

Abstract
Chronic hypoxia can be observed in the heart under physiological or pathophysiological states, including embryonic development or cyanotic congenital heart disease. The aim of the present study was to examine gene expression profiles of chronically hypoxic myocardium and to explore the pathophysiological mechanisms by which the heart adapts to chronic hypoxia. Raw data from the next-generation sequencing data set GSE36761 were downloaded from the Gene Expression Omnibus database. The data set comprised 30 specimens, including 8 healthy myocardia and 22 tetralogy of Fallot (TOF) congenital cardiac malformations; only 7 original data sets of healthy myocardia were obtained, and 5/22 TOFs were excluded. Gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses of differentially expressed genes (DEGs) were performed. Furthermore, network analysis of DEGs using Cytoscape software based on protein-protein interaction (PPI) data was also conducted. A total of 1,260 DEGs were selected, of which 926 DEGs were enriched in 83 GO biological process terms, including extracellular matrix organization, regeneration and monocyte chemotaxis. Furthermore, 406 DEGs were enriched in 13 KEGG pathways, including cytokine-cytokine receptor interaction, focal adhesion and apoptosis. PPI network analysis indicated that six hub genes with correlated degree scores >25 among nodes were identified, including G protein subunit β4, C-C motif chemokine receptor (CCR)1, CCR2, platelet factor 4, catenin β1 and Jun proto-oncogene (JUN). Of these, JUN was enriched in GO terms of regeneration and neuron projection regeneration, and in KEGG pathways of focal adhesion, apoptosis and Chagas disease (American trypanosomiasis). The present bioinformatics analysis of these DEGs and hub genes may provide a molecular insight to the role of diverse genes in the pathophysiology of chronically hypoxic myocardium and in myocardial adaptation to chronic hypoxia.
Keywords: bioinformatics analysis, chronic hypoxia, myocardium, next-generation sequencing, differentially expressed gene
Introduction
Chronic hypoxia is normal occurrence during embryonic development (1). In addition, it is the basic pathophysiological characteristic of individuals with cyanotic congenital heart disease (CHD) or those inhabitants who lived on the plateau for a long period of time (2,3). Circulation overload, chronic hypoxic stress and other complications on infants with cyanotic CHD results in mortality as high as 36.4% (4). Notably, it has been clinically observed that a number of infants with mild to moderate cyanosis have been able to survive for a prolonged period, including to adulthood. They tend to be well adapted to systematic hypoxia and typically demonstrate good cardiac function during the perioperative period of cardiac surgery (5). The observation suggested that the heart may respond to chronic hypoxia, with some protective adaptation. In addition, previous studies have demonstrated that the myocardium may develop a set of endogenous protective mechanisms to attenuate cardiomyocyte apoptosis and to promote potential proliferation during chronic hypoxia (6–8). However, this ability to replenish the lost cardiomyocytes during infarct or in cardiac overload disorders is extremely limited in adults (9). The molecular mechanisms underlying the hypoxic myocardial injury and adaptation remain unclear, which limits clinical treatment of myocardial hypoxia, ischemia and infarction. Therefore, exploring and understanding the molecular mechanisms may aid in providing protection against chronic hypoxic-ischemic heart disease, including coronary heart disease and cyanotic CHD.
Microarray and high-throughput sequencing have been widely used in the diagnosis and treatment of diseases. Among them, next-generation sequencing (NGS) technology is frequently applied to assess cardiovascular diseases. Notably, NGS can be used to identify potential genetic mutations in hereditary heart diseases, analyze transcriptome changes, screen biological targets of disease and identify molecular pathways involved in cardiovascular development and disease (10). The vast amount of data produced by the high-throughput platforms rely on analysis and interpretation using bioinformatics, which is a cutting-edge interdisciplinary approach that links biology, computer science and information technology. Accordingly, the combination of NGS technologies and bioinformatics has made data processing and data mining a reality, and has been extensively applied in the analysis of biological sequences, genome, transcriptome, proteome, metabolome and systems biology (11). Grunert et al (12) performed a comparative study between myocardial specimens of healthy organ donors and patients with tetralogy of Fallot (TOF) congenital cardiac malformations by DNA-targeted resequencing and mRNA-sequencing technology. The study examined the genetic origin and variation of TOF, but did not elucidate the biological significance of these differential expressed genes (DEGs) under chronically hypoxic conditions.
TOF is the most common cyanotic CHD, and chronic hypoxia is an important pathophysiological feature of this disease, in addition to genetic variations and hemodynamic abnormalities (13). Thus, based on the raw NGS data (GSE36761) uploaded by Grunert et al (12) to the Gene Expression Omnibus (GEO) database, the present study identified DEGs in TOF myocardium compared with healthy specimens. Furthermore, Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses on these DEGs were performed. The present findings may provide indications to molecular mechanisms underlying the injury and adaptation of chronically hypoxic cardiomyocytes.
Materials and methods
NGS data
Raw NGS data (GSE36761) were downloaded from GEO (http://www.ncbi.nlm.nih.gov/geo; downloaded on April 15, 2017); the data are in the Sequence Read Archive (SRA) format. These data, which included 8 healthy myocardia and 22 TOFs, were subjected to single-end read sequencing, which was performed on the GPL9052 platform [Illumina Genome Analyzer (Homo sapiens)]. Only 7 original data sets of the healthy specimens were obtained from the GEO database.
