

Abstract
Background
Ubiquitous in natural and engineered ecosystems, microbial immigration is one of the mechanisms shaping community assemblage. However, quantifying immigration impact remains challenging especially at individual population level. The activities of immigrants in the receiving community are often inadequately considered, leading to potential bias in identifying the relationship between community composition and environmental parameters.
Results
This study quantified microbial immigration from an upstream full-scale anaerobic reactor to downstream activated sludge reactors. A mass balance was applied to 16S rRNA gene amplicon sequencing data to calculate the net growth rates of individual populations in the activated sludge reactors. Among the 1178 observed operational taxonomic units (OTUs), 582 had a positive growth rate, including all the populations with abundance > 0.1%. These active populations collectively accounted for 99% of the total sequences in activated sludge. The remaining 596 OTUs with a growth rate ≤ 0 were classified as inactive populations. All the abundant populations in the upstream anaerobic reactor were inactive in the activated sludge process, indicating a negligible immigration impact. We used a supervised learning regressor to predict environmental parameters based on community composition and compared the prediction accuracy based on either the entire community or the active populations. Temperature was the most predictable parameter, and the prediction accuracy was improved when only active populations were used to train the regressor.
Conclusions
Calculating growth rate of individual microbial populations in the downstream system provides an effective approach to determine microbial activity and quantify immigration impact. For the studied biological process, a marginal immigration impact was observed, likely due to the significant differences in the growth environments between the upstream and downstream processes. Excluding inactive populations as a result of immigration further enhanced the prediction of key environmental parameters affecting process performance.
Electronic supplementary material
The online version of this article (10.1186/s40168-019-0682-x) contains supplementary material, which is available to authorized users.
Keywords: Immigration impact, Active population, Machine learning
Background
Microbial immigration is a widespread process in natural and engineered environments and plays important roles in shaping the downstream microbial community. In natural lake environments, bacterioplankton assemblage compositions were influenced by bacteria in the inlet running water [1], treated or untreated sewage discharge [2], and even atmospheric deposition [3]. In rivers, planktonic and sedimentary microbial communities can be influenced by the immigration between each other [4]. Microbial immigration in engineered systems has also been reported in tap water bacterial community where the composition after stagnation was strongly affected by microbes in city water and pipe biofilms [5]. In wastewater treatment processes, feed sludge affected lab-scale anaerobic digester community structure and functional stability by sustainably introducing specific taxa [6]. In a lab-scale three-stage moving bed biofilm reactor, an increase in species richness was observed in the last stage as the result of immigration from the influent and first two stages [7]. In full-scale systems, significant immigration of ammonia-oxidizing bacteria from the upstream nitrifying trickling filter to the downstream activated sludge reactor was observed using NO2− as a measurement of transportation [8]. In full-scale anaerobic digesters, feed microbial biomass was observed to account for a significant portion of the digester communities [9, 10]. Albeit weak, immigration impact was observed from the influent wastewater to the downstream activated sludge based on community diversity and composition [11].
While microbial immigration is widely observed, it is difficult to quantify its impact on the downstream microbial community assembly with currently available approaches. The neutral community model fits the observation frequency of different taxa as a function of mean relative abundance and calculates an immigration probability m of the entire community [12]. Although been applied in various environments [4, 5, 13], the model does not provide population-level resolution on how a specific downstream community member is affected by immigration. Another method, microbial source tracking, estimates the proportion of taxa in the downstream community that come from multiple upstream environments [14–16]. However, this method assumes that all the observed microbial populations in the downstream community come from upstream environments and ignores the fact that some active microorganisms will undergo rapid reproduction independent from immigration. Other studies simply count shared species between upstream and downstream communities, which is usually visualized with a Venn diagram [11, 17]. This approach could only provide a numerical summary of potential immigrants and ignores their different fates in the downstream environment. While acting as a continuous seed to inoculate active players to the downstream environment [8], the upstream immigrants can also introduce a significant amount of seemingly abundant but inactive organisms [9]. These challenges call for a better evaluation of the in situ activities of individual immigrants in the downstream environment during quantifying microbial immigration impacts.
Differentiating immigrant populations that actively or inactively occupy the downstream system can better predict the relationship between the microbial community and environmental parameters, which is the key mission in many ecological studies. Various methods have been applied to identify the key environmental parameters. Clustering methods such as unweighted pair group method with arithmetic mean and k-means clustering were used to group communities based on population abundance and correlate different clusters with distinct parameters in anaerobic digester [10] or human intestinal microbiome [18]. Ordination methods such as principal components analysis, non-metric multidimensional scaling, correspondence analysis, and redundancy analysis treat population abundance as multiple variables and characterize similar communities based on shared parameters in anaerobic enrichment cultures [19], fish gut [20], human throat [21], and bioreactors [22]. Compared to clustering and ordination methods, supervised learning methods (including classification and regression that can predict unlabeled samples based on labeled microbial communities) are less widely used. Overall, all those methods rely on the assumption that abundant populations are active and contributing to system function. Such community-environment correlation can be biased if the system contains inactive populations that immigrate from the upstream, which can be reduced by assessing immigration impact with consideration of microbial activity.
