

Abstract
Burn-site infections, commonly due to Pseudomonas aeruginosa, have been associated with deranged intestinal integrity, allowing bacteria and their products to translocate from the gut to the circulatory system. The P. aeruginosa quorum sensing (QS) transcription factor MvfR (PqsR) controls the expression of numerous virulence factors, and the synthesis of several toxic products. However, the role of QS in intestinal integrity alterations, to the best of our knowledge, has not been previously investigated. Using a proven anti-MvfR, anti-virulence agent, the in vivo results of the present study revealed that inhibition of MvfR function significantly decreased Fluorescein Isothiocyanate-Dextran (FITC-Dextran) flow from the intestine to the systemic circulation, diminished bacterial translocation from the intestine to mesenteric lymph nodes (MLNs), and improved tight junction integrity in thermally injured and infected mice. In addition, the MvfR antagonist administration alleviates the intestinal inflammation, as demonstrated by reduced ileal TNF-α and fecal lipocalin-2 concentrations. In addition, it is associated with lower levels of circulating endotoxin and decreased P. aeruginosa dissemination from the burn wound to the ileum. Collectively, these results hold great promise that the inhibition of this QS system mitigates gut hyperpermeability by attenuating the derangement of morphological and immune aspects of the intestinal barrier, suggesting that MvfR function is crucial in the deterioration of intestinal integrity following P. aeruginosa burn-site infection. Therefore, an anti-virulence approach targeting MvfR, could potentially offer a novel therapeutic approach against multi-drug resistant P. aeruginosa infections following thermal injuries. Since this approach is targeting virulence pathways that are non-essential for growth or viability, our strategy is hypothesized to minimize the development of bacterial resistance, and preserve the beneficial enteric microbes, while improving intestinal integrity that is deranged as a result of burn and infection.
Keywords: dorsal burn infection, pseudomonas aeruginosa, intestinal permeability, MvfR, PqsR, quorum-sensing and anti-virulence therapeutics
Introduction
The innermost layer of the intestinal mucosa, the simple columnar epithelium lining the intestinal tract, confers the barrier/filter function to the intestinal tissue, thus allowing an optimum exchange of elements between the body and the external environment. This exchange takes place either through the epithelial cells (transcellular permeability), or via the interepithelial space (paracellular transport), with the latter mainly being regulated by multi-protein complexes, known as tight junctions (TJs) (1). The maintenance of an optimal intestinal permeability is further supported by two auxiliary elements; first, the gel-like layer on the luminal surface of the intestinal epithelium, consisting of mucins and antimicrobial peptides, secreted by the epithelial cells (2), and serving as a potent chemical barrier (3); second, the immune modulation of the intestinal integrity, initially by microfold and antigen presenting cells (3) and ultimately by lymphocytes and cytokines (1).
Thermal injury, one of the leading causes of mortality and morbidity worldwide (4), has long been linked to alterations in intestinal permeability (5). More specifically, following severe burn, there is an initially increased systemic inflammatory and metabolic response, marked by elevated levels of reactive oxygen species (ROS) and cytokines, such as tumor necrosis factor α (TNF-α), that eventually affect secondary organs (6). In the gut, research has revealed increased intestinal epithelial apoptosis (7), as well as TJ breakdown secondary to thermal injury (8), thus attenuating the intestinal integrity. Furthermore, burn affects the gut immune homeostasis, and therefore the immune aspect of the intestinal barrier, as indicated by alterations in T-cell populations of intestinal origin (9). To add an extra layer of complexity to the dysregulation of the intestinal permeability following thermal injury, the aforementioned post-burn barrier alterations have been linked to dramatic shifts in the bacterial gut microbiome (10). In turn, the composition of the intestinal microbial population has been related to potent changes in the gut permeability (11), which then predisposes to bacterial translocation to extra-intestinal tissues and subsequent sepsis (10).
In addition to thermal injury, infection of the burn eschar is another major determinant of the intestinal barrier dysfunction. Subsequent to burn wound infection, systemic dissemination of the infectious agent, facilitated by the immunosuppression that follows the initial post-burn hyper-inflammatory state (6), gives rise to sepsis (12). Both the wound infection itself and the ensuing septic state have, in turn, been shown to affect the intestinal barrier function. Studies in animals (13) and humans (14) indicate that post-burn infection significantly increases the gut permeability compared to burn alone. What is more, it has previously been suggested that when superimposed on burn, wound infection and the resultant sepsis can prolong the duration of the intestinal hyperpermeability in rats (15).
Despite adequate resuscitation and current antibiotic treatment, wound infection and subsequent sepsis jeopardize the function of secondary organs, thus representing a compelling clinical challenge (16). In this setting, P. aeruginosa, one of the most common causative microorganisms, poses a major threat to burn patients (17). This opportunistic Gram negative bacterial pathogen displays a wide spectrum of antibiotic resistance (18). We have developed a promising strategy against this nosocomial pathogen, by targeting bacterial virulence regulatory systems. More specifically, we have developed several potent agents targeting MvfR (also known as PqsR), one of the quorum sensing (QS) cell-cell bacterial communication systems in P. aeruginosa. MvfR controls the expression of multiple virulence factors directly and indirectly, including the expression of the other QS system regulators LasR and RhlR (19,20). We have previously shown that our benzamine benzimidazole (BB) inhibitors specifically bind to the MvfR protein and that targeting MvfR with non-ligand based antagonists, effectively halts P. aeruginosa acute and chronic phenotypes, including the formation of antibiotic-tolerant/persister cells (21–24). The BB family of anti-MvfR agents we have developed and tested in vivo, alleviates P. aeruginosa acute infection, and averts infection relapse after the cessation of the antibiotic course (21–24). Importantly, these compounds are expected to thrive where traditional antibiotics fail; by targeting virulence functions that are not essential for bacterial growth or survival, they diminish bacterial infectivity and invasiveness, without imposing a strong selective pressure on the pathogens, thus potentially reducing the likelihood to generate resistant strains, while preserving the beneficial enteric flora. In view of our success in targeting P. aeruginosa virulence functions in vivo, the present study describes our effort to target the MvfR QS in the setting of P. aeruginosa burn-wound infection, aiming to ameliorate the subsequent intestinal barrier dysfunction, which we found to be significantly affected in our burn-infection mouse model.
Materials and methods
Mice
Eight-week-old male C57BL/6 mice were purchased from the Jackson Laboratories. Mice were maintained in a specific pathogen-free (SPF) environment at the Massachusetts General Hospital (MGH; Boston, USA), in a 12-h light 12-h dark photoperiod at an ambient temperature of 22±1°C, with food and water access ad libitum.
