

Abstract
Organelle genome fragmentation has been found in a wide range of eukaryotic lineages; however, its use in phylogenetic reconstruction has not been demonstrated. We explored the use of mitochondrial (mt) genome fragmentation in resolving the controversial suborder-level phylogeny of parasitic lice (order Phthiraptera). There are approximately 5000 species of parasitic lice in four suborders (Amblycera, Ischnocera, Rhynchophthirina, and Anoplura), which infest mammals and birds. The phylogenetic relationships among these suborders are unresolved despite decades of studies. We sequenced the mt genomes of eight species of parasitic lice and compared them with 17 other species of parasitic lice sequenced previously. We found that the typical single-chromosome mt genome is retained in the lice of birds but fragmented into many minichromosomes in the lice of eutherian mammals. The shared derived feature of mt genome fragmentation unites the eutherian mammal lice of Ischnocera (family Trichodectidae) with Anoplura and Rhynchophthirina to the exclusion of the bird lice of Ischnocera (family Philopteridae). The novel clade, namely Mitodivisia, is also supported by phylogenetic analysis of mt genome and cox1 gene sequences. Our results demonstrate, for the first time, that organelle genome fragmentation is informative for resolving controversial high-level phylogenies.
Keywords: Genome fragmentation, mitochondrial genome, parasitic lice, phylogeny
Eukaryotic organelles, such as mitochondria and chloroplasts, have their own genomes that encode genes for cellular energy production (Wallace 1982; Keeling and Archibald 2008). Organelle genomes typically consist of a single, circular chromosome; however, fragmented organelle genomes that comprise multiple chromosomes have been found in Dinoflagellata (Zhang et al. 1999; Koumandou et al. 2004; Howe et al. 2008), Mesozoa (Watanabe et al. 1999), Nematoda (Armstrong et al. 2000; Gibson et al. 2007), Cnidaria (Voigt et al. 2008; Smith et al. 2011a), Ichthyosporea (Burger et al. 2003), Euglenozoa (Marande and Burger 2007), Porifera (Lavrov et al. 2012), and Insecta (Shao et al. 2009; Wei et al. 2012; Dickey et al. 2015). Changes in organelle genome organization such as fragmentation provide a valuable source of information, independent of nucleotide sequences, for molecular phylogenetic reconstruction, in particular for resolving controversial high-level relationships (Boore and Brown 1998; Rokas and Holland 2000; Dowton et al. 2009; Cameron 2014). Although organelle genome fragmentation has been found in a wide range of lineages, its use in phylogenetic reconstruction has not been demonstrated. Here, we show that mitochondrial (mt) genome fragmentation unites the parasitic lice of eutherian mammals and is informative for resolving the controversial suborder-level phylogeny of parasitic lice (order Phthiraptera).
There are approximately 5000 species of parasitic lice in 303 genera, 24 families, and four suborders, parasitizing approximately 4000 species of birds and approximately 1000 species of mammals (Kim 1988; Price et al. 2003). Most of the parasitic lice are highly host specific; each species of louse is commonly found only on a single or only a few closely related host species. Parasitic lice are divided into chewing lice and sucking lice by mouthparts. Chewing lice are in three suborders: Amblycera, Ischnocera, and Rhynchophthirina, whereas sucking lice are in the suborder Anoplura (Barker et al. 2003; Price et al. 2003). Most of the lice (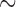 88%) in the Amblycera and the Ischnocera parasitize birds; the rest (
88%) in the Amblycera and the Ischnocera parasitize birds; the rest (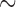 12%) parasitize mammals. All of the lice in the Rhynchophthirina and the Anoplura parasitize only eutherian mammals (Marshall 1981; Price et al. 2003). Of the four suborders, Ischnocera is the most species-rich (3120 species) and is divided into two families: Trichodectidae (lice of eutherian mammals) and Philopteridae (lice of birds except for the lemur louse, Trichophilopterus babakotophilus, which was almost certain host-switched from birds (Cruickshank et al. 2001; Price et al. 2003).
12%) parasitize mammals. All of the lice in the Rhynchophthirina and the Anoplura parasitize only eutherian mammals (Marshall 1981; Price et al. 2003). Of the four suborders, Ischnocera is the most species-rich (3120 species) and is divided into two families: Trichodectidae (lice of eutherian mammals) and Philopteridae (lice of birds except for the lemur louse, Trichophilopterus babakotophilus, which was almost certain host-switched from birds (Cruickshank et al. 2001; Price et al. 2003).
The suborder-level phylogeny of the parasitic lice has not been resolved despite decades of studies. Initially, all of the chewing lice from the three suborders Amblycera, Ischnocera, and Rhynchophthirina were grouped together as Mallophaga (Ferris 1951; Kim and Ludwig 1982). Subsequent studies, however, showed that Mallophaga was paraphyletic as the chewing mouthpart was plesiomorphic, that is, an ancestral character for all parasitic lice (Harrison 1928; Hopkins 1949; Clay 1970). Lyal (1985) analyzed extensively the morphology of Phthiraptera and concluded that: (i) Rhynchophthirina was more closely related to the sucking lice in the Anoplura than to the chewing lice in the Amblycera and Ischnocera; (ii) Ischnocera was the sister group to Rhynchophthirina + Anoplura, and (iii) Amblycera was the sister group to Ischnocera + (Rhynchophthirina + Anoplura).
Despite recognizing the Ischnocera as a suborder, Lyal (1985) could not find any synapomorphies to demonstrate the Ischnocera as a monophyletic group. Lyal (1985) accepted five families in the Ischnocera, which were later merged into two families by hosts: lice of eutherian mammals are in the Trichodectidae; lice of birds are in the Philopteridae (Price et al. 2003). There is evidence that Trichodectidae is monophyletic, whereas Philopteridae is paraphyletic or polyphyletic. After Lyal (1985), several studies used molecular data to address the suborder-level phylogeny of parasitic lice. Cruickshank et al. (2001) analyzed EF1-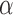 sequences of 127 species from the four suborders and showed the Ischnocera to be paraphyletic. Johnson and Whiting (2002) analyzed the sequences of three genes (EF1-
sequences of 127 species from the four suborders and showed the Ischnocera to be paraphyletic. Johnson and Whiting (2002) analyzed the sequences of three genes (EF1-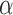 ,
, 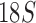 , and cox1) of 21 species from the four suborders and showed the Ischnocera to be monophyletic. Yoshizawa and Johnson (2003) analyzed mt rrnS and rrnL sequences of 18 species and showed the Ischnocera to be paraphyletic as species from the Trichodectidae and the Anoplura were grouped together to the exclusion of species from the Philopteridae. Barker et al. (2003) analyzed
, and cox1) of 21 species from the four suborders and showed the Ischnocera to be monophyletic. Yoshizawa and Johnson (2003) analyzed mt rrnS and rrnL sequences of 18 species and showed the Ischnocera to be paraphyletic as species from the Trichodectidae and the Anoplura were grouped together to the exclusion of species from the Philopteridae. Barker et al. (2003) analyzed 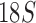 rRNA sequences of 33 species and showed the Ischnocera to be monophyletic. Yoshizawa and Johnson (2010) analyzed five genes (
rRNA sequences of 33 species and showed the Ischnocera to be monophyletic. Yoshizawa and Johnson (2010) analyzed five genes (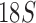 , histone 3, wingless, rrnL, and cox1) and showed the Ischnocera to be paraphyletic. Recently, Johnson et al. (2018) analyzed 1107 nuclear genes of 46 species and showed that Ischnocera to be paraphyletic as two species of the Trichodectidae were grouped with species of the Anoplura and the Rhynchophthirina to the exclusion of species from the Philopteridae. Although the suborder-level phylogeny of parasitic lice established by Lyal (1985) was flawed due to the questionable monophyly of Ischnocera as a suborder, the molecular studies in the past three decades advanced very little in this regard.
, histone 3, wingless, rrnL, and cox1) and showed the Ischnocera to be paraphyletic. Recently, Johnson et al. (2018) analyzed 1107 nuclear genes of 46 species and showed that Ischnocera to be paraphyletic as two species of the Trichodectidae were grouped with species of the Anoplura and the Rhynchophthirina to the exclusion of species from the Philopteridae. Although the suborder-level phylogeny of parasitic lice established by Lyal (1985) was flawed due to the questionable monophyly of Ischnocera as a suborder, the molecular studies in the past three decades advanced very little in this regard.
Extensive fragmentation of the mt genome was first discovered in the human body louse, Pediculus humanus (Shao et al. 2009). Since then, 11 other species of sucking lice (Anoplura) and the elephant louse, Haematomyzus elephantis (Rhynchophthirina), have been found with fragmented mt genomes (Shao et al. 2012, 2015, 2017; Jiang et al. 2013; Song et al. 2014; Dong et al. 2014a,b; Herd et al. 2015). The mt genomes of these lice have 9–20 minichromosomes; each minichromosome is 1.5–4 kb in size and has 1–8 genes (Shao et al. 2017). In contrast, the typical single-chromosome mt genomes have been found in three species of bird lice in the Philopteridae (Ischnocera) and the wallaby louse (Boopidae, Amblycera) (Shao et al. 2001; Covacin et al. 2006; Cameron et al. 2007; Cameron et al. 2011). Cameron et al. (2011) sequenced the partial mt genome of the deer louse, Damalinia meyeri (Ischnocera, Trichodectidae) and found three minichromosomes. Although Cameron et al. (2011) also suggested that three bird lice (Ischnocera, Philopteridae) might have multi-chromosomal mt genomes, no complete mt genome data from any bird louse is available to date to support this suggestion.