Data processing and identification of DEGs
The SRA files were converted into fastq using fastq-dump in the sratoolkit version 2.8.2 (http://www.ncbi.nlm.nih.gov/sra). The data were subjected to quality control using fastqc (http://www.bioinformatics.babraham.ac.uk/projects/fastqc) in the Ubuntu version 16.04 LTS system (http://www.ubuntu.com). The adapter and null base sequences were removed with fastx_clipper and the low-quality reads were filtered using a fastq_quality_filter in the FASTX toolkit version 0.0.14 (http://hannonlab.cshl.edu/fastx_toolkit), followed by further quality control checks. In addition, the differential analysis of the preprocessed data was conducted using the pipeline of hierarchical indexing for spliced alignment of transcripts (HISAT2)-high-throughput sequencing (HTSeq)-differential expression sequencing (DESeq2) (Fig. 1) (14). First, raw reads were aligned to the University of California Santa Cruz (UCSC; http://genome.ucsc.edu) human reference genome (hg38) using the splicing aligner HISAT2 v2.0.5 (http://ccb.jhu.edu/software/hisat2/index.shtml), which generated Sequence Alignment Map (SAM) formatted data. HISAT2 is a highly efficient system for aligning reads from RNA sequencing experiments. It is the fastest system currently available, with less memory usage and equal or better accuracy than other methods. The command ‘grep’ was used to select the uniquely aligned reads, and the SAM data was subsequently converted to Binary Alignment Map (BAM) format using samtools version 1.4 (http://www.htslib.org). The aligned reads were counted using HTSeq version 0.7.2 (http://htseq.readthedocs.io). The DESeq2 package in R version 3.5.0 (http://www.r-project.org) was used to convert the above count data to the data frame of gene expression in a Windows 7 system. Pearson's correlation coefficient was calculated, and the command ‘hclust’ was applied in the hierarchical clustering analysis of the unweighted pair group method with arithmetic mean (UPGMA). DESeq2 was subsequently used for differential expression analysis (P<0.05; fold changes >2), following the removal of the discrete specimens. The heat map and volcano plot of the DEGs were plotted using pheatmap and ggplot2, respectively.
Figure 1.
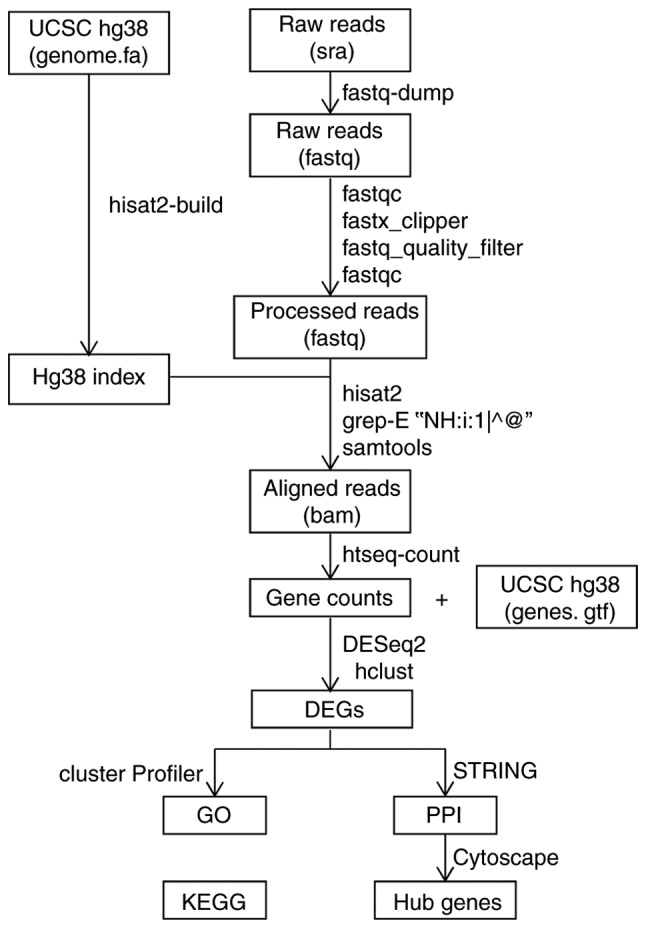
Bioinformatics pipeline for next-generation sequencing. BAM, Binary Alignment Map data format; DEGs, differentially expressed genes; DESeq2, differential expression sequencing; GO, Gene Ontology; hg38, human reference genome; HISAT2, hierarchical indexing for spliced alignment of transcripts; HTSeq, high-throughput sequencing; KEGG, Kyoto Encyclopedia of Genes and Genomes; PPI, protein-protein interaction; SAM, Sequence Alignment Map data format; SAR, Sequence Read Archive; UCSC, University of California Santa Cruz.
GO and KEGG pathway enrichment analyses of DEGs
GO is a bioinformatics program that develops a common language to annotate and classify the biological functions of genes and their products of all species (15). It is divided into three categories: Molecular function, cellular component and biological process (BP). The present study focused on the biological process of these DEGs, thus only the BP terms were analyzed. KEGG is a database that systematically analyzes gene functions and associates genes with different biological signaling pathways for annotation (16). Gene symbols were converted into Entrez IDs by the ‘org.Hs.eg.db’ package in R, and GO_BP and KEGG pathway enrichment analyses of these DEGs were performed using ‘clusterProfiler’ package in R using the following parameters: Adjusted P-values and q-values, both <0.05, and the minimum number of genes enriched for each signal pathway was >10 (17).
Protein-protein interaction (PPI) and network analyses of DEGs
Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) is a database and online analysis software for studying known and predicted PPIs (18). STRING (version 10.0; accessed on May 15, 2018) contains 9.6 million pieces of protein information from 2,031 species, and information on 1.38 billion PPIs. To evaluate the interactions between DEGs, gene symbols of DEGs were inputted to STRING, the confidence was set at 0.7 and the source of PPI was set as default, including text mining, experiments, databases, co-expression, neighborhood, gene fusion and co-occurrence. The obtained PPI data were entered into the Cytoscape version 3.5.0 (http://cytoscape.org) network analyzer, and the degrees of association between a node (gene) and neighboring nodes (genes) were calculated with the NetworkAnalyzer Tool.
Results
Identification of DEGs
A total of 29 myocardial specimens were analyzed, including 7 healthy myocardia (Control) and 22 TOFs (Fig. 2A). Following data cleaning, all specimens qualified, and through hierarchical clustering analysis, five discrete specimens from the TOF group were excluded, including SRR448095, SRR448104, SRR448105, SRR448112 and SRR448113. Following the two cluster analyses, 7 specimens were included in the Control group and 17 specimens were included in TOF group (Fig. 2B). Differential analysis using DESeq2 revealed that a total of 1,260 genes, including 494 upregulated and 766 downregulated genes, exhibited differential expression by over 2-fold. The top 25 upregulated and downregulated genes were indicated on the heat map according to the fold changes (Fig. 3), and all DEGs were plotted on a volcano plot (Fig. 4).
Figure 2.