Ethics statement
Animal protocols were reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) at the MGH (protocol no. 2006N000093) and are in strict accordance to the guidelines of the Committee on Animals of the MGH, Harvard Medical School (Boston, USA), and the regulations of the Subcommittee on Research Animal Care of the MGH and the National Institutes of Health. Animals were euthanized according to the guidelines of the Animal Veterinary Medical Association. All efforts were made to minimize suffering.
Bacterial strains and inoculum growth conditions
UCBPP-PA14 (PA14) is a rifampicin resistant P. aeruginosa human clinical isolate (Rahme laboratory). The mvfR mutant is isogenic to UCBPP-PA14 (Rahme laboratory) (25). Unless otherwise indicated, bacteria were grown in Luria Bertani (LB) broth, LB agar plates, or LB agar plates containing 100 µg/ml rifampicin.
Animal experiments
All mice were anesthetized using one 500 µl intraperitoneal (IP) injection of ketamine (125 mg/kg) and xylazine (12.5 mg/kg) in normal saline (N/S) and the dorsal fur was subsequently removed with an electric clipper. A 30% total body surface area (TBSA) dorsal burn was induced by immersion in 90°C water for 8 sec, using a polystyrene foam template, as in the well-established burn model described by Walker and Mason (1968), with some modifications (26). Spinal protection from the thermal injury was achieved by a dorsal subcutaneous injection of 500 µl N/S, prior to the induction of the burn injury. Fluid resuscitation and pain prevention following burn were achieved by a 100 µl subcutaneous injection of buprenorphine in N/S (0.3 mg/ml), in a non-burnt area. Sham animals underwent all procedures except for the thermal injury.
Immediately after burn, 100 µl of 10 mM MgSO4 containing approximately 105 colony forming units (CFUs) of P. aeruginosa clinical isolate PA14 culture, or isogenic mvfR mutant culture, were intradermally injected at the burn eschar of mice in the burn plus infection (BI) group. Mice in the sham and burn alone groups received an equivalent injection of 100 µl phosphate-buffered saline (PBS). After the experiment, all animals were returned to their cages, to allow recovery from anesthesia. During this period, all cages were kept on heating pads to prevent hypothermia. Food and hydrogel on the cage floor were provided ad libitum.
For the group supplemented with our MvfR-inhibiting compound [(M64-structure fully described and depicted in the publication by Starkey et al (24)], mice received four intravenous (tail vein) injections at 2, 4, 8 and 16 h post-BI, at a dose of 4 mg/kg body weight. Control groups received equivalent doses of dimethylsulfoxide (DMSO) vehicle.
In vivo intestinal permeability assay
For the assessment of the intestinal barrier function, 4 h prior to euthanasia, mice were gavaged with 0.2 ml of Fluorescein Isothiocyanate-Dextran (FITC-Dextran; 3–5 kDa; cat. no. FD4; Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) in PBS, so that a dose of 440 mg/kg body weight was achieved. 18–20 h post BI, mice were euthanized. Aseptic cardiac puncture was performed to obtain blood samples. The collected blood was kept on ice and then centrifuged at 21,000 × g for 10 min. The serum was removed and was used to assess the FITC levels with fluorescent spectrophotometry (excitation, 480 nm and emission, 520 nm). Mice used to assess the intestinal permeability changes over time, as shown in Fig. 1, were euthanized prior to, or at 4, 10 and 18 h post BI.
Figure 1.
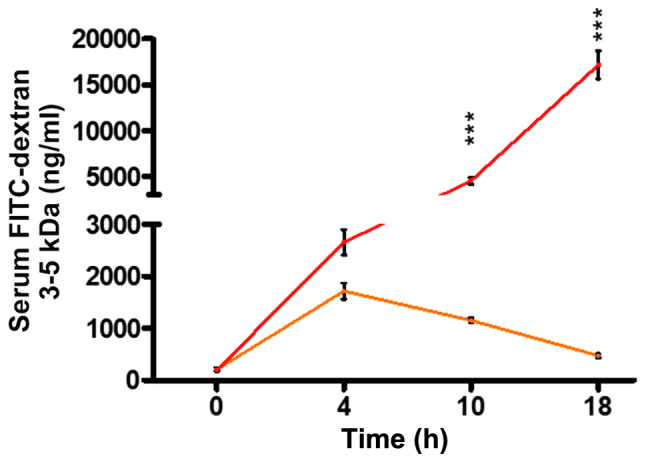
P. aeruginosa infection dramatically increases intestinal permeability following burn and infection. Mice were burnt (orange line) or burnt and infected with P. aeruginosa strain PA14 (red line). FITC-dextran 3–5 kDa flow from the intestinal lumen to the systemic circulation increases following burn alone, with a peak at 4 h, reaching 1,700 ng/ml and a gradual drop thereafter. FITC levels following BI show a continuous rise, reaching a concentration of over 17,000 ng/ml at 18 h following insult. The difference between the two groups becomes statistically significant at 10 h (P<0.001) and further rises at 18 h (P<0.001). Mice at 0 h demonstrate the sham FITC levels. PA14 burn-site infection was induced by intradermal administration of 105 CFUs/animal. FITC-dextran 3–5 kDa levels were assessed in the serum with fluorescent spectrophotometry (excitation, 480 nm and emission, 520 nm). Data show the average +/- SEM (n=5). Statistical significance was assessed using two-way ANOVA + Bonferroni correction. FITC-dextran, Fluorescein Isothiocyanate-Dextran. ***P<0.001.
Tissue harvesting
Immediately after euthanasia, the mesenteric lymph nodes (MLNs), ileum and colon were aseptically harvested through midline laparotomy. The intestine samples were flushed three times with sterile PBS. The samples were then snap frozen in liquid nitrogen or stored in RNA-Later and were then frozen at −80°C (Qiagen Ltd., Manchester, UK) for future analysis.
Stool samples
Stool pellets were collected at 0, 4, 10 and 20 h post infection. The samples were homogenized in sterile PBS and were processed as below for fecal lipocalin-2 (Lcn-2) measurements.
CFU assessment
The MLN tissue was used to assess bacterial translocation from the intestinal lumen. MLNs were homogenized in sterile PBS using metallic beads and were serially diluted 1/10-1/1000 and plated on LB agar plates.
Distal ileum was used to assess the PA14 dissemination to the intestinal lumen. The ileal tissue was homogenized in sterile PBS using metallic beads and was serially diluted 1/10-1/1000 and plated on LB agar plates containing 100 µg/ml rifampicin.
Systemic blood obtained via cardiac puncture, as described above, was used to assess bacteremia and was serially diluted 1/10-1/1000 in sterile PBS and plated on LB agar plates.
Following inoculation, all plates were incubated at 37°C and CFUs were quantified after 24 h.
