Concept. Annu. Rev. Biophys. 2008;37:215–246. doi: 10.1146/annurev.biophys.37.032807.125924. [DOI] [PubMed] [Google Scholar]
- 12.Uversky V.N., Gillespie J.R., Fink A.L. Why are “natively unfolded” proteins unstructured under physiologic conditions? Proteins Struct. Funct. Bioinform. 2000;41:415–427. doi: 10.1002/1097-0134(20001115)41:3<415::AID-PROT130>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 13.Uversky V.N., Dunker A.K. Understanding protein non-folding. Biochim. Biophys. Acta. 2010;1804:1231–1264. doi: 10.1016/j.bbapap.2010.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dunker A.K., Oldfield C.J., Meng J., Romero P., Yang J.Y., Chen J.W., Vacic V., Obradovic Z., Uversky V.N. The unfoldomics decade: An update on intrinsically disordered proteins. BMC Genom. 2008;26:S1. doi: 10.1186/1471-2164-9-S2-S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hartl F.U. Molecular chaperones in cellular protein folding. Nature. 1996;381:571–580. doi: 10.1038/381571a0. [DOI] [PubMed] [Google Scholar]
- 16.Uversky V.N. The triple power of D3: Protein intrinsic disorder in degenerative diseases. Front. Biosci. 2014;19:181–258. doi: 10.2741/4204. [DOI] [PubMed] [Google Scholar]
- 17.Kovacs G.G. Concepts and classification of neurodegenerative diseases. Handb. Clin. Neurol. 2017;145:301–307. doi: 10.1016/B978-0-12-802395-2.00021-3. [DOI] [PubMed] [Google Scholar]
- 18.Kransnoslobodtsev A.V., Shlyakhtenko L.S., Ukraintsev E., Zaikova T.O., Keana J.F.W., Lyubchenko Y.L. Nanomedicine and protein misfolding diseases. Nanomedicine. 2005;1:300–305. doi: 10.1016/j.nano.2005.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Uversky V.N. Targeting intrinsically disordered proteins in neurodegenerative and protein dysfunction diseases: Another illustration of the D2 concept. Expert Rev. Proteom. 2010;7:543–564. doi: 10.1586/epr.10.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Breydo L., Uversky V.N. Role of metal ions in aggregation of intrinsically disordered proteins in neurodegenerative diseases. Metallomics. 2011;3:1163–1180. doi: 10.1039/c1mt00106j. [DOI] [PubMed] [Google Scholar]
- 21.Eftekharzadeh B., Hyman B.T., Wegmann S. Structural studies on the mechanism of protein aggregation in age related neurodegenerative diseases. Mech. Ageing Dev. 2016;156:1–13. doi: 10.1016/j.mad.2016.03.001. [DOI] [PubMed] [Google Scholar]
- 22.Eisele Y.S., Monteiro C., Fearns C., Encalada S.E., Wiseman R.L., Powers E.T., Kelly J.W. Targeting protein aggregation for the treatment of degenerative diseases. Nat. Rev. Drug Discov. 2015;14:759–780. doi: 10.1038/nrd4593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goedert M., Jakes R., Spillantini M.G. The Synucleinopathies: Twenty Years On. J. Parkinsons Dis. 2017;7:S51–S69. doi: 10.3233/JPD-179005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Spillantini M., Crowther R., Jakes R., Hasegawa M., Goedert M. alpha-synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with Lewy bodies. Proc. Natl. Acad. Sci. USA. 1998;95:6469–6473. doi: 10.1073/pnas.95.11.6469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jakes R., Spillantini M.G., Goedert M. Identification of two distinct synucleins from human brain. FEBS Lett. 1994;345:27–32. doi: 10.1016/0014-5793(94)00395-5. [DOI] [PubMed] [Google Scholar]
- 26.Theillet F.X., Binolfi A., Bekei B., Martorana A., Rose H.M., Stuiver M., Verzini S., Lorenz D., van Rossum M., Goldfarb D., et al. Structural disorder of monomeric α-synuclein persists in mammalian cells. Nature. 2016;530:45–50. doi: 10.1038/nature16531. [DOI] [PubMed] [Google Scholar]
- 27.Ferreon A.C., Gambin Y., Lemke E.A., Deniz A.A. Interplay of alpha-synuclein binding and conformational switching probed by single-molecule fluorescence. Proc. Natl. Acad. Sci. USA. 2009;106:5645–5650. doi: 10.1073/pnas.0809232106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Choi T.S., Han J.Y., Heo C.E., Lee S.W., Kim H.I. Electrostatic and hydrophobic interactions of lipid-associated α-synuclein: The role of a water-limited interfaces in amyloid fibrillation. Biochim. Biophys. Acta Biomembr. 2018;1860:1854–1862. doi: 10.1016/j.bbamem.2018.02.007. [DOI] [PubMed] [Google Scholar]
- 29.Spillantini M.G., Goedert M. Neurodegeneration and the ordered assembly of α-synuclein. Cell Tissue Res. 2018;373:137–148. doi: 10.1007/s00441-017-2706-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Greten-Harrison B., Polydoro M., Morimoto-Tomita M., Diao L., Williams A.M., Nie E.H., Makani S., Tian N., Castillo P.E., Buchman V.L., et al. αβγ-Synuclein triple knockout mice reveal age-dependent neuronal dysfunction. Proc. Natl. Acad. Sci. USA. 2010;107:19573–19578. doi: 10.1073/pnas.1005005107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fortin D.L., Troyer M.D., Nakamura K., Kubo S., Anthony M.D., Edwards R.H. Lipid rafts mediate the synaptic localization of alpha-synuclein. J. Neurosci. 2004;24:6715–6723. doi: 10.1523/JNEUROSCI.1594-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Burré J., Sharma M., Südhof T.C. α-Synuclein assembles into higher-order multimers upon membrane binding to promote SNARE complex formation. Proc. Natl. Acad. Sci. USA. 2014;111:E4274–E4283. doi: 10.1073/pnas.1416598111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Logan T., Bendor J., Toupin C., Thorn K., Edwards R.H. α-Synuclein promotes dilation of the exocytotic fusion pore. Nat. Neurosci. 2017;20:681–689. doi: 10.1038/nn.4529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gai W.P., Yuan H.X., Li X.Q., Power J.T., Blumbergs P.C., Jensen P.H. In situ and in vitro study of colocalization and segregation of alpha-synuclein, ubiquitin, and lipids in Lewy bodies. Exp. Neurol. 2000;166:324–333. doi: 10.1006/exnr.2000.7527. [DOI] [PubMed] [Google Scholar]
- 35.Tuttle M.D., Comellas G., Nieuwkoop A.J., Covell D.J., Berthold D.A., Kloepper K.D., Courtney J.M., Kim J.K., Barclay A.M., Kendall A., et al. Solid-state NMR structure of a pathogenic fibril of full-length human α-synuclein. Nat. Struct. Mol. Biol. 2016;23:409–415. doi: 10.1038/nsmb.3194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Choi W., Zibaee S., Jakes R., Serpell L.C., Davletov B., Crowther R.A., Goedert M. Mutation E46K increases phospholipid binding and assembly into filaments of human alpha-synuclein. FEBS Lett. 2004;576:363–368. doi: 10.1016/j.febslet.2004.09.038. [DOI] [PubMed] [Google Scholar]
- 37.Tofaris G.K., Goedert M., Spillantini M.G. The Transcellular Propagation and Intracellular Trafficking of α-Synuclein. Cold Spring Harb. Perspect. Med. 2017;7:a024380. doi: 10.1101/cshperspect.a024380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Osterberg V.R., Spinelli K.J., Weston L.J., Luk K.C., Woltjer R.L., Unni V.K. Progressive aggregation of alpha-synuclein and selective degeneration of Lewy inclusion-bearing neurons in a mouse model of parkinsonism. Cell Rep. 2015;10:1252–1260. doi: 10.1016/j.celrep.2015.01.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fusco G., Chen S.W., Williamson P.T.F., Cascella R., Perni M., Jarvis J.A., Cecchi C., Vendruscolo M., Chiti F., Cremades N., et al. Structural basis of membrane disruption and cellular toxicity by α-synuclein oligomers. Science. 2017;358:1440–1443. doi: 10.1126/science.aan6160. [DOI] [PubMed] [Google Scholar]
- 40.Varela J.A., Rodrigues M., De S., Flagmeier P., Gandhi S., Dobson C.M., Klenerman D., Lee S.F. Optical Structural Analysis of Individual α-Synuclein Oligomers. Angew. Chem. Int. Ed. Engl. 2018;57:4886–4890. doi: 10.1002/anie.201710779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mattson M.P. Pathways towards and away from Alzheimer’s disease. Nature. 2004;430:631–639. doi: 10.1038/nature02621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Aleksis R., Oleskovs F., Jaudzems K., Pahnke J., Biverstål H. Structural studies of amyloid-β peptides: Unlocking the mechanism of aggregation and the associated toxicity. Biochimie. 2017;140:176–192. doi: 10.1016/j.biochi.2017.07.011. [DOI] [PubMed] [Google Scholar]
- 43.Selkoe D.J. Alzheimer’s disease: Genes, proteins, and therapy. Physiol. Rev. 2001;81:741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- 44.Kumari A., Rajput R., Shrivastava N., Somvanshi P., Grover A. Synergistic approaches unraveling regulation and aggregation of intrinsically disordered β-amyloids implicated in Alzheimer’s disease. Int. J. Biochem. Cell Biol. 2018;99:19–27. doi: 10.1016/j.biocel.2018.03.014. [DOI] [PubMed] [Google Scholar]
- 45.Kang J., Lemaire H.G., Unterbeck A., Salbaum J.M., Masters C.L., Grzeschik K.H., Multhaup G., Beyreuther K., Müller-Hill B. The precursor of Alzheimer’s disease amyloid A4 protein resembles a cell-surface receptor. Nature. 1987;325:733–736. doi: 10.1038/325733a0. [DOI] [PubMed] [Google Scholar]
- 46.Iwatsubo T., Odaka A., Suzuki N., Mizusawa H., Nukina N., Ihara Y. Visualization of Aβ42(43) and Aβ40 in senile plaques with end-specific Aβ monoclonals: Evidence that an initially deposited species is Aβ42(43) Neuron. 1994;13:45–53. doi: 10.1016/0896-6273(94)90458-8. [DOI] [PubMed] [Google Scholar]
- 47.De Strooper B., Saftig P., Craessaerts K., Vanderstichele H., Guhde G., Annaert W., Von Figura K., Van Leuven F. Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature. 1998;391:387–390. doi: 10.1038/34910. [DOI] [PubMed] [Google Scholar]
- 48.Stokin G.B., Lillo C., Falzone T.L., Brusch R.G., Rockenstein E., Mount S.L., Raman R., Davies P., Masliah E., Williams D.S., et al. Axonopathy and transport deficits early in the pathogenesis of Alzheimer’s disease. Science. 2005;307:1282–1288. doi: 10.1126/science.1105681. [DOI] [PubMed] [Google Scholar]
- 49.Lewis H., Beher D., Cookson N., Oakley A., Piggott M., Morris C.M., Jaros E., Perry R., Ince P., Kenny R.A., et al. Quantification of Alzheimer pathology in ageing and dementia: Age-related accumulation of amyloid-beta(42) peptide in vascular dementia. Neuropathol. Appl. Neurobiol. 2006;32:103–118. doi: 10.1111/j.1365-2990.2006.00696.x. [DOI] [PubMed] [Google Scholar]
- 50.Sosa L.J., Caceres A., Dupraz S., Oksdath M., Quiroga S., Lorenzo A. The physiological role of the amyloid precursor protein as an adhesion molecule in the developing nervous system. J. Neurochem. 2017;143:11–29. doi: 10.1111/jnc.14122. [DOI] [PubMed] [Google Scholar]
- 51.Goldgaber D., Lerman M.I., McBride O.W., Saffiotti U., Gajdusek D.C. Characterization and chromosomal localization of a cDNA encoding brain amyloid of Alzheimer’s disease. Science. 1987;235:877–880. doi: 10.1126/science.3810169. [DOI] [PubMed] [Google Scholar]
- 52.Tanzi R.E., Gusella J.F., Watkins P.C., Bruns G.A., St George-Hyslop P., Van Keuren M.L., Patterson D., Pagan S., Kurnit D.M., Neve R.L. Amyloid beta protein gene: cDNA, mRNA distribution, and genetic linkage near the Alzheimer locus. Science. 1987;235:880–884. doi: 10.1126/science.2949367. [DOI] [PubMed] [Google Scholar]
