

Abstract
Creutzfeldt–Jakob disease (CJD) belongs to a group of prion disease that is caused by abnormally folded proteins and is clinically characterized by rapidly progressive cognitive decline, gait abnormalities, and myoclonus. Familial CJD is very rare and is described only in few families around the world. We report a case with rapidly progressive cognitive decline, ataxia, and myoclonus, with a history of several members of his family developing similar symptoms and succumbing to it. Clinical presentation and neuroimaging were suggestive of CJD. On genetic analysis, our index case and two of his family members (younger brother and younger son) were found to have D178N mutation in PRNP gene. The polymorphism of the 129th amino acid was V/V. We report the first kindred familial CJD from South-East Asia with genetically proven D178N-129V haplotype.
Keywords: Creutzfeldt–Jakob disease, familial, prion, PRNP gene
INTRODUCTION
Creutzfeldt–Jakob disease (CJD) is classified into sporadic, familial, and iatrogenic forms. More than 85% of CJD cases are sporadic in nature. Classic symptoms are rapidly progressive cognitive decline, gait abnormalities, and myoclonus.[1] In comparison to sporadic CJD, familial CJD usually presents at a younger age with longer duration of survival.[1,2,3,4] We report the first kindred of familial CJD from South-East Asia with genetically proven D178N-129V haplotype.
CASE REPORT
A 42 year old man presented with a history of forgetfulness, behavioral abnormalities, and difficulty in walking for 6 months. The first symptom was gradual onset of forgetfulness to recent events which gradually progressed to difficulty in carrying out office duties. He developed a change in behavior in the form of decreased interest, irritability, and agitation. Over the next 2 months, his family members noted progressive unsteadiness while walking. Over the next 3 months, he developed sudden shock-like jerky movements which started in the upper limbs and gradually progressed to involve the lower limbs as well. These jerky movements were precipitated by a sudden noise. During the course of illness, he developed features of bulbar dysfunction and features of emotional lability. He had a significant family history on his maternal ancestry of multiple family members developing similar symptoms.
At presentation, he was conscious, and higher mental function examination revealed reduced attention span and impairment of recent memory and executive functions. Remote memory was relatively preserved. He had a moist voice with features of bulbar dysfunction. All deep tendon reflexes were normal with bilateral flexor plantar responses. He had cogwheel rigidity involving all the four limbs and signs of cerebellar dysfunction in the form of gait ataxia and intention tremor. Multifocal stimulus-sensitive myoclonic jerks were present with a frequency of 3–5/min. Given these clinical symptoms and his family history, a possibility of familial CJD was entertained.
Investigations
The patient's hemogram; liver, kidney, and thyroid function tests; serum Vitamin B12 levels; and cerebrospinal fluid (CSF) studies were normal. Thyroid peroxidase antibody test was negative. CSF protein 14-3-3 was negative. Diffusion-weighted imaging magnetic resonance imaging sequence of the brain revealed bilateral symmetric hyperintensities in caudate nuclei and insular cortices [Figure 1]. His electroencephalogram (EEG) revealed continuous generalized slowing without any periodic discharges. Blood samples of the patient and his younger brother and children were sent to the Centers for Disease Control and Prevention, China, for evaluation.
Figure 1.
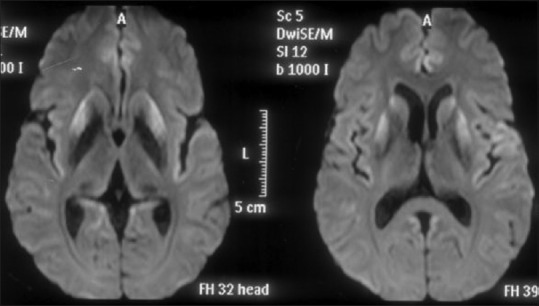
Magnetic resonance imaging diffusion weighted sequence showing diffusion restriction in bilateral caudate nuclei, putamen, and insular cortices
He and two of his family members (younger brother and younger son) were found to have D178N mutation in PRNP gene. The polymorphism of the 129th amino acid was V/V, while the polymorphism of 219th amino acid was E/E in all of them. The samples from other three family members (daughters and elder son) were found normal with normal sequencing of PNRP gene. The polymorphism of the 129th amino acid was M/V in all.
Outcome and follow-up
The patient was treated with supportive care. He died after 13 months of symptom onset.