To explore whether or not mt genome fragmentation is informative in resolving controversial high-level phylogenies, we sequenced the mt genomes of another eight species of parasitic lice: four species of eutherian mammal lice from the Trichodectidae (Ischnocera), two species of bird lice from the Philopteridae (Ischnocera), and two species of bird lice from the Menoponidae (Amblycera). We found that the bird lice in both the Ischnocera and the Amblycera have the typical single-chromosome mt genomes, whereas eutherian mammal lice in the Trichodectidae have fragmented mt genomes, as do the sucking lice (Anoplura) and the elephant louse (Rhynchophthirina). We show that mt genome fragmentation unites the lice of eutherian mammals from three suborders, Anoplura, Ischnocera, and Rhynchophthirina and is informative for resolving controversial high-level phylogenies that cannot be resolved reliably by morphological data and sequences of single genes or a few genes.
Materials and Methods
Louse Collection, DNA Extraction, Mitochondrial Genome Sequencing and Assembly
The eight species of parasitic lice sequenced in this study were: cattle louse, Bovicola bovis, goat louse, Bovicola caprae, sheep louse, Bovicola ovis, dog louse, Trichodectes canis, vulture lice, Colpocephalum griffoneae and Falcolipeurus quadripustulatus, peafowl louse, Amyrsidea minuta, and pigeon louse, Campanulotes compar (Table 1). Lice were preserved in ethanol after removal from their hosts until DNA extraction. For each louse species, total DNA was extracted from 20 to 30 individual lice with DNeasy Tissue kit (QIAGEN). Fragments of two mt genes of each species, rrnS (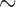 310 bp) and cox1 (
310 bp) and cox1 (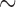 530 bp), were amplified by polymerase chain reaction (PCR) with primer pairs 12SA–12SB and mtd6–mtd11 and sequenced using the Sanger method; these primers target the conserved motifs among arthropods (Supplementary Table S1 available on Dryad at http://dx.doi.org/10.5061/dryad.qg8vf45). A library was prepared using Truseq nano DNA kit (Illumina) for each species using genomic DNA with an insert size of 450 bp and was sequenced on the Illumina Hiseq 2500 platform at Berry Genomics, Beijing; 4 Gb clean data (250 bp pair-end reads) was obtained for each louse species. Raw reads were filtered by removing reads containing adaptor contamination (with
530 bp), were amplified by polymerase chain reaction (PCR) with primer pairs 12SA–12SB and mtd6–mtd11 and sequenced using the Sanger method; these primers target the conserved motifs among arthropods (Supplementary Table S1 available on Dryad at http://dx.doi.org/10.5061/dryad.qg8vf45). A library was prepared using Truseq nano DNA kit (Illumina) for each species using genomic DNA with an insert size of 450 bp and was sequenced on the Illumina Hiseq 2500 platform at Berry Genomics, Beijing; 4 Gb clean data (250 bp pair-end reads) was obtained for each louse species. Raw reads were filtered by removing reads containing adaptor contamination (with 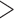 15 bp matched to the adaptor sequence), poly-Ns (
15 bp matched to the adaptor sequence), poly-Ns (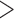 15 bp Ns), or
15 bp Ns), or 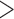 75 bp bases with quality score
75 bp bases with quality score 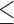 3. The number of clean reads for each library was shown in Supplementary Table S2 available on Dryad. Clean reads were assembled de novo using IDBA-UD (Peng et al. 2012) with the parameters: similarity threshold 98%, minimum
3. The number of clean reads for each library was shown in Supplementary Table S2 available on Dryad. Clean reads were assembled de novo using IDBA-UD (Peng et al. 2012) with the parameters: similarity threshold 98%, minimum 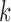 value 80, and maximum
value 80, and maximum 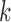 value 240. To isolate mt genome sequences, the contigs obtained were searched for the presence of rrnS (
value 240. To isolate mt genome sequences, the contigs obtained were searched for the presence of rrnS (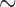 310 bp) and cox1 (
310 bp) and cox1 (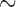 530 bp) sequences, and against records in GenBank using BLAST (Altschup et al. 1990). For the louse species that have a single mt chromosome, a single contig was obtained with the full set of mt genes; the mt genome was then manually checked and annotated in Geneious v9.0.4 (Kearse et al. 2012). For the louse species that have fragmented mt genomes, a contig that had mt rrnS but not any other protein-coding or rRNA genes was obtained; another contig that had mt cox1 but not any other protein-coding or rRNA genes was obtained. These two contigs were also obtained when rrnS (
530 bp) sequences, and against records in GenBank using BLAST (Altschup et al. 1990). For the louse species that have a single mt chromosome, a single contig was obtained with the full set of mt genes; the mt genome was then manually checked and annotated in Geneious v9.0.4 (Kearse et al. 2012). For the louse species that have fragmented mt genomes, a contig that had mt rrnS but not any other protein-coding or rRNA genes was obtained; another contig that had mt cox1 but not any other protein-coding or rRNA genes was obtained. These two contigs were also obtained when rrnS (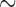 310 bp) and cox1 (
310 bp) and cox1 (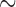 530 bp) sequences of each louse species were used respectively as references to assemble the 4 Gb clean data with Geneious v9.0.4 using “Map to reference” option. The assembly parameters were: minimum overlap identity 98%; no gaps; maximum mismatches per read 2%; maximum ambiguity 2; and minimum overlap 200 bp. When a minichromosome was assembled in full length, the two ends of the contig overlapped, indicating circular organization of the minichromosome. We observed in previous studies that each mt minichromosome has a distinct coding region but a well-conserved non-coding region, in particular the sequences immediately upstream and downstream the coding region (Shao et al. 2009, 2012, 2017; Jiang et al. 2013; Dong et al. 2014a,b; Song et al. 2014). Once the rrnS and cox1 minichromosomes were assembled, we identified the conserved non-coding region sequences between the two minichromosomes and used the conserved sequences as references to align with the 4 Gb clean data obtained for each louse species. In this way, we identified sequence reads from the two ends of the coding regions of all other mt minichromosomes. We then assembled these minichromosomes in full length using the same method stated above for rrnS and cox1 minichromosome assembly. The mean coverage for the mt genomes of the eight louse species sequenced in this study ranges from 397 to 3211 (Supplementary Table S2 available on Dryad). To verify the size and circular organization of each assembled mt minichromosome, specific primers were designed from the coding region of each minichromosome; PCRs with these primers amplified each circular minichromosome in full or nearly full length (Supplementary Table S1 available on Dryad).
530 bp) sequences of each louse species were used respectively as references to assemble the 4 Gb clean data with Geneious v9.0.4 using “Map to reference” option. The assembly parameters were: minimum overlap identity 98%; no gaps; maximum mismatches per read 2%; maximum ambiguity 2; and minimum overlap 200 bp. When a minichromosome was assembled in full length, the two ends of the contig overlapped, indicating circular organization of the minichromosome. We observed in previous studies that each mt minichromosome has a distinct coding region but a well-conserved non-coding region, in particular the sequences immediately upstream and downstream the coding region (Shao et al. 2009, 2012, 2017; Jiang et al. 2013; Dong et al. 2014a,b; Song et al. 2014). Once the rrnS and cox1 minichromosomes were assembled, we identified the conserved non-coding region sequences between the two minichromosomes and used the conserved sequences as references to align with the 4 Gb clean data obtained for each louse species. In this way, we identified sequence reads from the two ends of the coding regions of all other mt minichromosomes. We then assembled these minichromosomes in full length using the same method stated above for rrnS and cox1 minichromosome assembly. The mean coverage for the mt genomes of the eight louse species sequenced in this study ranges from 397 to 3211 (Supplementary Table S2 available on Dryad). To verify the size and circular organization of each assembled mt minichromosome, specific primers were designed from the coding region of each minichromosome; PCRs with these primers amplified each circular minichromosome in full or nearly full length (Supplementary Table S1 available on Dryad).
Table 1.
The parasitic lice and other insects included in this study
| Suborder | Family | Species | GenBank accession number | |
|---|---|---|---|---|
| Hemiptera | Heteroptera | Nabidae | Alloeorhynchus bakeri | NC_016432 |
| Psocoptera | Trogiomorpha | Lepidopsocidae | Lepidopsocid sp. | NC_004816 |
| Psocomorpha | Psocidae | Longivalvus hyalospilus | JQ910986 | |
| Psococerastis albimaculata | NC_021400 | |||
| Troctomorpha | Liposcelidae | Liposcelis bostrychophila | JN645275 | |
| Phthiraptera | Amblycera | Menoponidae | Colpocephalum griffoneae |
MH001228
|
| Amyrsidea minuta |
MH001227
|
|||
| Boopidae | Heterodoxus macropus | NC_002651 | ||
| Ischnocera | Trichodectidae | Bovicola bovis |
MH001189–200
|
|
| Bovicola ovis |