In this study, we attempted to address two key questions: “can we quantitatively assess the activities of immigrants after entering the downstream ecosystem?” and “can we better predict key environmental parameters from microbial community composition?” To answer these questions, we analyzed a full-scale system that couples an upstream upflow anaerobic sludge blanket (UASB) reactor and downstream activated sludge reactors to treat wastewater containing purified terephthalic acid (PTA), an important petrochemical product. We used a mass balance model with amplicon sequencing [23] to calculate the in situ activity of each community member and quantify the intensity of immigration from the upstream anaerobic reactor to the downstream activated sludge. A supervised learning regressor [24] was used to predict important environmental parameters after considering immigration impact.
Methods
Sampling the activated sludge system
Sludge samples were taken from a full-scale PTA-wastewater treatment facility that operated an upstream UASB reactor and three downstream parallel activated sludge processes (including aeration tanks and clarifiers) (Additional file 1: Figure S1). Six sampling events were conducted during the spring and summer of 2017. To profile the microbial community as a continuum, 11 samples were taken along the process at each sampling event (Additional file 1: Figure S1): one sample from the middle of the UASB reactor sludge bed, one sample from the UASB effluent before being split and fed into the three aeration tanks, two samples from each of the three parallel aeration tanks (at different locations as replicates), and one sample from each of the three clarifier underflows. After collection, the samples were shipped to the laboratory at the University of Illinois at Urbana-Champaign (UIUC) on ice overnight and stored at − 80 °C prior to further analyses. Operation or physicochemical parameters, including temperature, dissolved oxygen (DO), ammonia, phosphate, and total organic carbon (TOC), were monitored and provided by the facility managers (detailed methods are available in Additional file 1). Phosphate measurement at one third of the sampling locations was not available. Terephthalic acid concentration of the samples was measured in UIUC lab using a HPLC with Agilent ZORBAX eclipse XDB-C18 column [25]. Total suspended solids (TSS) and volatile suspended solids (VSS) of the samples were measured according to the standard protocol [26].
DNA extraction, PCR, and 16S rRNA gene sequencing
16S rRNA gene amplicon sequencing was performed as described previously [27]. Briefly, DNA was extracted using the FastDNA SPIN Kit for Soil, and 16S rRNA gene was amplified with the Bacteria/Archaea universal primer sets Univ515F/Univ909R that target V4-V5 region [28]. Purified and pooled PCR amplicons were sequenced on an Illumina Miseq platform using the v3 chemistry at the Roy J. Carver Biotechnology Center at UIUC.
Sequence analysis and supervised learning regression
16S rRNA gene sequences were analyzed with the QIIME 2 platform (v2018.6) [29]. Raw sequences were first processed using DADA2 [30], including quality filtering, denoising, paired-end sequence merging, and chimera filtering. DADA2 generated unique amplicon sequence variants that were equivalent to 100% similarity operational taxonomic units (OTU) in the conventional practice. In this publication, we still use the term OTU for the purpose of simplicity. Taxonomy was assigned using q2-feature-classifier [31] customized for the primer set used in this study with Silva SSU database release 132 [32]. Multiple sequence alignment and phylogenetic tree construction were performed using the QIIME 2 plugin q2-phylogeny. For downstream diversity analysis, the OTU table was rarefied to 18,578 sequences per sample determined by the sample with least sequences. Alpha and beta diversity analyses were performed using the QIIME 2 plugin q2-diversity. Supervised regression of operation parameters on community compositions was performed using the QIIME2 plugin q2-sample-classifier with default settings [24]. Eighty percent of the samples were randomly picked to train the regressor. The remaining 20% of the samples were used to validate classification accuracy of the optimized regressor. The neutral model that fit OTU frequency to mean relative abundance [12] was applied to calculate immigration probability using the nlsLM function in R package minpack.lm. During calculation, OTU frequency was counted among all the activated sludge samples and mean relative abundance was averaged. Redundancy analysis was performed using R package vegan, and the significance of constraints was tested with permutation tests (999 permutations) [33].