- 53.Haass C. Take five—BACE and the γ-secretase quartet conduct Alzheimer’s amyloid β-peptide generation. EMBO J. 2004;23:483–488. doi: 10.1038/sj.emboj.7600061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Haass C., Schlossmacher M.G., Hung A.Y., Vigo-Pelfrey C., Mellon A., Ostaszewski B.L., Lieberburg I., Koo E.H., Schenk D., Teplow D.B., et al. Amyloid β-peptide is produced by cultured cells during normal metabolism. Nature. 1992;359:322–325. doi: 10.1038/359322a0. [DOI] [PubMed] [Google Scholar]
- 55.Gouras G.K., Tsai J., Naslund J., Vincent B., Edgar M., Checler F., Greenfield J.P., Haroutunian V., Buxbaum J.D., Xu H., et al. Intraneuronal Aβ42 accumulation in human brain. Am. J. Pathol. 2000;156:15–20. doi: 10.1016/S0002-9440(10)64700-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bitan G., Vollers S.S., Teplow D.B. Elucidation of primary structure elements controlling early amyloid β-protein oligomerization. J. Biol. Chem. 2003;12:34882–34889. doi: 10.1074/jbc.M300825200. [DOI] [PubMed] [Google Scholar]
- 57.Suzuki N., Cheung T.T., Cai X.-D., Odaka A., Otvos L., Eckman C., Golde T.E., Younkin S.G. An increased percentage of long amyloid beta protein secreted by familial amyloid beta protein precursor (beta APP717) mutants. Science. 1994;264:1336–1340. doi: 10.1126/science.8191290. [DOI] [PubMed] [Google Scholar]
- 58.Lovell M.A., Robertson J.D., Teesdale W.J., Campbell J.L., Markesbery W.R. Copper, iron and zinc in Alzheimer’s disease senile plaques. J. Neurol. Sci. 1998;158:47–52. doi: 10.1016/S0022-510X(98)00092-6. [DOI] [PubMed] [Google Scholar]
- 59.Alexandrescu A.T. Amyloid accomplices and enforcers. Protein Sci. 2005;14:1–12. doi: 10.1110/ps.04887005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Scheuner D., Eckman C., Jensen M., Song X., Citron M., Suzuki N., Bird T.D., Hardy J., Hutton M., Kukull W., et al. Secreted amyloid beta-protein similar to that in the senile plaques of Alzheimer’s disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer’s disease. Nat. Med. 1996;2:864–870. doi: 10.1038/nm0896-864. [DOI] [PubMed] [Google Scholar]
- 61.Citron M., Westaway D., Xia W., Carlson G., Diehl T., Levesque G., Johnson-Wood K., Lee M., Seubert P., Davis A., et al. Mutant presenilins of Alzheimer’s disease increase production of 42-residue amyloid beta-protein in both transfected cells and transgenic mice. Nat. Med. 1997;3:67–72. doi: 10.1038/nm0197-67. [DOI] [PubMed] [Google Scholar]
- 62.Mullan M., Crawford F., Axelman K., Houlden H., Lilius L., Winblad B., Lannfelt L. A pathogenic mutation for probable Alzheimer’s disease in the APP gene at the N-terminus of β-amyloid. Nat. Genet. 1992;1:345–347. doi: 10.1038/ng0892-345. [DOI] [PubMed] [Google Scholar]
- 63.Sherrington R., Rogaev E.I., Liang Y., Rogaeva E.A., Levesque G., Ikeda M., Chi H., Lin C., Li G., Holman K., et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature. 1995;375:754–760. doi: 10.1038/375754a0. [DOI] [PubMed] [Google Scholar]
- 64.Selkoe D.J., Hardy J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016;8:595–608. doi: 10.15252/emmm.201606210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lanoiselée H.M., Nicolas G., Wallon D., Rovelet-Lecrux A., Lacour M., Rousseau S., Richard A.C., Pasquier F., Rollin-Sillaire A., Martinaud O., et al. APP, PSEN1, and PSEN2 mutations in early-onset Alzheimer disease: A genetic screening study of familial and sporadic cases. PLoS Med. 2017;14:e1002270. doi: 10.1371/journal.pmed.1002270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Simmons L.K., May P.C., Tomaselli K.J., Rydel R.E., Fuson K.S., Brigham E.F., Wright S., Lieberburg I., Becker G.W., Brems D.N. Secondary structure of amyloid beta peptide correlates with neurotoxic activity in vitro. Mol. Pharmacol. 1994;45:373–379. [PubMed] [Google Scholar]
- 67.Kirkitadze M.D., Condron M.M., Teplow D.B. Identification and characterization of key kinetic intermediates in amyloid beta-protein fibrillogenesis. J. Mol. Biol. 2001;312:1103–1119. doi: 10.1006/jmbi.2001.4970. [DOI] [PubMed] [Google Scholar]
- 68.Yan Y., Wang C. Aβ42 is More Rigid than Aβ40 at the C Terminus: Implications for Aβ Aggregation and Toxicity. J. Mol. Biol. 2006;364:853–862. doi: 10.1016/j.jmb.2006.09.046. [DOI] [PubMed] [Google Scholar]
- 69.Petkova A.T., Ishii Y., Balbach J.J., Antzutkin O.N., Leapman R.D., Delaglio F., Tycko R. A structural model for Alzheimer’s beta-amyloid fibrils based on experimental constraints from solid state NMR. Proc. Natl. Acad. Sci. USA. 2002;99:16742–16747. doi: 10.1073/pnas.262663499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Petkova A.T., Leapman R.D., Guo Z., Yau W.M., Mattson M.P., Tycko R. Self-propagating, molecular-level polymorphism in Alzheimer’s β-amyloid fibrils. Science. 2005;307:262–265. doi: 10.1126/science.1105850. [DOI] [PubMed] [Google Scholar]
- 71.Bertini I., Gonnelli L., Luchinat C., Mao J., Nesi A. A new structural model of Aβ40 fibrils. J. Am. Chem. Soc. 2011;133:16013–16022. doi: 10.1021/ja2035859. [DOI] [PubMed] [Google Scholar]
- 72.Parthasarathy S., Long F., Miller Y., Xiao Y., McElheny D., Thurber K., Ma B., Nussinov R., Ishii Y. Molecular-level examination of Cu2+ binding structure for amyloid fibrils of 40-residue Alzheimer’s β by solid-state NMR spectroscopy. J. Am. Chem. Soc. 2011;133:3390–3400. doi: 10.1021/ja1072178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sgourakis N.G., Yau W.M., Qiang W. Modeling an in-register, parallel “Iowa” Aβ fibril structure using solid-state NMR data from labeled samples with Rosetta. Structure. 2015;23:216–227. doi: 10.1016/j.str.2014.10.022. [DOI] [PubMed] [Google Scholar]
- 74.Xiao Y., Ma B., McElheny D., Parthasarathy S., Long F., Hoshi M., Nussinov R., Ishii Y. Aβ(1-42) fibril structure illuminates self-recognition and replication of amyloid in Alzheimer’s disease. Nat. Struct. Mol. Biol. 2015;22:499–505. doi: 10.1038/nsmb.2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Colvin M.T., Silvers R., Ni Q.Z., Can T.V., Sergeyev I., Rosay M., Donovan K.J., Michael B., Wall J., Linse S., et al. Atomic Resolution Structure of Monomorphic Aβ42 Amyloid Fibrils. J. Am. Chem. Soc. 2016;138:9663–9674. doi: 10.1021/jacs.6b05129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wälti M.A., Ravotti F., Arai H., Glabe C.G., Wall J.S., Böckmann A., Güntert P., Meier B.H., Riek R. Atomic-resolution structure of a disease-relevant Aβ(1–42) amyloid fibril. Proc. Natl. Acad. Sci. USA. 2016;113:E4976–E4984. doi: 10.1073/pnas.1600749113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Masters C.L., Multhaup G., Simms G., Martins R.N., Beyreuther K. Neuronal origin of a cerebral amyloid: Neurofibriliary tangles of Alzheimer’s disease contain the same protein as the amyloid of plaque cores and blood vessels. EMBO J. 1985;4:2757–2763. doi: 10.1002/j.1460-2075.1985.tb04000.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Portelius E., Bogdanovic N., Gustavsson M.K., Volkmann I., Brinkmalm G., Zetterberg H., Winblad B., Blennow K. Mass spectrometric characterization of brain amyloid beta isoform signatures in familial and sporadic Alzheimer’s disease. Acta Neuropathol. 2010;120:185–193. doi: 10.1007/s00401-010-0690-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Weingarten M.D., Lockwood A.H., Hwo S.Y., Kirschner M.W. A protein factor essential for microtubule assembly. Proc. Natl. Acad. Sci. USA. 1975;72:1858–1862. doi: 10.1073/pnas.72.5.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ebneth A., Godemann R., Stamer K., Illenberger S., Trinczek B., Mandelkow E. Overexpression of tau protein inhibits kinesin-dependent trafficking of vesicles, mitochondria, and endoplasmic reticulum: Implications for Alzheimer’s disease. J. Cell Biol. 1998;143:777–794. doi: 10.1083/jcb.143.3.777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Borna H., Assadoulahei K., Riazi G., Harchegani A.B., Shahriary A. Structure, Function and Interactions of Tau: Particular Focus on Potential Drug Targets for the Treatment of Tauopathies. CNS Neurol. Disord Drug Targets. 2018;17:325–337. doi: 10.2174/1871527317666180525112008. [DOI] [PubMed] [Google Scholar]
- 82.Bakota L., Ussif A., Jeserich G., Brandt R. Systemic and network functions of the microtubule-associated protein tau: Implications for tau-based therapies. Mol. Cell. Neurosci. 2017;84:132–141. doi: 10.1016/j.mcn.2017.03.003. [DOI] [PubMed] [Google Scholar]
- 83.Kosik K.S., Joachim C.L., Selkoe D.J. Microtubule-associated protein tau (tau) is a major antigenic component of paired helical filaments in Alzheimer disease. Proc. Natl. Acad. Sci. USA. 1986;83:4044–4448. doi: 10.1073/pnas.83.11.4044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Andreadis A., Brown W.M., Kosik K.S. Structure and novel exons of the human tau gene. Biochemistry. 1992;31:10626–10633. doi: 10.1021/bi00158a027. [DOI] [PubMed] [Google Scholar]
- 85.Goedert M., Spillantini M.G., Jakes R., Rutherford D., Crowther R.A. Multiple isoforms of human microtubule-associated protein tau: Sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron. 1989;3:519–526. doi: 10.1016/0896-6273(89)90210-9. [DOI] [PubMed] [Google Scholar]
- 86.Goedert M., Spillantini M.G., Potier M.C., Ulrich J., Crowther R.A. Cloning and sequencing of the cDNA encoding an isoform of microtubule-associated protein tau containing four tandem repeats: Differential expression of tau protein mRNAs in human brain. EMBO J. 1989;8:393–399. doi: 10.1002/j.1460-2075.1989.tb03390.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Goedert M., Wischik C.M., Crowther R.A., Walker J.E., Klug A. Cloning and sequencing of the cDNA encoding a core protein of the paired helical filament of Alzheimer disease: Identification as the microtubule-associated protein tau. Proc. Natl. Acad. Sci. USA. 1988;85:4051–4055. doi: 10.1073/pnas.85.11.4051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lee V.M., Balin B.J., Otvos L., Jr., Trojanowski J.Q. A68: A major subunit of paired helical filaments and derivatized forms of normal Tau. Science. 1991;251:675–678. doi: 10.1126/science.1899488. [DOI] [PubMed] [Google Scholar]
- 89.Santa-Maria I., Varghese M., Ksiezak-Reding H., Dzhun A., Wang J., Pasinetti G.M. Paired helical filaments from Alzheimer disease brain induce intracellular accumulation of Tau protein in aggresomes. J. Biol. Chem. 2012;287:20522–20533. doi: 10.1074/jbc.M111.323279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bhattacharya K., Rank K.B., Evans D.B., Sharma S.K. Role of cysteine-291 and cysteine-322 in the polymerization of human tau into Alzheimer-like filaments. Biochem. Biophys. Res. Commun. 2001;285:20–26. doi: 10.1006/bbrc.2001.5116. [DOI] [PubMed] [Google Scholar]
- 91.Novak P., Cehlar O., Skrabana R., Novak M. Tau Conformation as a Target for Disease-Modifying Therapy: The Role of Truncation. J. Alzheimers Dis. 2018;64:S535–S546. doi: 10.3233/JAD-179942. [DOI] [PubMed] [Google Scholar]
- 92.Florenzano F., Veronica C., Ciasca G., Ciotti M.T., Pittaluga A., Olivero G., Feligioni M., Iannuzzi F., Latina V., Maria Sciacca M.F., et al. Extracellular truncated tau causes early presynaptic dysfunction associated with Alzheimer’s disease and other tauopathies. Oncotarget. 2017;8:64745–64778. doi: 10.18632/oncotarget.17371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Schedin-Weiss S., Winblad B., Tjernberg L.O. The role of protein glycosylation in Alzheimer disease. FEBS J. 2014;281:46–62. doi: 10.1111/febs.12590. [DOI] [PubMed] [Google Scholar]