Cluster analysis. (A) The first cluster analysis was performed in all the 29 original specimens. (B) A second cluster analysis was performed following exclusion of five discrete specimens by the first clustering.
Figure 3.

Heat map of the top 50 differentially expressed genes. In the heatmap, red indicates upregulation and green indicates downregulation of gene expression levels.
Figure 4.

Volcano plot of 1,260 differentially expressed genes. Red, FC >2 and P<0.05; black, FC <2 and P>0.05. FC, fold change.
GO and KEGG pathway enrichment analyses
GO_BP and KEGG pathway enrichment analyses were performed on the 1,260 genes using ‘clusterProfiler’ to understand the primary enrichment of these DEGs in BPs and signaling pathways. A total of 926 DEGs were enriched in 83 GO_BP terms, including extracellular matrix (ECM) organization, neutrophil activation, extracellular structure organization, neutrophil degranulation, neutrophil mediated immunity, neutrophil activation involved in the immune response, leukocyte migration, regulation of blood circulation, negative regulation of cell adhesion, cell-substrate adhesion, mononuclear cell migration, regeneration, monocyte chemotaxis, neuron projection regeneration and respiratory burst (Table I). Furthermore, a total of 406 DEGs were enriched in 13 KEGG pathways, including cytokine-cytokine receptor interaction, focal adhesion, chemokine signaling pathway, phagosome, Hippo signaling pathway, apoptosis, Chagas disease (American trypanosomiasis), ECM-receptor interaction, protein digestion and absorption, pathogenic Escherichia coli infection, complement and coagulation cascades, mineral absorption and basal cell carcinoma (Table II). The top 15 GO_BP terms and all 13 KEGG pathways were graphically demonstrated according to the P- and q-values (Figs. 5 and 6, respectively).
Table I.
GO BP analysis of differentially expressed genes in hypoxic myocardium.
| GO_BP ID | Description | Gene ratio | P-value |
|---|---|---|---|
| GO:0030198 | Extracellular matrix organization | 42/926 | 2.40×10−07 |
| GO:0042119 | Neutrophil activation | 54/926 | 3.35×10−07 |
| GO:0043062 | extracellular structure organization | 46/926 | 3.36×10−07 |
| GO:0043312 | Neutrophil degranulation | 53/926 | 3.37×10−07 |
| GO:0002446 | Neutrophil mediated immunity | 54/926 | 3.80×10−07 |
| GO:0002283 | Neutrophil activation involved in immune response | 53/926 | 4.09×10−07 |
| GO:0050900 | Leukocyte migration | 48/926 | 7.90×10−06 |
| GO:1903522 | Regulation of blood circulation | 34/926 | 9.80×10−06 |
| GO:0007162 | Negative regulation of cell adhesion | 31/926 | 1.16×10−05 |
| GO:0031589 | Cell-substrate adhesion | 36/926 | 1.27×10−05 |
| GO:0071674 | Mononuclear cell migration | 15/926 | 1.29×10−05 |
| GO:0031099 | Regeneration | 25/926 | 1.45×10−05 |
| GO:0002548 | Monocyte chemotaxis | 13/926 | 1.58×10−05 |
| GO:0031102 | Neuron projection regeneration | 12/926 | 2.66×10−05 |
| GO:0045730 | Respiratory burst | 9/926 | 2.72×10−05 |
BP, biological proces; GO, Gene Ontology.
Table II.
KEGG pathway analysis of differentially expressed genes in hypoxic myocardium.
| KEGG ID | Description | Gene ratio | P-value | Genes |
|---|---|---|---|---|
| hsa05130 | Pathogenic Escherichia coli infection | 12/406 | 3.09×10−5 | WAS, ACTB, TUBA4A, CTNNB1, TLR5, ARPC1B, TUBA3C, TUBA1B, TUBB4B, TUBA1C, TUBA3D and TUBA3E |
| hsa04510 | Focal adhesion | 24/406 | 2.00×10−4 | COL1A1, COL4A3, BCL2, VTN, ITGA11, ACTB, COL6A6, BIRC3, CAV2, PIK3R1, CTNNB1, COL9A2, TNC, ITGA3, JUN, MAP2K1, RAC2, VASP, ITGA10, LAMC3, SHC3, MYL7, TNN and COL9A1 |
| hsa04145 | Phagosome | 20/406 | 2.11×10−4 | C3, CYBA, MPO, CYBB, ACTB, TUBA4A, ATP6V1B1, C1R, MRC1, MSR1, SFTPD, CTSS, TUBA3C, TLR6, TUBA1B, TUBB4B, MARCO, TUBA1C, TUBA3D and TUBA3E |
| hsa04978 | Mineral absorption | 10/406 | 3.60×10−4 | FTL, ATP1B3, SLC31A1, HMOX1, MT1A, MT1X, MT2A, SLC26A9, ATP1A4 and MT1M |
| hsa04062 | Chemokine signaling pathway | 22/406 | 4.46×10−4 | JAK3, WAS, CCR2, PIK3R1, CCR1, PF4, PPBP, MAP2K1, RAC2, CCL2, CCL11, CCL17, CCL21, CXCL14, DOCK2, CCL13, CCL8, CCL26, CCL19, SHC3, NFKBIA and GNB4 |
| hsa04512 | ECM-receptor interaction | 13/406 | 4.52×10−4 | COL1A1, COL4A3, VTN, ITGA11, COL6A6, COL9A2, TNC, ITGA3, ITGA10, LAMC3, SV2C, TNN and COL9A1 |
| hsa04060 | Cytokine-cytokine receptor interaction | 30/406 | 6.49×10−4 | ACVRL1, AMH, IL10, GDF6, TNFRSF1B, CCR2, IL15RA, CCR1, BMP6, BMP8B, NGFR, TNFRSF11B, PF4, PPBP, CCL2, CCL11, CCL17, CCL21, TNFSF9, IL18RAP, CXCL14, CCL13, CCL8, CCL26, CCL19, BMP10, TNFRSF12A, IL17RB, BMP5 and CSF1 |
| hsa04610 | Complement and coagulation cascades | 12/406 | 1.10×10−3 | C3, C6, F13A1, F5, SERPIND1, C1QB, SERPINE1, BDKRB2, SERPINA5, VTN, C1R and F3 |
| hsa04974 | Protein digestion and absorption | 13/406 | 1.12×10−3 | COL1A1, COL3A1, COL4A3, COL6A6, ATP1B3, COL9A2, CPA3, XPNPEP2, KCNE3, SLC7A8, COL14A1, COL9A1 and ATP1A4 |
| hsa04210 | Apoptosis | 17/406 | 1.12×10−3 | BCL2, ACTB, BIRC3, PIK3R1, TUBA4A, CTSH, CTSC, JUN, MAP2K1, CTSS, TUBA3C, TUBA1B, GADD45G, NFKBIA, TUBA1C, TUBA3D and TUBA3E |
| hsa05142 | Chagas disease (American trypanosomiasis) | 14/406 | 1.22×10−3 | C3, C1QB, IL10, SERPINE1, BDKRB2, ACE, PIK3R1, GNA15, JUN, CCL2, TLR6, PPP2R2C, NFKBIA and GNAO1 |
| hsa04390 | Hippo signaling pathway | 18/406 | 1.76×10−3 | AMH, SERPINE1, GDF6, ACTB, DLG2, DVL1, CTNNB1, FZD2, BMP6, BMP8B, SNAI2, WNT9A, FZD1, FZD7, WNT6, PPP2R2C, BMP5 and NKD1 |
| hsa05217 | Basal cell carcinoma | 10/406 | 2.00×10−3 | DVL1, CTNNB1, FZD2, WNT9A, FZD1, FZD7, SMO, WNT6, GADD45G and HHIP |
ECM, extracellular matrix; Hsa, Homo sapiens; KEGG, Kyoto Encyclopedia of Genes and Genomes.
