

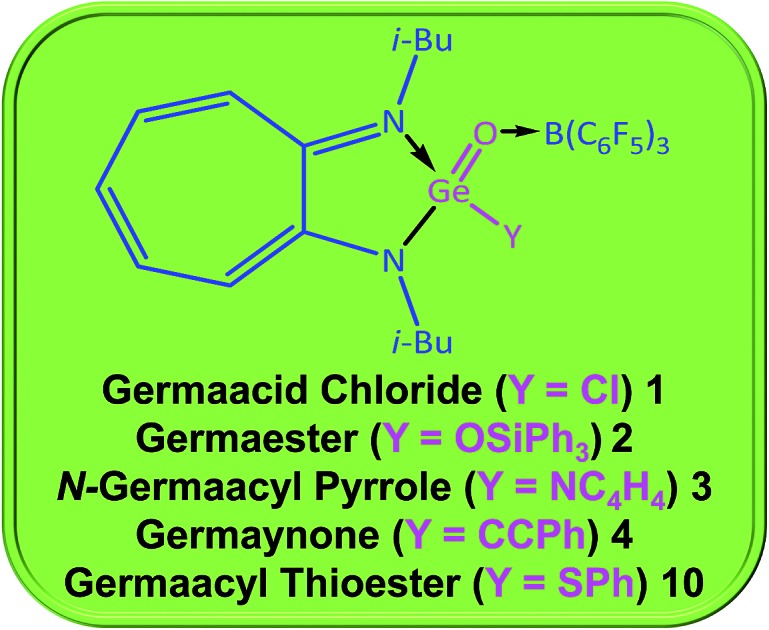
 Hitherto unknown germaacid chloride (1), germaester (2), and Ngermaacyl pyrrole (3) compounds are stabilised as Lewis acid complexes. Reactivity studies on them yielded compounds such as germaynone (4) and germaacyl thioester (10).
Hitherto unknown germaacid chloride (1), germaester (2), and Ngermaacyl pyrrole (3) compounds are stabilised as Lewis acid complexes. Reactivity studies on them yielded compounds such as germaynone (4) and germaacyl thioester (10).
Abstract
Germaacid chloride, germaester, and N-germaacyl pyrrole compounds were not known previously. Therefore, donor–acceptor-stabilised germaacid chloride (i-Bu)2ATIGe(O)(Cl) → B(C6F5)3 (1), germaester (i-Bu)2ATIGe(O)(OSiPh3) → B(C6F5)3 (2), and N-germaacyl pyrrole (i-Bu)2ATIGe(O)(NC4H4) → B(C6F5)3 (3) compounds, with Cl–Ge O, Ph3SiO–Ge O, and C4H4N–Ge O moieties, respectively, are reported here. Germaacid chloride 1 reacts with PhCCLi, KOt-Bu, and RLi (R = Ph, Me) to afford donor–acceptor-stabilised germaynone (i-Bu)2ATIGe(O)(CCPh) → B(C6F5)3 (4), germaester (i-Bu)2ATIGe(O)(Ot-Bu) → B(C6F5)3 (5), and germanone (i-Bu)2ATIGe(O)(R) → B(C6F5)3 (R = Ph 6, Me 7) compounds, respectively. Interconversion between a germaester and a germaacid chloride is achieved; reaction of germaesters 2 and 5 with TMSCl gave germaacid chloride 1, and 1 reacted with Ph3SiOLi and KOt-Bu to produce germaesters 2 and 5. Reaction of N-germaacyl pyrrole 3 with thiophenol produced a donor–acceptor-stabilised germaacyl thioester (i-Bu)2ATIGe(O)(SPh) → B(C6F5)3 (10). Furthermore, the attempted syntheses of germaamides and germacarboxylic acids are also discussed.
Introduction
The carbonyl group (C O) in organic compounds such as ketones [RC(O)R], aldehydes [RC(O)H], acid halides [RC(O)X], esters [RC(O)OR], amides [RC(O)NR2], carboxylic acids [RC(O)OH], and acid anhydrides [RC(O)OC(O)R] is of great importance in organic chemistry (R = alkyl/aryl group; X = halogen). The significance of these carbon compounds provides inspiration for the synthesis of their heavier analogues,1–3 but the synthetic efforts are typically hampered by the lability of the M O bond (M = Si, Ge, Sn, Pb). The instability of this bond stems from the σ-bond polarisation and poor π-type overlap between M and O atoms, which usually leads to oligomerisation/polymerisation of compounds containing such M O bonds.4–6 Strategies that utilise tailor-made ligands and/or provide donor–acceptor stabilisation to M/O atoms have been applied to address the aforementioned problems and have yielded various stable compounds containing M O bonds.7–11 Thus, silanones (silaketones) and germanones (germaketones) with formal Si O and Ge O bonds, respectively, were successfully isolated, and the variety of silanones exceeds that of the germanones.7–10 In addition to silanones, silicon analogues of aldehyde, ester, amide, formyl chloride, carboxylic acid, and acid anhydride compounds were also synthesised via various methods mainly by the groups of Driess, Roesky, Baceiredo, and Kato.12 Very recently, Aldridge and co-workers reported the generation of a silicon analogue of an acid chloride [(N-nacnac)ArSi(Cl) O (I)] through the reaction of the silylene (N-nacnac)ArSiCl with N2O (Chart 1) [(N-nacnac)Ar = HC{(Me2N)C(Ar)N}2; Ar = 2,6-i-Pr2C6H3]. The metathesis reactions of I with K[Et3BH] and KOt-Bu afforded a silaaldehyde [(N-nacnac)ArSi(H) O → BEt3 (II)] and a silaester [(N-nacnac)ArSi(Ot-Bu) O (III)], respectively (Chart 1).12a Surprisingly, such analogues of germanium [LGe(O)Y] [L = a monoanionic ligand; Y = H (germaaldehyde), Cl (germaacid chloride), OR (germaester), NR2 (germaamide), OH (germacarboxylic acid), and (OGe(O)L) germaacid anhydride] are not yet known, perhaps due to the difficulty in adding an electron-withdrawing Y atom/group to the germanium atom in light of the already heavily polarised Ge O bond. Owing to our continued interest in the chemistry of germanium, we were able to isolate the Lewis acid (LA) complexes (i-Bu)2ATIGe(i-Pr) O → LA (LA = B(C6F5)3 (IV), ZnCl2 (V), SnCl2 (VI), and GeCl2 (VII)) of a germanone10 starting from a germanium-μ-oxo dimer [ATI = aminotroponiminate, a monoanionic bidentate ligand]. We now understand that this synthetic protocol is exploitable for the synthesis of hitherto unknown germaacid chlorides and germaesters. Consequently, we report in this article the isolation and reactivity of the first examples of a donor–acceptor-stabilised germaacid chloride (i-Bu)2ATIGe(O)(Cl) → B(C6F5)3 (1), germaester (i-Bu)2ATIGe(O)(OSiPh3) → B(C6F5)3 (2), and N-germaacyl pyrrole (i-Bu)2ATIGe(O)(NC4H4) → B(C6F5)3 (3). Compound 3 was obtained during our search for stable germaamides.
Chart 1. Silicon analogues of an acid chloride I, aldehyde II, and ester III.