- 94.Ligabue-Braun R., Carlini C.R. Moonlighting Toxins: Ureases and Beyond. In: Gopalakrishnakone P., Carlini C.R., Ligabue-Braun R., editors. Plant Toxins. Springer; Dordrecht, The Netherlands: 2015. pp. 199–219. [Google Scholar]
- 95.Oliveira J.M., Henriques A.G., Martins F., Rebelo S., da Cruz e Silva O.A. Amyloid-beta Modulates Both AbetaPP and Tau Phosphorylation. J. Alzheimers Dis. 2015;45:495–507. doi: 10.3233/JAD-142664. [DOI] [PubMed] [Google Scholar]
- 96.Ittner L.M., Ke Y.D., Delerue F., Bi M., Gladbach A., van Eersel J., Wölfing H., Chieng B.C., Christie M.J., Napier I.A., et al. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell. 2010;142:387–397. doi: 10.1016/j.cell.2010.06.036. [DOI] [PubMed] [Google Scholar]
- 97.Prusiner S.B. Novel proteinaceous infectious particles cause scrapie. Science. 1982;216:136–144. doi: 10.1126/science.6801762. [DOI] [PubMed] [Google Scholar]
- 98.Prusiner S.B., Mckinley M.P., Groth D.F., Bowman K.A., Mock N.I., Cochran S.P., Masiarz F.R. Scrapie agent contains a hydrophobic protein. Proc. Natl. Acad. Sci. USA. 1981;78:6675–6679. doi: 10.1073/pnas.78.11.6675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Prusiner S.B., Groth D.F., Bolton D.C., Kent S.B., Hood L.E. Purification and structural studies of a major scrapie prion protein. Cell. 1984;38:127–134. doi: 10.1016/0092-8674(84)90533-6. [DOI] [PubMed] [Google Scholar]
- 100.Westergard L., Christensen H.M., Harris D.A. The cellular prion protein (PrP(C)): Its physiological function and role in Disease. Biochim. Biophys. Acta. 2007;1772:629–644. doi: 10.1016/j.bbadis.2007.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bremer J., Baumann F., Tiberi C., Wessig C., Fischer H., Schwarz P., Steele A.D., Toyka K.V., Nave K.A., Weis J., et al. Axonal prion protein is required for peripheral myelin maintenance. Nat. Neurosci. 2010;13:310–318. doi: 10.1038/nn.2483. [DOI] [PubMed] [Google Scholar]
- 102.Chakravarty A.K., Jarosz D.F. More than Just a Phase: Prions at the Crossroads of Epigenetic Inheritance and Evolutionary Change. J. Mol. Biol. 2018;430:4607–4618. doi: 10.1016/j.jmb.2018.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Riek R., Hornemann S., Wider G., Glockshuber R., Wüthrich K. NMR characterization of the full-length recombinant murine prion protein, mPrP(23-231) FEBS Lett. 1997;413:282–288. doi: 10.1016/S0014-5793(97)00920-4. [DOI] [PubMed] [Google Scholar]
- 104.Donne D.G., Viles J.H., Groth D., Mehlhorn I., James T.L., Cohen F.E., Prusiner S.B., Wright P.E., Dyson H.J. Structure of the recombinant full-length hamster prion protein PrP (29-231): The N terminus is highly flexible. Proc. Natl. Acad. Sci. USA. 1997;94:13452–13457. doi: 10.1073/pnas.94.25.13452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zahn R., Liu A., Luhrs T., Riek R., von Schroetter C., Lopez-Garcia F., Billeter M., Calzolai L., Wider G., Wuthrich K. NMR solution structure of the human prion protein. Proc. Natl. Acad. Sci. USA. 2000;97:145–150. doi: 10.1073/pnas.97.1.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lopez-García F.L., Zahn R., Riek R., Wüthrich K. NMR structure of the bovine prion protein. Proc. Natl. Acad. Sci. USA. 2000;97:8334–8339. doi: 10.1073/pnas.97.15.8334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sabate R., Rousseau F., Schymkowitz J., Ventura S. What makes a protein sequence a prion? PLoS Comput. Biol. 2015;11:e1004013. doi: 10.1371/journal.pcbi.1004013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Cong X., Casiraghi N., Rossetti G., Mohanty S., Giachin G., Legname G., Carloni P. Role of Prion Disease-Linked Mutations in the Intrinsically Disordered N-Terminal Domain of the Prion Protein. J. Chem. Theory Comput. 2013;9:5158–5167. doi: 10.1021/ct400534k. [DOI] [PubMed] [Google Scholar]
- 109.Pan K.-M., Baldwin M., Nguyen J., Gasset M., Serban A.N.A., Groth D., Mehlhorn I., Huang Z., Fletterick R.J., Cohen F.E. Conversion of alpha-helices into beta-sheets features in the formation of the scrapie prion proteins. Proc. Natl. Acad. Sci. USA. 1993;90:10962–10966. doi: 10.1073/pnas.90.23.10962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Prusiner S.B. Molecular biology of prion diseases. Science. 1991;252:1515–1522. doi: 10.1126/science.1675487. [DOI] [PubMed] [Google Scholar]
- 111.Prusiner S.B. Chemistry and biology of prions. Biochemistry. 1992;31:12277–12288. doi: 10.1021/bi00164a001. [DOI] [PubMed] [Google Scholar]
- 112.Brown P., Gajdusek D.C. The Human Spongiform Encephalopathies: Kuru, Creutzfeldt-Jakob Disease, and the Gerstmann-Sträussler-Scheinker Syndrome. In: Chesebro B.W., editor. Transmissible Spongiform Encephalopathies: Current Topics in Microbiology and Immunology. Volume 172. Springer; Berlin, Germany: 1991. pp. 1–20. [DOI] [PubMed] [Google Scholar]
- 113.Nathanson N., Wilesmith J., Griot C. Bovine spongiform encephalopathy (BSE): Causes and consequences of a common source epidemic. Am. J. Epidemiol. 1997;145:959–969. doi: 10.1093/oxfordjournals.aje.a009064. [DOI] [PubMed] [Google Scholar]
- 114.Pattison I.H. The relative susceptibility of sheep, goats and mice to two types of the goat scrapie agent. Res. Vet. Sci. 1966;7:207–212. doi: 10.1016/S0034-5288(18)34700-3. [DOI] [PubMed] [Google Scholar]
- 115.Scott M., Foster D., Mirenda C., Serban D., Coufal F., Wälchli M., Torchia M., Groth D., Carlson G., DeArmond S.J., et al. Transgenic mice expressing hamster prion protein produce species-specific scrapie infectivity and amyloid plaques. Cell. 1989;59:847–857. doi: 10.1016/0092-8674(89)90608-9. [DOI] [PubMed] [Google Scholar]
- 116.Prusiner S.B., Scott M., Foster D., Pan K.-M., Groth D., Mirenda C., Torchia M., Yang S.-L., Serban D., Carlson G.A., et al. Transgenetic studies implicate interactions between homologous PrP isoforms in scrapie prion replication. Cell. 1990;63:673–686. doi: 10.1016/0092-8674(90)90134-Z. [DOI] [PubMed] [Google Scholar]
- 117.Bartz J.C., McKenzie D.I., Bessen R.A., Marsh R.F., Aiken J.M. Transmissible mink encephalopathy species barrier effect between ferret and mink: PrP gene and protein analysis. J. Gen. Virol. 1994;75:2947–2953. doi: 10.1099/0022-1317-75-11-2947. [DOI] [PubMed] [Google Scholar]
- 118.Bian J., Khaychuk V., Angers R.C., Fernández-Borges N., Vidal E., Meyerett-Reid C., Kim S., Calvi C.L., Bartz J.C., Hoover E.A., et al. Prion replication without host adaptation during interspecies transmissions. Proc. Natl. Acad. Sci. USA. 2017;114:1141–1146. doi: 10.1073/pnas.1611891114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Prusiner S.B. Molecular biology and pathogenesis of prion diseases. Trends Biochem. Sci. 1996;21:482–487. doi: 10.1016/S0968-0004(96)10063-3. [DOI] [PubMed] [Google Scholar]
- 120.Colby D.W., Prusiner S.B. Prions. Cold Spring Harb. Perspect. Biol. 2011;3:a006833. doi: 10.1101/cshperspect.a006833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Makarava N., Kovacs G.G., Bocharova O., Savtchenko R., Alexeeva I., Budka H., Rohwer R.G., Baskakov I.V. Recombinant prion protein induces a new transmissible prion disease in wild-type animals. Acta Neuropathol. 2010;119:177–187. doi: 10.1007/s00401-009-0633-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Stahl N., Baldwin M.A., Prusiner S.B., Teplow D.B., Hood L., Gibson B.W., Burlingame A.L. Structural Studies of the Scrapie Prion Protein Using Mass Spectrometry and Amino Acid Sequencing. Biochemistry. 1993;32:1991–2002. doi: 10.1021/bi00059a016. [DOI] [PubMed] [Google Scholar]
- 123.Meyer R.K., McKinley M.P., Bowman K.A., Braunfeld M.B., Barry R.A., Prusiner S.B. Separation and properties of cellular and scrapie prion proteins. Proc. Natl. Acad. Sci. USA. 1986;83:2310–2314. doi: 10.1073/pnas.83.8.2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Saverioni D., Notari S., Capellari S., Poggiolini I., Giese A., Kretzschmar H.A., Parchi P. Analyses of protease resistance and aggregation state of abnormal prion protein across the spectrum of human prions. J. Biol. Chem. 2013;288:27972–27985. doi: 10.1074/jbc.M113.477547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Safar J., Wille H., Itri V., Groth D., Serban H., Torchia M., Cohen F.E., Prusiner S.B. Eight prion strains have PrP Sc molecules with different conformations. Nat. Med. 1998;4:1157–1165. doi: 10.1038/2654. [DOI] [PubMed] [Google Scholar]
- 126.Tzaban S., Friedlander G., Schonberger O., Horonchik L., Yedidia Y., Shaked G., Gabizon R., Taraboulos A. Protease-sensitive scrapie prion protein in aggregates of heterogeneous sizes. Biochemistry. 2002;41:12868–12875. doi: 10.1021/bi025958g. [DOI] [PubMed] [Google Scholar]
- 127.DeArmond S.J., Sánchez H., Yehiely F., Qiu Y., Ninchak-Casey A., Daggett V., Camerino A.P., Cayetano J., Rogers M., Groth D., et al. Selective neuronal targeting in prion disease. Neuron. 1997;19:1337–1348. doi: 10.1016/S0896-6273(00)80424-9. [DOI] [PubMed] [Google Scholar]
- 128.Colby D.W., Zhang Q., Wang S., Groth D., Legname G., Riesner D., Prusiner S.B. Prion detection by an amyloid seeding assay. Proc. Natl. Acad. Sci. USA. 2007;104:20914–20919. doi: 10.1073/pnas.0710152105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Wille H., Prusiner S.B., Cohen F.E. Scrapie infectivity is independent of amyloid staining properties of the N-Terminally truncated prion protein. J. Struct. Biol. 2000;130:323–338. doi: 10.1006/jsbi.2000.4242. [DOI] [PubMed] [Google Scholar]
- 130.Uversky V.N., Davé V., Iakoucheva L.M., Malaney P., Metallo S.J., Pathak R.R., Joerger A.C. Pathological unfoldomics of uncontrolled chaos: Intrinsically disordered proteins and human diseases. Chem. Rev. 2014;114:6844–6879. doi: 10.1021/cr400713r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Mol P.R. Oncogenes as Therapeutic Targets in Cancer: A Review. IOSR J. Dent. Med. Sci. 2013;5:46–56. doi: 10.9790/0853-0524656. [DOI] [Google Scholar]
- 132.Dunker A.K., Uversky V.N. Drugs for “protein clouds”: Targeting intrinsically disordered transcription factors. Curr. Opin. Pharmacol. 2010;10:782–788. doi: 10.1016/j.coph.2010.09.005. [DOI] [PubMed] [Google Scholar]
- 133.Uversky V.N. p53 Proteoforms and Intrinsic Disorder: An Illustration of the Protein Structure-Function Continuum Concept. Int. J. Mol. Sci. 2016;17:1874. doi: 10.3390/ijms17111874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Levine A.J. Targeting therapies for the p53 protein in cancer treatments. Annu. Rev. Cancer Biol. 2019 doi: 10.1146/annurev-cancerbio-030518-055455. [DOI] [Google Scholar]
- 135.Hollstein M., Sidransky D., Vogelstein B., Curtis C. p53 Mutation Human Cancers. Science. 1991;253:49–53. doi: 10.1126/science.1905840. [DOI] [PubMed] [Google Scholar]
- 136.Muller P.A.J., Vousden K.H. P53 mutations in cancer. Nat. Cell Biol. 2013;15:2–8. doi: 10.1038/ncb2641. [DOI] [PubMed] [Google Scholar]
- 137.Dawson R., Müller L., Dehner A., Klein C., Kessler H., Buchner J. The N-terminal domain of p53 is natively unfolded. J. Mol. Biol. 2003;332:1131–1141. doi: 10.1016/j.jmb.2003.08.008. [DOI] [PubMed] [Google Scholar]