MH001201–12
|
|||
| Bovicola caprae |
MH001176–88
|
|||
| Trichodectes canis |
MH001213–24 , MH823541 , MH823541
|
|||
| Philopteridae | Ibidoecus bisignatus | NC_015999 | ||
| Campanulotes bidentatus compar | NC_007884 | |||
| Bothriometopus macrocnemis | NC_009983 | |||
| Falcolipeurus quadripustulatus |
MH001226
|
|||
| Campanulotes compar |
MH001225
|
|||
| Rhyncophthirina | Haematomyzidae | Haematomyzus elephantis | KF933032–41 | |
| Anoplura | Polyplacidae | Polyplax spinulosa | KF647762–72 | |
| Polyplax asiatica | KF647751–61 | |||
| Hoplopleuridae | Hoplopleura akanezumi | KJ648922–32 | ||
| Hoplopleura kitti | KJ648933–43 | |||
| Microthoraciidae | Microthoracius praelongiceps | KX090378–89 | ||
| Haematopinidae | Haematopinus asini | KF939318, KF939322, KF939324, KF939326, KJ434034–38 | ||
| Haematopinus apri | KC814611–19 | |||
| Haematopinus suis | KC814602–10 | |||
| Pthiridae | Pthirus pubis | EU219988–95, HM241895–98 | ||
| Pediculidae | Pediculus schaeffi | KC241882–97, KR706168–69 | ||
| Pediculus capitis | JX080388–407 | |||
| Pediculus humanus | FJ499473–90 |
 These sequences were produced by the current study.
These sequences were produced by the current study.
Ex Taq (Takara) was used in the initial short PCRs with the following cycling conditions: 94 C for 1 min; 35 cycles of 98
C for 1 min; 35 cycles of 98 C for 10 s, 45
C for 10 s, 45 C for 30 s, 72
C for 30 s, 72 C for 1 min; and a final extension of 72
C for 1 min; and a final extension of 72 C for 2 min. LA Taq (Takara) was used in the long PCRs with the cycling conditions: 94
C for 2 min. LA Taq (Takara) was used in the long PCRs with the cycling conditions: 94 C for 1 min; 35 cycles of 98
C for 1 min; 35 cycles of 98 C for 10 s, 50–65
C for 10 s, 50–65 C (depending on primers) for 40 s, 68
C (depending on primers) for 40 s, 68 C for 4 min; and a final extension of 72
C for 4 min; and a final extension of 72 C for 8 min. Negative controls were run with each PCR experiment. PCR amplicons were checked with agarose gel electrophoresis (1%); the sizes of amplicons were estimated by comparison with DNA markers. For Sanger sequencing, PCR products were purified with Wizard SV Gel/PCR clean-up system (Promega).
C for 8 min. Negative controls were run with each PCR experiment. PCR amplicons were checked with agarose gel electrophoresis (1%); the sizes of amplicons were estimated by comparison with DNA markers. For Sanger sequencing, PCR products were purified with Wizard SV Gel/PCR clean-up system (Promega).
To annotate the assembled mt genomes, protein-coding and rRNA genes were identified using BLAST searches in GenBank and subsequently by alignment with sequences of other parasitic lice available in GenBank. tRNA genes were identified with tRNAscan-SE Search Server v1.21(Lowe and Eddy 1997) and ARWEN (Laslett and Canback 2008). tRNA genes that could not be identified by these programs were identified by sequence alignment and comparison with those of the lice available in GenBank. The annotated mt genomes of the eight species of parasitic lice sequenced in this study have been deposited in GenBank under accession numbers MH001176-MH001228 and MH823541.
Phylogenetic Analyses
We used two approaches to reconstruct the high-level phylogeny of parasitic lice. Twenty-five species of parasitic lice were included in our analyses: (i) the wallaby louse, Heterodoxus macropus; (ii) four species of chewing lice of eutherian mammals from the family Trichodectidae; (iii) seven species of chewing lice of birds; (iv) the elephant louse, Haematomyzus elephantis; and (v) 12 species of sucking lice (Table 1). In the first approach, we formed species into clades using shared derived features (i.e., synapomorphies) in mt genome organization. For each species of parasitic lice, we explored three types of features: (i) whether the species has the typical single-chromosome mt genome or a fragmented mt genome with multiple minichromosomes; (ii) whether the species shares any type of minichromosome with other species; and (iii) whether the species shares any derived gene clusters with other species. This approach is based on traditional cladistics principles, which are used widely in morphology-based phylogenetic analyses, and in a small number of cases, used in genome-structure-based phylogenetic analyses (Boore and Brown 1998; Rokas and Holland 2000; Gissi et al. 2008; Babbucci et al. 2014). In the second approach, we inferred the phylogeny using mt genome sequences. We retrieved the sequences of eight mt protein-coding genes (atp6, atp8, cox1, cox2, cox3, cob, nad4L, nad6) and two rRNA genes (rrnS and rrnL) of 22 species of parasitic lice, book lice and bark lice available in the NCBI database and combined these sequences with those we generated in this study for the eight species of parasitic lice. Sequences of five protein-coding genes (nad1, nad2, nad3, nad4, and nad5) were only available for some but not all of the 30 species and thus were not included in our analysis. Each protein-coding gene was aligned individually based on codons for amino acids using the MAFFT algorithm implemented in TranslatorX with the L-INS-i strategy (Abascal et al. 2010). Two rRNA genes were individually aligned using the MAFFT 7.0 online server with the G-INS-i strategy (Katoh and Standley 2013). Individual gene alignments were concatenated after removing poorly aligned sites using GBlocks v0.91b (Talavera and Castresana 2007). Two concatenated alignments were used in subsequent phylogenetic analyses: (i) PCGRNA matrix, which contains all of the three codon positions of the eight protein-coding genes and the two rRNA genes (5677 bp in total) and (ii) PCG12RNA matrix, which contains only the first and the second codon positions of the eight protein-coding genes and the two rRNA genes (4182 bp in total). In order to maximize the number of species of parasitic lice in our phylogenetic analysis, we also used cox1 matrix, which contained cox1 sequences (1395 bp) of 41 species of parasitic lice (Supplementary Table S3 available on Dryad). All matrices were analyzed using maximum likelihood (ML) and Bayesian inference (BI) on IQ-TREE web server (Trifinopoulos et al. 2016) and PhyloBayes MPI 1.7a (Lartillot et al. 2013), respectively. For ML analyses, optimal evolutionary models were selected with the “Auto” option and “TIM+F+I+G4” model was chosen for all of the three matrices according to Bayesian Information Criterion. Maximum-likelihood phylogenetic trees were constructed using an ultrafast bootstrap approximation approach with 10,000 replicates. For Bayesian analyses, the site-heterogeneous mixture model (CAT+GTR) was used (Li et al. 2015; Song et al. 2016). Two independent chains starting from a random tree were run for 60,000 cycles, with trees being sampled every 10 cycles. The initial 1500 trees of each MCMC run were discarded as burn-in. A consensus tree was computed from the remaining 9000 trees combined from two runs, and the two runs converged at a maxdiff of less than 0.1. All phylogenetic analyses were carried out on the CIPRES Science Gateway (Miller et al. 2010) and resources from the National Computational Infrastructure (NCI).
Results
Chewing Lice of Eutherian Mammals but not Birds in the Suborder Ischnocera (Family Trichodectidae) have Fragmented Mitochondrial Genomes
Of the eight species of parasitic lice sequenced in this study, four species were chewing lice of eutherian mammals (Ischnocera, Trichodectidae): cattle louse, Bovicola bovis, goat louse, B. caprae, sheep louse, B. ovis, and dog louse, Trichodectes canis (Table 1). All of these four species of chewing lice of eutherian mammals have fragmented mt genomes, whereas the four species of bird lice in both Ischnocera and Amblycera have the typical single-chromosome mt genomes (Supplementary Fig. S1 available on Dryad). The partially sequenced mt genome of the deer louse Damalinia meyeri (Ischnocera, Trichodectidae) had three minichromosomes, indicating that this louse might have a fragmented mt genome too (Cameron et al. 2011). The cattle louse, B. bovis, and the sheep louse, B. ovis, have the same mt karyotype consisting of 12 minichromosomes, whereas the goat louse, B. caprae, and the dog louse, T. canis, have 13 minichromosomes (Fig. 1). Compared with the cattle louse and the sheep louse, the goat louse has 11 minichromosomes in common and differs only in one minichromosome (Fig. 1). The five genes in the trnG-nad3-trnE-cob-trnS minichromosome of the cattle louse and the sheep louse are in two separate minichromosomes in the goat lice: trnG-nad3 and trnE-cob-trnS
minichromosome of the cattle louse and the sheep louse are in two separate minichromosomes in the goat lice: trnG-nad3 and trnE-cob-trnS (note: minichromosomes are named after their gene content and arrangement). The dog louse, T. canis, has 10 minichromosomes in common with Bovicola species and differ in three minichromosomes. In the dog louse, nad3 and nad6 are on the same minichromosome, trnM-trnG-nad3-trnF-nad6-trnT (Fig. 1). Moreover, the dog louse has a minichromosome consisting of only two tRNA genes, trnY-trnP; minichromosomes with only tRNA genes were not found in the three Bovicola species above but were found previously in lice of the horse, human and chimpanzee (Shao et al. 2009; Shao and Barker 2011; Song et al. 2014).
(note: minichromosomes are named after their gene content and arrangement). The dog louse, T. canis, has 10 minichromosomes in common with Bovicola species and differ in three minichromosomes. In the dog louse, nad3 and nad6 are on the same minichromosome, trnM-trnG-nad3-trnF-nad6-trnT (Fig. 1). Moreover, the dog louse has a minichromosome consisting of only two tRNA genes, trnY-trnP; minichromosomes with only tRNA genes were not found in the three Bovicola species above but were found previously in lice of the horse, human and chimpanzee (Shao et al. 2009; Shao and Barker 2011; Song et al. 2014).
Figure 1.