Reactor kinetics
The mass balance calculation was performed as described previously [34]. The control volume was defined as one aeration tank and its downstream classifier. The change of mass (cell number) of a given microbial population x in this control volume was contributed by incoming biomass from the UASB effluent, outgoing biomass in the wasted sludge and clarifier effluent, and net growth in the aeration tank. The mass balance can be described as:
where Nx, AS is the cell number of population x in the aeration tank; nx, UASB is the number of x in the UASB effluent entering aeration tank per day [d−1]; nx, waste is the number of x in the wasted sludge leaving the system per day [d−1]; nx, eff is the number of x in the clarifier leaving the system per day [d−1]; and μx is net growth rate constant of x [d−1]. The number of x is obtained by multiplying the total cell number in the sample, which was approximated by concentration of volatile suspended solids [34], and the relative abundance of x in the microbial community, which was calculated based on 16S rRNA gene sequences. Detailed steps for calculation are available in Additional file 1. Growth rates of individual populations were calculated for the three activated sludge systems on different dates separately and averaged for further analyses.
Results
Mass balance identified active populations and indicated marginal immigration impact
In total, 1178 OTUs were observed and rarefaction curves indicated that sufficient sequencing depth was achieved (Additional file 1: Figure S2). To calculate the growth rate of individual populations, three assumptions were made and validated based on system performance data. First, the system was assumed to be operated at steady state at the time of sampling, which was supported by the stable TOC concentrations in the aeration tanks (Additional file 1: Figure S3a–c). The first sampling event in tank A was excluded for the analysis due to unstable TOC concentrations during the sampling period (Additional file 1: Figure S3a). Second, the biological activity in the clarifiers was negligible, and all the growth was assumed to occur in the aeration tank, which was supported by the similar TOC concentrations in the aeration tank and clarifier (Additional file 1: Figure S3a–c). Third, the biomass in the clarifier effluent was negligible, which was supported by the significantly low TSS concentrations in the clarifier effluent compared to the TSS of wasted sludge (Additional file 1: Figure S3d).
The growth rate calculation results are presented in Fig. 1. In total, 586 OTUs had a positive growth rate in activated sludge and accounted for 99% of the total sequences. OTUs with abundance higher than 0.1% all had a positive growth rate (Fig. 1a). This active subset of the community (i.e., active community) included the most abundant populations that were commonly detected in activated sludge taxa such as Zoogloea, Chitinophagales, Flavobacteriales, and Burkholderiaceae. The remaining 592 OTUs had a growth rate ≤ 0. This inactive subset of the community included populations that were abundant in the upstream UASB reactor, such as Syntrophus, Syntrophorhabdus, Pelotomaculum, and Methanosaeta, which were obligate anaerobes (Fig. 1b). They exhibited a significant decrease in abundance in aeration tanks and collectively contributed to 1% of the total sequences in activated sludge. Such marginal immigration impact by the upstream UASB community was supported by the small immigration probability m (0.028) for the entire activated sludge community calculated by fitting OTU frequency as a function of mean relative abundance using the neutral community model (Additional file 1: Figure S4). This suggested that local reproduction was the dominant process during community assemblage with a high probability at 0.972. A Venn diagram also revealed a similar result that only 78 OTUs were shared between UASB and activated sludge community and accounted for 0.4% of total sequences in obtained activated sludge (Additional file 1: Figure S5).
Fig. 1.

Growth rate of populations observed in the activated sludge reactors. The y-axis in a denotes abundance in aeration tank, and in b abundance in UASB reactor
Active populations better predicted environmental parameters
We applied a supervised learning regressor to predict environmental parameters based on microbial community composition (i.e., OTU abundance). A selected fraction of the samples was used to train the regressor, and the remaining samples were used to assess the prediction accuracy by comparing observed and predicted values. Intuitively, we used samples from two of the three parallel aeration tanks for training and used the third tank for testing. However, poor prediction accuracy was observed, suggested by the large discrepancies between observed and predicted values (Additional file 1: Figure S6). None of the squared linear least-squares regression coefficients (R) exceeded 0.7, except for temperature.
Instead of using two thirds of the samples for training the regressor, we pooled all the samples from the three tanks and tested the optimal fraction of samples for training the regressor. Based on the prediction accuracy, including mean squared error, R squared, and slope (Additional file 1: Table S1), it was shown that using 80% of the samples for regressor training and 20% samples for accuracy testing produced the optimal results. When the entire microbial community was used to train the regressor, temperature had higher predictability than other parameters. The fitted line of the predicted value versus observed value had a R squared of 0.780 and a slope of 0.848 (Fig. 2a). The prediction of phosphate also had high R squared (0.910) and slope (0.761), but the 95% confidence interval was comparably large due to missing one third of the phosphate concentration data (Additional file 1: Figure S7). Other parameters, i.e., TOC, DO, ammonia, and TA concentration, had relatively poor predictability with R squared and slope lower than 0.7.
Fig. 2.