- 138.Kubbutat M.H.G., Jones S.N., Vousden K.H. Regulation of p53 stability by Mdm2. Nature. 1997;387:299–303. doi: 10.1038/387299a0. [DOI] [PubMed] [Google Scholar]
- 139.Nag S., Qin J., Srivenugopal K.S., Wang M., Zhang R. The MDM2-p53 pathway revisited. J. Biomed. Res. 2013;27:254–271. doi: 10.7555/JBR.27.20130030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Williams A.B., Schumacher B. p53 in the DNA-damage-repair process. Cold Spring Harb. Perspect. Med. 2016;6:a026070. doi: 10.1101/cshperspect.a026070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Cho Y., Gorina S., Jeffrey P.D., Pavletich N.P. Crystal structure of a p53 tumor suppressor-DNA complex: Understanding tumorigenic mutations. Science. 1994;265:346–355. doi: 10.1126/science.8023157. [DOI] [PubMed] [Google Scholar]
- 142.Clore G.M., Ernst J., Clubb R., Omichinski J.G., Kennedy W.M.P., Sakaguchi K., Appella E., Gronenborn A.M. Refined solution structure of the oligomerization domain of the tumour suppressor p53. Nat. Struct. Mol. Biol. 1995;2:321–333. doi: 10.1038/nsb0495-321. [DOI] [PubMed] [Google Scholar]
- 143.Fields S., Jang S.K. Presence of a potent transcription activating sequence in the p53 protein. Science. 1990;249:1046–1049. doi: 10.1126/science.2144363. [DOI] [PubMed] [Google Scholar]
- 144.Haupt Y., Maya R., Kazaz A., Oren M. Mdm2 promotes the rapid degradation of p53. Nature. 1997;387:296–299. doi: 10.1038/387296a0. [DOI] [PubMed] [Google Scholar]
- 145.Honda R., Tanaka H., Yasuda H. Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett. 1997;420:25–27. doi: 10.1016/S0014-5793(97)01480-4. [DOI] [PubMed] [Google Scholar]
- 146.Joerger A.C., Fersht A.R. Structural Biology of the Tumor Suppressor p53. Annu. Rev. Biochem. 2008;77:557–582. doi: 10.1146/annurev.biochem.77.060806.091238. [DOI] [PubMed] [Google Scholar]
- 147.Lee W., Harvey T.S., Yin Y., Yau P., Litchfield D., Arrowsmith C.H. Solution structure of the tetrameric minimum transforming domain of p53. Nat. Struct. Mol. Biol. 1994;1:877–890. doi: 10.1038/nsb1294-877. [DOI] [PubMed] [Google Scholar]
- 148.Uversky A.V., Xue B., Peng Z., Kurgan L., Uversky V.N. On the intrinsic disorder status of the major players in programmed cell death pathways. F1000Research. 2013;2:190. doi: 10.12688/f1000research.2-190.v1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Kussie P.H., Gorina S., Marechal V., Elenbaas B., Moreau J., Levine A.J., Pavletich N.P. Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science. 1996;274:948–953. doi: 10.1126/science.274.5289.948. [DOI] [PubMed] [Google Scholar]
- 150.Marine J.-C., Jochemsen A.G. Mdmx as an essential regulator of p53 activity. Biochem. Biophys. Res. Commun. 2005;331:750–760. doi: 10.1016/j.bbrc.2005.03.151. [DOI] [PubMed] [Google Scholar]
- 151.Schon O., Friedler A., Bycroft M., Freund S.M.V., Fersht A.R. Molecular mechanism of the interaction between MDM2 and p53. J. Mol. Biol. 2002;323:491–501. doi: 10.1016/S0022-2836(02)00852-5. [DOI] [PubMed] [Google Scholar]
- 152.Borcherds W., Kashtanov S., Wu H., Daughdrill G.W. Structural divergence is more extensive than sequence divergence for a family of intrinsically disordered proteins. Proteins Struct. Funct. Bioinform. 2013;81:1686–1698. doi: 10.1002/prot.24303. [DOI] [PubMed] [Google Scholar]
- 153.Chi S.W., Lee S.H., Kim D.H., Ahn M.J., Kim J.S., Woo J.Y., Torizawa T., Kainosho M., Han K.H. Structural details on mdm2-p53 interaction. J. Biol. Chem. 2005;280:38795–38802. doi: 10.1074/jbc.M508578200. [DOI] [PubMed] [Google Scholar]
- 154.Popowicz G.M., Czarna A., Rothweiler U., Szwagierczak A., Krajewski M., Weber L., Holak T.A. Molecular basis for the inhibition of p53 by Mdmx. Cell Cycle. 2007;6:2386–2392. doi: 10.4161/cc.6.19.4740. [DOI] [PubMed] [Google Scholar]
- 155.Vise P.D., Baral B., Latos A.J., Daughdrill G.W. NMR chemical shift and relaxation measurements provide evidence for the coupled folding and binding of the p53 transactivation domain. Nucleic Acids Res. 2005;33:2061–2077. doi: 10.1093/nar/gki336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Chene P. The role of tetramerization in p53 function. Oncogene. 2001;20:2611–2617. doi: 10.1038/sj.onc.1204373. [DOI] [PubMed] [Google Scholar]
- 157.Pelengaris S., Khan M., Evan G. c-MYC: More than just a matter of life and death. Nat. Rev. Cancer. 2002;2:764–776. doi: 10.1038/nrc904. [DOI] [PubMed] [Google Scholar]
- 158.Dang C.V. c-Myc Target Genes Involved in Cell Growth, Apoptosis, and Metabolism. Mol. Cell. Biol. 1999;19:1–11. doi: 10.1128/MCB.19.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Nesbit C.E., Tersak J.M., Prochownik E.V. MYC oncogenes and human neoplastic disease. Oncogene. 1999;18:3004–3016. doi: 10.1038/sj.onc.1202746. [DOI] [PubMed] [Google Scholar]
- 160.Schlagbauer-Wadl H., Griffioen M., Van Elsas A., Schrier P.I., Pustelnik T., Eichler H., Wolff K., Pehamberger H., Jansen B. Influence of Increased c-Myc Expression on the Growth Characteristics of Human Melanoma. J. Investig. Dermatol. 1999;112:332–336. doi: 10.1046/j.1523-1747.1999.00506.x. [DOI] [PubMed] [Google Scholar]
- 161.Kumar D., Sharma N., Giri R. Therapeutic interventions of cancers using intrinsically disordered proteins as drug targets: C-myc as model system. Cancer Inform. 2017;16 doi: 10.1177/1176935117699408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Amati B., Dalton S., Brooks M.W., Littlewood T.D., Evan G.I., Land H. Transcriptional activation by the human c-Myc oncoprotein in yeast requires interaction with Max. Nature. 1992;359:423–426. doi: 10.1038/359423a0. [DOI] [PubMed] [Google Scholar]
- 163.Blackwell T.K., Kretzner L., Blackwood E.M., Eisenman R.N., Weintraub H. Sequence-specific DNA binding by the c-Myc protein. Science. 1990;250:1149–1151. doi: 10.1126/science.2251503. [DOI] [PubMed] [Google Scholar]
- 164.Blackwood E.M., Eisenman R.N. Max: A helix-loop-helix zipper protein that forms a sequence-specific DNA-binding complex with Myc. Science. 1991;251:1211–1217. doi: 10.1126/science.2006410. [DOI] [PubMed] [Google Scholar]
- 165.Kato G.J., Lee W.M., Chen L.L., Dang C.V. Max: Functional domains and interaction with c-Myc. Genes Dev. 1992;6:81–92. doi: 10.1101/gad.6.1.81. [DOI] [PubMed] [Google Scholar]
- 166.Andresen C., Helander S., Lemak A., Farès C., Csizmok V., Carlsson J., Penn L.Z., Forman-Kay J.D., Arrowsmith C.H., Lundström P., et al. Transient structure and dynamics in the disordered c-Myc transactivation domain affect Bin1 binding. Nucleic Acids Res. 2012;40:6353–6366. doi: 10.1093/nar/gks263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Mao D.Y.L., Watson J.D., Yan P.S., Barsyte-Lovejoy D., Khosravi F., Wong W.W.-L., Farnham P.J., Huang T.H.-M., Penn L.Z. Analysis of Myc bound loci identified by CpG island arrays shows that Max is essential for Myc-dependent repression. Curr. Biol. 2003;13:882–886. doi: 10.1016/S0960-9822(03)00297-5. [DOI] [PubMed] [Google Scholar]
- 168.Clausen D.M., Guo J., Parise R.A., Beumer J.H., Egorin M.J., Lazo J.S., Prochownik E.V., Eiseman J.L. In vitro cytotoxicity and in vivo efficacy, pharmacokinetics, and metabolism of 10074-G5, a novel small-molecule inhibitor of c-Myc/Max dimerization. J. Pharmacol. Exp. Ther. 2010;335:715–727. doi: 10.1124/jpet.110.170555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Raffeiner P., Röck R., Schraffl A., Hartl M., Hart J.R., Janda K.D., Vogt P.K., Stefan E., Bister K. In vivo quantification and perturbation of Myc-Max interactions and the impact on oncogenic potential. Oncotarget. 2014;5:8869–8878. doi: 10.18632/oncotarget.2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Ferrannini E. Insulin resistance versus insulin deficiency in non-insulin-dependent diabetes mellitus: Problems and prospects. Endocr. Rev. 1998;19:477–490. doi: 10.1210/edrv.19.4.0336. [DOI] [PubMed] [Google Scholar]
- 171.Cooper G.J., Willis A.C., Clark A., Turner R.C., Sim R.B., Reid K.B. Purification and characterization of a peptide from amyloid-rich pancreases of type 2 diabetic patients. Proc. Natl. Acad. Sci. USA. 1987;84:8628–8632. doi: 10.1073/pnas.84.23.8628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Westermark P., Wernstedt C., Wilander E., Hayden D.W., O’Brien T.D., Johnson K.H. Amyloid fibrils in human insulinoma and islets of Langerhans of the diabetic cat are derived from a neuropeptide-like protein also present in normal islet cells. Proc. Natl. Acad. Sci. USA. 1987;84:3881–3885. doi: 10.1073/pnas.84.11.3881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Mosselman S., Höppener J.W., Zandberg J., van Mansfeld A.D., Geurts van Kessel A.H., Lips C.J., Jansz H.S. Islet amyloid polypeptide: Identification and chromosomal localization of the human gene. FEBS Lett. 1988;239:227–232. doi: 10.1016/0014-5793(88)80922-0. [DOI] [PubMed] [Google Scholar]
- 174.Kapurniotu A. Amyloidogenicity and cytotoxicity of islet amyloid polypeptide. Biopolymers. 2001;60:438–459. doi: 10.1002/1097-0282(2001)60:6<438::AID-BIP10182>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 175.Moore S.J., Sonar K., Bharadwaj P., Deplazes E., Mancera R.L. Characterisation of the Structure and Oligomerisation of Islet Amyloid Polypeptides (IAPP): A Review of Molecular Dynamics Simulation Studies. Molecules. 2018;23:2142. doi: 10.3390/molecules23092142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Goldsbury C., Goldie K., Pellaud J., Seelig J., Frey P., Müller S.A., Kistler J., Cooper G.J., Aebi U. Amyloid fibril formation from full-length and fragments of amylin. J. Struct. Biol. 2000;130:352–362. doi: 10.1006/jsbi.2000.4268. [DOI] [PubMed] [Google Scholar]
- 177.Yonemoto I.T., Kroon G.J., Dyson H.J., Balch W.E., Kelly J.W. Amylin proprotein processing generates progressively more amyloidogenic peptides that initially sample the helical state. Biochemistry. 2008;47:9900–9910. doi: 10.1021/bi800828u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Reddy A.S., Wang L., Singh S., Ling Y.L., Buchanan L., Zanni M.T., Skinner J.L., de Pablo J.J. Stable and metastable states of human amylin in solution. Biophys. J. 2010;99:2208–2216. doi: 10.1016/j.bpj.2010.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Qiao Q., Bowman G.R., Huang X. Dynamics of an intrinsically disordered protein reveal metastable conformations that potentially seed aggregation. J. Am. Chem. Soc. 2013;135:16092–16101. doi: 10.1021/ja403147m. [DOI] [PubMed] [Google Scholar]
- 180.Höppener J.W., Ahrén B., Lips C.J. Islet amyloid and type 2 diabetes mellitus. N. Engl. J. Med. 2000;343:411–419. doi: 10.1056/NEJM200008103430607. [DOI] [PubMed] [Google Scholar]
- 181.Höppener J.W., Lips C.J. Role of islet amyloid in type 2 diabetes mellitus. Int. J. Biochem. Cell Biol. 2006;38:726–736. doi: 10.1016/j.biocel.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 182.Dong X., Qiao Q., Qian Z., Wei G. Recent computational studies of membrane interaction and disruption of human islet amyloid polypeptide: Monomers, oligomers and protofibrils. Biochim. Biophys. Acta Biomembr. 2018;1860:1826–1839. doi: 10.1016/j.bbamem.2018.03.006. [DOI] [PubMed] [Google Scholar]