Mitochondrial genome organization of the eutherian mammal lice in the family Trichodectidae (Ischnocera): cattle louse, Bovicola bovis, goat louse, B. caprae, sheep louse, B. ovis, and dog louse, Trichodectes canis. Gene name, transcription orientation and length (bp) are indicated in the coding region; non-coding regions are in black. Gene names are: atp6 and atp8 (for ATP synthase subunits 6 and 8), cox1-3 (for cytochrome c oxidase subunits 1-3), cob (for cytochrome b), nad1-6 and nad4L (for NADH dehydrogenase subunits 1-6 and 4L), rrnS and rrnL (for small and large subunits of ribosomal RNA). tRNA genes are indicated with their single-letter abbreviations of the corresponding amino acids.
The length of minichromosomes varies from 800 bp for trnY-trnP minichromosome in the dog louse to 2413 bp for trnG-nad3-trnE-cob-trnS minichromosome in the sheep louse. Regardless of the length, each minichromosome contains a coding region and a non-coding region (Fig. 1; Table 2). In the coding region, most minichromosomes have a single protein-coding or rRNA gene and 1–3 tRNA genes. No pseudo gene was found in any of the minichromosomes, nor was the same gene found on two different minichromosomes (Fig. 1). All of the mt genes in each species have the same orientation of transcription relative to the non-coding region except for trnT, which has an opposite orientation to that of all other genes (Fig. 1). The non-coding regions range from 286 bp for rrnL minichromosome in the goat louse to 1014 bp for cox3 minichromosome in the sheep louse (Table 2). Minichromosomes that have longer coding regions (such as nad5 and cox1) tend to have shorter non-coding regions, and vice versa. The size contrast between coding and non-coding regions seems common among the parasitic lice that have fragmented mt genomes and indicates a selective pressure for the overall size of minichromosomes in these lice (Shao et al. 2017).
minichromosome in the sheep louse. Regardless of the length, each minichromosome contains a coding region and a non-coding region (Fig. 1; Table 2). In the coding region, most minichromosomes have a single protein-coding or rRNA gene and 1–3 tRNA genes. No pseudo gene was found in any of the minichromosomes, nor was the same gene found on two different minichromosomes (Fig. 1). All of the mt genes in each species have the same orientation of transcription relative to the non-coding region except for trnT, which has an opposite orientation to that of all other genes (Fig. 1). The non-coding regions range from 286 bp for rrnL minichromosome in the goat louse to 1014 bp for cox3 minichromosome in the sheep louse (Table 2). Minichromosomes that have longer coding regions (such as nad5 and cox1) tend to have shorter non-coding regions, and vice versa. The size contrast between coding and non-coding regions seems common among the parasitic lice that have fragmented mt genomes and indicates a selective pressure for the overall size of minichromosomes in these lice (Shao et al. 2017).
Table 2.
Size of mitochondrial minichromosomes of the eutherian mammal lice in the Trichodectidae (Ischnocera) identified by Illumina sequencing
| Minichromosome | Minichromosome size (bp) | Size of coding region (bp) | Size of non-coding region (bp) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Bb | Bc | Bo | Tc | Bb | Bc | Bo | Tc | Bb | Bc | Bo | Tc | |
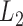 -atp8-atp6
-atp8-atp6
|
1368 | 1522 | 1891 | 1262 | 979 | 962 | 967 | 931 | 389 | 560 | 924 | 331 |
| I-cox1 | 1946 | 2038 | 2107 | 2014 | 1608 | 1604 | 1602 | 1599 | 338 | 434 | 505 | 415 |
| Y-cox2-R-nad4L | 1447 | 1535 | 1807 | 1525 | 1086 | 1080 | 1069 | 1045 | 361 | 455 | 738 | 480 |
| M-cox3 | 1252 | 1441 | 1913 | 1218 | 837 | 828 | 899 | 865 | 415 | 613 | 1014 | 353 |
G-nad3-E-cob-S