TJ assays
Immunofluorescence
Samples of distal ileum were fixed with 4% paraformaldehyde. Sections then were cut and mounted on microscope slides. After deparaffinization and antigen retrieval (Antigen Retrieval reagent; R&D Systems, Minneapolis, MN, USA), tissue sections were immersed in PBS/0.1 tween for 10 min and were blocked by Normal Goat Serum. They were then incubated with primary antibody rabbit polyclonal anti-claudin-1 [ab15098; RRID: AB_301644; final concentration, 1:100; Abcam, Cambridge, MA, USA (27)] overnight in a humid chamber at 4°C. The sections were washed three times with PBS, and secondary antibody goat anti-rabbit [ab150077; RRID: AB_2630356; final concentration: 1:500; (28)] and DAPI (both Abcam, Cambridge, MA, USA) were added. This was followed by a 1-h incubation at room temperature. The sections were washed three times with PBS, dried and mounted and images were collected using a confocal fluorescent microscope (magnification, ×400) (Nikon A1; Nikon Corp., Tokyo, Japan).
Reverse transcription-quantitative polymerase chain reaction (RT-qPCR)
The distal ileum was dissected and stored in RNA-Later at −80°C as described above. RNA was isolated using the RNeay Plus Mini kit (250) (Qiagen Ltd.), according to the manufacturer's protocol. cDNA was generated using the iScript TM Reverse Transcription Supermix for RT-qPCR (Bio-Rad Laboratories, Inc., Hercules, CA, USA), according to the manufacturer's instructions. The sequences of the primers used were Claudin 1 forward, 5′-AGTCTTCGACTCCTTGCTGA-3′ and reverse, 5′-ACAAAGATTGCGATCAGCCC-3. Expression of mRNA was normalized with the reference gene TATA-box Binding Protein (TBP) mRNA expression (29). RT-qPCR was performed using the Brilliant II SYBR-Green qPCR Master Mix with Low ROX (Agilent 600830; Agilent Technologies; Santa Clara, CA, USA). The total volume per reaction was 12 ul, consisting of 6 µl SYBR-Green qPCR Master Mix with Low ROX, 0.6 µl forward primer (5 µM), 0.6 reverse primer (5 µM), 3.8 µl distilled water amd 1 µl cDNA sample (pre-diluted to 1/5). The cycling conditions were as it follows: 95°C for 10 min (1 cycle); 95°C for 30 min (40 cycles); 58°C for 1 min (40 cycles); 72°C for 1 min (40 cycles).
Intestinal inflammation assessment
Serum and distal ileal TNF-α, and fecal Lcn-2 were quantified using the mouse TNF-α ELISA Ready-SET-Go kit (eBioscience; San Diego, CA, USA) and the Mouse Lipocalin-2/NGAL DuoSet ELISA (R&D Systems) respectively, as per the manufacturer's instructions.
Endotoxin quantification
Serum lipopolysaccharide (LPS) levels were determined using a commercially available Limulus Amebocyte Lysate (LAL) assay kit (ToxinSensor™ Chromogenic LAL Endotoxin Assay kit; GenScript, Piscataway, NJ, USA), following the manufacturer's protocol. Absorbance for each sample was measured at 545 nm and the LPS concentration was determined as per the manufacturer's instructions (LPS concentration =0.2618 × absorbance at 545 nm −0.0012).
Statistical analysis
Triplicate samples were used for all assays and all experiments were repeated at least three times, using 5 mice in each group. The statistical significance among groups was determined using one-way analysis of variance (ANOVA), with multiple post-hoc comparisons using Tukey's test, or two-way ANOVA analysis, using Bonferroni post-hoc test, as indicated (Graphpad Software, La Jolla, CA, USA). A P<0.05 was considered statistically significant.
Results
P. aeruginosa infection dramatically increases the intestinal permeability following thermal injury
FITC-dextran has long and broadly been used as a reliable marker of intestinal permeability in mice (30,31). In our study, we used this macromolecule to determine the contribution of P. aeruginosa in the functional status of the intestinal barrier in the context of thermal injury. First, using a burn mouse model, we assessed the intestinal permeability of mice following burn, or burn plus P. aeruginosa infection (burn-infection, BI), at several time points, by assessing FITC-dextran 3–5 kDa flux from the intestinal lumen to the systemic circulation. Fig. 1 shows that in our model, gut permeability increases over time, reaching a peak of 1,714 ng/ml at 4 h following the induction of thermal injury, and then gradually returns to almost sham levels (472 ng/ml) by 18 h. However, mice that underwent post-burn infection with the P. aeruginosa clinical isolate PA14, exhibited a dramatically increased intestinal permeability compared to burn alone, thus, allowing larger volumes of FITC to flow out of the intestine (4,539 ng/ml in BI vs. 1,151 ng/ml in burn alone at 10 h following injury; P<0.001). Furthermore, BI mice exhibited a prolonged rise in gut permeability levels over time, with FITC concentration reaching 17,166 ng/ml by 18 h, indicating the strong impact of infection on the intestinal barrier dysfunction.
Inhibition of the P
aeruginosa QS transcription factor MvfR alleviates the deranged intestinal integrity following burn-site infection
Inhibition of MvfR activity in vivo has previously been linked to decreased virulence (24). To determine whether the in vivo inactivation of MvfR can also ameliorate the intestinal barrier dysfunction promoted by PA14 burn-site infection, we sought to investigate the FITC-dextran 3–5 kDa flux changes when using one of our original well-established MvfR antagonist, M64 (22–24). We assessed the FITC flux out of the intestine at 18–20 h, when the burn impact on gut permeability largely returned to the sham levels (as indicated by animals euthanized at 0 h, ie prior to burn, or BI), while strong impact of infection on the intestinal barrier dysfunction is observed (Fig. 1). Fig. 2A shows that FITC flow from the intestinal lumen to the systemic circulation was increased by PA14 in the BI group, compared to burn alone (P<0.001). Notably, FITC concentration in the serum was significantly decreased in mice treated with the anti-MvfR compound M64 (10,348 ng/ml), compared to non-treated animals (17,166 ng/ml; P<0.05). Thus, MvfR antagonist administration significantly protected the intestinal integrity in the setting of PA14 burn-site infection, rendering the gut barrier less permeable (Fig. 2A). The MvfR contribution on the intestinal integrity was validated in mice infected with the isogenic mvfR mutant, which we have previously reported that exerts reduced virulence and mortality in burnt and infected mice (19,24). Mice infected with the mvfR mutant exhibited considerably reduced FITC flux outside the intestinal lumen, compared to that promoted by the isogenic parental strain (P<0.001) (data not shown; available upon request).
Figure 2.