- 183.Longhena F., Spano P., Bellucci A. Targeting of Disordered Proteins by Small Molecules in Neurodegenerative Diseases. Handb. Exp. Pharmacol. 2018;245:85–110. doi: 10.1007/164_2017_60. [DOI] [PubMed] [Google Scholar]
- 184.Babu M.M., van der Lee R., de Groot N.S., Gsponer J. Intrinsically disordered proteins: Regulation and disease. Curr. Opin. Struct. Biol. 2011;21:432–440. doi: 10.1016/j.sbi.2011.03.011. [DOI] [PubMed] [Google Scholar]
- 185.Wright P.E., Dyson H.J. Intrinsically disordered proteins in cellular signalling and regulation. Nat. Rev. Mol. Cell Biol. 2015;16:18–29. doi: 10.1038/nrm3920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Bah A., Forman-Kay J.D. Modulation of intrinsically disordered protein function by post-translational modifications. J. Biol. Chem. 2016;291:6696–6705. doi: 10.1074/jbc.R115.695056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Dyson H.J. Making Sense of Intrinsically Disordered Proteins. Biophys. J. 2016;110:1013–1016. doi: 10.1016/j.bpj.2016.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Van Der Lee R., Buljan M., Lang B., Weatheritt R.J., Daughdrill G.W., Dunker A.K., Fuxreiter M., Gough J., Gsponer J., Jones D.T., et al. Classification of intrinsically disordered regions and proteins. Chem. Rev. 2014;114:6589–6631. doi: 10.1021/cr400525m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Babu M.M. The contribution of intrinsically disordered regions to protein function, cellular complexity, and human disease. Biochem. Soc. Trans. 2016;44:1185–1200. doi: 10.1042/BST20160172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Strome B., Hsu I.S., Li Cheong Man M., Zarin T., Nguyen Ba A., Moses A.M. Short linear motifs in intrinsically disordered regions modulate HOG signaling capacity. BMC Syst. Biol. 2018;12:75. doi: 10.1186/s12918-018-0597-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Van Roey K., Uyar B., Weatheritt R.J., Dinkel H., Seiler M., Budd A., Gibson T.J., Davey N.E. Short linear motifs: Ubiquitous and functionally diverse protein interaction modules directing cell regulation. Chem. Rev. 2014;114:6733–6778. doi: 10.1021/cr400585q. [DOI] [PubMed] [Google Scholar]
- 192.Tompa P. Unstructural biology coming of age. Curr. Opin. Struct. Biol. 2011;21:419–425. doi: 10.1016/j.sbi.2011.03.012. [DOI] [PubMed] [Google Scholar]
- 193.Hu G., Wu Z., Uversky V.N., Kurgan L. Functional analysis of human hub proteins and their interactors involved in the intrinsic disorder-enriched interactions. Int. J. Mol. Sci. 2017;18:2761. doi: 10.3390/ijms18122761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Tompa P., Kovacs D. Intrinsically disordered chaperones in plants and animals. Biochem. Cell Biol. 2010;88:167–174. doi: 10.1139/O09-163. [DOI] [PubMed] [Google Scholar]
- 195.Sharma R., Raduly Z., Miskei M., Fuxreiter M. Fuzzy complexes: Specific binding without complete folding. FEBS Lett. 2015;589:2533–2542. doi: 10.1016/j.febslet.2015.07.022. [DOI] [PubMed] [Google Scholar]
- 196.Weiss M.A., Ellenberger T., Wobbe C.R., Lee J.P., Harrison S.C., Struhl K. Folding transition in the DMA-binding domain of GCN4 on specific binding to DNA. Nature. 1990;347:575–578. doi: 10.1038/347575a0. [DOI] [PubMed] [Google Scholar]
- 197.Bracken C., Carr P.A., Cavanagh J., Palmer A.G. Temperature dependence of intramolecular dynamics of the basic leucine zipper of GCN4: Implications for the entropy of association with DNA. J. Mol. Biol. 1999;285:2133–2146. doi: 10.1006/jmbi.1998.2429. [DOI] [PubMed] [Google Scholar]
- 198.Shammas S.L., Crabtree M.D., Dahal L., Wicky B.I.M., Clarke J. Insights into coupled folding and binding mechanisms from kinetic studies. J. Biol. Chem. 2016;291:6689–6695. doi: 10.1074/jbc.R115.692715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Hammes G.G., Chang Y.-C., Oas T.G. Conformational selection or induced fit: A flux description of reaction mechanism. Proc. Natl. Acad. Sci. USA. 2009;106:13737–13741. doi: 10.1073/pnas.0907195106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.DeForte S., Uversky V.N. Order, disorder, and everything in between. Molecules. 2016;21:1090. doi: 10.3390/molecules21081090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Xie H., Vucetic S., Iakoucheva L.M., Oldfield C.J., Dunker A.K., Obradovic Z., Uversky V.N. Functional anthology of intrinsic disorder. 3. Ligands, post-translational modifications, and diseases associated with intrinsically disordered proteins. J. Proteome Res. 2007;6:1917–1932. doi: 10.1021/pr060394e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Schwalbe M., Biernat J., Bibow S., Ozenne V., Jensen M.R., Kadavath H., Blackledge M., Mandelkow E., Zweckstetter M. Phosphorylation of human tau protein by microtubule affinity-regulating kinase 2. Biochemistry. 2013;52:9068–9079. doi: 10.1021/bi401266n. [DOI] [PubMed] [Google Scholar]
- 203.Ou L., Ferreira A.M., Otieno S., Xiao L., Bashford D., Kriwacki R.W. Incomplete folding upon binding mediates Cdk4/cyclin D complex activation by tyrosine phosphorylation of inhibitor p27 protein. J. Biol. Chem. 2011;286:30142–30151. doi: 10.1074/jbc.M111.244095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Zeng Y., He Y., Yang F., Mooney S.M., Getzenberg R.H., Orban J., Kulkarni P. The cancer/testis antigen prostate-associated gene 4 (PAGE4) is a highly intrinsically disordered protein. J. Biol. Chem. 2011;286:13985–13994. doi: 10.1074/jbc.M110.210765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Coskuner-Weber O., Uversky V.N. Insights into the molecular mechanisms of Alzheimer’s and Parkinson’s diseases with molecular simulations: Understanding the roles of artificial and pathological missense mutations in intrinsically disordered proteins related to pathology. Int. J. Mol. Sci. 2018;19:336. doi: 10.3390/ijms19020336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Edwards Y.J.K., Lobley A.E., Pentony M.M., Jones D.T. Insights into the regulation of intrinsically disordered proteins in the human proteome by analyzing sequence and gene expression data. Genome Biol. 2009;10:R50. doi: 10.1186/gb-2009-10-5-r50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Gsponer J., Futschik M.E., Teichmann S.A., Babu M.M. Tight regulation of unstructured proteins: From transcript synthesis to protein degradation. Science. 2008;322:1365–1368. doi: 10.1126/science.1163581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Forbes J.G., Jin A.J., Ma K., Gutierrez-Cruz G., Tsai W.L., Wang K. Titin PEVK segment: Charge-driven elasticity of the open and flexible polyampholyte. J. Muscle Res. Cell Motil. 2005;26:291–301. doi: 10.1007/s10974-005-9035-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Boothby T.C., Tapia H., Brozena A.H., Piszkiewicz S., Smith A.E., Giovannini I., Rebecchi L., Pielak G.J., Koshland D., Goldstein B. Tardigrades Use Intrinsically Disordered Proteins to Survive Desiccation. Mol. Cell. 2017;65:975.e5–984.e5. doi: 10.1016/j.molcel.2017.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Russo J., Olivas W.M. Conditional regulation of Puf1p, Puf4p, and Puf5p activity alters YHB1 mRNA stability for a rapid response to toxic nitric oxide stress in yeast. Mol. Biol. Cell. 2015;26:1015–1029. doi: 10.1091/mbc.E14-10-1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Wang M., Ogé L., Perez-Garcia M.D., Hamama L., Sakr S. The PUF protein family: Overview on PUF RNA targets, biological functions, and post transcriptional regulation. Int. J. Mol. Sci. 2018;19:410. doi: 10.3390/ijms19020410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Tsvetkov P., Reuven N., Shaul Y. The nanny model for IDPs. Nat. Chem. Biol. 2009;5:778–781. doi: 10.1038/nchembio.233. [DOI] [PubMed] [Google Scholar]
- 213.Inobe T., Matouschek A. Paradigms of protein degradation by the proteasome. Curr. Opin. Struct. Biol. 2014;24:156–164. doi: 10.1016/j.sbi.2014.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Hagai T., Azia A., Tóth-Petróczy Á., Levy Y. Intrinsic disorder in ubiquitination substrates. J. Mol. Biol. 2011;412:319–324. doi: 10.1016/j.jmb.2011.07.024. [DOI] [PubMed] [Google Scholar]
- 215.Wenzel T., Baumeister W. Conformational constraints in protein degradation by the 20S proteasome. Nat. Struct. Biol. 1995;2:199–204. doi: 10.1038/nsb0395-199. [DOI] [PubMed] [Google Scholar]
- 216.Theillet F.-X., Binolfi A., Frembgen-Kesner T., Hingorani K., Sarkar M., Kyne C., Li C., Crowley P.B., Gierasch L., Pielak G.J., et al. Physicochemical Properties of Cells and Their Effects on Intrinsically Disordered Proteins (IDPs) Chem. Rev. 2014;114:6661–6714. doi: 10.1021/cr400695p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Dou F., Netzer W.J., Tanemura K., Li F., Hartl F.U., Takashima A., Gouras G.K., Greengard P., Xu H. Chaperones increase association of tau protein with microtubules. Proc. Natl. Acad. Sci. USA. 2003;100:721–726. doi: 10.1073/pnas.242720499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Wang H., Tan M.S., Lu R.C., Yu J.T., Tan L. Heat shock proteins at the crossroads between cancer and Alzheimer’s disease. BioMed Res. Int. 2014;2014:239164. doi: 10.1155/2014/239164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Tortosa E., Santa-Maria I., Moreno F., Lim F., Perez M., Avila J. Binding of Hsp90 to tau promotes a conformational change and aggregation of tau protein. J. Alzheimers Dis. 2009;17:319–325. doi: 10.3233/JAD-2009-1049. [DOI] [PubMed] [Google Scholar]
- 220.Dou F., Chang X., Ma D. Hsp90 maintains the stability and function of the tau phosphorylating kinase GSK3β. Int. J. Mol. Sci. 2007;8:51–60. doi: 10.3390/i8010060. [DOI] [Google Scholar]
- 221.Shelton L.B., Baker J.D., Zheng D., Sullivan L.E., Solanki P.K., Webster J.M., Sun Z., Sabbagh J.J., Nordhues B.A., Koren J., et al. Hsp90 activator Aha1 drives production of pathological tau aggregates. Proc. Natl. Acad. Sci. USA. 2017;114:9707–9712. doi: 10.1073/pnas.1707039114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Mayer M.P. Hsp70 chaperone dynamics and molecular mechanism. Trends Biochem. Sci. 2013;38:507–514. doi: 10.1016/j.tibs.2013.08.001. [DOI] [PubMed] [Google Scholar]
- 223.Young Z.T., Rauch J.N., Assimon V.A., Jinwal U.K., Ahn M., Li X., Dunyak B.M., Ahmad A., Carlson G.A., Srinivasan S.R., et al. Stabilizing the Hsp70-Tau complex promotes turnover in models of Tauopathy. Cell Chem. Biol. 2016;23:992–1001. doi: 10.1016/j.chembiol.2016.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Westhoff B., Chapple J.P., van der Spuy J., Höhfeld J., Cheetham M.E. HSJ1 is a neuronal shuttling factor for the sorting of chaperone clients to the proteasome. Curr. Biol. 2005;15:1058–1064. doi: 10.1016/j.cub.2005.04.058. [DOI] [PubMed] [Google Scholar]
- 225.Shin Y., Klucken J., Patterson C., Hyman B.T., McLean P.J. The co-chaperone carboxyl terminus of Hsp70-interacting protein (CHIP) mediates alpha-synuclein degradation decisions between proteasomal and lysosomal pathways. J. Biol. Chem. 2005;280:23727–23734. doi: 10.1074/jbc.M503326200. [DOI] [PubMed] [Google Scholar]