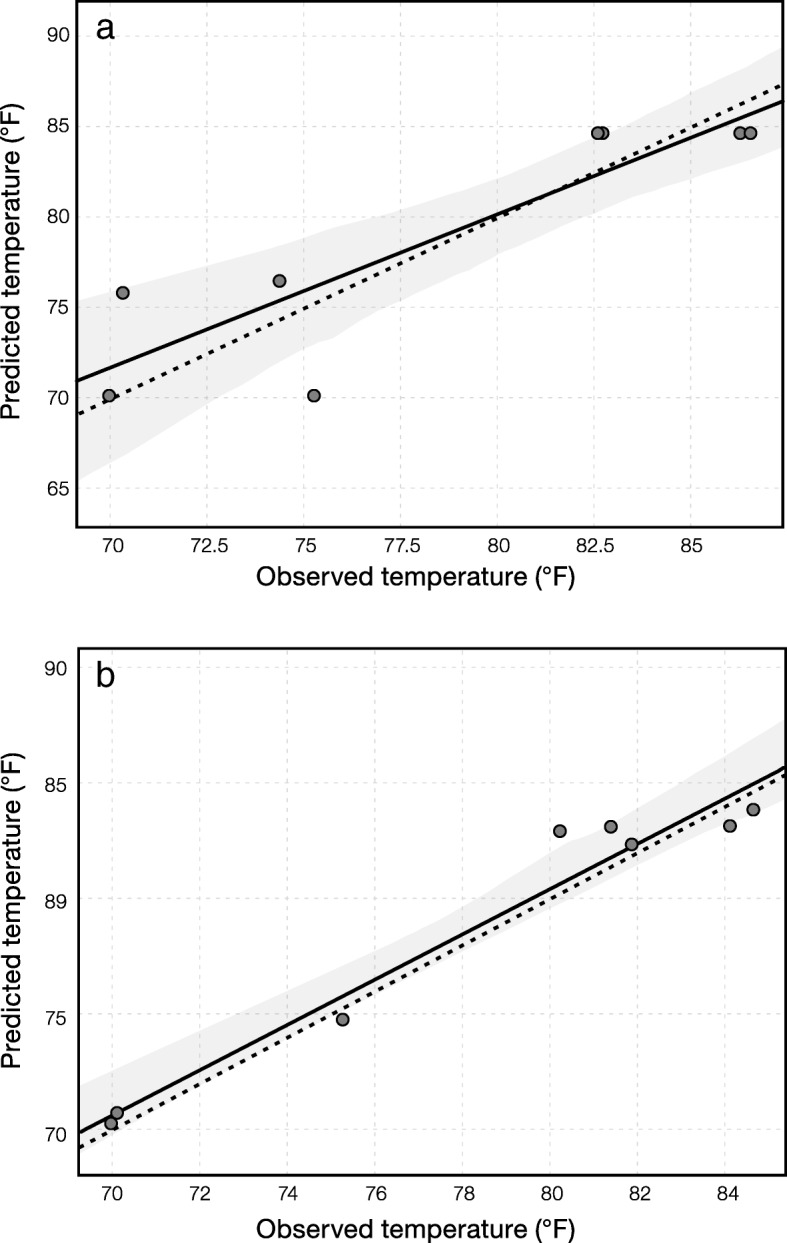
Prediction of temperature based on a the entire community and b the active community. The x- and y-axis denote observed and predicted temperature, respectively. The dashed line represents 1:1 ratio of observed and predicted value. The solid line represents the fitted trend of the observed and predicted value. The gray shadow represents 95% confidence interval with 100 times of estimation
We further used the active community (i.e., OTUs with positive growth rates) in the activated sludge reactor to train and test the regressor (Additional file 1: Figure S8). Temperature was still the most predictable parameter with almost perfect accuracy. The R squared of 0.955 and slope of 0.980 were very close to the 1:1 observed/predicted line and were both higher than the values when the entire community was used (Fig. 2b). An improved confidence interval was also observed throughout the entire prediction zone. For other parameters, the prediction also improved as suggested by lower mean squared error, higher R squared, and higher slope in most cases (Table 1).
Table 1.