|
1979 | NA | 2413 | NA | 1614 | NA | 1638 | NA | 365 | NA | 775 | NA |
| W-nad1-Q | 1485 | 1620 | 1840 | 1427 | 1047 | 1050 | 1048 | 994 | 438 | 570 | 792 | 433 |
| N-nad2-C | 1565 | 1647 | 1979 | 1521 | 1190 | 1177 | 1142 | 1161 | 375 | 470 | 837 | 360 |
K-nad4-A-S
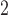
|
1926 | 2090 | 2106 | 1858 | 1535 | 1531 | 1527 | 1518 | 391 | 559 | 579 | 340 |
| D-nad5 | 2081 | 2202 | 2196 | 2001 | 1728 | 1742 | 1760 | 1660 | 353 | 460 | 436 | 341 |
| L1-F-nad6-T | 1335 | 1674 | 1504 | NA | 698 | 684 | 681 | NA | 637 | 990 | 823 | NA |
| P-rrnS | 1280 | 1338 | 1646 | 1206 | 839 | 793 | 853 | 782 | 441 | 545 | 793 | 424 |
| rrnL-V | 1655 | 1680 | 1891 | 1631 | 1172 | 1394 | 1380 | 1226 | 483 | 286 | 511 | 405 |
E-cob-S

|
NA | 1731 | NA | 1579 | NA | 1214 | NA | 1224 | NA | 517 | NA | 355 |
| G-nad3 | NA | 1237 | NA | NA | NA | 402 | NA | NA | NA | 835 | NA | NA |
| M-G-nad3-F-nad6-T | NA | NA | NA | 1573 | NA | NA | NA | 1095 | NA | NA | NA | 478 |