DISCUSSION
Familial CJD belongs to a broad group of genetic prion diseases, which also includes Gerstmann–Straussler–Scheinker syndrome and fatal familial insomnia (FFI).[2,4,5,6,7,8,9,10] Familial CJD may have an unusual onset of symptoms and an atypical clinical course.[11]
The D178N mutation was first reported in Finnish families with a CJD-like illness. Familial CJD associated with D178N mutation presents as progressive memory loss and abnormal behavior followed by ataxia, dysarthria, and myoclonus [Table 1 and Figure 2].[8] It has an earlier age of onset and slower disease course in comparison to sporadic CJD. The disease typically presents between third and fifth decades. The disease course ranges from few months to 4 years.[1,6] Different studies of familial CJD D178N-129V haplotype (CJDD178N–129V)have reported cognitive abnormalities (80%–83% of patients), cerebellar signs (43%–55%), visual signs (19%), and myoclonus (12%) at the time of presentation.[1,6,7] Cognitive impairment initially starts with mild confusion progressing to global dementia. Ataxia, either truncal or appendicular, develops in 79% of cases in the course of illness.[1,7] Stimulus-sensitive myoclonus develops in about 75% of the patients.
Table 1.
Clinical characteristics of the affected family members
| Relation to patient [as per the pedigree chart Figure 2] | Sex | Age at onset of symptoms (years) | Clinical features | Investigations (MRI brain/EEG/genetic study) | Time death from onset of symptoms (months) |
|---|---|---|---|---|---|
| II-1 | Female | 65 years | Forgetfulness followed by ataxia and myoclonus | No investigation done | 14 months |
| II-2 | Male | 60 years | Forgetfulness followed by akinetic mutism and Myoclonus | No investigation done | 12 months |
| II-4 | Female | 55 years | Forgetfulness and ataxia followed by myoclonus | No investigation done | 11 months |
| III-1 | Female | 53 years | Forgetfulness, ataxia, slurring of speech followed by akinetic mutism and myoclonus | MRI brain: Not done | 3 months |
| EEG: Not done | |||||
| Genetic study: Not done | |||||
| III-2 | Female | 48 years | Forgetfulness, insomnia, behavioral changes, ataxia, myoclonus, and akinetic mutism in terminal stages | MRI: Done | 11 months |
| EEG: Not done | |||||
| Genetic study: Not done | |||||
| III-5 | Male | 45 years | Forgetfulness, behavioral changes, extrapyramidal symptoms followed by myoclonus | MRI: Not done | 14 months |
| EEG: Not done | |||||
| Genetic study: Not done | |||||
| IV-2 (index case) | Male | 42 years | Described in detail in the text | MRI: Suggestive of CJD | 13 months |
| EEG: Done | |||||
| Genetic study: Positive for D178N mutation | |||||
| IV-4 | Male | Now aged 32 years | Asymptomatic | MRI/EEG: Not done | - |
| Genetic study: Positive for D178N mutation. Currently asymptomatic | |||||
| V-3 | Male | Now aged 17 years | Asymptomatic | MRI/EEG: Not done | - |
| Genetic study: Positive for D178N mutation. Currently asymptomatic |
MRI=Magnetic resonance imaging, EEG=Electroencephalogram, CJD=Creutzfeldt-Jakob disease
Figure 2.
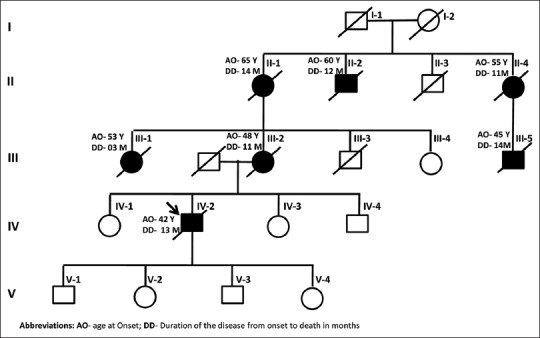
Pedigree chart of the affected family
In our case, all the affected family members had a progressive cognitive decline, behavioral abnormality, ataxia, and myoclonus. The genetic analysis of the index case revealed D178N mutation in the PNRP gene, and polymorphism at the 129th amino acid was V/V. Two of the asymptomatic family members also demonstrated the same mutation. All the symptomatic cases died within 3–15 months. The available neuroimaging of the case III-2 and of the index case IV revealed diffusion restriction in bilateral caudate and insular cortices, suggestive of CJD. The EEG of our patient revealed slowing but no triphasic periodic discharges. This is in accordance with other studies which have also found no periodic discharges in D178N-affected patients.[6] CSF 14-3-3 was negative in our patient. This too was in accordance with previous studies which have found that CSF 14-3-3 is negative in D178N patients.