MvfR antagonists reduce the flow from the intestinal lumen to the systemic circulation, as well as the bacterial translocation to the MLNs. (A) FITC-dextran 3–5 kDa flow, and (B) bacterial translocation from the intestinal lumen to the systemic circulation at 18 h following burn and infection are considerably elevated in the BI group, while MvfR antagonist administration significantly reduces both FITC and bacterial flux out of the gut. FITC levels were assessed in the serum with fluorescent spectrophotometry (excitation, 480 nm and emission, 520 nm). LB agar plates were used for the bacterial CFUs assessment in the MLNs. Black bars, sham; Orange bars, burn; Red bars, PA14 burn-site infection (intradermal administration of 105 CFUs/animal); Blue bars, MvfR antagonist intravenous administration at 2, 4, 8 and 16 h following burn and infection. Data show the average +/- SEM (n=5). Statistical significance was assessed using one-way ANOVA + Tukey's post-hoc test. FITC-dextran, Fluorescein Isothiocyanate-Dextran; MLNs, mesenteric lymph nodes; BI, burn plus infection; CFU, colony forming units; LB, Luria Bertani; ANOVA, one-way analysis of variance. *P<0.05; ***P<0.001.
Intestinal hyperpermeability after burn injury has previously been correlated with increased bacterial translocation from the intestinal lumen to the MLNs (10). Therefore, we interrogated the impediment of this translocation after MvfR antagonist treatment, by assessing the bacterial CFUs in the MLNs. Fig. 2B shows that burn alone is responsible for an increase in bacterial translocation from the intestine compared to sham (P<0.001), while Pseudomonas burn-site infection further deregulates the function of the already defective intestinal barrier, thus doubling the number of bacterial colonies in the MLNs (P<0.0001). On the contrary, the MvfR antagonist demonstrates a marked reduction in bacterial MLN translocation compared to the BI group, with the number of CFUs being 1.5 log lower in the animals treated with the antagonist (P<0.001). These results corroborate our findings regarding the MvfR role in the functional status of the intestinal barrier, and further indicate that inhibition of MvfR confers significant protection to the intestinal integrity (Fig. 2B).
MvfR silencing improves morphological intestinal barrier features in BI mice
Key regulators of the intestinal barrier function are the TJ protein families. Burn-mediated dysregulation of these multiple-protein complexes that orchestrate the paracellular gut permeability, leads to barrier defects (8). Claudins, a family of transmembrane junctional proteins, seem to be crucial modulators of the intestinal barrier integrity (32). More specifically, some claudin family members are considered to be protective ‘tightening’ proteins, while others mostly contribute to intestinal permeability functions (33). We sought to evaluate the qualitative and quantitative changes of one of the ‘tightening’ claudin proteins, claudin-1, by exposure-matched confocal microscopy images and RT-qPCR analysis, respectively. Fig. 3Ab shows a marked decrease in staining for claudin-1 in the BI group, compared to sham animals (Fig. 3Aa), with a less even distribution at the areas of cell-cell contact and eliminated delineation of the cell periphery. On the contrary, MvfR inhibition attenuates this effect, as can be appreciated in Fig. 3Ac, where claudin-1 staining exhibits a more organized appearance at the periphery of the cells, with a more uniform localization at the sites of cell-cell interaction. Quantification of these results is shown in Fig. 3Ad, and it correlates well with our RT-qPCR data, as shown in Fig. 3B. PA14 burn wound infection significantly reduces claudin-1 mRNA levels, as compared to the sham and burn groups (P<0.01). Conversely, mice treated with MvfR antagonist display an augmented claudin-1 mRNA expression, in comparison to the BI group (P<0.05). These data indicate a considerable improvement in the morphology of the intestinal paracellular transport following MvfR silencing. Hence, the role of MvfR in the fate of this key intestinal permeability regulator becomes apparent in the setting of Pseudomonas burn wound infection.
Figure 3.

MvfR antagonists ameliorate the tight junction downregulation in BI mice. (A) Confocal microscopy images of distal ileum immunostaining for claudin-1 at 18 h following BI; (Aa). Representative image from sham animal depicting a uniform, organized pattern of claudin-1 staining at the periphery of the cells; (Ab). PA14 burn-site infection dysregulates the smooth, continuous distribution of claudin-1 and eliminates the cell periphery delineation; (Ac). Treatment with MvfR antagonist exhibits a more even localization of claudin-1 at the sites of cell-cell interaction, with less areas of claudin-1 breakdown. Size bar, 50 µm. (B) Quantification of confocal microscopy images. (C) RT-qPCR analysis of claudin-1 mRNA expression levels at 18 h post BI validates that PA14 burn wound infection downregulates claudin-1 expression, while MvfR inhibition alleviates this effect. Normalization of mRNA expression was performed for the reference gene TBP. Black bar, sham; Orange bar, burn; Red bar, PA14 burn-site infection (intradermal administration of 105 CFUs/animal); Blue bar, MvfR antagonist intravenous administration at 2, 4, 8 and 16 h following burn and infection. Data show the average +/- SEM (n=5). Statistical significance was assessed using one-way ANOVA + Tukey's post-hoc test. TBP, TATA-box Binding Protein; BI, burn plus infection; CFU, colony forming units; RT-qPCR, reverse transcription-quantitative polymerase chain reaction. *P<0.05; **P<0.01; ***P<0.001.
MvfR inhibition alleviates intestinal inflammation in BI mice
Following deregulation of the intestinal barrier integrity, microbial paracellular transport out of the lumen cues an inflammatory response from the intestinal mucosa (34). Similarly, mucosal inflammation is known to increase TJ permeability, further deranging the paracellular transport (35), thus leading to a vicious cycle of defective intestinal integrity. We therefore sought to determine changes in the levels of intestinal inflammation following BI, with and without MvfR inhibition. Fig. 4A demonstrates a sharp rise of the ileal TNF-α in the BI group (the mean TNF-α level was 58 pg/mg of ileal tissue compared to 11 pg/mg in the burn group; P<0.001), with a significant reduction achieved after treatment with the MvfR antagonist, going down to a mean TNF-α concentration of 38 pg/mg (P<0.01). Similarly, we investigated the changes in fecal Lcn-2, an acute phase protein that has proven to be a highly reliable marker of the inflammatory status in the intestinal lumen (36). Fecal Lcn-2 levels displayed a gradual time-related rise in both the BI and the antagonist groups (Fig. 4B), with the levels of this protein being significantly lower in the burn alone group at all three time points (4, 10 and 18 h following insult). Interestingly, there is a clearly disproportionate elevation of Lcn-2 levels in the BI animals over time, compared to mice treated with our MvfR antagonist. More specifically, while the two groups do not significantly differ at 4 h, there is a progressive difference at 10 (mean for the BI group was 13 ng/mg; mean for the antagonist group was 10 ng/mg; P<0.01) and 18 h (mean for the BI group was 19 ng/mg; mean for the antagonist group was 15 ng/mg; P<0.001). These variations in fecal Lcn-2 levels further support our observation that P. aeruginosa burn-site infection promotes, while MvfR inhibition significantly diminishes inflammation within the intestinal lumen, as shown in Fig. 4B. Taken together, our data support the contribution of MvfR in the elevated levels of intestinal inflammation post BI, as well as the significance of the inhibition of this QS system in this setting.