To synthesise a germaacid chloride, oxidation of the germylene monochloride13 (i-Bu)2ATIGeCl (G1) with N2O was carried out in tetrahydrofuran at room temperature. However, germylene G1 did not react with N2O at room temperature, and therefore, this reaction was performed at higher temperatures. Germylene G1 reacted with N2O at 60 °C in tetrahydrofuran and afforded the germanium μ-oxo dimer {(i-Bu)2ATIGe(Cl)(μ-O)}2 (D1) after 2 h as a yellow solid in 60% yield (Scheme 1).5,14 It appears that 60 °C is the optimum temperature for this reaction; higher temperatures afforded the ATI ligand salt [ATIH]+(Cl)–, and lower temperatures resulted in lower yields of μ-oxo dimer D1. Based on the successful conversion of a germanium μ-oxo dimer {(i-Bu)2ATIGe(i-Pr)(μ-O)}2 (D) containing Ge–C bonds into donor–acceptor-stabilised germanones IV–VII through the reaction of D with Lewis acids, we planned to react germanium μ-oxo dimer D1 containing Ge–Cl bonds with B(C6F5)3. To our surprise, treatment of μ-oxo dimer D1 with two equivalents of B(C6F5)3 in toluene for 2 h at room temperature yielded the first example of a donor–acceptor-stabilised germaacid chloride (i-Bu)2ATIGe(O)(Cl) → B(C6F5)3 (1) in quantitative yield (Scheme 1). This accomplishment inspired us to determine whether hitherto unknown germaesters and germaamides could also be isolated using this synthetic strategy of reacting suitable germanium μ-oxo dimers with Lewis acids. Thus, to synthesise a germaester, a germylene siloxide15 (i-Bu)2ATIGeOSiPh3 (G2) was reacted with N2O in tetrahydrofuran at 60 °C for 2 h to obtain the germanium μ-oxo dimer {(i-Bu)2ATIGe(OSiPh3)(μ-O)}2 (D2). The reaction of μ-oxo dimer D2 containing Ge–OSiPh3 bonds with two equivalents of B(C6F5)3 in toluene at room temperature afforded the first example of a donor–acceptor-stabilised germaester, namely, (i-Bu)2ATIGe(O)(OSiPh3) → B(C6F5)3 (2) (Scheme 2), and demonstrated the suitability of the germanium μ-oxo dimer route for the preparation of germaesters. To extend this route for the synthesis of germaamides, a germanium μ-oxo dimer with Ge–NR2 moieties is required. Two such germanium μ-oxo dimers, {(i-Bu)2ATIGeN(H)Ph(μ-O)}2 (D3) and {(i-Bu)2ATIGeN(Me)Ph(μ-O)}2 (D4), were obtained through the reaction of the amidogermylenes (i-Bu)2ATIGeN(H)Ph (G3) and (i-Bu)2ATIGeN(Me)Ph (G4) with N2O at 60 °C for 2 h in tetrahydrofuran (Scheme 3). However, the reaction of μ-oxo dimers D3 and D4 with two equivalents of B(C6F5)3 resulted in the amine → borane adducts PhNH2 → B(C6F5)3 and Ph(Me)NH → B(C6F5)3, respectively, along with an unidentified oily material instead of the expected germaamides (Scheme 3). These reactions suggest that the synthetic route discussed above is not suitable for the isolation of donor–acceptor-stabilised germaamides. On the basis of the products obtained, it was thought that the lone pairs of electrons on the nitrogen atoms of the NR2 moieties in D3 and D4 interfered with the expected reaction of these compounds (D3 and D4) with B(C6F5)3. To confirm this hypothesis, a germanium μ-oxo dimer containing amino functional groups with nitrogen atoms that cannot donate lone pairs of electrons to Lewis acids was synthesised and used. As a pyrrole substituent (Py; NC4H4) can satisfy the required criterion, the germanium μ-oxo dimer {(i-Bu)2ATIGe(NC4H4)(μ-O)}2 (D5) with two Ge–NC4H4 moieties was synthesised in quantitative yield by the reaction of the N-germylene pyrrole (i-Bu)2ATIGe(NC4H4) (G5) with N2O in tetrahydrofuran at 60 °C for 2 h (Scheme 4).16 Treatment of μ-oxo dimer D5 with two equivalents of B(C6F5)3 in toluene at room temperature resulted in the first donor–acceptor-stabilised N-germaacyl pyrrole, (i-Bu)2ATIGe(O)(NC4H4) → B(C6F5)3 (3) in quantitative yield (Scheme 4). The feasibility of isolating N-germaacyl pyrrole 3 as a stable species proves that the aforementioned hypothesis of the interference of lone pairs of electrons on the nitrogen atoms of the NR2 moieties in μ-oxo dimers D3 and D4 is factually valid.
Scheme 1. Synthesis of donor–acceptor-stabilised germaacid chloride 1. Notes: (a) in the alphanumerical numbering pattern, G denotes germylene, and D denotes germanium μ-oxo dimer, and (b) products with a Ge O → B(C6F5)3/Ge-OTMS → B(C6F5)3 moiety are given a linear/arbitrary numerical numbering pattern (starting from 1).

Scheme 2. Synthesis of donor–acceptor-stabilised germaester 2.

Scheme 3. Attempted synthesis of donor–acceptor-stabilised germaamides that resulted in amine → borane adducts.

Scheme 4. Synthesis of donor–acceptor-stabilised N-germaacyl pyrrole 3.

In all the reactions, germanium μ-oxo dimers D1–D5 were reacted with the Lewis acid B(C6F5)3.17 To understand the utility of other Lewis acids for the successful conversion of germanium μ-oxo dimers D1, D2, and D5 to the corresponding donor–acceptor-stabilised germaacid chloride, germaester, and N-germaacyl pyrrole, a range of Lewis acids (such as BF3, GeCl2, and SnCl2) were screened. However, all of these reactions were typically unsuccessful until now (see the ESI‡ for details). Surprisingly, the germanium-μ-oxo dimer {(i-Bu)2ATIGe(i-Pr)(μ-O)}2 (D) with Ge–i-Pr bonds was insensitive to the nature of the Lewis acid used.10 Thus, it reacted smoothly with B(C6F5)3, ZnCl2, SnCl2, and GeCl2 to afford the donor–acceptor-stabilised germanones IV, V, VI, and VII, respectively.10
As the germanium analogues of acid halides, esters, and amides were previously unknown, there has been no reactivity study on them. Therefore, the reactivity of the donor–acceptor-stabilised germaacid chloride 1, germaester 2, and N-germaacyl pyrrole 3 was studied with great interest to understand how these compounds behave chemically. It was found that germaacid chloride 1 can react with various lithium salts and afford clean products. Thus, through reaction of 1 with lithium phenylacetylide in toluene for 12 h, a unique example of a germaynone (i-Bu)2ATIGe(O)(CCPh) → B(C6F5)3 (4) was obtained (Scheme 5). Notably, until now, there has been no example of a silaynone. Furthermore, this reaction reveals that the chloride attached to the germaacyl moiety can be replaced with other functional groups, a reactivity omnipresent among acid chlorides in organic chemistry. Germaacid chloride 1, a heavier analogue of acid halides, exhibits reactivity similar to that of acid halides and silaacid chloride;12a therefore, this reactivity of 1 was further exploited. The lithium and potassium salts of triphenylsilanol and t-butanol reacted with 1 to result in germaesters 2 and (i-Bu)2ATIGe(O)(Ot-Bu) → B(C6F5)3 (5), respectively (Scheme 5), which is another route for the isolation of germaesters in addition to that shown in Scheme 2.
Scheme 5. Reactions of germaacid chloride 1 with various lithium/potassium salts.