|
NA | NA | NA | 800 | NA | NA | NA | 137 | NA | NA | NA | 663 |
Note: Bb is for Bovicola bovis, Bc for Bovicolacaprae, Bo for Bovicola ovis and  c for Trichodectes canis. NA: not applicable.
c for Trichodectes canis. NA: not applicable.
Shared Derived Features in Mitochondrial Karyotype among Eutherian Mammal Lice in Three Suborders: Anoplura, Ischnocera, and Rhynchophthirina
Like species in the Anoplura and the Rhynchophthirina, the eutherian mammal lice in the Ischnocera (Trichodectidae) also have fragmented mt genomes. Of the two families of Ischnocera, only the mammal lice (Trichodectidae) have fragmented mt genomes, whereas the bird lice (Philopteridae) have the typical single-chromosome mt genomes, as do the species of the suborder Amblycera. The mt karyotypes are highly diverse among the mammal lice of the Anoplura, Rhynchophthirina, and Ischnocera. No two species have identical mt karyotypes except the human head louse and human body louse (Shao et al. 2012), the domestic pig louse and wild pig louse (Jiang et al. 2013), and the sheep louse and cattle louse in this study. However, when we only consider mt protein-coding and rRNA genes, which are much less mobile than tRNA genes, the four species of mammal lice of Ischnocera share five minichromosomes with the elephant louse and the inferred most recent common ancestor (MRCA) of sucking lice (Shao et al. 2017; Table 3). When the arrangement of all genes including tRNA genes are compared, a gene cluster, trnE-cob-trnS , is unique among the MRCA of sucking lice (Anoplura) and the mammal lice of Ischnocera and is not be found in any other parasitic lice (Table 4).
, is unique among the MRCA of sucking lice (Anoplura) and the mammal lice of Ischnocera and is not be found in any other parasitic lice (Table 4).
Table 3.
The mitochondrial minichromosomes shared among parasitic lice (Phthiraptera) when only protein-coding and rRNA genes are considered
| Fragmented | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Suborder | Family | Species | mt genome | cox1 | nad4 | nad5 | rrnS | rrnL | atp6-atp8 | nad1 | nad2 |
| Amblycera | Boopidae | Heterodoxus macropus |

|

|

|

|

|

|

|

|

|
| Menoponidae | Colpocephalum griffoneae |

|

|

|

|

|

|

|

|

|
|
| Amyrsidea minuta |

|

|

|

|

|

|

|

|

|
||
| Ischnocera | Philopteridae | Campanulotes bidentatus compar |

|

|

|

|

|

|

|

|

|
| Campanulotes compar |

|

|

|

|

|

|

|

|

|
||
| Ibidoecus bisignatus |

|

|

|

|

|

|

|

|

|
||
| Falcolipeurus quadripustulatus |

|

|

|

|

|

|

|

|

|
||
| Bothriometopus macrocnemis |

|

|

|

|

|

|

|

|

|
||
| Trichodectidae | Trichodectes canis |
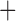
|
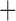
|
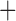
|
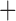
|
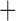
|
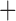
|
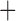
|
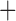
|
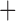
|
|
| Bovicola ovis |
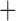
|
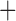
|
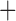
|
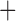
|
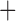
|
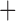
|
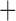
|
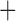
|
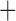
|
||
| Bovicola caprae |
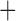
|
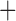
|
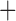
|
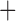
|
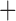
|
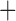
|
|
|
|
||
| Bovicola bovis |
|
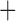
|
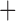
|
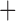
|
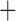
|
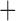
|
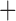
|
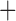
|
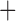
|
||
| Rhyncophthirina | Haematomyzidae | Haematomyzus elephantis |
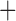
|
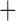
|
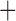
|
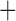
|
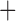
|
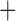
|

|
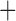
|
NA |
| Anoplura | Ancestral mitochondrial karyotype |
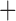
|
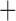
|
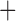
|
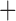
|
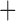
|
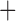
|
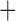
|

|
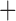
|
Note: Plus ( ) indicates presence; minus (
) indicates presence; minus ( ) indicates absence. NA: not applicable.
) indicates absence. NA: not applicable.
Table 4.
The derived mitochondrial gene clusters shared by parasitic lice (Phthiraptera)
| Suborder | Family | Species | trnI-cox1 | trnY-cox2 |
trnE-cob-trnS

|
nad1-trnQ | trnG-nad3 | trnR-nad4L | trnK-nad4 | rrnL-trnV |
|---|---|---|---|---|---|---|---|---|---|---|
| Amblycera | Boopidae | Heterodoxus macropus |