Comparison of prediction accuracy between the entire community and active community. MSE stands for mean square error
| Parameter | Entire community | Active community | Active/Entire | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| MSE | R squared | p value | Slope | MSE | R squared | p value | Slope | MSE | R squared | Slope | |
| Temp. | 9.375 | 0.780 | 0.004 | 0.848 | 3.608 | 0.955 | 0.000 | 0.980 | 0.385 | 1.225 | 1.155 |
| TOC | 7.203 | 0.671 | 0.013 | 0.425 | 2.606 | 0.843 | 0.001 | 0.615 | 0.362 | 1.255 | 1.445 |
| DO | 1.037 | 0.378 | 0.105 | 0.187 | 0.464 | 0.195 | 0.274 | 0.537 | 0.447 | 0.515 | 2.877 |
| NH4+ | 0.006 | 0.659 | 0.014 | 0.587 | 0.006 | 0.541 | 0.038 | 0.726 | 0.997 | 0.820 | 1.238 |
| PO43− | 0.116 | 0.910 | 0.012 | 0.761 | 0.059 | 0.946 | 0.005 | 0.720 | 0.505 | 1.041 | 0.946 |
| TA | 0.000 | 0.120 | 0.400 | 0.125 | 0.000 | 0.531 | 0.040 | 0.153 | 1.750 | 4.416 | 1.219 |
Redundancy analysis identified active populations associated with environmental parameters
Redundancy analysis was performed on the active community to identify which populations were associated with individual environmental parameters (Fig. 3). Permutation tests indicated statistical significance (p < 0.05) of the associations for temperature, NH4+, TOC, and TA, but not for DO (p = 0.259). In terms of community structure, all the three parallel aeration tanks followed similar evolution trajectory along sampling time from spring to summer (light to dark symbols on the left panel). This trend was consistent with the direction of increasing temperature and agreed with the aforementioned result that temperature was the most predictable environmental parameters to describe the community structure. The impact of temperature was further observed on individual OTUs, where the most dominant OTUs were distributed in the direction of increasing temperature (right panel). The only dominant OTU that was located in the opposite direction of temperature was related to Cytophagales, which might prefer lower temperature. The direction of increasing temperature opposed that of TOC, implying higher temperature was associated with higher consumptions of organic carbon. TA concentration was independent from the temperature effect, and abundant OTUs related to Methyloversatilis and Acidobacteria were associated with higher TA concentration.
Fig. 3.

Redundancy analysis based on the abundance of active populations in activated sludge. The left panel is a tri-plot of samples, OTUs, and performance parameters. Stars, hexagons, and diamonds represent samples from the three parallel tanks. Gray scale of the fill color represents time scale of the sampling from February to June. Small dots in the middle represent OTUs, which are zoomed in on the right panel. Size of the circle represents relative abundance in activated sludge. The length of an environmental parameter arrow in the resulting ordination indicates the strength of the relationship of that parameter to community composition
Discussion
Microbial immigration constantly contributes to community assemblage in natural and engineered microbial ecosystems. However, the intensity of its impact varies. Abundant populations in the upstream process can play active or inactive roles in the downstream. This could not be effectively addressed by many previous studies [4, 5, 11, 13–17]. Coupling a mass balance model with amplicon sequencing data allows the calculation of the net growth rate of individual community members, enabling the differentiation between actively growing populations and non-growing ones. This further allows to evaluate the impact of microbial immigration on the downstream system by addressing whether the upstream process can introduce microbial populations that will remain active in the downstream system. This strategy was first used to reveal that influent wastewater introduced populations that could actively grow in the receiving activated sludge system as well as non-growing populations [23]. A more significant immigration impact was observed in anaerobic digesters where 25% of the sequences were associated with non-growing populations introduced by the feed microbial biomass [34]. In contrast to the previous two cases of immigration from raw wastewater (aerobic) to activated sludge (aerobic) environment and from activated sludge (aerobic) to anaerobic digester (anaerobic) environment, the present study examined another distinct immigration case from UASB reactor (anaerobic) to activated sludge (aerobic) environment. Non-growing populations only occupied 1% of the activated sludge community, and abundant populations in the upstream anaerobic reactor did not grow in activated sludge due to a significant change in environmental condition (anaerobic vs. aerobic). The marginal immigration impact observed could also be reflected by the small migration probability in the Sloan model and small number of shared OTUs in Venn diagram (Additional file 1: Figure S4 and Additional file 1: Figure S5).
The active populations in activated sludge processes, which are not influenced by upstream immigration, can be viewed as the key players of important ecological functions. Methanol was one of the major components in PTA wastewater [35], and an active population related to Methyloversatilis that could utilize methanol as carbon and energy source [36] was observed. Populations associated with Cytophaga, Flavobacterium, and Chitinophaga have been reported to degrade soluble microbial products in anaerobic reactor effluent [37]. OTUs related to these taxa that might scavenge anaerobic by-products that were present activated sludge given that TA and BA accounted for only 31–69% of all TOC (Additional file 1: Table S2).
The inactive populations, although not abundant, were also detected and accounted for 50.2% of the 1178 OTUs observed in activated sludge. This suggested that functionally active populations only accounted for half of the observed species richness, which could not be revealed by using approaches currently available. Analyzing 16S rRNA gene sequences alone could only provide evidences of detectable microorganisms but not necessarily active ones and could be influenced by the rRNA copy numbers associated with different microorganisms [38]. Meta-omics approaches (e.g., metagenomic and metatranscriptomic) could shed lights on metabolic function at multiple levels, but the information obtained relies on the availability of reference genomes with high-quality annotations, which are still scarce [39]. Stable-isotope probing coupled with molecular biomarker sequencing can associate microbial identity with function [40], but is limited by its throughput to monitor all populations. Also, cross-feeding phenomena can sometimes restrict its application. Fluorescence in situ hybridization can provide evidence of presence and activity, especially by coupling with microautoradiography to visualize uptake of specific substrates [41]. However, applications of such target-specific methods are limited to a few pre-determined microorganisms. Our findings showed that the mass balance model coupled with amplicon sequencing provides a novel and high-throughput approach to effectively characterize microbial activity compared to the aforementioned methods, and can be widely applied in environmental and applied microbiology.