In a similar fashion, alternate synthetic protocols can be suggested for N-germaacyl pyrrole 3 and germanones. For example, treatment of 1 with lithium pyrrol-1-ide and phenyl/methyl lithium yielded N-germaacyl pyrrole 3 and the germanones (i-Bu)2ATIGe(O)(Ph) → B(C6F5)3 (6)/(i-Bu)2ATIGe(O)(Me) → B(C6F5)3 (7) as products, respectively (Scheme 5). Thus, from germaacid chloride 1, germaesters, N-germaacyl pyrrole, and germanones can be derived without the need to isolate the corresponding germanium-μ-oxo dimers. This route was also attempted for the possible isolation of germaamides, and the reactions of germaacid chloride 1 with the lithium salts PhN(H)Li and PhN(Me)Li were carried out. However, these reactions faced the same fate as that of the abovementioned reactions carried out for the isolation of germaamides (shown in Scheme 3) by yielding amine → borane adducts only.
However, another reaction of germaacid chloride 1 with lithium bis(trimethylsilyl)amide, which aimed again at obtaining the elusive germaamide, occurred differently and resulted in the germaimine (i-Bu)2ATIGe(NTMS)(OTMS) → B(C6F5)3 (9) in quantitative yield (Scheme 6). This result reveals that the desired germaamide [8] was formed as an intermediate, which then underwent 1,3-silyl migration to form the stable compound 9 (Scheme 6).
Scheme 6. Reaction of germaacid chloride 1 with lithium bis(trimethylsilyl)amide.

Reactivity studies with donor–acceptor-stabilised germaesters 2 and 5 demonstrated that an interconversion between these germaesters and germaacid chloride 1 is achievable. Germaesters 2 and 5 reacted with a slight excess of Me3SiCl in toluene at room temperature and offered germaacid chloride 1 (Scheme 7). As mentioned above (Scheme 5), reactions of germaacid chloride 1 with one equivalent of LiOSiPh3 and KOt-Bu in toluene at room temperature generated the germaesters 2 and 5, respectively (Scheme 7). This type of interconversion is not known among the analogous silicon compounds.
Scheme 7. Interconversion between germaesters 2/5 and germaacid chloride 1.

The reactivity studies on N-germaacyl pyrrole 3 demonstrated that the thiophenoxide moiety of thiophenol can substitute the pyrrolide of 3. Accordingly, the reaction of N-germaacyl pyrrole 3 with thiophenol at room temperature in toluene for 6 h resulted in the first example of a germaacyl thioester (i-Bu)2ATIGe(O)(SPh) → B(C6F5)3 (10) in quantitative yield (Scheme 8).
Scheme 8. Reaction of N-germaacyl pyrrole 3 with thiophenol.

Considering this reaction, the feasibility of substituting the pyrrolide of 3 with hydroxide from a suitable precursor was investigated, as this might lead to the first example of a donor–acceptor-stabilised germacarboxylic acid. However, the reaction of 3 with water in a 1 : 1 molar ratio for 2 h in toluene resulted in [ATIH]+[(OH)(B(C6F5)3)]– and not the expected germacarboxylic acid (Scheme S1; see the ESI‡). The commonality in all of the abovementioned reactions of donor–acceptor-stabilised germaacid chloride 1, germaester 2, and N-germaacyl pyrrole 3 is that these reactants undergo nucleophilic substitution in the presence of suitable substrates without any damage to the Ge O → B(C6F5)3 moiety.
The germanium-μ-oxo dimers D1 and D3–D5, germaacid chloride 1, germaesters 2 and 5, N-germaacyl pyrrole 3, germaynone 4, germanones 6 and 7, and germaacyl thioester 10 are stable at room temperature in an inert atmosphere of dinitrogen. All these compounds are freely soluble in common organic solvents, such as toluene, chloroform, and dichloromethane. Though the germanium-μ-oxo dimers D1–D5 are also freely soluble in tetrahydrofuran, products 1–7 and 10, containing a Ge O → B(C6F5)3 moiety, decompose even in tetrahydrofuran dried over a potassium mirror to afford [ATIH]+[(OH)(B(C6F5)3)]–.
Compounds D1, D3–D5, 1–7, and 10 were characterised through multinuclear NMR spectroscopic (1H, 11B, 13C, 19F, and 29Si) and single-crystal X-ray diffraction studies in the solution and solid states, respectively (see the ESI‡ for details). In the 1H NMR spectra of D1 and D5, all the resonances are shifted slightly downfield in comparison to those of the precursor molecules, germylene monochloride G1 and N-germylene pyrrole G5, respectively. This shifting is due to the attachment of germanium atoms to electronegative oxygen atoms and the concomitant increase in the formal oxidation state of germanium atoms from +2 to +4. The resonances of the seven-membered ring protons in 1–7 and 10 are shifted downfield in comparison to the corresponding protons in germanium-μ-oxo dimer D1. Owing to the increased electrophilicity of the germanium atom in the Ge O → B(C6F5)3 moiety (of 1–7 and 10) in comparison to the germanium atoms in the Ge(μ-O)2Ge moiety of D1, these shifts are expected. In the 13C NMR spectra of D1, D3–D5, 1–7, and 10, the expected numbers of signals were observed. In the 11B NMR spectra of 1–6, and 10, singlet resonances at –2.46, –2.61, –2.72, –2.79, –2.44, –3.12, and –2.73 ppm were observed, respectively (Table 1). In comparison, B(C6F5)3 and the donor–acceptor-stabilised germanone (i-Bu)2ATIGe(O)(i-Pr) → B(C6F5)3 (IV) showed singlet resonances at –2.30 ppm18,19 and –4.52 ppm,10 respectively. These data reveal that the resonances in 1–6 and 10 are in between the resonances of B(C6F5)3 and IV. These results suggest that the electron donation by the germaacyl oxygen atom to the boron atom in 1–6, and 10 is reduced relative to that in IV due to the electron-withdrawing effect of the Cl, OSiPh3, NC4H4, CCPh, Ot-Bu, Ph, and SPh atom/group on the germanium atom, respectively (IV has an electron-donating i-Pr group on the germanium atom). The donor–acceptor-stabilised silaaldehyde L′Si(H) O → B(C6F5)3 (VIII),12g silaformyl chloride IPr·SiH(Cl) O → B(C6F5)3 (IX),12c silaacid anhydride [{PhC(t-BuN)2}Si{ O→B(C6F5)3}O–Si(H){ O→B(C6F5)3}(Nt-Bu)(HNt-Bu)CPh] (X),12d monoalumoxane L*Al O → B(C6F5)3 (XI),20 and boraacid chloride IPr → B(Cl) O → B(C6F5)3 (XII)21 have B(C6F5)3 as the acceptor in the M O → B(C6F5)3 moiety (M = Si VIII, IX, X; Al XI; B XII) [L′ = HC[CMeN(Ar)]2 IPr = 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene, L* = Et2NCH2CH2NC(Me)CHC(Me)NCH2CH2NEt2]. It may therefore be appropriate to compare the boron and fluorine resonances of these compounds with those of 1–6 and 10 (Table 1). These resonances in compounds VIII, IX, X, XI, and XII are shifted upfield with respect to the corresponding resonances of B(C6F5)3 (Table 1), which indicates the shielding of boron and fluorine atoms due to electron donation by oxygen atoms. This result is similar to that observed for compounds 1–6 and 10, containing a Ge O → B(C6F5)3 moiety (Table 1), but as revealed by the 11B NMR spectral data (Table 1), the magnitude of the shielding in these compounds is lower than that in compounds VIII, IX, X, and XI. In the 29Si NMR spectra of germaester 2, a signal at –13.62 ppm for the SiPh3 group is shifted downfield in comparison to that in germylene G2 (–24.72 ppm).15
Table 1. Comparison of the 11B and 19F NMR spectral resonances of boron and fluorine atoms and the O–B bond distances in compounds 1–6 and 10 with B(C6F5)3 and other related compounds of group 13–14 elements with an M O → B(C6F5)3 moiety(s) (M = Ge, Si, Al, B).
| S. no. | Compound | 11B NMR chemical shift (ppm) | 19F NMR chemical shift (ppm) | O–B bond length (Å) | Reference |
| 1 | Germanone, (i-Bu)2ATIGe(i-Pr)(O) → B(C6F5)3 (IV) | –4.52 a | (–134, –161, and –166) a | 1.473(4) | 10 |
| 2 | Silaaldehyde, L′Si(H) O → B(C6F5)3 (VIII) | –4.70 b | (–132, –162, and –165) b | 1.503(3) | 12g |
| 3 | Silaformyl chloride, IPr·SiH(Cl) O → B(C6F5)3 (IX) | –5.28 c | (–134, –163, and –168) c | 1.492(3) | 12c |
| 4 | Silaacid anhydride, [{PhC(t-BuN)2}Si{ O→B(C6F5)3}O–Si(H) { O→B(C6F5)3}(Nt-Bu)(HNt-Bu)CPh] (X) | (–3.99, and –5.46) c | (–134, –135, –164, –165, –167, and –168) c | 1.493(3), and 1.488(3) | 12d |
| 5 | Monoalumoxane, L*Al O → B(C6F5) (XI) | -4.83 d | (-134, -164, and -166) d | 1.444(3) | 20 |
| 6 | Boraacid chloride, IPr → B(Cl) O → B(C6F5) (XII) | -2.7 e | (-131, -160, and -165) e | 1.518(3) | 21 |
| 7 | B(C6F5)3 | –2.30 a | (–127, –143, and –160) a | — | 19 |
| 8 | Germaacid chloride, (i-Bu)2ATIGe(O)(Cl) → B(C6F5)3 (1) | –2.46 a | (-133, -159, and -165) a | 1.493(5) | This work |
| 9 | Germaester, (i-Bu)2ATIGe(O)(OSiPh3) → B(C6F5)3 (2) | –2.61 a | (-132, -160, and -165) a | 1.497(3) | This work |
| 10 | N-Germaacyl pyrrole, (i-Bu)2ATIGe(O)(NC4H4) → B(C6F5)3 (3) | –2.72 a | (–133, –159, and –165) a | 1.494(6) | This work |
| 11 | Germaynone, (i-Bu)2ATIGe(O)(CCPh) → B(C6F5)3 (4) | –2.79 a | (–133, –161, and –165) a | 1.489(4) | This work |
| 12 | Germaester, (i-Bu)2ATIGe(O)(Ot-Bu) → B(C6F5)3 (5) | –2.44 a | (–132, –160, and –165) a | 1.505(3) and 1.502(3) | This work |
| 13 | Germanone, (i-Bu)2ATIGe(O)(Ph) → B(C6F5)3 (6) | –3.12 a | (–133, –160, and –165) a | 1.481(3) | This work |
| 14 | Germaacyl thioester, (i-Bu)2ATIGe(O)(SPh) → B(C6F5)3 (10) | –2.73 a | (–133, –160, and –165) a | 1.501(5) | This work |
aIn CDCl3.
bIn CD2Cl2.
cIn THF-d8.
dIn C6D6/THF-d8.
eIn C6D6.
In a preliminary study of optical properties, the UV-vis spectra of compounds 1, 2, and 10 were recorded in toluene at room temperature. Compounds 1, 2, and 10 showed an absorption maximum in the visible region at approximately 420 nm (Fig. 1). Theoretical studies suggested that these absorptions in compounds 1, 2, and 10 are essentially due to 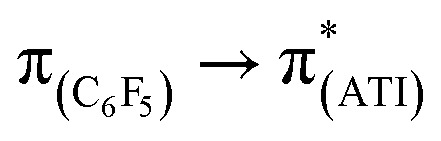 ,
, 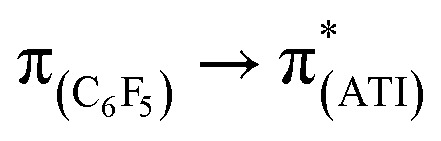 , and
, and 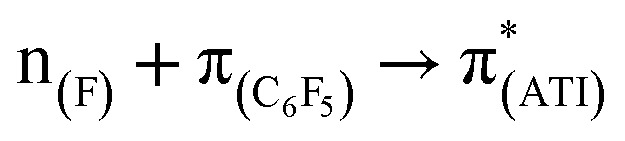 transitions, respectively (Table S1; see the ESI‡ for details). Furthermore, there are two high-energy transitions in each of these compounds with λmax values of approximately 350 and 285 nm (Fig. 1), which are due to multiple transitions (Table S1; see the ESI‡ for details). The optical properties of compounds with formal M
O → LA moieties (M = Ge, Si) have rarely been studied. For germanone VII with a Ge
O → GeCl2 moiety, optical properties have been reported. In comparison to compounds 1, 2, and 10, the absorption maximum of VII in the visible region (437 nm) is slightly redshifted, and this absorption is due to a
transitions, respectively (Table S1; see the ESI‡ for details). Furthermore, there are two high-energy transitions in each of these compounds with λmax values of approximately 350 and 285 nm (Fig. 1), which are due to multiple transitions (Table S1; see the ESI‡ for details). The optical properties of compounds with formal M
O → LA moieties (M = Ge, Si) have rarely been studied. For germanone VII with a Ge
O → GeCl2 moiety, optical properties have been reported. In comparison to compounds 1, 2, and 10, the absorption maximum of VII in the visible region (437 nm) is slightly redshifted, and this absorption is due to a 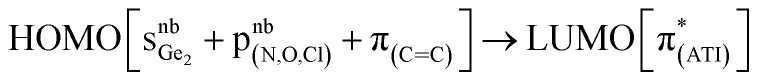 transition. Most likely, a different Lewis acid in compound VII altered the composition of the HOMO.
transition. Most likely, a different Lewis acid in compound VII altered the composition of the HOMO.
Fig. 1. UV-vis spectra of compounds 1, 2, and 10 (30 μM solution) in toluene.