Figure 5.

Enrichment analysis of Gene Ontology BP terms. p.adjust (adjusted P-value): Red < purple < blue. Red boxes indicate the enrichment of JUN. BP, biological process.
Figure 6.

KEGG pathway enrichment analysis. Dot size represents the number of genes in each KEGG pathway; p.adjust (adjusted P-value): Red < purple < blue. Red boxes indicate the enrichment of JUN. ECM, extracellular matrix; KEGG, Kyoto Encyclopedia of Genes and Genomes.
Network analysis from the PPI data
Based on the data from PPI analysis, six hub genes with correlated degree scores >25 were identified: G protein subunit β 4 (GNB4), C-C motif chemokine receptor (CCR)2, CCR1, platelet factor 4 (PF4), catenin β1 (CTNNB1) and Jun proto-oncogene (JUN), although the fold changes of these genes did not reach the top 25 genes in the heat map (Fig. 3). Notably, CTNNB1 and JUN were upregulated (Table III; Fig. 7). Following GO and KEGG analyses, JUN was indicated to be primarily enriched in the GO_BP terms of regeneration and neuronal projection regeneration, and in the signaling pathways of focal adhesion, apoptosis and Chagas disease (American trypanosomiasis; Figs. 5 and 6).
Table III.
Hub genes identified by Cytoscape network analysis.
| Hub gene | Degree | Regulation |
|---|---|---|
| JUN | 31 | Up |
| CTNNB1 | 26 | Up |
| GNB4 | 32 | Down |
| CCR2 | 27 | Down |
| CCR1 | 25 | Down |
| PF4 | 25 | Down |
CCR, C-C motif chemokine receptor; CTNNB1, catenin β1; GNB4, G protein subunit β4; JUN, Jun proto-oncogene; PF4, platelet factor 4.
Figure 7.

Protein-protein interaction data network of JUN and CTNNB1. Red, upregulation; green, downregulation. Color depth and size of the dots were proportional to the fold change and correlated degree, respectively; thickness of the line between two nodes indicated the connection strength positively. CTNNB1, catenin β1; JUN, Jun proto-oncogene.
Discussion
Chronic hypoxia is the basic pathophysiological state of cyanotic CHD; the heart forms endogenous mechanisms to protect cardiomyocytes against injuries caused by hypoxia (6–8). To identify the effects of chronic hypoxia on the heart, the present study analyzed DEGs in chronically hypoxic myocardium. NGS data was extracted from the GSE36761 data set, and 1,260 DEGs between healthy and chronically hypoxic myocardium were identified using bioinformatics. The DEGs that co-existed or were co-expressed under chronically hypoxic conditions were likely to be involved in similar biological processes (19). Therefore, for better understanding of the interactions between these DEGs, enrichment analysis was performed, which suggested that they were predominantly enriched in GO_BPs such as inflammatory response, cell regeneration and apoptosis. Further analysis of the PPI network also identified six hub genes underlying the myocardial adaptation to chronic hypoxia.
GO analysis indicated that hypoxia in the myocardium was associated with inflammatory responses activated by neutrophils and monocytes, which involved changes in ECM organization, cell substrate adhesion, monocyte migration and chemotaxis. Consistent with previous studies (20–24), this indicated that chronic hypoxia was associated with inflammation. In accordance with the findings of hypoxia-activated cardiomyocyte mitosis and regeneration (25), GO analysis revealed that the DEGs in hypoxic myocardium were also enriched in cell regeneration.
KEGG pathway analysis indicated that the DEGs identified in the present study were predominantly enriched in cytokine-cytokine receptor interactions, chemokine signaling pathways, ECM-receptor interaction and signaling pathways, such as focal adhesion and apoptosis. Focal adhesions are multi-protein structures that contain integrin that can mechanically connect the cytoplasmic actin skeleton of different cells to the ECM (26,27). Thus, ECM signals can be transmitted by focal adhesion kinase (FAK) into the cell, thereby mediating cell adhesion, migration, survival, cell cycle regulation, proliferation and apoptosis (28). Notably, hypoxia induces the activation of the FAK signaling pathway in cardiomyocytes, promotes the adhesive interaction between cardiomyocytes and ECMs (29) and may result in cell proliferation and survival. However, the DEGs in the present study were also enriched in the apoptosis pathway, which also supports the findings that hypoxia may induce cardiomyocyte apoptosis (30). Therefore, it is notable that these DEGs were associated with regeneration, focal adhesion and apoptosis, which was indicative of the homeostasis between loss and renewal of cardiomyocytes exposed to hypoxia.