|
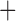
|

|

|

|

|

|

|
| Menoponidae | Colpocephalum griffoneae |

|

|

|

|

|

|

|

|
|
| Amyrsidea minuta |

|

|

|

|

|

|

|

|
||
| Ischnocera | Philopteridae | Campanulotes bidentatus compar |
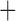
|

|

|

|
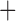
|

|

|

|
| Campanulotes compar |
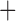
|

|

|

|
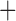
|

|

|

|
||
| Ibidoecus bisignatus |
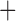
|
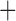
|

|

|

|

|

|

|
||
| Falcolipeurus quadripustulatus |
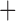
|
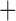
|

|

|

|

|
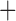
|

|
||
| Bothriometopus macrocnemis |

|
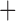
|

|
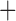
|
|
|
|
|
||
| Trichodectidae | Trichodectes canis |
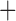
|

|
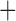
|
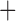
|

|
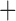
|
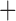
|
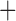
|
|
| Bovicola ovis |
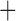
|
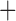
|
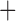
|
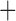
|
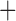
|
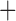
|
|
|
||
| Bovicola caprae |
|
|
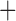
|
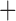
|
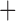
|
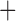
|
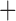
|
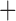
|
||
| Bovicola bovis |
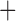
|
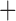
|
|
|
|
|
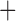
|
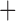
|
||
| Rhyncophthirina | Haematomyzidae | Haematomyzus elephantis |
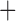
|
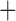
|

|
|
|
|
|
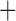
|
| Anoplura | Ancestral mitochondrial karyotype |
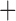
|
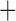
|
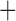
|
|
|
|
|
|
Note: Plus ( ) indicates presence; minus (
) indicates presence; minus ( ) indicates absence.
) indicates absence.
The Suborder-level Phylogeny of Parasitic Lice Based on Shared Derived Characters in Mitochondrial Genome Organization
The shared derived characters in mt genome organization identify a novel clade that comprises the sucking lice (Anoplura), the elephant louse (Rhynchophthirina), and the mammal lice of Ischnocera (Trichodectidae). First, all of the 17 species of eutherian mammal lice from these three suborders have fragmented mt genomes with 10–20 minichromosomes, whereas other parasitic lice have the ancestral single-chromosome mt genomes (Fig. 2, Table 3; Shao et al. 2012; Herd et al. 2015; Shao et al. 2015; Shao et al. 2017). Second, five minichromosomes are found only in the mammal lice of Ischnocera (Trichodectidae), the elephant louse (Rhynchophthirina), and the MRCA of sucking lice (Anoplura), not in any other parasitic lice (Fig. 2, Table 3). Third, a gene cluster, trnE-cob-trnS , is found only in the MRCA of sucking lice (Anoplura) and the mammal lice of Ischnocera (Trichodectidae), not in any other parasitic lice (Table 4).
, is found only in the MRCA of sucking lice (Anoplura) and the mammal lice of Ischnocera (Trichodectidae), not in any other parasitic lice (Table 4).
Figure 2.