- 226.Lang K., Schmid F.X., Fischer G. Catalysis of protein folding by prolyl isomerase. Nature. 1987;329:268–270. doi: 10.1038/329268a0. [DOI] [PubMed] [Google Scholar]
- 227.Nigro P., Pompilio G., Capogrossi M.C. Cyclophilin A: A key player for human disease. Cell Death Dis. 2013;4:e888. doi: 10.1038/cddis.2013.410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Torbeev V.Y., Hilvert D. Both the cis-trans equilibrium and isomerization dynamics of a single proline amide modulate β2-microglobulin amyloid assembly. Proc. Natl. Acad. Sci. USA. 2013;110:20051–20056. doi: 10.1073/pnas.1310414110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Baker J.D., Shelton L.B., Zheng D., Favretto F., Nordhues B.A., Darling A., Sullivan L.E., Sun Z., Solanki P.K., Martin M.D., et al. Human cyclophilin 40 unravels neurotoxic amyloids. PLoS Biol. 2017;15:e2001336. doi: 10.1371/journal.pbio.2001336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Tsvetkov P., Reuven N., Shaul Y. Ubiquitin-independent p53 proteasomal degradation. Cell Death Differ. 2010;17:103–108. doi: 10.1038/cdd.2009.67. [DOI] [PubMed] [Google Scholar]
- 231.Cheng Y., LeGall T., Oldfield C.J., Mueller J.P., Van Y.Y.J., Romero P., Cortese M.S., Uversky V.N., Dunker A.K. Rational drug design via intrinsically disordered protein. Trends Biotechnol. 2006;24:435–442. doi: 10.1016/j.tibtech.2006.07.005. [DOI] [PubMed] [Google Scholar]
- 232.Metallo S.J. Intrinsically disordered proteins are potential drug targets. Curr. Opin. Chem. Biol. 2010;14:481–488. doi: 10.1016/j.cbpa.2010.06.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Wang J.H., Cao Z.X., Zhao L.L., Li S.Q. Novel strategies for drug discovery based on intrinsically disordered proteins (IDPs) Int. J. Mol. Sci. 2011;12:3205–3219. doi: 10.3390/ijms12053205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Zhang Y., Cao H., Liu Z. Binding cavities and druggability of intrinsically disordered proteins. Protein Sci. 2015;24:688–705. doi: 10.1002/pro.2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Liu Z., Huang Y. Advantages of proteins being disordered. Protein Sci. 2014;23:539–550. doi: 10.1002/pro.2443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Zhu M., De Simone A., Schenk D., Toth G., Dobson C.M., Vendruscolo M. Identification of small-molecule binding pockets in the soluble monomeric form of the Aβ42 peptide. J. Chem. Phys. 2013;139:035101. doi: 10.1063/1.4811831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Fokkens M., Schrader T., Klärner F.G. A molecular tweezer for lysine and arginine. J. Am. Chem. Soc. 2005;127:14415–14421. doi: 10.1021/ja052806a. [DOI] [PubMed] [Google Scholar]
- 238.Sinha S., Lopes D.H., Du Z., Pang E., Shanmugam A., Lomakin A., Talbiersky P., Tennstaedt A., McDaniel K., Bakshi R., et al. Lysine-specific molecular tweezers are broad-spectrum inhibitors of assembly and toxicity of amyloid proteins. J. Am. Chem. Soc. 2011;133:16958–16969. doi: 10.1021/ja206279b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.O’Hare E., Scopes D.I., Kim E.M., Palmer P., Spanswick D., McMahon B., Amijee H., Nerou E., Treherne J.M., Jeggo R. Novel 5-aryloxypyrimidine SEN1576 as a candidate for the treatment of Alzheimer’s disease. Int. J. Neuropsychopharmacol. 2014;17:117–126. doi: 10.1017/S1461145713000886. [DOI] [PubMed] [Google Scholar]
- 240.Prabhudesai S., Sinha S., Attar A., Kotagiri A., Fitzmaurice A.G., Lakshmanan R., Ivanova M.I., Loo J.A., Klärner F.G., Schrader T., et al. A novel “molecular tweezer” inhibitor of α-synuclein neurotoxicity in vitro and in vivo. Neurotherapeutics. 2012;9:464–476. doi: 10.1007/s13311-012-0105-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Sievers S.A., Karanicolas J., Chang H.W., Zhao A., Jiang L., Zirafi O., Stevens J.T., Münch J., Baker D., Eisenberg D. Structure-based design of non-natural amino-acid inhibitors of amyloid fibril formation. Nature. 2011;475:96–100. doi: 10.1038/nature10154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Frenkel-Pinter M., Tal S., Scherzer-Attali R., Abu-Hussien M., Alyagor I., Eisenbaum T., Gazit E., Segal D. Naphthoquinone-Tryptophan Hybrid Inhibits Aggregation of the Tau-Derived Peptide PHF6 and Reduces Neurotoxicity. J. Alzheimers Dis. 2016;51:165–178. doi: 10.3233/JAD-150927. [DOI] [PubMed] [Google Scholar]
- 243.Jones C.L., Njomen E., Sjögren B., Dexheimer T.S., Tepe J.J. Small Molecule Enhancement of 20S Proteasome Activity Targets Intrinsically Disordered Proteins. ACS Chem. Biol. 2017;12:2240–2247. doi: 10.1021/acschembio.7b00489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Joerger A.C., Fersht A.R. The tumor suppressor p53: From structures to drug discovery. Cold Spring Harb. Perspect. Biol. 2010;2:a000919. doi: 10.1101/cshperspect.a000919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Li Z.Y., Ni M., Li J.K., Zhang Y.P., Ouyang Q., Tang C. Decision making of the p53 network: Death by integration. J. Theor. Biol. 2011;271:205–211. doi: 10.1016/j.jtbi.2010.11.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Huang Y.Q., Liu Z.R. Anchoring intrinsically disordered proteins to multiple targets: Lessons from N terminus of the p53 protein. Int. J. Mol. Sci. 2011;12:1410–1430. doi: 10.3390/ijms12021410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Vassilev L.T., Vu B.T., Graves B., Carvajal D., Podlaski F., Filipovic Z., Kong N., Kammlott U., Lukacs C., Klein C., et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303:844–848. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- 248.Tovar C., Rosinski J., Filipovic Z., Higgins B., Kolinsky K., Hilton H., Zhao X.L., Vu B.T., Qing W.G., Packman K., et al. Small-molecule MDM2 antagonists reveal aberrant p53 signaling in cancer: Implications for therapy. Proc. Natl. Acad. Sci. USA. 2006;103:1888–1893. doi: 10.1073/pnas.0507493103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Yu X., Narayanan S., Vazquez A., Carpizo D.R. Small molecule compounds targeting the p53 pathway: Are we finally making progress? Apoptosis. 2014;19:1055–1068. doi: 10.1007/s10495-014-0990-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Burgess A., Chia K.M., Haupt S., Thomas D., Haupt Y., Lim E. Clinical Overview of MDM2/X-Targeted Therapies. Front. Oncol. 2016;6:7. doi: 10.3389/fonc.2016.00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Hammoudeh D.I., Follis A.V., Prochownik E.V., Metallo S.J. Multiple independent binding sites for small molecule inhibitors on the oncoprotein c-Myc. J. Am. Chem. Soc. 2009;131:7390–7401. doi: 10.1021/ja900616b. [DOI] [PubMed] [Google Scholar]
- 252.Berg T., Cohen S.B., Desharnais J., Sonderegger C., Maslyar D.J., Goldberg J., Boger D.L., Vogt P.K. Small-molecule antagonists of Myc/Max dimerization inhibit Myc-induced transformation of chicken embryo fibroblasts. Proc. Natl. Acad. Sci. USA. 2002;99:3830–3835. doi: 10.1073/pnas.062036999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Shi J., Stover J.S., Whitby L.R., Vogt P.K., Boger D.L. Small molecule inhibitors of Myc/Max dimerization and Myc-induced cell transformation. Bioorg. Med. Chem. Lett. 2009;19:6038–6041. doi: 10.1016/j.bmcl.2009.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Yin X.Y., Giap C., Lazo J.S., Prochownik E.V. Low molecular weight inhibitors of Myc-Max interaction and function. Oncogene. 2003;22:6151–6159. doi: 10.1038/sj.onc.1206641. [DOI] [PubMed] [Google Scholar]
- 255.Zirath H., Frenzel A., Oliynyk G., Segerstrom L., Westermark U.K., Larsson K., Persson M.M., Hultenby K., Lehtio J., Einvik C., et al. MYC inhibition induces metabolic changes leading to accumulation of lipid droplets in tumor cells. Proc. Natl. Acad. Sci. USA. 2013;110:10258–10263. doi: 10.1073/pnas.1222404110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Fletcher S., Prochownik E.V. Small-molecule inhibitors of the Myc oncoprotein. Biochim. Biophys. Acta. 2015;1849:525–543. doi: 10.1016/j.bbagrm.2014.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Yu C., Niu X., Jin F., Liu Z., Jin C., Lai L. Structure-based Inhibitor Design for the Intrinsically Disordered Protein c-Myc. Sci. Rep. 2016;6:22298. doi: 10.1038/srep22298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Erkizan H.V., Kong Y.L., Merchant M., Schlottmann S., Barber-Rotenberg J.S., Yuan L.S., Abaan O.D., Chou T.H., Dakshanamurthy S., Brown M.L., et al. A small molecule blocking oncogenic protein EWS-FLI1 interaction with RNA helicase A inhibits growth of Ewing’s sarcoma. Nat. Med. 2009;15:750–757. doi: 10.1038/nm.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Hong S.H., Youbi S.E., Hong S.P., Kallakury B., Monroe P., Erkizan H.V., Barber-Rotenberg J.S., Houghton P., Uren A., Toretsky J.A. Pharmacokinetic modeling optimizes inhibition of the ’undruggable’ EWS-FLI1 transcription factor in Ewing Sarcoma. Oncotarget. 2014;5:338–350. doi: 10.18632/oncotarget.1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Hegyi H., Buday L., Tompa P. Intrinsic structural disorder confers cellular viability on oncogenic fusion proteins. PLoS Comput. Biol. 2009;5:e1000552. doi: 10.1371/journal.pcbi.1000552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Huang Y.Q., Liu Z.R. Do intrinsically disordered proteins possess high specificity in protein-protein interactions? Chem.-Eur. J. 2013;19:4462–4467. doi: 10.1002/chem.201203100. [DOI] [PubMed] [Google Scholar]
- 262.Srinivasan R.S., Nesbit J.B., Marrero L., Erfurth F., LaRussa V.F., Hemenway C.S. The synthetic peptide PFWT disrupts AF4-AF9 protein complexes and induces apoptosis in t(4;11) leukemia cells. Leukemia. 2004;18:1364–1372. doi: 10.1038/sj.leu.2403415. [DOI] [PubMed] [Google Scholar]
- 263.Palermo C.M., Bennett C.A., Winters A.C., Hemenway C.S. The AF4-mimetic peptide, PFWT, induces necrotic cell death in MV4–11 leukemia cells. Leuk. Res. 2008;32:633–642. doi: 10.1016/j.leukres.2007.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Watson V.G., Drake K.M., Peng Y., Napper A.D. Development of a high-throughput screening-compatible assay for the discovery of inhibitors of the AF4-AF9 interaction using AlphaScreen technology. Assay Drug Dev. Technol. 2013;11:253–268. doi: 10.1089/adt.2012.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Johnson T.O., Ermolieff J., Jirousek M.R. Protein tyrosine phosphatase 1B inhibitors for diabetes. Nat. Rev. Drug Discov. 2002;1:696–709. doi: 10.1038/nrd895. [DOI] [PubMed] [Google Scholar]