To further understand the intrinsic significance of the 1,260 DEGs, that is, the interactions between them, a PPI network was constructed. The data identified six hub genes with correlated degree scores of >25 through the network correlation analysis, including GNB4, CCR2, CCR1, PF4, CTNNB1 and JUN. Notably, JUN and CTNNB1 were identified to be upregulated. GNB4 is the β-4 subunit of guanine nucleotide-binding protein (G protein), an important regulator of the α subunit of G protein and some signal transduction receptors and effectors (31). It is typically co-expressed with the γ subunit to form dimers that regulate important effector systems, including the N-type Ca2+ channel, the G protein-gated inwardly-rectifying K+ channel and phospholipase (32,33). Downregulation of GNB4 in chronically hypoxic myocardium is associated with ion channel-associated heart rate and contractility (34), reduction of G-protein-coupled receptors such as chemokine receptors (20), and apoptosis (35). CCR2 and CCR1 are predominantly expressed on the surface of inflammatory cells, including lymphocytes, monocytes and macrophages; they are types of G protein-coupled chemokine receptors that mediate the infiltration of neutrophils following ischemia (36). The downregulation of CCR2 and CCR1 in response to hypoxia was in accord with a previous study (20). The reduction of chemokine receptor expression seemed to imply that less inflammatory cells (such as mononuclear macrophages and neutrophils) migrated into the hypoxic myocardium, thereby alleviating inflammation, improving adverse cardiac remodeling and reducing apoptosis (37,38). This may possibly be considered a protective adaptation of the myocardium to chronic hypoxia (39,40). PF4 is released from α-granules of activated platelets; not only are the activated platelets associated with cell apoptosis and survival (41), but also PF4 itself has been linked to apoptosis. Notably, PF4 inhibits tumor growth and tumor angiogenesis (42) and induces tumor cell apoptosis (43). Furthermore, as the small CXC chemokine and agonist of CCR1, PF4 promotes chemotaxis in inflammatory cells, including neutrophils and monocytes (44,45). Downregulation of PF4 under chronic hypoxia may be connected with apoptosis and the reduction of chemokine receptors in myocardium. Cytoplasmic β-catenin, encoded by the CTNNB1 gene, is a key protein in the Wnt signaling pathway; this is a conserved pathway during animal evolution and development, yet the specific genes it regulates depend on the cell type and background (46). In adult hearts, the Wnt signaling pathway is quiescent under normal conditions but is activated following myocardial injury and participates in cardiac remodeling (47). Once exposed to hypoxia, hypoxia inducible factor-1α (HIF-1α) competitively inhibits the formation of β-catenin/T-cell factor-4 complex and the downstream transcriptional activities in colorectal tumor cells, resulting in G1 arrest in the cell cycle. However, β-catenin also enhances HIF-1α-mediated transcription and helps the tumor cells with survival and adaptation to hypoxia (48). A previous study has reported that the Wnt/β-catenin signaling pathway may promote proliferation and protect cardiomyocytes from apoptosis, though little direct evidence exists to date (49). Therefore, the present study hypothesized that activation of the Wnt/β-catenin pathway may have an inhibitory effect on apoptosis under chronic hypoxia.
In the present study, the role of JUN in hypoxic myocardium became the primary focus. The transcription factor c-Jun, which is encoded by the oncogene JUN and belongs to the basic-region leucine zippers (bZIP) family, constitutes the activator protein-1 (AP-1) in the form of homodimers or heterodimers with other bZIPs, thereby regulating transcription and cellular activities, including proliferation, apoptosis, tumorigenesis and tissue morphogenesis (50). Through the GO and KEGG pathway enrichment analyses, it was identified that c-Jun was enriched in the biological function of ‘regeneration’ and the signaling pathways of ‘apoptosis’ and ‘focal adhesion’. It seemed paradoxical that c-Jun functioned in promoting survival as well as apoptosis in hypoxic myocardium. As an important endpoint of mitogen-activated protein kinase (MAPK) cascade activation, the DNA binding and transcriptional activity of c-Jun are enhanced following activation by JNK phosphorylation at Ser63/73. Following this, it forms AP-1, transcribes the downstream proteins (51) and mediates the expression or activation of pro-apoptotic genes, including Fas ligand, insulin-like growth factor binding protein 4, tumor necrosis factor (TNF)-α, B cell lymphoma-2 homologous antagonist/killer (52), p53 upregulated modulator of apoptosis (53) and Bim (54). Conversely, c-Jun has also been demonstrated to have anti-apoptotic properties. Endogenous c-Jun could upregulate survivin, counteract apoptosis, maintain mammary epithelial cell survival (55) and prevent TNF-α-induced hepatocyte apoptosis by antagonizing the activity of the pro-apoptotic gene p53 (56). Additionally, it works with signal transducer and activator of transcription 3 to inhibit Fas transcription, thereby reducing the sensitivity of tumor cells to apoptosis (57). Another study identified that c-Jun transcriptionally inhibits the tumor suppressor phosphatase and tensin homolog, which in turn activates the Akt signaling pathway and promotes fibroblast survival (58). In addition to its functions in apoptosis, c-Jun also promotes cell cycle progression through direct transcriptional activation of cyclin D1 (59) and repression of p53 (60). Previous studies have demonstrated that the MAPK subfamily JNK and its target c-Jun are activated by phosphorylation, which induces cardiomyocyte apoptosis under hypoxia stress (6,7). However, the downstream mechanisms underlying apoptosis caused by c-Jun remains to be elucidated. Therefore, whether c-Jun takes part in anti-apoptotic effects during hypoxic myocardial apoptosis is worthy of further investigation. One possible explanation for the opposing nature of pro- and anti-apoptosis (or pro-survival) is that c-Jun may activate anti-apoptotic programs under hypoxia to inhibit the excessive cardiomyocyte apoptosis caused by c-Jun itself and other factors. In addition, c-Jun may even promote cell cycle progression and proliferation to replenish the lost cardiomyocytes subjected to hypoxia. In this way, a new cellular homeostasis under hypoxia could be maintained. Therefore, the role of c-Jun in promoting survival and apoptosis may be the adaptation of myocardial tissue to chronic hypoxia, that is, some mechanism of self-protection and self-repair.