The structures of compounds D1, D3–D5, 1–7, 9, and 10 in the solid state were determined by single-crystal X-ray diffraction analysis (Fig. 2–4 and S53–S62, Tables S2–S5, and Experimental section; see the ESI‡).22 Compounds 1–4 and 6 crystallised in the triclinic space group P1[combining macron] (Tables S3 and S4; see the ESI‡). Compounds 5, 7, and 10 crystallised in the monoclinic, orthorhombic, and monoclinic space groups P21/n, P212121, and P21/c, respectively (Table S4; see the ESI‡).
Fig. 2. Molecular structure of germaacid chloride 1 with thermal ellipsoids at the 50% probability level. All hydrogen atoms and a solvent molecule (dichloromethane) are omitted for clarity. Selected bond lengths (Å) and angles (deg): Ge1–O1 1.698(2), O1–B1 1.493(5), Ge1–Cl1 2.117(1), Ge1–N1 1.831(3), Ge1–N2 1.846(3); O1–Ge1–N1 111.60(1), O1–Ge1–N2 116.79(1), O1–Ge1–Cl1 112.25(9), B1–O1–Ge1 134.6(2), N2–Ge1–N1 87.46(1), N1–Ge1–Cl1 116.19(1), N2–Ge1–Cl1 110.52(1). Data collection temperature: 100 K.

Fig. 3. Molecular structure of germaynone 4 with thermal ellipsoids at the 50% probability level. All hydrogen atoms and a solvent molecule (dichloromethane) are omitted for clarity. Selected bond lengths (Å) and angles (deg): Ge1–O1 1.708(2), O1–B1 1.489(4), Ge1–C16 1.856(3), Ge1–N1 1.860(2), Ge1–N2 1.845(2); O1–Ge1–N1 114.10(1), O1–Ge1–N2 110.17(1), O1–Ge1–C16 113.63(12), B1–O1–Ge1 131.46(2), N2–Ge1–N1 86.91(1), N1–Ge1–C16 112.42(2), N2–Ge1–C16 116.98(1). Data collection temperature: 100 K.