Figure 4.

The role of MvfR in augmenting the intestinal inflammation post BI. (A) shows TNF-α concentration from distal ileal tissue at 18 h post BI. Animals that received MvfR antagonist exhibit significantly reduced TNF-α levels in the ileum. (B) shows Lcn-2 concentrations from animal feces at 4, 10 and 18 h following BI. Lcn-2 levels increase with time in both BI and MvfR antagonist groups, however there is a disproportionate rise in the Lcn-2 levels compared to the MvfR inhibition group at 18 h. TNF-α and Lcn-2 levels were assessed using ELISA. Black bars, sham; Orange bars, burn; Red bars, PA14 burn-site infection (intradermal administration of 105 CFUs/animal); Blue bars, MvfR antagonist intravenous administration at 2, 4, 8 and 16 h following burn and infection. Data show the average +/- SEM (n=5). Statistical significance was assessed using one-way ANOVA + Tukey's post-hoc test in Fig. 5A and B, and two-way ANOVA + Bonferroni correction in Fig. 5C. TNF-α, tumor necrosis factor α; Lcn-2, lipocalin-2; BI, burn plus infection; ANOVA, one-way analysis of variance; CFU, colony formation unit. ***P<0.001.
MvfR inhibition mitigates the post-BI augmented systemic inflammation and bacterial systemic dissemination
The surge of the systemic inflammatory response to burn has been linked to circulating damage-associated molecular patterns (DAMPs) following thermal injury (37). Besides this, P. aeruginosa burn wound infection in rodent models has long been recognized to progress from wound colonization to hematogenous dissemination (38,39). Consistent with these data, in our study, superimposition of PA14 infection on thermal injury led to a significant rise of the systemic inflammatory response, as well as P. aeruginosa systemic dissemination. More specifically, all three systemic inflammatory indicators we studied, serum endotoxin levels, bacterial load in the systemic circulation, and TNF-α levels in the serum, were substantially elevated in the BI group compared to burn alone (all P-values were <0.001) (Fig. 5A-C). MvfR antagonist administration led to a significant reduction in both endotoxin levels (P<0.01) and bacterial CFUs number (P<0.001) (Fig. 5A and B). TNF-α reduction following MvfR inhibition failed to reach statistical significance (Fig. 5C). Nevertheless, TNF-α concentration still follows the same downward trend in the antagonist group, as observed in the case of endotoxin levels and bacterial load in the systemic circulation. The above findings were accompanied by a substantial rise in visceral PA14 load in the BI group compared to burn alone (P<0.001), as shown by the number of colonies in the ileal tissue in Fig. 5D. Notably, MvfR inhibition demonstrated a marked reduction of the PA14 CFUs in the ileum (P<0.001), indicating that inactivation of MvfR confers a reduced systemic bacterial dissemination, as expected. Taken together, these results suggest that this QS system is highly important for the rise of systemic inflammation and P. aeruginosa systemic dissemination following burn wound infection.
Figure 5.

MvfR inactivation ameliorates the post-BI elevated systemic inflammation and PA14 systemic dissemination. (A) Systemic serum endotoxin levels, (B) bacterial CFUs number, and (C) TNF-α concentration respectively, at 18 h following BI. A marked elevation of all three systemic inflammation indicators is demonstrated in the BI group. MvfR inhibition achieves a significant decrease of endotoxin levels and CFUs number in the serum. TNF-α concentration also follows the same downward trend in the MvfR inhibition group, even though it does not reach statistical significance. (D) Treatment with MvfR antagonist results in a significant decrease in PA14 CFUs in the ileum, compared to the BI group, thus representing the respective trends in PA14 dissemination from the burn wound to the intestine. Endotoxin levels were determined using a LAL assay; TNF-α was assessed using ELISA; LB agar plates were used for the bacterial CFUs assessment in the serum; LB agar plates containing 100 µg/ml rifampicin were used for the PA14 CFUs assessment in the ileal tissue. Black bars, sham; Orange bars, burn; Red bars, PA14 burn-site infection (intradermal administration of 105 CFUs/animal); Blue bars, MvfR antagonist intravenous administration at 2, 4, 8 and 16 h following burn and infection. Data show the average +/- SEM (n=5). Statistical significance was assessed using one-way ANOVA + Tukey's post-hoc test. EU, endotoxin units; CFU, colony formation unit; TNF-α, tumor necrosis factor α; LAL, Limulus Amebocyte Lysate; ANOVA, analysis of variance. **P<0.01; ***P<0.001.
Discussion
Despite advances in post-burn resuscitation, P. aeruginosa burn wound infection, often resistant to the typical antibiotic regimens, remains a constraining challenge following thermal injury. Identification of specific P. aeruginosa traits that contribute to the observed increase in mortality and morbidity following BI will expedite the development of novel, more effective therapeutic approaches. Previous studies have demonstrated that MvfR is a key regulator of multiple P. aeruginosa virulence factors (19,20,24). Our team has formulated a promising strategy against this opportunistic nosocomial pathogen, by developing and utilizing several potent anti-MvfR agents (21,22,24). We have previously reported that these robust MvfR-targeting agents successfully impede P. aeruginosa acute and chronic phenotypes, both in vitro and in vivo, including the mortality in thermally injured and infected mice and the ability of this pathogen to cause relapsing and persistent infection (21–24). The current study provides novel insights in the understanding of the MvfR role in the context of intestinal barrier dysregulation following P. aeruginosa burn wound infection. Indeed, the data presented here suggest that MvfR functions, in the context of BI, significantly contribute to the derangement of functional and morphological traits of the intestinal barrier. We also provide evidence that MvfR QS plays an important role in the post-BI modulation of intestinal and systemic inflammation, as well as in the bacterial systemic dissemination. In agreement with these findings, we have previously shown that P. aeruginosa dissemination in the blood stream is decreased as a result of MvfR regulon inhibition using anthranilic acid analogs (40). Importantly, we herein provide strong evidence that inhibition of MvfR QS with one of our potent anti-MvfR BB agents can ameliorate the aforementioned intestinal and systemic alterations following thermal injury and infection.