In conclusion, data mining was used in the present study to reveal the biological significance of DEGs in hypoxic myocardium, and genes associated with the life or death of myocardium were screened. The results provided a series of potential targets for studying mechanisms underlying the cellular adaptation to chronic hypoxia. However, the specific molecular mechanisms require further biological validation in in vitro or in in vivo experiments.
Acknowledgements
Not applicable.
Funding
The present study was supported by The National Science Foundation of China (grant nos. 81700277 and 81270228).
Availability of data and materials
The original data and program codes are available from the corresponding author on reasonable request or from the research data repositories Figshare (https://figshare.com/s/d7d700afae1d4194c391).
Authors' contributions
YX conceived and supervised the study; FW performed the primary bioinformatics analysis and was a major contributor in writing the manuscript; FG mainly contributed in the analysis of the biological significance of hub genes and picture editing; and SH was involved in interpretation of parts of the hub genes and English language editing. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Compernolle V, Brusselmans K, Franco D, Moorman A, Dewerchin M, Collen D, Carmeliet P. Cardia bifida, defective heart development and abnormal neural crest migration in embryos lacking hypoxia-inducible factor-1alpha. Cardiovasc Res. 2003;60:569–579. doi: 10.1016/j.cardiores.2003.07.003. [DOI] [PubMed] [Google Scholar]
- 2.Burchell HB, Taylor BE, et al. Circulatory adjustments to the hypoxemia of congenital heart disease of the cyanotic type. Circulation. 1950;1:404–414. doi: 10.1161/01.CIR.1.3.404. [DOI] [PubMed] [Google Scholar]
- 3.Fan X, Ma L, Zhang Z, Li Y, Hao M, Zhao Z, Zhao Y, Liu F, Liu L, Luo X, et al. Associations of high-altitude polycythemia with polymorphisms in PIK3CD and COL4A3 in Tibetan populations. Hum Genomics. 2018;12:37. doi: 10.1186/s40246-018-0169-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Humayun KN, Atiq M. Clinical profile and outcome of cyanotic congenital heart disease in neonates. J Coll Physicians Surg Pak. 2008;18:290–293. [PubMed] [Google Scholar]
- 5.Rafiee P, Shi Y, Kong X, Pritchard KA, Jr, Tweddell JS, Litwin SB, Mussatto K, Jaquiss RD, Su J, Baker JE. Activation of protein kinases in chronically hypoxic infant human and rabbit hearts: Role in cardioprotection. Circulation. 2002;106:239–245. doi: 10.1161/01.CIR.0000022018.68965.6D. [DOI] [PubMed] [Google Scholar]
- 6.He S, Liu P, Jian Z, Li J, Zhu Y, Feng Z, Xiao Y. miR-138 protects cardiomyocytes from hypoxia-induced apoptosis via MLK3/JNK/c-jun pathway. Biochem Biophys Res Commun. 2013;441:763–769. doi: 10.1016/j.bbrc.2013.10.151. [DOI] [PubMed] [Google Scholar]
- 7.He S, Liu S, Wu X, Xin M, Ding S, Xin D, Ouyang H, Zhang J. Protective role of downregulated MLK3 in myocardial adaptation to chronic hypoxia. J Physiol Biochem. 2016;73:371–380. doi: 10.1007/s13105-017-0561-5. [DOI] [PubMed] [Google Scholar]
- 8.Liu B, Zhang HG, Zhu Y, Jiang YH, Luo GP, Tang FQ, Jian Z, Xiao YB. Cardiac resident macrophages are involved in hypoxiainduced postnatal cardiomyocyte proliferation. Mol Med Rep. 2017;15:3541–3548. doi: 10.3892/mmr.2017.6432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Laflamme MA, Murry CE. Heart regeneration. Nature. 2011;473:326–335. doi: 10.1038/nature10147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Churko JM, Mantalas GL, Snyder MP, Wu JC. Overview of high throughput sequencing technologies to elucidate molecular pathways in cardiovascular diseases. Circ Res. 2013;112:1613–1623. doi: 10.1161/CIRCRESAHA.113.300939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Manzoni C, Kia DA, Vandrovcova J, Hardy J, Wood NW, Lewis PA, Ferrari R. Genome, transcriptome and proteome: The rise of omics data and their integration in biomedical sciences. Brief Bioinform. 2018;19:286–302. doi: 10.1093/bib/bbw114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grunert M, Dorn C, Schueler M, Dunkel I, Schlesinger J, Mebus S, Alexi-Meskishvili V, Perrot A, Wassilew K, Timmermann B, et al. Rare and private variations in neural crest, apoptosis and sarcomere genes define the polygenic background of isolated Tetralogy of Fallot. Hum Mol Genet. 2014;23:3115–3128. doi: 10.1093/hmg/ddu021. [DOI] [PubMed] [Google Scholar]
- 13.Ghorbel MT, Cherif M, Jenkins E, Mokhtari A, Kenny D, Angelini GD, Caputo M. Transcriptomic analysis of patients with tetralogy of Fallot reveals the effect of chronic hypoxia on myocardial gene expression. J Thorac Cardiovasc Surg. 2010;140:337–345.e26. doi: 10.1016/j.jtcvs.2009.12.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ahmed M, Nguyen HQ, Hwang JS, Zada S, Lai TH, Kang SS, Kim DR. Systematic characterization of autophagy-related genes during the adipocyte differentiation using public-access data. Oncotarget. 2018;9:15526–15541. doi: 10.18632/oncotarget.24506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kanehisa M, Goto S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yu G, Wang LG, Han Y, He QY. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS. 2012;16:284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos A, Tsafou KP, et al. STRING v10: Protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015;43:D447–D452. doi: 10.1093/nar/gku1003. Database Issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roy S, Bhattacharyya DK, Kalita JK. Reconstruction of gene co-expression network from microarray data using local expression patterns. BMC Bioinformatics. 2014;15(Suppl 7):S10. doi: 10.1186/1471-2105-15-S7-S10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bosco MC, Puppo M, Santangelo C, Anfosso L, Pfeffer U, Fardin P, Battaglia F, Varesio L. Hypoxia modifies the transcriptome of primary human monocytes: Modulation of novel immune-related genes and identification of CC-chemokine ligand 20 as a new hypoxia-inducible gene. J Immunol. 2006;177:1941–1955. doi: 10.4049/jimmunol.177.3.1941. [DOI] [PubMed] [Google Scholar]