Fig. 4. Molecular structure of germaacyl thioester 10 with thermal ellipsoids at the 50% probability level. All hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and angles (deg): Ge1–O1 1.698(3), O1–B1 1.501(5), Ge1–S1 2.199(2), Ge1–N1 1.864(4), Ge1–N2 1.866(4); O1–Ge1–S1 116.19(1), B1–O1–Ge1 144.0(3), N2–Ge1–N1 85.72(2). Data collection temperature: 100 K.
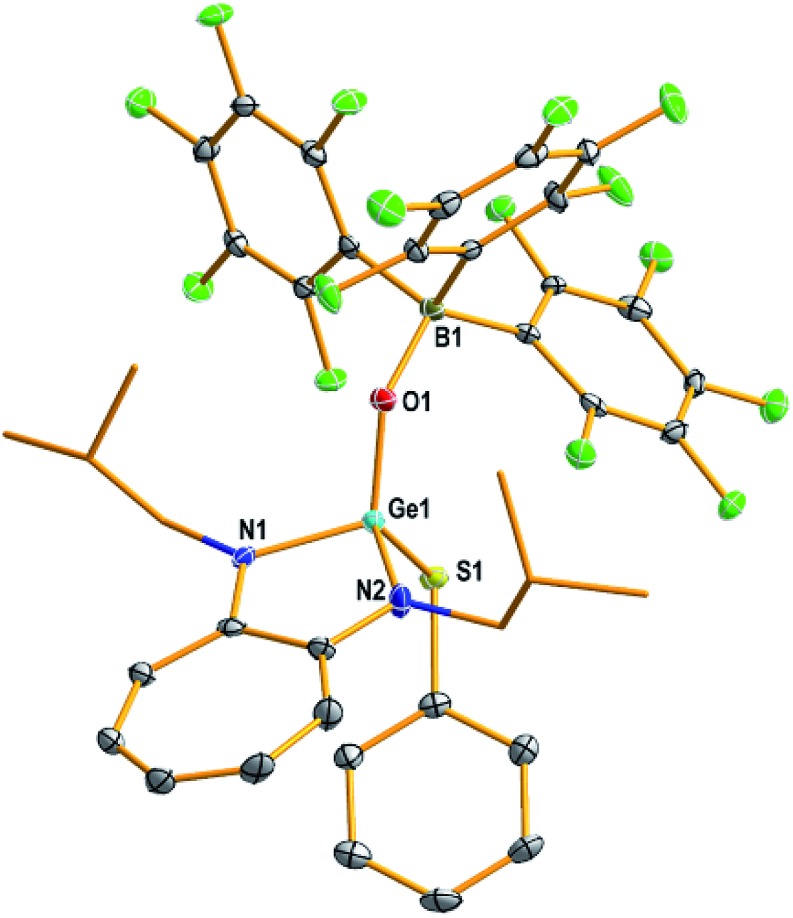
The molecular structures of compounds 1–7 and 10 [Fig. 2 (1), 3 (4), 4 (10), S57 (2), S58 (3), S59 (5), S60 (6), and S61 (7)‡] confirmed the presence of a (Y)Ge O → B(C6F5)3 moiety [Y = Cl (1), OSiPh3 (2), NC4H4 (3), CCPh (4), Ot-Bu (5), Ph (6), Me (7), and SPh (10)]. In these compounds, the germanium atom has a distorted tetrahedral geometry with two ATI ligand nitrogens, one germaacyl oxygen, and one Cl (1), O (2), N (3), C (4), O (5), C (6), C (7), or S (10) atom. The average length of the Ge–Nligand bonds in compounds 1 (1.838 Å), 2 (1.848 Å), and 3 (1.843 Å) is shorter than that in their precursors D1 (1.931 Å), D2 (1.946 Å), and D5 (1.942 Å), respectively. Similarly, the Ge–Y bond in compounds 1 (2.117(1) Å; Y = Cl), 2 (1.719(2) Å; Y = OSiPh3), and 3 (1.820(4) Å; Y = NC4H4) is also shorter than that in compounds D1 (2.20(8) Å), D2 (1.767(3) Å), and D5 (1.892(3) Å), respectively. These differences are due to the electrophilicity of the oxygen atom in the Ge O → B(C6F5)3 moiety of compounds 1, 2, and 3 being higher than that of the oxygen atoms in the Ge(μ-O)2Ge moiety of D1, D2, and D5, which makes the germanium atom in the former set of compounds more electrophilic than that in the latter set. Though these effects are observed in germanone IV, in comparison to the electron-donating i-Pr group bound to the germanium atom of germanone IV, the Cl, OSiPh3, NC4H4, CCPh, and SPh atom/group bound to the germanium atom in germaacid chloride 1, germaester 2, N-germaacyl pyrrole 3, germaynone 4, and germaacyl thioester 10, respectively, exert electron-withdrawing (+I) effects and compete for the germanium atom's electron density, thus increasing the interaction between the germanium and oxygen atoms of the Ge O bond. Therefore, the length of the formal Ge O bond in compounds 1 (1.698(2) Å), 2 (1.696(2) Å), 3 (1.695(3) Å), 4 (1.708(2) Å), and 10 (1.698(3) Å) is shorter than that in germanones IV (1.718(2) Å), V (1.724(2) and 1.728(2) Å), VI (1.728(5) Å), and VII (1.718(2) Å).10 These data also reveal that relative to the polarisation of the Ge O bond in germanone IV,10 the same bonds in germaacid chloride 1, germaester 2, N-germaacyl pyrrole 3, germaynone 4, and germaacyl thioester 10 are less polarised due to the electron-withdrawing effect of the Cl, OSiPh3, NC4H4, CCPh, and SPh atoms/groups bound to the germanium atom, respectively. A consequence of the increased interaction between the germanium and oxygen atoms of the germaacyl bond in these compounds is the reduced Lewis basicity of the oxygen atom. This result is reflected in the interaction of this oxygen atom with the Lewis acid B(C6F5)3, where the O → B bond in compounds 1 (1.493(5) Å), 2 (1.497(3) Å), 3 (1.494(6) Å), 4 (1.489(4) Å), and 10 (1.501(5) Å) is longer than the corresponding bond in germanone IV (1.473(4) Å).10 The O → B bond lengths observed in these compounds are similar to those observed in analogous silicon derivatives (VIII 1.503(3), IX 1.492(3), and X 1.493(3) and 1.488(3); M = Si) and boraacid chloride (XII 1.518(3); M = B) with an M O → B(C6F5)3 bond (Table 1).12g,12c,12d,21 However, in the monoalumoxane20XI with an Al O → B(C6F5)3 bond, the O → B bond is shorter (1.444(3) Å) than those in compounds 1–4, 10, VIII, IX, X, and XII. All the bonding aspects discussed here are supported by theoretical studies (vide infra). Furthermore, the Ge O bond (vide supra) in compounds 1–4 and 10 is slightly longer than the Ge O bond in the base-stabilised germanones[L′′LDGe O] (L′′ = [CH{(C CH2)(CMe)(NAr)2}]; LD = [{(Me)CN(Me)}2C] (XIII), [{(Me)CN(i-Pr)}2C] (XIV), 4-(Me2N)–C5H4N (XV)) without an acceptor at an oxygen atom (1.646(2)-1.672(3) Å)8 and shorter than the Ge–O single bonds in germanium-μ-oxo dimers D1, D2, and D5 (1.848(2)–1.787(3) Å).
The nature of the Ge O bond in compounds 1–3 and 10 was analysed through natural bond orbital (NBO)23,24 studies, and the details are provided in Table S6 (see the ESI‡). The Ge–O σ-bond in compounds 1 and 10 is formed by the overlap of the sp2.59 and sp2 hybrid orbitals of germanium with the sp1.62 and sp2.66 hybrid orbitals of oxygen, respectively (Fig. 5 and Table S6; see the ESI‡). In compounds 2 and 3, the sp2.53 and sp2.43 hybrid orbitals of germanium overlap with the sp2.89 and sp2.57 hybrid orbitals of oxygen to form the Ge–O bond, respectively (Fig. 5 and Table S6; see the ESI‡). MO calculations also reveal the presence of Ge–O bonds in compounds 1–3 and 10, and these bonds are deeply buried (Figure S63, see the ESI‡).
Fig. 5. NBO calculated Ge–O σ-bond in germaacid chloride 1, N-germaacyl pyrrole 3, and germaacyl thioester 10. The hybridisations of the germanium and oxygen orbitals involved in the overlap are mentioned along with the percentage contributions of the constituent atoms to the Ge–O bond.