However, there are two novel points in our kindred:
-
Usually, familial CJD with D178N mutation tends to have a more protracted course as compared to sporadic CJD. Average survival after symptom onset is 14 ± 4 months (range, 9–18 months) for 129VV patients and 27 ± 14 months (P < 0.05; range, 7–51 months) for the 129VM patients.[1] The shortest survival period recorded after the onset of symptoms is 7 months[1,6]
However, one of the patients in our kindred (III-1) had very rapid onset of symptoms and died in 3 months after symptom onset. Whether she was 129V/V or 129 V/M could not be determined as her genetic analysis was not done. This shows that occasionally familial CJD D178N mutation can be associated with a rapid progression of symptoms
Another patient in our kindred (III-2) had insomnia as a prominent complaint. However, insomnia was not the presenting complaint, and there were no overt autonomic symptoms. Nevertheless, this suggests that there may be some phenotypic overlap between FFI and familial CJD D178N type. Unfortunately, genetic study of this patient could not be carried out, so whether the haplotype was 129M or 129V could not be determined.
CONCLUSION
This case report represents the first familial CJD with D178N mutation reported from South-East Asian region.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
- 1.Gambetti P, Kong Q, Zou W, Parchi P, Chen SG. Sporadic and familial CJD: Classification and characterisation. Br Med Bull. 2003;66:213–39. doi: 10.1093/bmb/66.1.213. [DOI] [PubMed] [Google Scholar]
- 2.Haltia M, Kovanen J, Van Crevel H, Bots GT, Stefanko S. Familial Creutzfeldt-Jakob disease. J Neurol Sci. 1979;42:381–9. doi: 10.1016/0022-510x(79)90171-0. [DOI] [PubMed] [Google Scholar]
- 3.Gass CS, Luis CA, Meyers TL, Kuljis RO. Familial Creutzfeldt-Jakob disease: A neuropsychological case study. Arch Clin Neuropsychol. 2000;15:165–75. [PubMed] [Google Scholar]
- 4.Kovács GG, Puopolo M, Ladogana A, Pocchiari M, Budka H, van Duijn C, et al. Genetic prion disease: The EUROCJD experience. Hum Genet. 2005;118:166–74. doi: 10.1007/s00439-005-0020-1. [DOI] [PubMed] [Google Scholar]
- 5.Kretzschmar HA, Neumann M, Stavrou D. Codon 178 mutation of the human prion protein gene in a German family (Backer family): Sequencing data from 72-year-old celloidin-embedded brain tissue. Acta Neuropathol. 1995;89:96–8. doi: 10.1007/BF00294264. [DOI] [PubMed] [Google Scholar]
- 6.Goldfarb LG, Haltia M, Brown P, Nieto A, Kovanen J, McCombie WR, et al. New mutation in scrapie amyloid precursor gene (at codon 178) in Finnish Creutzfeldt-Jakob kindred. Lancet. 1991;337:425. doi: 10.1016/0140-6736(91)91198-4. [DOI] [PubMed] [Google Scholar]
- 7.Brown P, Goldfarb LG, Kovanen J, Haltia M, Cathala F, Sulima M, et al. Phenotypic characteristics of familial Creutzfeldt-Jakob disease associated with the codon 178Asn PRNP mutation. Ann Neurol. 1992;31:282–5. doi: 10.1002/ana.410310309. [DOI] [PubMed] [Google Scholar]
- 8.McLean CA, Storey E, Gardner RJ, Tannenberg AE, Cervenáková L, Brown P, et al. The D178N (cis-129M) “fatal familial insomnia” mutation associated with diverse clinicopathologic phenotypes in an Australian kindred. Neurology. 1997;49:552–8. doi: 10.1212/wnl.49.2.552. [DOI] [PubMed] [Google Scholar]
- 9.Medori R, Tritschler HJ, LeBlanc AC. In: Chapter 18. Prusiner SB, Collinge J, Powell J, Anderton B, editors. London: Ellis Horwood; 1992. [Google Scholar]
- 10.Goldfarb LG, Petersen RB, Tabaton M, Brown P, LeBlanc AC, Montagna P, et al. Fatal familial insomnia and familial Creutzfeldt-Jakob disease: Disease phenotype determined by a DNA polymorphism. Science. 1992;258:806–8. doi: 10.1126/science.1439789. [DOI] [PubMed] [Google Scholar]
- 11.Waliszewska-Prosół M, Obara K, Szewczyk P, Śniatowska M, Budrewicz S. Cerebellar ataxia as a first manifestation of Creutzfeldt- Jakob disease in two cousins. Postgrad Med J. 2018 doi: 10.1136/postgradmedj-2018-135566. doi:10.1136/postgradmedj-2018-13556. [DOI] [PubMed] [Google Scholar]