- 21.Murdoch C, Muthana M, Lewis CE. Hypoxia regulates macrophage functions in inflammation. J Immunol. 2005;175:6257–6263. doi: 10.4049/jimmunol.175.10.6257. [DOI] [PubMed] [Google Scholar]
- 22.Hoenderdos K, Lodge KM, Hirst RA, Chen C, Palazzo SG, Emerenciana A, Summers C, Angyal A, Porter L, Juss JK, et al. Hypoxia upregulates neutrophil degranulation and potential for tissue injury. Thorax. 2016;71:1030–1038. doi: 10.1136/thoraxjnl-2015-207604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bartels K, Grenz A, Eltzschig HK. Hypoxia and inflammation are two sides of the same coin. Proc Natl Acad Sci USA. 2013;110:18351–18352. doi: 10.1073/pnas.1318345110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Taylor CT, Colgan SP. Regulation of immunity and inflammation by hypoxia in immunological niches. Nat Rev Immunol. 2017;17:774–785. doi: 10.1038/nri.2017.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nakada Y, Canseco DC, Thet S, Abdisalaam S, Asaithamby A, Santos CX, Shah AM, Zhang H, Faber JE, Kinter MT, et al. Hypoxia induces heart regeneration in adult mice. Nature. 2017;541:222–227. doi: 10.1038/nature20173. [DOI] [PubMed] [Google Scholar]
- 26.Chen CS, Alonso JL, Ostuni E, Whitesides GM, Ingber DE. Cell shape provides global control of focal adhesion assembly. Biochem Biophys Res Commun. 2003;307:355–361. doi: 10.1016/S0006-291X(03)01165-3. [DOI] [PubMed] [Google Scholar]
- 27.Zaidel-Bar R, Cohen M, Addadi L, Geiger B. Hierarchical assembly of cell-matrix adhesion complexes. Biochem Soc Trans. 2004;32:416–420. doi: 10.1042/bst0320416. [DOI] [PubMed] [Google Scholar]
- 28.Vadali K, Cai X, Schaller MD. Focal adhesion kinase: An essential kinase in the regulation of cardiovascular functions. IUBMB Life. 2007;59:709–716. doi: 10.1080/15216540701694245. [DOI] [PubMed] [Google Scholar]
- 29.Seko Y, Takahashi N, Sabe H, Tobe K, Kadowaki T, Nagai R. Hypoxia induces activation and subcellular translocation of focal adhesion kinase (p125(FAK)) in cultured rat cardiac myocytes. Biochem Biophys Res Commun. 1999;262:290–296. doi: 10.1006/bbrc.1999.1185. [DOI] [PubMed] [Google Scholar]
- 30.Jung F, Weiland U, Johns RA, Ihling C, Dimmeler S. Chronic hypoxia induces apoptosis in cardiac myocytes: A possible role for Bcl-2-like proteins. Biochem Biophys Res Commun. 2001;286:419–425. doi: 10.1006/bbrc.2001.5406. [DOI] [PubMed] [Google Scholar]
- 31.Smrcka AV. G protein β subunits: Central mediators of G protein-coupled receptor signaling. Cell Mol Life Sci. 2008;65:2191–2214. doi: 10.1007/s00018-008-8006-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ruiz-Velasco V, Ikeda SR, Puhl HL. Cloning, tissue distribution, and functional expression of the human G protein beta 4-subunit. Physiol Genomics. 2002;8:41–50. doi: 10.1152/physiolgenomics.00085.2001. [DOI] [PubMed] [Google Scholar]
- 33.Rosskopf D, Nikula C, Manthey I, Joisten M, Frey U, Kohnen S, Siffert W. The human G protein beta4 subunit: Gene structure, expression, Ggamma and effector interaction. FEBS Lett. 2003;544:27–32. doi: 10.1016/S0014-5793(03)00441-1. [DOI] [PubMed] [Google Scholar]
- 34.Clapham DE, Neer EJ. G protein beta gamma subunits. Annu Rev Pharmacol Toxicol. 1997;37:167–203. doi: 10.1146/annurev.pharmtox.37.1.167. [DOI] [PubMed] [Google Scholar]
- 35.Adams JW, Brown JH. G-proteins in growth and apoptosis: Lessons from the heart. Oncogene. 2001;20:1626–1634. doi: 10.1038/sj.onc.1204275. [DOI] [PubMed] [Google Scholar]
- 36.Reichel CA, Khandoga A, Anders HJ, Schlondorff D, Luckow B, Krombach F. Chemokine receptors Ccr1, Ccr2, and Ccr5 mediate neutrophil migration to postischemic tissue. J Leukoc Biol. 2006;79:114–122. doi: 10.1189/jlb.0605337. [DOI] [PubMed] [Google Scholar]
- 37.Liehn EA, Merx MW, Postea O, Becher S, Djalali-Talab Y, Shagdarsuren E, Kelm M, Zernecke A, Weber C. Ccr1 deficiency reduces inflammatory remodelling and preserves left ventricular function after myocardial infarction. J Cell Mol Med. 2008;12:496–506. doi: 10.1111/j.1582-4934.2007.00194.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Majmudar MD, Keliher EJ, Heidt T, Leuschner F, Truelove J, Sena BF, Gorbatov R, Iwamoto Y, Dutta P, Wojtkiewicz G, et al. Monocyte-directed RNAi targeting CCR2 improves infarct healing in atherosclerosis-prone mice. Circulation. 2013;127:2038–2046. doi: 10.1161/CIRCULATIONAHA.112.000116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hayasaki T, Kaikita K, Okuma T, Yamamoto E, Kuziel WA, Ogawa H, Takeya M. CC chemokine receptor-2 deficiency attenuates oxidative stress and infarct size caused by myocardial ischemia-reperfusion in mice. Circ J. 2006;70:342–351. doi: 10.1253/circj.70.342. [DOI] [PubMed] [Google Scholar]