Our study also highlighted that the application of a supervised learning regressor could identify predictive and important environmental parameters. When the regressor was trained on the active subset of the community instead of the entire community, the prediction accuracy of environmental parameters was greatly improved. This is reasonable because active populations are primarily responsible for important ecological functions. Inactive populations likely did not contribute to key ecological functions and thus could be regarded as baseline noise of the community. If the immigration impact is strong, for example, if a large fraction of the community is occupied by non-growing populations introduced from the upstream process, such an approach can be even more effective. Last, we demonstrated that sufficient sample numbers and comprehensive environmental parameters monitoring were critical to ensure good performance of regression, which emphasizes the necessity to perform holistic sampling and monitoring for engineered processes in future studies.
Additional file
Supplementary figures and tables. This file contains supplementary Figures S1–S8. and Tables S1–S2. (PDF 4054 kb)
Acknowledgements
The staff at the Roy J. Carver Biotechnology Center in University of Illinois at Urbana-Champaign are gratefully acknowledged for the high throughput sequencing assists.
Funding
This study was supported by a grant from Energy Biosciences Institute (EBI) at UIUC to WTL.
Availability of data and materials
The paired-end sequences obtained in this study have been deposited under DDBJ/EMBL/GenBank accession no. DRA007328.
Authors’ contributions
RM designed the study, performed the experiment, analyzed the data, and wrote the manuscript. JK performed the experiment. FW designed the study. BB designed the study, performed the experiment, and revised the manuscript. WTL designed the study and revised the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Ran Mei, Email: ranmei2@illinois.edu.
Jinha Kim, Email: jinhahk2@illinois.edu.
Fernanda P. Wilson, Email: fernandapaes@gmail.com
Benjamin T. W. Bocher, Email: BENJAMIN.BOCHER@bp.com
Wen-Tso Liu, Phone: +1-217-333-8442, Email: wtliu@illinois.edu.
References
- 1.Lindström ES, Bergström A-K. Influence of inlet bacteria on bacterioplankton assemblage composition in lakes of different hydraulic retention time. Limnol Oceanogr. 2004;49:125–136. doi: 10.4319/lo.2004.49.1.0125. [DOI] [Google Scholar]
- 2.Czekalski N, Berthold T, Caucci S, Egli A, Bürgmann H. Increased levels of multiresistant bacteria and resistance genes after wastewater treatment and their dissemination into Lake Geneva, Switzerland. Front Microbiol. 2012;3:106. doi: 10.3389/fmicb.2012.00106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jones SE, Newton RJ, McMahon KD. Potential for atmospheric deposition of bacteria to influence bacterioplankton communities. FEMS Microbiol Ecol. 2008;64:388–394. doi: 10.1111/j.1574-6941.2008.00476.x. [DOI] [PubMed] [Google Scholar]
- 4.Liu T, Zhang AN, Wang J, Liu S, Jiang X, Dang C, Ma T, Liu S, Chen Q, Xie S. Integrated biogeography of planktonic and sedimentary bacterial communities in the Yangtze River. Microbiome. 2018;6:16. doi: 10.1186/s40168-017-0388-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ling F, Whitaker R, LeChevallier MW, Liu W-T. Drinking water microbiome assembly induced by water stagnation. ISME J. 2018;12:1520-31. [DOI] [PMC free article] [PubMed]
- 6.Ju F, Lau F, Zhang T. Linking microbial community, environmental variables, and methanogenesis in anaerobic biogas digesters of chemically enhanced primary treatment sludge. Environ Sci Technol. 2017;51:3982–3992. doi: 10.1021/acs.est.6b06344. [DOI] [PubMed] [Google Scholar]
- 7.Walsh EA, Kirkpatrick JB, Rutherford SD, Smith DC, Sogin M, D’Hondt S. Bacterial diversity and community composition from seasurface to subseafloor. ISME J. 2016;10:979-89. [DOI] [PMC free article] [PubMed]
- 8.Wells GF, Wu CH, Piceno YM, Eggleston B, Brodie EL, DeSantis TZ, Andersen GL, Hazen TC, Francis CA, Criddle CS. Microbial biogeography across a full-scale wastewater treatment plant transect: evidence for immigration between coupled processes. Appl Microbiol Biotechnol. 2014;98:4723–4736. doi: 10.1007/s00253-014-5564-3. [DOI] [PubMed] [Google Scholar]