NBO second-order perturbation theory analysis reveals that in germaacid chloride 1, the sigma bond between germanium and oxygen is formed by the donation of the lone pair of electrons on the oxygen atom to the σ* orbital of the Ge–Cl bond (Fig. 6a; 79.3 kcal mol–1). The lone pair of electrons on the oxygen atom also interacts with the π* orbitals of the Ge–NATI bonds (Fig. 6b; 100.3 kcal mol–1 and Fig. 6c; 52.8 kcal mol–1). However, in addition to these interactions, there are two strong stabilising interactions between the sp3.82 (Fig. 6d; 44.8 kcal mol–1) and sp0.29 (Fig. 6e; 43.6 kcal mol–1) orbitals of oxygen and the π* orbital of the Ge–N4 bond. Compounds 2, 3, and 10, instead of showing the aforementioned n (lone pair of electrons on oxygen) to σ*/π* orbital interactions, showed strong NBO donor–acceptor interactions from the s, p or spx orbitals of oxygen atoms to vacant s, p or spx orbitals of the germanium atoms [Fig. 6f–h (2), Fig. 6i–l (3), and Fig. 6m–p (10)]. However, in compound 10, a moderately strong NBO donor–acceptor interaction was found between the p orbital of oxygen and the σ* orbital of the Ge–S bond (27.9 kcal mol–1) (Fig. 6q). In comparison, germanone IV showed three σ interactions: two O → Ge interactions and one O → σ*(Ge–Ci-Pr) interaction; these interactions result in a total stabilisation energy of 236.3 kcal mol–1.10 Thus, the total stabilisation energy due to the donor–acceptor interactions in compounds 1 (320.8 kcal mol–1), 2 (284.7 kcal mol–1), 3 (303.7 kcal mol–1), and 10 (329.2 kcal mol–1) is higher than that in germanone IV, which is due to the difference in the nature of the atoms/moieties bound to germanium atom in these compounds (–Cl, –OSiPh3, –NC4H4, and –SPh, respectively) instead of an i-Pr group. The Wiberg bond index (WBI) calculations for compounds 1, 3, and 10 also showed a slightly increased bond order for the Ge O bond (0.74–0.76) relative to that in germanone IV (0.70)10 (Table S6; see the ESI‡). A similar bond order (0.7955) was calculated for silaaldehyde II (with BEt3 as an acceptor bound to the oxygen atom); for silaacid chloride I and silaester III (without any acceptor bound to the oxygen atom), the calculated WBI values are 1.0993 and 1.0441, respectively.12a In compounds 1, 2, and 10, the HOMO is localised on the phenyl ring of the B(C6F5)3 moiety (Fig. S64; see the ESI‡), and in compound 3, it is localised on the pyrrole ring, which also reveals the stabilisation of the formal Ge O bonds in these compounds (Fig. S64; see the ESI‡). Furthermore, NBO donor–acceptor interactions between oxygen and boron atoms can be observed in all these compounds (Fig. S65; see the ESI‡); the stabilisation energies due to these interactions are 280.3 kcal mol–1, 315.6 kcal mol–1, 296.3 kcal mol–1, and 294.6 kcal mol–1 in compounds 1 (Fig. S65a‡), 2 (Fig. S65b‡), 3 (Fig. S65c‡), and 10 (Fig. S65d‡), respectively. All these stabilisation energies are lower than that observed in germanone IV (334.9 kcal mol–1),10 indicating the reduced electron donation from oxygen atoms to boron atoms in compounds 1–3, and 10.
Fig. 6. Pictorial view of NBO donor–acceptor interactions between p or spx (x = 3.82, 0.29) orbitals of oxygen and the σ* orbital of the Ge–Cl bond/π* orbitals of the Ge–NATI bonds in compound 1 (a–e), s or p orbitals of oxygen and s or p orbitals of germanium in compound 2 (f–h), p or spx (x = 0.32, 4.59) orbitals of oxygen and s or p orbitals of germanium in compound 3 (i–l), s or p orbitals of oxygen and p or sp1.45 orbitals of germanium in compound 10 (m–p), and p orbital of oxygen and σ* orbital of Ge–S bond in compound 10 (q). Energy values are given in kcal mol–1. Hydrogen atoms are omitted for clarity. The cut–off interaction energies for LP → LP* and LP → BD* are ≥30 kcal mol–1 and 20 kcal mol–1, respectively.