- 266.Krishnan N., Koveal D., Miller D.H., Xue B., Akshinthala S.D., Kragelj J., Jensen M.R., Gauss C.M., Page R., Blackledge M., et al. Targeting the disordered C terminus of PTP1B with an allosteric inhibitor. Nat. Chem. Biol. 2014;10:558–566. doi: 10.1038/nchembio.1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Bieschke J., Russ J., Friedrich R.P., Ehrnhoefer D.E., Wobst H., Neugebauer K., Wanker E.E. EGCG remodels mature alpha-synuclein and amyloid-beta fibrils and reduces cellular toxicity. Proc. Natl. Acad. Sci. USA. 2010;107:7710–7715. doi: 10.1073/pnas.0910723107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Martin-Bastida A., Ward R.J., Newbould R., Piccini P., Sharp D., Kabba C., Patel M.C., Spino M., Connelly J., Tricta F., et al. Brain iron chelation by deferiprone in a phase 2 randomised double-blinded placebo controlled clinical trial in Parkinson’s disease. Sci. Rep. 2017;7:1398. doi: 10.1038/s41598-017-01402-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Hung L.W., Villemagne V.L., Cheng L., Sherratt N.A., Ayton S., White A.R., Crouch P.J., Lim S., Leong S.L., Wilkins S., et al. The hypoxia imaging agent CuII(atsm) is neuroprotective and improves motor and cognitive functions in multiple animal models of Parkinson’s disease. J. Exp. Med. 2012;209:837–854. doi: 10.1084/jem.20112285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Savolainen M.H., Richie C.T., Harvey B.K., Männistö B.T., Maguire-Zeiss K.A., Myöhänen T.T. The beneficial effect of a prolyl oligopeptidase inhibitor, KYP-2047, on alpha-synuclein clearance and autophagy in A30P transgenic mouse. Neurobiol. Dis. 2014;68:1–15. doi: 10.1016/j.nbd.2014.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Svarcbahs R., Julku U.H., Myöhänen T.T. Inhibition of Prolyl Oligopeptidase Restores Spontaneous Motor Behavior in the α-Synuclein Virus Vector-Based Parkinson’s Disease Mouse Model by Decreasing α-Synuclein Oligomeric Species in Mouse Brain. J. Neurosci. 2016;36:12485–12497. doi: 10.1523/JNEUROSCI.2309-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Levin J., Schmidt F., Boehm C., Prix C., Bötzel K., Ryazanov S., Leonov A., Griesinger C., Giese A. The oligomer modulator anle138b inhibits disease progression in a Parkinson mouse model even with treatment started after disease onset. Acta Neuropathol. 2014;127:779–780. doi: 10.1007/s00401-014-1265-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Price D.L., Koike M.A., Khan A., Wrasidlo W., Rockenstein E., Masliah E., Bonhaus D. The small molecule alpha-synuclein misfolding inhibitor, NPT200-11, produces multiple benefits in an animal model of Parkinson’s disease. Sci. Rep. 2018;8:16165. doi: 10.1038/s41598-018-34490-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Krishnan R., Hefti F., Tsubery H., Lulu M., Proschitsky M., Fisher R. Conformation as the Therapeutic Target for Neurodegenerative Diseases. Curr. Alzheimer Res. 2017;14:393–402. doi: 10.2174/1567205014666170116152622. [DOI] [PubMed] [Google Scholar]
- 275.Levenson J.M., Schroeter S., Carroll J.C., Cullen V., Asp E., Proschitsky M., Chung C.H., Gilead S., Nadeem M., Dodiya H.B., et al. NPT088 reduces both amyloid-β and tau pathologies in transgenic mice. Alzheimers Dement. 2016;2:141–155. doi: 10.1016/j.trci.2016.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Yuan B., Sierks M.R. Intracellular targeting and clearance of oligomeric alpha-synuclein alleviates toxicity in mammalian cells. Neurosci. Lett. 2009;459:16–18. doi: 10.1016/j.neulet.2009.04.046. [DOI] [PubMed] [Google Scholar]
- 277.Bhatt M.A., Messer A., Kordower J.H. Can intrabodies serve as neuroprotective therapies for Parkinson’s disease? Beginning thoughts. J. Parkinsons Dis. 2013;3:581–591. doi: 10.3233/JPD-130252. [DOI] [PubMed] [Google Scholar]
- 278.Emadi S., Liu R., Yuan B., Schulz P., McAllister C., Lyubchenko Y., Messer A., Sierks M.R. Inhibiting aggregation of alpha-synuclein with human single chain antibody fragments. Biochemistry. 2004;43:2871–2878. doi: 10.1021/bi036281f. [DOI] [PubMed] [Google Scholar]
- 279.Butler D.C., Joshi S.N., Genst E., Baghel A.S., Dobson C.M., Messer A. Bifunctional Anti-Non-Amyloid Component α-Synuclein Nanobodies Are Protective In Situ. PLoS ONE. 2016;11:e0165964. doi: 10.1371/journal.pone.0165964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Chatterjee D., Bhatt M., Butler D., De Genst E., Dobson C.M., Messer A., Kordower J.H. Proteasome-targeted nanobodies alleviate pathology and functional decline in an α-synuclein-based Parkinson’s disease model. NPJ Parkinsons Dis. 2018;4:25. doi: 10.1038/s41531-018-0062-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Dehay B., Bourdenx M., Gorry P., Przedborski S., Vila M., Hunot S., Singleton A., Olanow C., Merchant K., Bezard E., et al. Targeting alpha-synuclein for treatment of Parkinson’s disease: Mechanistic and therapeutic considerations. Lancet Neurol. 2015;14:855–866. doi: 10.1016/S1474-4422(15)00006-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Schenk D.B., Koller M., Ness D.K., Griffith S.G., Grundman M., Zago W., Soto J., Atiee G., Ostrowitzki S., Kinney G.G. First-in-human assessment of PRX002, an anti-α-synuclein monoclonal antibody, in healthy volunteers. Mov. Disord. 2017;32:211–218. doi: 10.1002/mds.26878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Jankovic J., Goodman I., Safirstein B., Marmon T.K., Schenk D.B., Koller M., Zago W., Ness D.K., Griffith S.G., Grundman M., et al. Safety and Tolerability of Multiple Ascending Doses of PRX002/RG7935, an Anti-α-Synuclein Monoclonal Antibody, in Patients With Parkinson Disease: A Randomized Clinical Trial. JAMA Neurol. 2018;75:1206–1214. doi: 10.1001/jamaneurol.2018.1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Mandler M., Valera E., Rockenstein E., Weninger H., Patrick C., Adame A., Santic R., Meindl S., Vigl B., Smrzka O., et al. Next-generation active immunization approach for synucleinopathies: Implications for Parkinson’s disease clinical trials. Acta Neuropathol. 2014;127:861–879. doi: 10.1007/s00401-014-1256-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.O’Hara D.M., Kalia S.K., Kalia L.V. Emerging disease-modifying strategies targeting alpha-synuclein for the treatment of Parkinson’s disease. Br. J. Pharmacol. 2018;175:3080–3089. doi: 10.1111/bph.14345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Kiss R., Csizmadia G., Solti K., Keresztes A., Zhu M., Pickhardt M., Mandelkow E., Toth G. Structural Basis of Small Molecule Targetability of Monomeric Tau Protein. ACS Chem. Neurosci. 2018;9:2997–3006. doi: 10.1021/acschemneuro.8b00182. [DOI] [PubMed] [Google Scholar]
- 287.Jouanne M., Rault S., Voisin-Chiret A.S. Tau protein aggregation in Alzheimer’s disease: An attractive target for the development of novel therapeutic agents. Eur. J. Med. Chem. 2017;139:153–167. doi: 10.1016/j.ejmech.2017.07.070. [DOI] [PubMed] [Google Scholar]
- 288.Pickhardt M., Neumann T., Schwizer D., Callaway K., Vendruscolo M., Schenk D., St George-Hyslop P., Mandelkow E.M., Dobson C.M., McConlogue L., et al. Identification of Small Molecule Inhibitors of Tau Aggregation by Targeting Monomeric Tau As a Potential Therapeutic Approach for Tauopathies. Curr. Alzheimer Res. 2015;12:814–828. doi: 10.2174/156720501209151019104951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Baggett D.W., Nath A. The Rational Discovery of a Tau Aggregation Inhibitor. Biochemistry. 2018;57:6099–6107. doi: 10.1021/acs.biochem.8b00581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Shiryaev N., Pikman R., Giladi E., Gozes I. Protection against tauopathy by the drug candidates NAP (davunetide) and D-SAL: Biochemical, cellular and behavioral aspects. Curr. Pharm. Des. 2011;17:2603–2612. doi: 10.2174/138161211797416093. [DOI] [PubMed] [Google Scholar]
- 291.Ivashko-Pachima Y., Gozes I. NAP protects against Tau hyperphosphorylation through GSK3. Curr. Pharm. Des. 2018;24:3868–3877. doi: 10.2174/1381612824666181112105954. [DOI] [PubMed] [Google Scholar]
- 292.Dammers C., Yolcu D., Kukuk L., Willbold D., Pickhardt M., Mandelkow E., Horn A.H., Sticht H., Malhis M.N., Will N., et al. Selection and Characterization of Tau Binding -Enantiomeric Peptides with Potential for Therapy of Alzheimer Disease. PLoS ONE. 2016;11:e0167432. doi: 10.1371/journal.pone.0167432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Kim J.H., Kim E., Choi W.H., Lee J., Lee J.H., Lee H., Kim D.E., Suh Y.H., Lee M.J. Inhibitory RNA Aptamers of Tau Oligomerization and Their Neuroprotective Roles against Proteotoxic Stress. Mol. Pharm. 2016;13:2039–2048. doi: 10.1021/acs.molpharmaceut.6b00165. [DOI] [PubMed] [Google Scholar]
- 294.Rafiee S., Asadollahi K., Riazi G., Ahmadian S., Saboury A.A. Vitamin B12 Inhibits Tau Fibrillization via Binding to Cysteine Residues of Tau. ACS Chem. Neurosci. 2017;8:2676–2682. doi: 10.1021/acschemneuro.7b00230. [DOI] [PubMed] [Google Scholar]
- 295.Yoshitake J., Soeda Y., Ida T., Sumioka A., Yoshikawa M., Matsushita K., Akaike T., Takashima A. Modification of Tau by 8-Nitroguanosine 3,5-Cyclic Monophosphate (8-Nitro-cGMP): Effects of nitric oxide-linked chemical modification on tau aggregation. J. Biol. Chem. 2016;291:22714–22720. doi: 10.1074/jbc.M116.734350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Sun W., Lee S., Huang X., Liu S., Inayathullah M., Kim K.-M., Tang H., Ashford J.W., Rajadas J. Attenuation of synaptic toxicity and MARK4/PAR1-mediated Tau phosphorylation by methylene blue for Alzheimer’s disease treatment. Sci. Rep. 2016;6:34784. doi: 10.1038/srep34784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.George R.C., Lew J., Graves D.J. Interaction of Cinnamaldehyde and Epicatechin with Tau: Implications of Beneficial Effects in Modulating Alzheimer’s Disease Pathogenesis. J. Alzheimers Dis. 2013;36:21–40. doi: 10.3233/JAD-122113. [DOI] [PubMed] [Google Scholar]
- 298.Gandini A., Bartolini M., Tedesco D., Martinez-Gonzalez L., Roca C., Campillo N.E., Zaldivar-Diez J., Perez C., Zuccheri G., Miti A., et al. Tau-Centric Multitarget Approach for Alzheimer’s Disease: Development of First-in-Class Dual Glycogen Synthase Kinase 3 beta and Tau-Aggregation Inhibitors. J. Med. Chem. 2018;61:7640–7656. doi: 10.1021/acs.jmedchem.8b00610. [DOI] [PubMed] [Google Scholar]
- 299.Llorach-Pares L., Nonell-Canals A., Avila C., Sanchez-Martinez M. Kororamides, Convolutamines, and Indole Derivatives as Possible Tau and Dual-Specificity Kinase Inhibitors for Alzheimer’s Disease: A Computational Study. Mar. Drugs. 2018;16:386. doi: 10.3390/md16100386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Moussa C.E. Beta-secretase inhibitors in phase I and phase II clinical trials for Alzheimer’s disease. Expert Opin. Investig. Drugs. 2017;26:1131–1136. doi: 10.1080/13543784.2017.1369527. [DOI] [PubMed] [Google Scholar]