- 9.Kirkegaard RH, McIlroy SJ, Kristensen JM, Nierychlo M, Karst SM, Dueholm MS, Albertsen M, Nielsen PH. The impact of immigration on microbial community composition in full-scale anaerobic digesters. Sci Rep. 2017;7:9343. doi: 10.1038/s41598-017-09303-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mei R, Nobu MK, Narihiro T, Kuroda K, Munoz Sierra J, Wu Z, Ye L, Lee PKH, Lee PH, van Lier JB, et al. Operation-driven heterogeneity and overlooked feed-associated populations in global anaerobic digester microbiome. Water Res. 2017;124:77–84. doi: 10.1016/j.watres.2017.07.050. [DOI] [PubMed] [Google Scholar]
- 11.Lee S-H, Kang H-J, Park H-D. Influence of influent wastewater communities on temporal variation of activated sludge communities. Water Res. 2015;73:132–144. doi: 10.1016/j.watres.2015.01.014. [DOI] [PubMed] [Google Scholar]
- 12.Sloan WT, Lunn M, Woodcock S, Head IM, Nee S, Curtis TP. Quantifying the roles of immigration and chance in shaping prokaryote community structure. Environ Microbiol. 2006;8:732–740. doi: 10.1111/j.1462-2920.2005.00956.x. [DOI] [PubMed] [Google Scholar]
- 13.Morris A, Beck JM, Schloss PD, Campbell TB, Crothers K, Curtis JL, Flores SC, Fontenot AP, Ghedin E, Huang L. Comparison of the respiratory microbiome in healthy nonsmokers and smokers. Am J Respir Crit Care Med. 2013;187:1067–1075. doi: 10.1164/rccm.201210-1913OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shanks OC, Newton RJ, Kelty CA, Huse SM, Sogin ML, McLellan SL. Comparison of the microbial community structures of untreated wastewaters from different geographic locales. Appl Environ Microb. 2013;79:2906. doi: 10.1128/AEM.03448-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Staley C, Gould TJ, Wang P, Phillips J, Cotner JB, Sadowsky MJ. Species sorting and seasonal dynamics primarily shape bacterial communities in the Upper Mississippi River. Sci Total Environ. 2015;505:435–445. doi: 10.1016/j.scitotenv.2014.10.012. [DOI] [PubMed] [Google Scholar]
- 16.Ahmed W, Staley C, Sadowsky MJ, Gyawali P, Sidhu JPS, Palmer A, Beale DJ, Toze S. Toolbox approaches using molecular markers and 16S rRNA gene amplicon data sets for identification of fecal pollution in surface water. Appl Environ Microb. 2015;81:7067. doi: 10.1128/AEM.02032-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liao J, Fang C, Yu J, Sathyagal A, Willman E, Liu W-T. Direct treatment of high-strength soft drink wastewater using a down-flow hanging sponge reactor: performance and microbial community dynamics. Appl Microbiol Biotechnol. 2017;101:5925–5936. doi: 10.1007/s00253-017-8326-1. [DOI] [PubMed] [Google Scholar]
- 18.McHardy IH, Goudarzi M, Tong M, Ruegger PM, Schwager E, Weger JR, Graeber TG, Sonnenburg JL, Horvath S, Huttenhower C. Integrative analysis of the microbiome and metabolome of the human intestinal mucosal surface reveals exquisite inter-relationships. Microbiome. 2013;1:17. doi: 10.1186/2049-2618-1-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mei R, Narihiro T, Nobu MK, Liu W-T. Effects of heat shocks on microbial community structure and microbial activity of a methanogenic enrichment degrading benzoate. Lett Appl Microbiol. 2016;63:356–362. doi: 10.1111/lam.12629. [DOI] [PubMed] [Google Scholar]
- 20.Ye L, Amberg J, Chapman D, Gaikowski M, Liu W-T. Fish gut microbiota analysis differentiates physiology and behavior of invasive Asian carp and indigenous American fish. ISME J. 2014;8:541–551. doi: 10.1038/ismej.2013.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jakobsson HE, Jernberg C, Andersson AF, Sjölund-Karlsson M, Jansson JK, Engstrand L. Short-term antibiotic treatment has differing long-term impacts on the human throat and gut microbiome. PLoS One. 2010;5:e9836. doi: 10.1371/journal.pone.0009836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Narihiro T, Kim NK, Mei R, Nobu MK, Liu WT. Microbial community analysis of anaerobic reactors treating soft drink wastewater. PLoS One. 2015;10:e0119131. doi: 10.1371/journal.pone.0119131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Saunders AM, Albertsen M, Vollertsen J, Nielsen PH. The activated sludge ecosystem contains a core community of abundant organisms. ISME J. 2016;10:11-20. [DOI] [PMC free article] [PubMed]