As none of the monoanionic ligands, such as β-diketiminate and amidinate ligands, are known to stabilise compounds with formal Ge O bonds, it is of interest to examine how the bulky monoanionic aminotroponiminate (ATI) ligand used in the present study helps to stabilise various compounds with formal Ge O bonds. NBO second-order perturbation theory analysis reveals the existence of donor–acceptor interactions between (a) spx orbitals of nitrogen atoms of the ATI ligand to vacant s, p or spx orbitals of germanium in compounds 1–3 and 10 (Fig. S66a, b, S67a–d, S68a–d, and S69a–d; see the ESI‡); (b) NATI orbitals to the σ* orbital of the Ge–Cl bond in compound 1 (Fig. S66c and d; see the ESI‡) and NATI orbitals to the σ* orbital of the Ge–S bond in compound 10 (Fig. S69e and f; see the ESI‡); and (c) s or p orbitals of the chlorine atom to π* orbitals of Ge–NATI bonds in compound 1 (Fig. S66e and f; see the ESI‡). Owing to the interactions of types (b) and (c), the energies of the σ* orbital of the Ge–Cl bond in compound 1, π* orbitals of the Ge–NATI bonds in compound 1, and the σ* orbital of the Ge–S bond in compound 10 are lower, and these orbitals are available for accepting electrons donated by the O atom of the Ge O bond. Further, energy decomposition analysis (EDA)25 was performed using {Y–Ge O → B(C6F5)3} (Y = Cl (1), OSiPh3 (2), NC4H4 (3), SPh (10)) as one fragment and the {ATI} ligand as another fragment with frozen geometries obtained from DFT calculations; the results are summarised in Table S7 (see the ESI‡). The large interaction energy (Eint) observed for these compounds arises essentially due to the favourable ΔEorb term that describes the stabilising interaction between the ATI ligand and the Y–Ge O → B(C6F5)3 moiety (Y = Cl (1), OSiPh3 (2), NC4H4 (3), SPh (10)).
Conclusions
Donor–acceptor-stabilised germaacyl chloride (i-Bu)2ATIGe(O)(Cl) → B(C6F5)3 (1), germaester (i-Bu)2ATIGe(O)(OSiPh3) → B(C6F5)3 (2), and N-germaacyl pyrrole (i-Bu)2ATIGe(O)(NC4H4) → B(C6F5)3 (3) compounds were successfully isolated as stable species for the first time. Compounds 1, 2, and 3 can undergo nucleophilic substitution reactions without any disturbance to the Ge O → B(C6F5)3 moiety to afford germaynone (i-Bu)2ATIGe(O)(CCPh) → B(C6F5)3 (4), germaester (i-Bu)2ATIGe(O)(Ot-Bu) → B(C6F5)3 (5), germanone (i-Bu)2ATIGe(O)(R) → B(C6F5)3 (R = Ph 6, Me 7), and germaacyl thioester (i-Bu)2ATIGe(O)(SPh) → B(C6F5)3 (10) compounds in good yields. Interestingly, through the reactivity of 1 and 2, the feasibility to interconvert germaesters and germaacid chlorides is exposed. Attempts were also made to synthesise germaamides and germacarboxylic acids, and it is anticipated that the wisdom obtained during these endeavours will offer new directions to the isolation of these compounds as stable species in the near future.
Author contributions
M. K. S. carried out all the experimental studies and drafted the manuscript. S. S. and P. M. helped M. K. S. with some of the experimental studies. The theoretical studies were carried out by G. M., who also wrote the theoretical section of the manuscript. B. P. assisted G. M. with some of the theoretical calculations/write-up. S. N. and G. R. corrected the experimental and theoretical write-ups of the manuscript, respectively.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
M. K. S., P. M. and S. S. thank the Indian Institute of Technology Delhi (IITD), New Delhi, India, and the University Grants Commission (UGC), New Delhi, India, for research fellowships. S. N. thanks the SERB, Department of Science and Technology (DST), New Delhi, India, for funding (EMR/2017/005519) and DST-FIST for establishing single-crystal X-ray diffraction (SR/FST/CSII-027/2014) and HRMS (SR/FST/CS-1-195/2008) facilities in the Department of Chemistry, IIT Delhi.
Footnotes
†Dedicated to Prof. V. Chandrasekhar on the occasion of his 60th birthday.
‡Electronic supplementary information (ESI) available: Experimental section, UV-vis spectra of compounds 1, 2, and 10; molecular structure determination of compounds D1, D3–D5, 1–7, 9, and 10; computational details (PDF). CIFs for compounds D1, D3–D5, 1–7, 9, and 10, are deposited with the Cambridge Structural Database (CSD). CCDC 1564828–1564834, 1564836, and 1851011–1851015. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc05380d
References
- and references cited therein; ; (a) Xiong Y., Yao S., Driess M. Angew. Chem., Int. Ed. 2013;52:4302. doi: 10.1002/anie.201209766. [DOI] [PubMed] [Google Scholar]; (b) Asay M., Jones C., Driess M. Chem. Rev. 2011;111:354. doi: 10.1021/cr100216y. [DOI] [PubMed] [Google Scholar]; (c) Fischer C. R., Power P. P. Chem. Rev. 2010;110:3877. doi: 10.1021/cr100133q. [DOI] [PubMed] [Google Scholar]; (d) Mizuhata Y., Sasamori T., Tokitoh N. Chem. Rev. 2009;109:3479. doi: 10.1021/cr900093s. [DOI] [PubMed] [Google Scholar]; (e) Nagendran S., Roesky H. W. Organometallics. 2008;27:457. [Google Scholar]; (f) Okazaki R., Tokitoh N. Acc. Chem. Res. 2000;33:625. doi: 10.1021/ar980073b. [DOI] [PubMed] [Google Scholar]; (g) Power P. P. Chem. Rev. 1999;99:3463. doi: 10.1021/cr9408989. [DOI] [PubMed] [Google Scholar]; (h) Barrau J., Rima G. Coord. Chem. Rev. 1998;178–180:593. [Google Scholar]
- (a) Bonnefille E., Mazières S., Bibal C., Saffon N., Gornitzka H., Couret C. Eur. J. Inorg. Chem. 2008:4242. [Google Scholar]; (b) Pu L., Hardman N. J., Power P. P. Organometallics. 2001;20:5105. [Google Scholar]; (c) Veith M., Rammo A. Z. Anorg. Allg. Chem. 1997;623:861. [Google Scholar]; (d) Jutzi P., Schmidt H., Neumann B., Stammler H.-G. Organometallics. 1996;15:741. [Google Scholar]; (e) Tokitoh N., Matsumoto T., Okazaki R. Chem. Lett. 1995:1087. [Google Scholar]
- Wegner G. L., Berger R. J. F., Schier A., Schmidbaur H. Organometallics. 2001;20:418. [Google Scholar]
- (a) Tacke R., Kobelt C., Baus J. A., Bertermann R., Burschka C. Dalton Trans. 2015;44:14959. doi: 10.1039/c5dt01581b. [DOI] [PubMed] [Google Scholar]; (b) Junold K., Nutz M., Baus J. A., Burschka C., Fonseca Guerra C., Bickelhaupt F. M., Tacke R. Chem.–Eur. J. 2014;20:9319. doi: 10.1002/chem.201402483. [DOI] [PubMed] [Google Scholar]; (c) Azhakar R., Ghadwal R. S., Roesky H. W., Wolf H., Stalke D. Chem. Commun. 2012;48:4561. doi: 10.1039/c2cc31041d. [DOI] [PubMed] [Google Scholar]; (d) Jana A., Azhakar R., Sarish S. P., Samuel P. P., Roesky H. W., Schulzke C., Koley D. Eur. J. Inorg. Chem. 2011:5006. [Google Scholar]; (e) Sen S. S., Tavčar G., Roesky H. W., Kratzert D., Hey J., Stalke D. Organometallics. 2010;29:2343. [Google Scholar]
- (a) Ellis D., Hitchcock P. B., Lappert M. F. J. Chem. Soc., Dalton Trans. 1992:3397. [Google Scholar]; (b) Wang H., Xie Z. Eur. J. Inorg. Chem. 2017:4430. [Google Scholar]
- Chlupatý T., Padělková Z., DeProft F., Willem R., Růzička A. Organometallics. 2012;31:2203. [Google Scholar]