- 40.Zhou L, Azfer A, Niu J, Graham S, Choudhury M, Adamski FM, Younce C, Binkley PF, Kolattukudy PE. Monocyte chemoattractant protein-1 induces a novel transcription factor that causes cardiac myocyte apoptosis and ventricular dysfunction. Circ Res. 2006;98:1177–1185. doi: 10.1161/01.RES.0000220106.64661.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gawaz M, Vogel S. Platelets in tissue repair: Control of apoptosis and interactions with regenerative cells. Blood. 2013;122:2550–2554. doi: 10.1182/blood-2013-05-468694. [DOI] [PubMed] [Google Scholar]
- 42.Tanaka T, Manome Y, Wen P, Kufe DW, Fine HA. Viral vector-mediated transduction of a modified platelet factor 4 cDNA inhibits angiogenesis and tumor growth. Nat Med. 1997;3:437–442. doi: 10.1038/nm0497-437. [DOI] [PubMed] [Google Scholar]
- 43.Liang P, Cheng SH, Cheng CK, Lau KM, Lin SY, Chow EY, Chan NP, Ip RK, Wong RS, Ng MH. Platelet factor 4 induces cell apoptosis by inhibition of STAT3 via up-regulation of SOCS3 expression in multiple myeloma. Haematologica. 2013;98:288–295. doi: 10.3324/haematol.2012.065607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Deuel TF, Senior RM, Chang D, Griffin GL, Heinrikson RL, Kaiser ET. Platelet factor 4 is chemotactic for neutrophils and monocytes. Proc Natl Acad Sci USA. 1981;78:4584–4587. doi: 10.1073/pnas.78.7.4584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fox JM, Kausar F, Day A, Osborne M, Hussain K, Mueller A, Lin J, Tsuchiya T, Kanegasaki S, Pease JE. CXCL4/Platelet Factor 4 is an agonist of CCR1 and drives human monocyte migration. Sci Rep. 2018;8:9466. doi: 10.1038/s41598-018-27710-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lam AP, Gottardi CJ. β-catenin signaling: A novel mediator of fibrosis and potential therapeutic target. Curr Opin Rheumatol. 2011;23:562–567. doi: 10.1097/BOR.0b013e32834b3309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bergmann MW. WNT signaling in adult cardiac hypertrophy and remodeling: Lessons learned from cardiac development. Circ Res. 2010;107:1198–1208. doi: 10.1161/CIRCRESAHA.110.223768. [DOI] [PubMed] [Google Scholar]
- 48.Kaidi A, Williams AC, Paraskeva C. Interaction between beta-catenin and HIF-1 promotes cellular adaptation to hypoxia. Nat Cell Biol. 2007;9:210–217. doi: 10.1038/ncb1534. [DOI] [PubMed] [Google Scholar]
- 49.Ozhan G, Weidinger G. Wnt/β-catenin signaling in heart regeneration. Cell Regen (Lond) 2015;4:3. doi: 10.1186/s13619-015-0017-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Meng Q, Xia Y. c-Jun, at the crossroad of the signaling network. Protein Cell. 2011;2:889–898. doi: 10.1007/s13238-011-1113-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell. 2000;103:239–252. doi: 10.1016/S0092-8674(00)00116-1. [DOI] [PubMed] [Google Scholar]
- 52.Fan M, Chambers TC. Role of mitogen-activated protein kinases in the response of tumor cells to chemotherapy. Drug Resist Updat. 2001;4:253–267. doi: 10.1054/drup.2001.0214. [DOI] [PubMed] [Google Scholar]
- 53.Zhao Z, Wang J, Tang J, Liu X, Zhong Q, Wang F, Hu W, Yuan Z, Nie C, Wei Y. JNK- and Akt-mediated Puma expression in the apoptosis of cisplatin-resistant ovarian cancer cells. Biochem J. 2012;444:291–301. doi: 10.1042/BJ20111855. [DOI] [PubMed] [Google Scholar]
- 54.Tomicic MT, Meise R, Aasland D, Berte N, Kitzinger R, Kramer OH, Kaina B, Christmann M. Apoptosis induced by temozolomide and nimustine in glioblastoma cells is supported by JNK/c-Jun-mediated induction of the BH3-only protein BIM. Oncotarget. 2015;6:33755–33768. doi: 10.18632/oncotarget.5274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Katiyar S, Casimiro MC, Dettin L, Ju X, Wagner EF, Tanaka H, Pestell RG. C-jun inhibits mammary apoptosis in vivo. Mol Biol Cell. 2010;21:4264–4274. doi: 10.1091/mbc.e10-08-0705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Eferl R, Ricci R, Kenner L, Zenz R, David JP, Rath M, Wagner EF. Liver tumor development. c-Jun antagonizes the proapoptotic activity of p53. Cell. 2003;112:181–192. doi: 10.1016/S0092-8674(03)00042-4. [DOI] [PubMed] [Google Scholar]
- 57.Ivanov VN, Bhoumik A, Krasilnikov M, Raz R, Owen-Schaub LB, Levy D, Horvath CM, Ronai Z. Cooperation between STAT3 and c-jun suppresses Fas transcription. Mol Cell. 2001;7:517–528. doi: 10.1016/S1097-2765(01)00199-X. [DOI] [PubMed] [Google Scholar]
- 58.Hettinger K, Vikhanskaya F, Poh MK, Lee MK, de Belle I, Zhang JT, Reddy SA, Sabapathy K. c-Jun promotes cellular survival by suppression of PTEN. Cell Death Differ. 2007;14:218–229. doi: 10.1038/sj.cdd.4401946. [DOI] [PubMed] [Google Scholar]
- 59.Wisdom R, Johnson RS, Moore C. c-Jun regulates cell cycle progression and apoptosis by distinct mechanisms. EMBO J. 1999;18:188–197. doi: 10.1093/emboj/18.1.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schreiber M, Kolbus A, Piu F, Szabowski A, Möhle-Steinlein U, Tian J, Karin M, Angel P, Wagner EF. Control of cell cycle progression by c-Jun is p53 dependent. Genes Dev. 1999;13:607–619. doi: 10.1101/gad.13.5.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The original data and program codes are available from the corresponding author on reasonable request or from the research data repositories Figshare (https://figshare.com/s/d7d700afae1d4194c391).