- 24.Bokulich N, Dillon M, Bolyen E, Kaehler BD, Huttley GA, Caporaso JG. q2-sample-classifier: machinelearning tools for microbiome classification and regression. J Open Source Software. 2018;3(30):934-7. [DOI] [PMC free article] [PubMed]
- 25.Narihiro T, Nobu MK, Bocher BTW, Mei R, Liu WT. Co-occurrence network analysis reveals thermodynamics-driven microbial interactions in methanogenic bioreactors. Environ Microbiol Rep. 2018;10(6):673-85. [DOI] [PubMed]
- 26.Federation WE, Association A . Standard methods for the examination of water and wastewater. Washington, DC: American Public Health Association (APHA); 2005. [Google Scholar]
- 27.Mei R, Nobu MK, Narihiro T, Yu J, Sathyagal A, Willman E, Liu W-T. Novel Geobacter species and diverse methanogens contribute to enhanced methane production in media-added methanogenic reactors. Water Res. 2018;147:403-12. [DOI] [PubMed]
- 28.Wang Y, Qian P-Y. Conservative fragments in bacterial 16S rRNA genes and primer design for 16S ribosomal DNA amplicons in metagenomic studies. 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pena AG, Goodrich JK, Gordon JI. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP. DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods. 2016;13:581. doi: 10.1038/nmeth.3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bokulich NA, Kaehler BD, Rideout JR, Dillon M, Bolyen E, Knight R, Huttley GA, Caporaso JG. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome. 2018;6:90. doi: 10.1186/s40168-018-0470-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glöckner FO. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 2012;41:D590–D596. doi: 10.1093/nar/gks1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Oksanen J, Blanchet FG, Kindt R, Legendre P, Minchin PR, O’hara R, Simpson GL, Solymos P, Stevens MHH, Wagner H. Package ‘vegan’. Community ecology package, version 2. 2013. [Google Scholar]
- 34.Mei R, Narihiro T, Nobu MK, Kuroda K, Liu W-T. Evaluating digestion efficiency in full-scale anaerobic digesters by identifying active microbial populations through the lens of microbial activity. Sci Rep. 2016;6:34090. doi: 10.1038/srep34090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kuroda K, Nobu MK, Mei R, Narihiro T, Bocher BTW, Yamaguchi T, Liu W-T. A single-granule-level approach reveals ecological heterogeneity in an upflow anaerobic sludge blanket reactor. PLoS One. 2016;11:e0167788. doi: 10.1371/journal.pone.0167788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kalyuzhnaya MG, De Marco P, Bowerman S, Pacheco CC, Lara JC, Lidstrom ME, Chistoserdova L. Methyloversatilis universalis gen. nov., sp. nov., a novel taxon within the Betaproteobacteria represented by three methylotrophic isolates. Int J Syst Evol Microbiol. 2006;56:2517–2522. doi: 10.1099/ijs.0.64422-0. [DOI] [PubMed] [Google Scholar]
- 37.Kim N-K, Oh S, Liu W-T. Enrichment and characterization of microbial consortia degrading soluble microbial products discharged from anaerobic methanogenic bioreactors. Water Res. 2016;90:395–404. doi: 10.1016/j.watres.2015.12.021. [DOI] [PubMed] [Google Scholar]
- 38.Blazewicz SJ, Barnard RL, Daly RA, Firestone MK. Evaluating rRNA as an indicator of microbial activity in environmental communities: limitations and uses. ISME J. 2013;7:2061–2068. doi: 10.1038/ismej.2013.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vanwonterghem I, Jensen PD, Ho DP, Batstone DJ, Tyson GW. Linking microbial community structure, interactions and function in anaerobic digesters using new molecular techniques. Curr Opin Biotechnol. 2014;27:55–64. doi: 10.1016/j.copbio.2013.11.004. [DOI] [PubMed] [Google Scholar]
- 40.Radajewski S, Ineson P, Parekh NR, Murrell JC. Stable-isotope probing as a tool in microbial ecology. Nature. 2000;403:646–649. doi: 10.1038/35001054. [DOI] [PubMed] [Google Scholar]
- 41.Ariesyady HD, Ito T, Okabe S. Functional bacterial and archaeal community structures of major trophic groups in a full-scale anaerobic sludge digester. Water Res. 2007;41:1554–1568. doi: 10.1016/j.watres.2006.12.036. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary figures and tables. This file contains supplementary Figures S1–S8. and Tables S1–S2. (PDF 4054 kb)
Data Availability Statement
The paired-end sequences obtained in this study have been deposited under DDBJ/EMBL/GenBank accession no. DRA007328.