We initially investigated the time-related variations in intestinal integrity of mice following a 30% dorsal burn, or after BI. Through evaluation of FITC-dextran flux out of the intestine at different time points, we observed that the increase of intestinal permeability exhibits different patterns depending on the type of the insult. A previous study demonstrated that burn wound infection with P. aeruginosa (PSA 59-1244) led to an extended period of intestinal barrier dysfunction, compared to burn alone in Wistar rats (15). This report is consistent with our finding that while intestinal permeability returns to almost pre-insult levels at 18 h following burn alone, BI leads to prolonged barrier dysfunction. Interestingly, our study further indicates that BI leads to a dramatic exacerbation of the gut hyperpermeable state compared to burn alone. This is in contrast to the findings of Jones et al (15) who reported that bacterial translocation was comparable in the burn and BI groups. This difference could possibly be attributed to the type of animals used, mice vs. rats, or to the different P. aeruginosa strain used in our study. Indeed, the findings of a later study (13), utilizing a different strain of P. aeruginosa (P037, Fisher Immunotype-1), indicate that BI prompts a further increase in the intestinal permeability compared to burn alone, and are thus in unison with our present results. Moreover, a clinical study assessing the gut barrier function following burn, also suggests that thermally injured and infected patients exhibited higher levels of intestinal permeability (14).
In agreement with the role of the MvfR-regulon in P. aeruginosa virulence, our results provide evidence that MvfR has a significant contribution to the intestinal permeability alterations following the dual BI insult. We herein demonstrate for the first time a robust relation between the inhibition of a crucial P. aeruginosa QS system, and the regulation of the intestinal integrity following thermal injury and burn wound infection. Our results provide strong evidence that MvfR silencing leads to a less prominent induction of gut barrier damage and lower levels of systemic inflammation. Therefore, these data further suggest a key role for MvfR in the regulation of intestinal permeability following burn and P. aeruginosa wound infection.
A plethora of mechanisms may be involved in the gut barrier function changes observed post BI. Alterations in morphological and functional elements of the barrier have been implicated. Pathological intestinal epithelial cell shedding and subsequent disruption of the intestinal barrier have previously been demonstrated in hyperinflammatory states (41), while systemic endotoxin seems to play a crucial role in the observed gut injury (42). Similarly, we found higher levels of systemic endotoxin to occur in association with augmented intestinal hyperpermeability following BI, with MvfR inhibition diminishing both of these findings. Additionally, disrupted TJ integrity following burn has been demonstrated in animal models (8,43,44). In our present study, claudin-1 was found to be significantly dysregulated following BI, while MvfR inhibition attenuated this effect. What is more, cytokine levels such as interferon-γ (INF-γ) and TNF-α have been implicated in the regulation of paracellular permeability in the gut (45,46). These data, along with our finding of elevated TNF-α levels in the BI group suggest that this cytokine may play a causative role in the deterioration of the intestinal barrier function. Finally, microbial dysbiosis in the gut following burn has recently been shown to precipitate the derangement of the intestinal permeability (10).
Whatever the mechanism may be, collectively, our study reveals an intriguing association between P. aeruginosa QS inhibition and post-BI intestinal barrier function. This finding highlights the importance of MvfR in this setting and offers the potential to enrich the therapeutic armamentarium against P. aeruginosa burn-site infections with novel and effective virulence-targeting antimicrobial compounds that we continue to improve, and that could either be used in combination with antibiotics or as monotherapy. This would be particularly important in the context of thermal injuries, where infections with multi-drug resistant P. aeruginosa strains are very frequent. Follow-up studies could interrogate whether our compounds attenuate the deranged intestinal function by exerting additional effects on the host, or on the commensal bacteria populations residing the gut, thus directly modulating the molecular components of intestinal dysfunction. Implementation of clinical investigations to translate our animal-study findings to patients will be an important future direction of our research efforts.
Acknowledgements
Not applicable.
Glossary
Abbreviations
- QS
quorum sensing
- FITC-Dextran
Fluorescein Isothiocyanate-Dextran
- MLNs
mesenteric lymph nodes
- TJs
tight junctions
- ROS
reactive oxygen species
- TNF-α
tumor necrosis factor α
- BB
benzamine benzimidazole
- SPF
specific pathogen-free
- MGH
Massachusetts General Hospital
- IACUC
Institutional Animal Care and Use Committee
- PA14
UCBPP-PA14
- LB
Luria Bertani
- IP
Intraperitoneal
- TBSA
total body surface area
- N/S
normal saline
- CFUs
colony forming units
- BI
burn plus infection
- PBS
phosphate-buffered saline
- DMSO
dimethylsulfoxide
- Lcn-2
lipocalin-2
- RT-qPCR
reverse transcription-quantitative polymerase chain reaction
- TBP
TATA-box Binding Protein
- LPS
lipopolysaccharide
- LAL
Limulus Amebocyte Lysate
- ANOVA
analysis of variance
- DAMPs
damage-associated molecular patterns
- INF-γ
interferon-γ
Funding
The present study was supported by NIAID grants AI063433 and AI105902 and was supported by a Shriners Postdoctoral Research Fellowship from the Shriners Hospitals for Children, Boston (grant no. 84313).
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Authors' contributions
FA undertook the acquisition and analysis/interpretation of data, critical revision and final approval of the manuscript. MA performed analysis/interpretation of data, manuscript preparation, critical revision and final approval of the manuscript. MHG was responsible for the acquisition and analysis/interpretation of data, critical revision and final approval of the manuscript. MN also undertook the acquisition and analysis/interpretation of data, critical revision and final approval of the manuscript. RAH made substantial contribution to study conception and design, critical revision and final approval of the manuscript, and LGR also made substantial contribution to study conception and design, critical revision and gave final approval of the manuscript.