- For selected references see: ; (a) Alvarado-Beltran I., Rosas-Sánchez A., Baceiredo A., Saffon-Merceron N., Branchadell V., Kato T. Angew. Chem., Int. Ed. 2017;56:10481. doi: 10.1002/anie.201705644. [DOI] [PubMed] [Google Scholar]; (b) Linden M. M., Reisenauer H. P., Gerbig D., Karni M., Schäfer A., Müller T., Apeloig Y., Schreiner P. R. Angew. Chem., Int. Ed. 2015;54:12404. doi: 10.1002/anie.201501844. [DOI] [PubMed] [Google Scholar]; (c) Ahmad S. U., Szilvási T., Irran E., Inoue S. J. Am. Chem. Soc. 2015;137:5828. doi: 10.1021/jacs.5b01853. [DOI] [PubMed] [Google Scholar]; (d) Muraoka T., Abe K., Kimura H., Haga Y., Ueno K., Sunada Y. Dalton Trans. 2014;43:16610. doi: 10.1039/c4dt02159b. [DOI] [PubMed] [Google Scholar]; (e) Filippou A. C., Baars B., Chernov O., Lebedev Y. N., Schnakenburg G. Angew. Chem., Int. Ed. 2014;53:565. doi: 10.1002/anie.201308433. [DOI] [PubMed] [Google Scholar]; (f) Sen S. S. Angew. Chem., Int. Ed. 2014;53:8820–8822. doi: 10.1002/anie.201404793. [DOI] [PubMed] [Google Scholar]; (g) Rodriguez R., Troadec T., Gau D., Saffon-Merceron N., Hashizume D., Miqueu K., Sotiropoulos J., Baceiredo A., Kato T. Angew. Chem., Int. Ed. 2013;52:4426. doi: 10.1002/anie.201210010. [DOI] [PubMed] [Google Scholar]; (h) Xiong Y., Yao S., Driess M. Angew. Chem., Int. Ed. 2013;52:4302. doi: 10.1002/anie.201209766. [DOI] [PubMed] [Google Scholar]; (i) Muraoka T., Abe K., Haga Y., Nakamura T., Ueno K. J. Am. Chem. Soc. 2011;133:15365. doi: 10.1021/ja207395w. [DOI] [PubMed] [Google Scholar]; (j) Gao Y., Hu H., Cui C. Chem.–Eur. J. 2011;17:8803. doi: 10.1002/chem.201100587. [DOI] [PubMed] [Google Scholar]; (k) Xiong Y., Yao S., Driess M. Dalton Trans. 2010;39:9282. doi: 10.1039/c0dt00148a. [DOI] [PubMed] [Google Scholar]; (l) Yao S., Xiong Y., Driess M. Chem.–Eur. J. 2010;16:1281. doi: 10.1002/chem.200902467. [DOI] [PubMed] [Google Scholar]; (m) Xiong Y., Yao S., Müller R., Kaupp M., Driess M. Nat. Chem. 2010;2:577. doi: 10.1038/nchem.666. [DOI] [PubMed] [Google Scholar]; (n) Epping J. D., Yao S., Karni M., Apeloig Y., Driess M. J. Am. Chem. Soc. 2010;132:5443. doi: 10.1021/ja1004812. [DOI] [PubMed] [Google Scholar]; (o) Xiong Y., Yao S., Driess M. J. Am. Chem. Soc. 2009;131:7562. doi: 10.1021/ja9031049. [DOI] [PubMed] [Google Scholar]
- (a) Yao S., Xiong Y., Wang W., Driess M. Chem.–Eur. J. 2011;17:4890. doi: 10.1002/chem.201003409. [DOI] [PubMed] [Google Scholar]; (b) Yao S., Xiong Y., Driess M. Chem. Commun. 2009:6466. doi: 10.1039/b914060c. [DOI] [PubMed] [Google Scholar]
- Li L., Fukawa T., Matsuo T., Hashizume D., Fueno H., Tanaka K., Tamao K. Nat. Chem. 2012;4:361. doi: 10.1038/nchem.1305. [DOI] [PubMed] [Google Scholar]
- Sinhababu S., Yadav D., Karwasara S., Sharma M. K., Mukherjee G., Rajaraman G., Nagendran S. Angew. Chem., Int. Ed. 2016;55:7742. doi: 10.1002/anie.201601445. [DOI] [PubMed] [Google Scholar]
- Zabula A. V., Pape T., Hepp A., Schappacher F. M., Rodewald U. C., Pöttgen R., Hahn F. E. J. Am. Chem. Soc. 2008;130:5648. doi: 10.1021/ja801000b. [DOI] [PubMed] [Google Scholar]
- (a) Do D. C. H., Protchenko A. V., Ángeles Fuentes M., Hicks J., Kolychev E. L., Vasko P., Aldridge S. Angew. Chem., Int. Ed. 2018;57:13907. doi: 10.1002/anie.201807543. [DOI] [PubMed] [Google Scholar]; (b) Rodriguez R., Gau D., Troadec T., Saffon-Merceron N., Branchadell V., Baceiredo A., Kato T. Angew. Chem., Int. Ed. 2013;52:8980. doi: 10.1002/anie.201304482. [DOI] [PubMed] [Google Scholar]; (c) Ghadwal R. S., Azhakar R., Roesky H. W., Pröpper K., Dittrich B., Goedecke C., Frenking G. Chem. Commun. 2012;48:8186. doi: 10.1039/c2cc32887a. [DOI] [PubMed] [Google Scholar]; (d) Ghadwal R. S., Azhakar R., Roesky H. W., Propper K., Dittrich B., Klein S., Frenking G. J. Am. Chem. Soc. 2011;133:17552. doi: 10.1021/ja206702e. [DOI] [PubMed] [Google Scholar]; (e) Xiong Y., Yao S., Müller R., Kaupp M., Driess M. J. Am. Chem. Soc. 2010;132:6912. doi: 10.1021/ja1031024. [DOI] [PubMed] [Google Scholar]; (f) Xiong Y., Yao S., Driess M. Angew. Chem., Int. Ed. 2010;49:6642. doi: 10.1002/anie.201002970. [DOI] [PubMed] [Google Scholar]; (g) Yao S., Brym M., Wullen C., Driess M. Angew. Chem., Int. Ed. 2007;46:4159. doi: 10.1002/anie.200700398. [DOI] [PubMed] [Google Scholar]; (h) Yao S., Xiong Y., Brym M., Driess M. J. Am. Chem. Soc. 2007;129:7268. doi: 10.1021/ja072425s. [DOI] [PubMed] [Google Scholar]
- (a) Sinhababu S., Siwatch R. K., Mukherjee G., Rajaraman G., Nagendran S. Inorg. Chem. 2012;51:9240. doi: 10.1021/ic300715y. [DOI] [PubMed] [Google Scholar]; (b) Siwatch R. K., Kundu S., Kumar D., Nagendran S. Organometallics. 2011;30:1998. [Google Scholar]; (c) Dias H. V. R., Wang Z., Jin W. Coord. Chem. Rev. 1998;176:67. [Google Scholar]; (d) Dias H. V. R., Wang Z. J. Am. Chem. Soc. 1997;119:4650. [Google Scholar]
- (a) Yang D., Guo J., Wu H., Ding Y., Zheng W. Dalton Trans. 2012;41:2187. doi: 10.1039/c1dt11774b. [DOI] [PubMed] [Google Scholar]; (b) Al-Rafia S. M. I., Lummis P. A., Ferguson M. J., McDonald R., Rivard E. Inorg. Chem. 2010;49:9709. doi: 10.1021/ic101485a. [DOI] [PubMed] [Google Scholar]; (c) Veith M., Becker S., Huch V. Angew. Chem., Int. Ed. 1989;28:1237. [Google Scholar]
- Karwasara S., Siwatch R. K., Jha C. K., Nagendran S. Organometallics. 2015;34:3246. [Google Scholar]
- Karwasara S., Sharma M. K., Tripathi R., Nagendran S. Organometallics. 2013;32:3830. [Google Scholar]
- (a) Beckett M. A., Brassington D. S., Coles S. J., Hursthouse M. B. Inorg. Chem. Commun. 2000;3(10):530. [Google Scholar]; (b) Beckett M. A., Strickland G. C. Polymer. 1996;37:4629. [Google Scholar]
- Hoshi M., Shirakawa K., Okimoto M. Tetrahedron Lett. 2007;48:8475. [Google Scholar]
- Bähr A., Wilkins L. C., Ollegott K., Kariuki B. M., Melen R. L. Molecules. 2015;20:4530. doi: 10.3390/molecules20034530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neculai D., Roesky H. W., Neculai A. M., Magull J., Walfort B., Stalke D. Angew. Chem., Int. Ed. 2002;41:4294. doi: 10.1002/1521-3773(20021115)41:22<4294::AID-ANIE4294>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- Swarnakar A. K., Hering-Junghans C., Ferguson M. J., McDonald R., Rivard E. Chem.–Eur. J. 2017;23:8628. doi: 10.1002/chem.201702154. [DOI] [PubMed] [Google Scholar]
- CCDC 1564828–1564834, ; 1564836, and ; 1851011–1851015 contains the crystallographic data for this paper.
- Wheinhold F. and Landis C., Valency and Bonding, Cambridge, 2005. [Google Scholar]
- (a) Reed A. E., Curtiss L. A., Weinhold F. Chem. Rev. 1988;88:899. [Google Scholar]; (b) Glendening E. D., Reed A. E., Carpenter J. E. and Weinhold F., NBO Version 3.1.
- (a) Gorelsky S. I., AOMix: Program for Molecular Orbital Analysis, version 6.6, University of Ottawa, Ottawa, 2010, http://www.sg-chem.net/.; (b) Gorelsky S. I., Ghosh S., Solomon E. I. J. Am. Chem. Soc. 2006;128:278. doi: 10.1021/ja055856o. [DOI] [PubMed] [Google Scholar]; (c) Ziegler T., Rauk A. Theor. Chem. Acc. 1977;46:1. [Google Scholar]; (d) Kitaura K., Morokuma K. Int. J. Quantum Chem. 1976;10:325. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.