- 301.Mead E., Kestoras D., Gibson Y., Hamilton L., Goodson R., Jones S., Eversden S., Davies P., O’Neill M., Hutton M., et al. Halting of Caspase Activity Protects Tau from MC1-Conformational Change and Aggregation. J. Alzheimers Dis. 2016;54:1521–1538. doi: 10.3233/JAD-150960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Rao M.V., McBrayer M.K., Campbell J., Kumar A., Hashim A., Sershen H., Stavrides P.H., Ohno M., Hutton M., Nixon R.A. Specific calpain inhibition by calpastatin prevents tauopathy and neurodegeneration and restores normal lifespan in tau P301L mice. J. Neurosci. 2014;34:9222–9234. doi: 10.1523/JNEUROSCI.1132-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Blair L.J., Sabbagh J.J., Dickey C.A. Targeting Hsp90 and its co-chaperones to treat Alzheimer’s disease. Expert Opin. Ther. Targets. 2014;18:1219–1232. doi: 10.1517/14728222.2014.943185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Kontsekova E., Zilka N., Kovacech B., Novak P., Novak M. First-in-man tau vaccine targeting structural determinants essential for pathological tau-tau interaction reduces tau oligomerisation and neurofibrillary degeneration in an Alzheimer’s disease model. Alzheimers Res. Ther. 2014;6:44. doi: 10.1186/alzrt278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Novak P., Kontsekova E., Zilka N., Novak M. Ten Years of Tau-Targeted Immunotherapy: The Path Walked and the Roads Ahead. Front. Neurosci. 2018;12:798. doi: 10.3389/fnins.2018.00798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Shahpasand K., Sepehri Shamloo A., Nabavi S.M., Lu K.P., Zhou X.Z. Tau immunotherapy: Hopes and hindrances. Hum. Vaccin Immunother. 2018;14:277–284. doi: 10.1080/21645515.2017.1393594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Cehlar O., Skrabana R., Kovac A., Kovacech B., Novak M. Crystallization and preliminary X-ray diffraction analysis of tau protein microtubule-binding motifs in complex with Tau5 and DC25 antibody Fab fragments. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2012;68:1181–1185. doi: 10.1107/S1744309112030382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Panza F., Solfrizzi V., Seripa D., Imbimbo B.P., Lozupone M., Santamato A., Tortelli R., Galizia I., Prete C., Daniele A., et al. Tau-based therapeutics for Alzheimer’s disease: Active and passive immunotherapy. Immunotherapy. 2016;8:1119–1134. doi: 10.2217/imt-2016-0019. [DOI] [PubMed] [Google Scholar]
- 309.Novak P., Zilka N., Zilkova M., Kovacech B., Skrabana R., Ondrus M., Fialova L., Kontsekova E., Otto M., Novak M. AADvac1, an Active Immunotherapy for Alzheimer’s Disease and Non Alzheimer Tauopathies: An Overview of Preclinical and Clinical Development. J. Prev. Alzheimers Dis. 2019;6:63–69. doi: 10.14283/jpad.2018.45. [DOI] [PubMed] [Google Scholar]
- 310.Novak P., Schmidt R., Kontsekova E., Zilka N., Kovacech B., Skrabana R., Vince-Kazmerova Z., Katina S., Fialova L., Prcina M., et al. Safety and immunogenicity of the tau vaccine AADvac1 in patients with Alzheimer’s disease: A randomised, double-blind, placebo-controlled, phase 1 trial. Lancet Neurol. 2017;16:123–134. doi: 10.1016/S1474-4422(16)30331-3. [DOI] [PubMed] [Google Scholar]
- 311.Novak P., Schmidt R., Kontsekova E., Kovacech B., Smolek T., Katina S., Fialova L., Prcina M., Parrak V., Dal-Bianco P., et al. FUNDAMANT: An interventional 72-week phase 1 follow-up study of AADvac1, an active immunotherapy against tau protein pathology in Alzheimer’s disease. Alzheimers Res. Ther. 2018;10:108. doi: 10.1186/s13195-018-0436-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Theunis C., Crespo-Biel N., Gafner V., Pihlgren M., Lopez-Deber M., Reis P., Hickman D., Adolfsson O., Chuard N., Ndao D., et al. Efficacy and Safety of A Liposome-Based Vaccine against Protein Tau, Assessed in Tau.P301L Mice That Model Tauopathy. PLoS ONE. 2013;8:e72301. doi: 10.1371/journal.pone.0072301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Yanamandra K., Jiang H., Mahan T., Maloney S., Wozniak D., Diamond M., Holtzmanm D. Anti-tau antibody reduces insoluble tau and decreases brain atrophy. Ann. Clin. Trans. Neurol. 2016;2:278–288. doi: 10.1002/acn3.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.West T., Hu Y., Verghese P.B., Bateman R.J., Braunstein J.B., Fogelman I., Budur K., Florian H., Mendonca N., Holtzman D.M. Preclinical and Clinical Development of ABBV-8E12, a Humanized Anti-Tau Antibody, for Treatment of Alzheimer’s Disease and Other Tauopathies. J. Prev. Alzheimers Dis. 2017;4:236–241. doi: 10.14283/jpad.2017.36. [DOI] [PubMed] [Google Scholar]
- 315.Rosenberg R.N., Fu M., Lambracht-Washington D. Active full-length DNA Aβ42 immunization in 3xTg-AD mice reduces not only amyloid deposition but also tau pathology. Alzheimers Res. Ther. 2018;10:115. doi: 10.1186/s13195-018-0441-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Dai C.L., Tung Y.C., Liu F., Gong C.X., Iqbal K. Tau passive immunization inhibits not only tau but also Aβ pathology. Alzheimers Res. Ther. 2017;9:1. doi: 10.1186/s13195-016-0227-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Hol W.G.J. Protein Crystallography and Computer Graphics—Toward Rational Drug Design. Angew. Chem. 1986;25:767–778. doi: 10.1002/anie.198607673. [DOI] [Google Scholar]
- 318.Roberts N.A., Martin J.A., Kinchington D., Broadhurst A.V., Craig J.C., Duncan I.B., Galpin S.A., Handa B.K., Kay J., Kröhn A., et al. Rational design of peptide-based HIV proteinase inhibitors. Science. 1990;248:358–361. doi: 10.1126/science.2183354. [DOI] [PubMed] [Google Scholar]
- 319.Von Itzstein M., Wu W.Y., Kok G.B., Pegg M.S., Dyason J.C., Jin B., Van Phan T., Smythe M.L., White H.F., Oliver S.W., et al. Rational design of potent sialidase-based inhibitors of influenza virus replication. Nature. 1993;363:418–423. doi: 10.1038/363418a0. [DOI] [PubMed] [Google Scholar]
- 320.Sliwoski G., Kothiwale S., Meiler J., Lowe E.W., Jr. Computational methods in drug discovery. Pharmacol. Rev. 2013;66:334–395. doi: 10.1124/pr.112.007336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Joshi P., Vendruscolo M. Druggability of Intrinsically Disordered Proteins. Adv. Exp. Med. Biol. 2015;870:383–400. doi: 10.1007/978-3-319-20164-1_13. [DOI] [PubMed] [Google Scholar]
- 322.Marasco D., Scognamiglio P.L. Identification of inhibitors of biological interactions involving intrinsically disordered proteins. Int. J. Mol. Sci. 2015;16:7394–7412. doi: 10.3390/ijms16047394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Tsafou K., Tiwari P.B., Forman-Kay J.D., Metallo S.J., Toretsky J.A. Targeting Intrinsically Disordered Transcription Factors: Changing the Paradigm. J. Mol. Biol. 2018;430:2321–2341. doi: 10.1016/j.jmb.2018.04.008. [DOI] [PubMed] [Google Scholar]
- 324.Rezaei-Ghaleh N., Blackledge M., Zweckstetter M. Intrinsically disordered proteins: From sequence and conformational properties toward drug discovery. ChemBioChem. 2012;13:930–950. doi: 10.1002/cbic.201200093. [DOI] [PubMed] [Google Scholar]
- 325.Uversky V.N. Dancing Protein Clouds: The Strange Biology and Chaotic Physics of Intrinsically Disordered Proteins. J. Biol. Chem. 2016;291:6681–6688. doi: 10.1074/jbc.R115.685859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Jin F., Yu C., Lai L., Liu Z. Ligand clouds around protein clouds: A scenario of ligand binding with intrinsically disordered proteins. PLoS Comput. Biol. 2013;9:e1003249. doi: 10.1371/journal.pcbi.1003249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Bier D., Thiel P., Briels J., Ottmann C. Stabilization of Protein-Protein Interactions in chemical biology and drug discovery. Prog. Biophys. Mol. Biol. 2015;119:10–19. doi: 10.1016/j.pbiomolbio.2015.05.002. [DOI] [PubMed] [Google Scholar]
- 328.Arkin M.R., Wells J.A. Small-molecule inhibitors of protein-protein interactions: Progressing towards the dream. Nat. Rev. Drug Discov. 2004;3:301–317. doi: 10.1038/nrd1343. [DOI] [PubMed] [Google Scholar]
- 329.Wells J.A., McClendon C.L. Reaching for high-hanging fruit in drug discovery at protein-protein interfaces. Nature. 2007;450:1001–1009. doi: 10.1038/nature06526. [DOI] [PubMed] [Google Scholar]
- 330.Hopkins A.L., Groom C.R. The druggable genome. Nat. Rev. Drug Discov. 2002;1:727–730. doi: 10.1038/nrd892. [DOI] [PubMed] [Google Scholar]
- 331.Drews J., Ryser S. The role of innovation in drug development. Nat. Biotechnol. 1997;15:1318–1319. doi: 10.1038/nbt1297-1318. [DOI] [PubMed] [Google Scholar]
- 332.Drews J. Drug discovery: A historical perspective. Science. 2000;287:1960–1964. doi: 10.1126/science.287.5460.1960. [DOI] [PubMed] [Google Scholar]
- 333.Li J., Feng Y., Wang X., Li J., Liu W., Rong L., Bao J. An Overview of Predictors for Intrinsically Disordered Proteins over 2010–2014. Int. J. Mol. Sci. 2015;16:23446–23562. doi: 10.3390/ijms161023446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Heller G.T., Aprile F.A., Vendruscolo M. Methods of probing the interactions between small molecules and disordered proteins. Cell. Mol. Life Sci. 2017;74:3225–3243. doi: 10.1007/s00018-017-2563-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Ambadipudi S., Zweckstetter M. Targeting intrinsically disordered proteins in rational drug discovery. Expert Opin. Drug Discov. 2016;11:65–77. doi: 10.1517/17460441.2016.1107041. [DOI] [PubMed] [Google Scholar]
- 336.Henriques J., Cragnell C., Skepö M. Molecular Dynamics Simulations of Intrinsically Disordered Proteins: Force Field Evaluation and Comparison with Experiment. J. Chem. Theory Comput. 2015;11:3420–3431. doi: 10.1021/ct501178z. [DOI] [PubMed] [Google Scholar]
- 337.Robustelli P., Piana S., Shaw D.E. Developing a molecular dynamics force field for both folded and disordered protein states. Proc. Natl. Acad. Sci. USA. 2018;115:E4758–E4766. doi: 10.1073/pnas.1800690115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338.Uversky V.N. Unusual biophysics of intrinsically disordered proteins. Biochim. Biophys. Acta. 2013;1834:932–951. doi: 10.1016/j.bbapap.2012.12.008. [DOI] [PubMed] [Google Scholar]
- 339.Dogan J., Gianni S., Jemth P. The binding mechanisms of intrinsically disordered proteins. Phys. Chem. Chem. Phys. 2014;16:6323–6331. doi: 10.1039/C3CP54226B. [DOI] [PubMed] [Google Scholar]
- 340.Shirai N.C., Kikuchi M. Structural flexibility of intrinsically disordered proteins induces stepwise target recognition. J. Chem. Phys. 2013;139:225103. doi: 10.1063/1.4838476. [DOI] [PubMed] [Google Scholar]
- 341.Sammak S., Zinzalla G. Targeting protein-protein interactions (PPIs) of transcription factors: Challenges of intrinsically disordered proteins (IDPs) and regions (IDRs) Prog. Biophys. Mol. Biol. 2015;119:41–46. doi: 10.1016/j.pbiomolbio.2015.06.004. [DOI] [PubMed] [Google Scholar]
- 342.Hausrath A.C., Kingston R.L. Conditionally disordered proteins: Bringing the environment back into the fold. Cell. Mol. Life Sci. 2017;74:3149–3162. doi: 10.1007/s00018-017-2558-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343.Hultqvist G., Åberg E., Camilloni C., Sundell G.N., Andersson E., Dogan J., Chi C.N., Vendruscolo M., Jemth P. Emergence and evolution of an interaction between intrinsically disordered proteins. eLife. 2017;6:e16059. doi: 10.7554/eLife.16059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344.De Cássia Ruy P., Torrieri R., Toledo J.S., de Souza Alves V., Cruz A.K., Ruiz J.C. Intrinsically disordered proteins (IDPs) in trypanosomatids. BMC Genom. 2014;15:1100. doi: 10.1186/1471-2164-15-1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345.Longhi S. Structural disorder within paramyxoviral nucleoproteins. FEBS Lett. 2015;589:2649–2659. doi: 10.1016/j.febslet.2015.05.055. [DOI] [PubMed] [Google Scholar]
- 346.Russo A., Manna S.L., Novellino E., Malfitano A.M., Marasco D. Molecular signaling involving intrinsically disordered proteins in prostate cancer. Asian J. Androl. 2016;18:673–681. doi: 10.4103/1008-682X.181817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347.Ruan H., Sun Q., Zhang W., Liu Y., Lai L. Targeting intrinsically disordered proteins at the edge of chaos. Drug Discov. Today. 2019;24:217–227. doi: 10.1016/j.drudis.2018.09.017. [DOI] [PubMed] [Google Scholar]