Ethics approval and consent to participate
Animal protocols were reviewed and approved by the IACUC at the MGH (protocol no. 2006N000093) and are in strict accordance to the guidelines of the Committee on Animals of the MGH, Harvard Medical School, and the regulations of the Subcommittee on Research Animal Care of the MGH and the National Institutes of Health. Animals were euthanized according to the guidelines of the Animal Veterinary Medical Association. All efforts were made to minimize suffering.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Groschwitz KR, Hogan SP. Intestinal barrier function: Molecular regulation and disease pathogenesis. J Allergy Clin Immunol. 2009;124:3–20. doi: 10.1016/j.jaci.2009.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Muniz LR, Knosp C, Yeretssian G. Intestinal antimicrobial peptides during homeostasis, infection, and disease. Front Immunol. 2012;3:310. doi: 10.3389/fimmu.2012.00310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Van Spaendonk H, Ceuleers H, Witters L, Patteet E, Joossens J, Augustyns K, Lambeir AM, De Meester I, De Man JG, De Winter BY. Regulation of intestinal permeability: The role of proteases. World J Gastroenterol. 2017;23:2106–2123. doi: 10.3748/wjg.v23.i12.2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Forjuoh SN. Burns in low- and middle-income countries: A review of available literature on descriptive epidemiology, risk factors, treatment, and prevention. Burns. 2006;32:529–537. doi: 10.1016/j.burns.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 5.Ryan CM, Yarmush ML, Burke JF, Tompkins RG. Increased gut permeability early after burns correlates with the extent of burn injury. Crit Care Med. 1992;20:1508–1512. doi: 10.1097/00003246-199211000-00005. [DOI] [PubMed] [Google Scholar]
- 6.Nielson CB, Duethman NC, Howard JM, Moncure M, Wood JG. Burns: Pathophysiology of systemic complications and current management. J Burn Care Res. 2017;38:e469–ee481. doi: 10.1097/BCR.0000000000000355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wolf SE, Ikeda H, Matin S, Debroy MA, Rajaraman S, Herndon DN, Thompson JC. Cutaneous burn increases apoptosis in the gut epithelium of mice. J Am Coll Surg. 1999;188:10–16. doi: 10.1016/S1072-7515(98)00260-9. [DOI] [PubMed] [Google Scholar]
- 8.Costantini TW, Loomis WH, Putnam JG, Drusinsky D, Deree J, Choi S, Wolf P, Baird A, Eliceiri B, Bansal V, Coimbra R. Burn-induced gut barrier injury is attenuated by phosphodiesterase inhibition: Effects on tight junction structural proteins. Shock. 2009;31:416–422. doi: 10.1097/SHK.0b013e3181863080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fazal N, Shelip A, Alzahrani AJ. Burn-injury affects gut-associated lymphoid tissues derived CD4+ T cells. Results Immunol. 2013;3:85–94. doi: 10.1016/j.rinim.2013.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Earley ZM, Akhtar S, Green SJ, Naqib A, Khan O, Cannon AR, Hammer AM, Morris NL, Li X, Eberhardt JM, et al. Burn injury alters the intestinal microbiome and increases gut permeability and bacterial translocation. PLoS One. 2015;10:e0129996. doi: 10.1371/journal.pone.0129996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Noble EE, Hsu TM, Kanoski SE. Gut to brain dysbiosis: Mechanisms linking western diet consumption, the microbiome, and cognitive impairment. Front Behav Neurosci. 2017;11:9. doi: 10.3389/fnbeh.2017.00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li N, Hu X, Liu Y, Wang Y, Wang Y, Liu J, Cai W, Bai X, Zhu X, Han J, Hu D. Systemic inflammatory responses and multiple organ dysfunction syndrome following skin burn wound and Pseudomonas aeruginosa infection in mice. Shock. 2013;40:152–159. doi: 10.1097/SHK.0b013e31829aef41. [DOI] [PubMed] [Google Scholar]
- 13.Ryan CM, Bailey SH, Carter EA, Schoenfeld DA, Tompkins RG. Additive effects of thermal injury and infection on gut permeability. Arch Surg. 1994;129:325–328. doi: 10.1001/archsurg.1994.01420270103021. [DOI] [PubMed] [Google Scholar]
- 14.Ziegler TR, Smith RJ, O'Dwyer ST, Demling RH, Wilmore DW. Increased intestinal permeability associated with infection in burn patients. Arch Surg. 1988;123:1313–1319. doi: 10.1001/archsurg.1988.01400350027003. [DOI] [PubMed] [Google Scholar]
- 15.Jones WG, II, Barber AE, Minei JP, Fahey TJ, III, Shires GT, III, Shires GT. Differential pathophysiology of bacterial translocation after thermal injury and sepsis. Ann Surg. 1991;214:24–30. doi: 10.1097/00000658-199107000-00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yan S, Tsurumi A, Que YA, Ryan CM, Bandyopadhaya A, Morgan AA, Flaherty PJ, Tompkins RG, Rahme LG. Prediction of multiple infections after severe burn trauma: A prospective cohort study. Ann Surg. 2015;261:781–792. doi: 10.1097/SLA.0000000000000759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alp E, Coruh A, Gunay GK, Yontar Y, Doganay M. Risk factors for nosocomial infection and mortality in burn patients: 10 years of experience at a university hospital. J Burn Care Res. 2012;33:379–385. doi: 10.1097/BCR.0b013e318234966c. [DOI] [PubMed] [Google Scholar]
- 18.Lister PD, Wolter DJ, Hanson ND. Antibacterial-resistant Pseudomonas aeruginosa: Clinical impact and complex regulation of chromosomally encoded resistance mechanisms. Clin Microbiol Rev. 2009;22:582–610. doi: 10.1128/CMR.00040-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Déziel E, Gopalan S, Tampakaki AP, Lépine F, Padfield KE, Saucier M, Xiao G, Rahme LG. The contribution of MvfR to Pseudomonas aeruginosa pathogenesis and quorum sensing circuitry regulation: Multiple quorum sensing-regulated genes are modulated without affecting lasRI, rhlRI or the production of N-acyl-L-homoserine lactones. Mol Microbiol. 2005;55:998–1014. doi: 10.1111/j.1365-2958.2004.04448.x. [DOI] [PubMed] [Google Scholar]
- 20.Maura D, Hazan R, Kitao T, Ballok AE, Rahme LG. Evidence for direct control of virulence and defense gene circuits by the Pseudomonas aeruginosa quorum sensing regulator, MvfR. Sci Rep. 2016;6:34083. doi: 10.1038/srep34083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Allegretta G, Maurer CK, Eberhard J, Maura D, Hartmann RW, Rahme L, Empting M. In-depth profiling of MvfR-regulated small molecules in Pseudomonas aeruginosa after quorum sensing inhibitor treatment. Front Microbiol. 2017;8:924. doi: 10.3389/fmicb.2017.00924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maura D, Drees SL, Bandyopadhaya A, Kitao T, Negri M, Starkey M, Lesic B, Milot S, Déziel E, Zahler R, et al. Polypharmacology approaches against the Pseudomonas aeruginosa MvfR regulon and their application in blocking virulence and antibiotic tolerance. ACS Chem Biol. 2017;12:1435–1443. doi: 10.1021/acschembio.6b01139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Maura D, Rahme LG. Pharmacological inhibition of the Pseudomonas aeruginosa MvfR Quorum-sensing system interferes with biofilm formation and potentiates antibiotic-mediated biofilm disruption. Antimicrob Agents Chemother. 2017;61(pii):e01362–17. doi: 10.1128/AAC.01362-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Starkey M, Lepine F, Maura D, Bandyopadhaya A, Lesic B, He J, Kitao T, Righi V, Milot S, Tzika A, Rahme L. Identification of anti-virulence compounds that disrupt quorum-sensing regulated acute and persistent pathogenicity. PLoS Pathog. 2014;10:e1004321. doi: 10.1371/journal.ppat.1004321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rahme LG, Stevens EJ, Wolfort SF, Shao J, Tompkins RG, Ausubel FM. Common virulence factors for bacterial pathogenicity in plants and animals. Science. 1995;268:1899–1902. doi: 10.1126/science.7604262. [DOI] [PubMed] [Google Scholar]
- 26.Walker HL, Mason AD., Jr A standard animal burn. J Trauma. 1968;8:1049–1051. doi: 10.1097/00005373-196811000-00006. [DOI] [PubMed] [Google Scholar]
- 27.Pasternak AJ, Hamonic GM, Van Kessel A, Wilson HL. Postnatal regulation of MAMDC4 in the porcine intestinal epithelium is influenced by bacterial colonization. Physiol Rep. 2016;4(pii):e13018. doi: 10.14814/phy2.13018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yu L, Zhai Q, Tian F, Liu X, Wang G, Zhao J, Zhang H, Narbad A, Chen W. Potential of Lactobacillus plantarum CCFM639 in protecting against aluminum toxicity mediated by intestinal barrier function and oxidative stress. Nutrients. 2016;8(pii):E783. doi: 10.3390/nu8120783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 30.Cani PD, Bibiloni R, Knauf C, Waget A, Neyrinck AM, Delzenne NM, Burcelin R. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes. 2008;57:1470–1481. doi: 10.2337/db07-1403. [DOI] [PubMed] [Google Scholar]
- 31.Volynets V, Reichold A, Bardos G, Rings A, Bleich A, Bischoff SC. Assessment of the intestinal barrier with five different permeability tests in healthy C57BL/6J and BALB/cJ mice. Dig Dis Sci. 2016;61:737–746. doi: 10.1007/s10620-015-3935-y. [DOI] [PubMed] [Google Scholar]
- 32.Gunzel D, Yu AS. Claudins and the modulation of tight junction permeability. Physiol Rev. 2013;93:525–569. doi: 10.1152/physrev.00019.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Markov AG, Veshnyakova A, Fromm M, Amasheh M, Amasheh S. Segmental expression of claudin proteins correlates with tight junction barrier properties in rat intestine. J Comp Physiol B. 2010;180:591–598. doi: 10.1007/s00360-009-0440-7. [DOI] [PubMed] [Google Scholar]
- 34.Michielan A, D'Inca R. Intestinal permeability in inflammatory bowel disease: Pathogenesis, clinical evaluation, and therapy of leaky gut. Mediators Inflamm. 2015;2015:628157. doi: 10.1155/2015/628157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bruewer M, Luegering A, Kucharzik T, Parkos CA, Madara JL, Hopkins AM, Nusrat A. Proinflammatory cytokines disrupt epithelial barrier function by apoptosis-independent mechanisms. J Immunol. 2003;171:6164–6172. doi: 10.4049/jimmunol.171.11.6164. [DOI] [PubMed] [Google Scholar]
- 36.Chassaing B, Srinivasan G, Delgado MA, Young AN, Gewirtz AT, Vijay-Kumar M. Fecal lipocalin 2, a sensitive and broadly dynamic non-invasive biomarker for intestinal inflammation. PLoS One. 2012;7:e44328. doi: 10.1371/journal.pone.0044328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, Brohi K, Itagaki K, Hauser CJ. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. 2010;464:104–107. doi: 10.1038/nature08780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Teplitz C, Davis D, Mason AD, Jr, Moncrief JA. Pseudomonas burn wound sepsis. I pathogenesis of experimental Pseudomonas burn wound sepsis. J Surg Res. 1964;4:200–216. doi: 10.1016/S0022-4804(64)80026-3. [DOI] [PubMed] [Google Scholar]
- 39.Teplitz C, Davis D, Walker HL, Raulston GL, Mason AD, Jr, Moncrief JA. Pseudomonas burn wound sepsis. Ii hematogenous infection at the junction of the burn wound and the unburned hypodermis. J Surg Res. 1964;4:217–222. doi: 10.1016/S0022-4804(64)80027-5. [DOI] [PubMed] [Google Scholar]
- 40.Lesic B, Lépine F, Déziel E, Zhang J, Zhang Q, Padfield K, Castonguay MH, Milot S, Stachel S, Tzika AA, et al. Inhibitors of pathogen intercellular signals as selective anti-infective compounds. PLoS Pathog. 2007;3:1229–1239. doi: 10.1371/journal.ppat.0030126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Williams JM, Duckworth CA, Burkitt MD, Watson AJ, Campbell BJ, Pritchard DM. Epithelial cell shedding and barrier function: A matter of life and death at the small intestinal villus tip. Vet Pathol. 2015;52:445–455. doi: 10.1177/0300985814559404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Williams JM, Duckworth CA, Watson AJ, Frey MR, Miguel JC, Burkitt MD, Sutton R, Hughes KR, Hall LJ, Caamaño JH, et al. A mouse model of pathological small intestinal epithelial cell apoptosis and shedding induced by systemic administration of lipopolysaccharide. Dis Model Mech. 2013;6:1388–1399. doi: 10.1242/dmm.013284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cannon AR, Akhtar S, Hammer AM, Morris NL, Javorski MJ, Li X, Kennedy RH, Gamelli RL, Choudhry MA. Effects of mesalamine treatment on gut barrier integrity after burn injury. J Burn Care Res. 2016;37:283–292. doi: 10.1097/BCR.0000000000000396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Samonte VA, Goto M, Ravindranath TM, Fazal N, Holloway VM, Goyal A, Gamelli RL, Sayeed MM. Exacerbation of intestinal permeability in rats after a two-hit injury: Burn and Enterococcus faecalis infection. Crit Care Med. 2004;32:2267–2273. doi: 10.1097/01.CCM.0000145579.66001.05. [DOI] [PubMed] [Google Scholar]
- 45.Capaldo CT, Nusrat A. Cytokine regulation of tight junctions. Biochim Biophys Acta. 2009;1788:864–871. doi: 10.1016/j.bbamem.2008.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ma TY, Iwamoto GK, Hoa NT, Akotia V, Pedram A, Boivin MA, Said HM. TNF-alpha-induced increase in intestinal epithelial tight junction permeability requires NF-kappa B activation. Am J Physiol Gastrointest Liver Physiol. 2004;286:G367–G376. doi: 10.1152/ajpgi.00173.2003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.