A novel clade of parasitic lice of eutherian mammals supported by shared derived characters in mitochondrial genome organization and phylogenetic analysis of mitochondrial genome sequences. In the upper broken-line box are the bird lice from the suborders Amblycera and Ischnocera and the wallaby louse from the Amblycera; these lice all have the typical single-chromosome mitochondrial genomes. In the lower broken-line box are the eutherian mammal lice from the suborders Ischnocera, Rhynchophthirina, and Anoplura; these lice all have fragmented mitochondrial genomes. Bird and mammal hosts are indicated after the names of parasitic lice. When only protein-coding and rRNA genes are considered, the eutherian mammal lice from the Ischnocera and the Rhynchophthirina and the most recent common ancestor of Anoplura have five minichromosomes in common. The phylogenetic tree was constructed with PCGRNA matrix (5677 bp in total) using PhyloBayes with the CAT+GTR model and IQTREE web server with the TIM+F+I+G4 model (see Materials and Methods for details). ML bootstrap values and Bayesian posterior probabilities for each grouping are indicated near the branch nodes.
The Suborder-level Phylogeny of Parasitic Lice Based on Mitochondrial Genome and Gene Sequences
To validate the novel clade identified above by shared derived characters in mt genome organization, we inferred the phylogeny of parasitic lice using mt genome sequences. We included 25 species of parasitic lice from all of the four suborders in our analysis (Table 1). The four trees reconstructed from two data sets (PCGRNA and PCG12RNA) with two methods (ML and BI) have similar topologies (Fig. 2 and Supplementary Figs. S2 and S3 available on Dryad). The monophyly of Phthiraptera was supported in all trees. The Anoplura (sucking lice) was monophyletic with strong support (BPP = 1 in BI trees and bootstrap 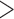 85 in ML trees; Fig. 2). A monophyletic Amblycera is also well supported in all of the four trees (BPP = 1 in BI trees and bootstrap = 100 in ML trees). The Ischnocera, however, was paraphyletic as the mammal lice in the family Trichodectidae were grouped with the sucking lice (Anoplura) and the elephant louse (Rhynchophthirina) to the exclusion of the bird lice in the family Philopteridae. The bird lice in the Ischnocera were paraphyletic in all of the four trees. Thus, the novel clade of Trichodectidae + (Anoplura + Rhynchophthirina), identified above by shared derived characters in mt genome organization was also revealed by mt genome sequence analyses with strong support (BPP = 1 in BI trees, bootstrap
85 in ML trees; Fig. 2). A monophyletic Amblycera is also well supported in all of the four trees (BPP = 1 in BI trees and bootstrap = 100 in ML trees). The Ischnocera, however, was paraphyletic as the mammal lice in the family Trichodectidae were grouped with the sucking lice (Anoplura) and the elephant louse (Rhynchophthirina) to the exclusion of the bird lice in the family Philopteridae. The bird lice in the Ischnocera were paraphyletic in all of the four trees. Thus, the novel clade of Trichodectidae + (Anoplura + Rhynchophthirina), identified above by shared derived characters in mt genome organization was also revealed by mt genome sequence analyses with strong support (BPP = 1 in BI trees, bootstrap 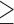 95 in ML tress).
95 in ML tress).
To maximize the number of species of lice in our analyses, we also reconstructed phylogenetic trees with cox1 sequences of 46 species of insects including 41 species of parasitic lice (Supplementary Table S3 available on Dryad). The two trees obtained from BI and ML methods consistently support the paraphyly of Ischnocera and the close relationship of Trichodectidae to Anoplura and Rhynchophthirina (Supplementary Fig. S4 available on Dryad). The monophylies of Amblycera and Anoplura were well supported in cox1 trees. The clade of eutherian mammal lice, Trichodectidae + Rhynchophthirina + Anoplura, was recovered although the support value dropped (BPP = 0.64, bootstrap = 33). Weaker support to the monophyly of the mammal lice from cox1 is expected due to the lack of information from a single gene; this is consistent with the previous studies that addressed this relationship with sequences of single or a few genes (Cruickshank et al. 2001; Johnson and Whiting 2002; Barker et al. 2003; Yoshizawa and Johnson 2003; Yoshizawa and Johnson 2010).
Discussion
Organelle genome fragmentation provides a source of information currently unexplored for phylogenetic reconstruction. Using parasitic lice as an example in this study, we show that mt genome fragmentation is valuable for resolving controversial high-level phylogeny. The currently accepted suborder-level phylogeny and classification of parasitic lice were based on the morphological studies by Clay (1970) and Lyal (1985). Of the four suborders of parasitic lice, Anoplura, Amblycera, and Rhynchophthirina are monophyletic. There is, however, no evidence for the monophyly of Ischnocera (Lyal 1985). Thus, the suborder-level phylogeny and classification of parasitic lice (order Phthiraptera) is questionable. The Ischnocera contains two families: Trichodectidae (eutherian mammal lice) and Philopteridae (bird lice except for the lemur louse, Trichophilopterus babakotophilus); Trichodectidae is monophyletic, whereas Philopteridae is almost certainly paraphyletic (Lyal 1985). Molecular phylogenetic studies on parasitic lice in the past two decades mostly rejected the monophyly of Ischnocera but have been inconsistent. Analyses of EF1-a, cox1, rrnS, and rrnL sequence showed the Ischnocera to be paraphyletic (Cruickshank et al. 2001; Yoshizawa and Johnson 2003). Barker et al. (2003) showed that the Ischnocera was monophyletic using 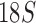 sequence, however, only two species from Trichodectidae were included in their analysis; analyses of combined sequences of EF1-a,
sequence, however, only two species from Trichodectidae were included in their analysis; analyses of combined sequences of EF1-a, 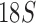 , and cox1 also provided weak support to the monophyly of Ischnocera (Johnson and Whiting 2002; Smith et al. 2011b). However, a recent study by Johnson et al. (2018) explored 1107 nuclear genes and showed that the Ischnocera to be paraphyletic.
, and cox1 also provided weak support to the monophyly of Ischnocera (Johnson and Whiting 2002; Smith et al. 2011b). However, a recent study by Johnson et al. (2018) explored 1107 nuclear genes and showed that the Ischnocera to be paraphyletic.
The shared derived characters from mt genome fragmentation provided strong evidence to the resolution of the long-standing controversial phylogeny of parasitic lice. These characters united the eutherian mammal lice of the Ischnocera (Trichodectidae) with the sucking lice (Anoplura) and the elephant louse (Rhynchophthirina) to the exclusion of the bird lice of the Ischnocera (Philopteridae). The parasitic lice of eutherian mammals in the Anoplura, Rhynchophthirina, and Ischnocera, thus, form a monophyletic group, which we name as Mitodivisia for having fragmented mitochondrial genomes. This novel clade was also supported by a recent study of Johnson et al. (2018) that analyzed the sequences of 1107 nuclear genes of 46 species of parasitic lice. The evidence available to date including that from the current study indicates strongly that the Ischnocera is paraphyletic. We thus propose that: (i) the Ischnocera as a suborder to be revised and (ii) the family Trichodectidae (eutherian mammal lice currently in the Ischnocera) to be moved to a new suborder Trichodectera, in parallel with the suborders Anoplura and Rhynchophthirina. There is also strong, consistent evidence, including that from the current study (but not Johnson et al. 2018), that the family Philopteridae (bird lice currently in the Ischnocera) is paraphyletic. However, how the bird lice in the Philopteridae are related to each other and to the mammal lice is still far from being resolved. For the sake of classification, Philopteridae may stay in the suborder Ischnocera and further investigation is needed to resolve its relationship with other parasitic lice.
It is noteworthy that fragmented mt genomes are found so far only in the parasitic lice of eutherian mammals, not in any of the eight species of bird lice that have been investigated (Fig. 1, Fig. 2, Supplementary Fig. S1 available on Dryad). Although it has been suggested that some bird lice (Ischnocera, Philopteridae) may have multi-chromosomal mt genomes (Cameron et al. 2011), no complete mt genome data from any bird louse is available to date to support this suggestion. Given the fact that only approximately 20% of the described species of parasitic lice are on mammals, whereas the vast majority (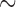 80%) of parasitic lice are on birds, it is likely and plausible that infestation of birds is ancestral to parasitic lice whereas infestation of mammals is secondary. If mt genome fragmentation is indeed only in mammal lice, it would indicate a link between mt genome fragmentation and the host switch of parasitic lice from birds to mammals. If so, would mt genome fragmentation have a role in the host switch and adaptation of parasitic lice to the new host niche? Alternatively, mt genome fragmentation may have occurred in bird lice as well but has not yet been revealed by our limited sampling. These two scenarios are equally likely and further studies are obviously needed.
80%) of parasitic lice are on birds, it is likely and plausible that infestation of birds is ancestral to parasitic lice whereas infestation of mammals is secondary. If mt genome fragmentation is indeed only in mammal lice, it would indicate a link between mt genome fragmentation and the host switch of parasitic lice from birds to mammals. If so, would mt genome fragmentation have a role in the host switch and adaptation of parasitic lice to the new host niche? Alternatively, mt genome fragmentation may have occurred in bird lice as well but has not yet been revealed by our limited sampling. These two scenarios are equally likely and further studies are obviously needed.
Supplementary Material
Data available from the Dryad Digital Repository: http://dx.doi.org/10.5061/dryad.qg8vf45.
Funding
This work was supported by the National Natural Science Foundation of China [31420103902, 31730086], the Beijing Natural Science Foundation [6152016, 6144027], the China Postdoctoral Science Foundation [2016M601178], the Training Program for Excellent Young Innovators of Changsha [KQ1707005], the Australia-China Science and Research Fund Group Mission [ACSRF00980], and the Australian Research Council [DP120100240].
Acknowledgements
We would like to thank the associate editor and the anonymous reviewers for comments and advice that improved the manuscript. We also thank Lance Durden, Ricardo Palma and Ian Beveridge for advice on naming taxonomic groups.
References
- Abascal F., Zardoya R., Telford M.J.. 2010. TranslatorX: multiple alignment of nucleotide sequences guided by amino acid translations. Nucleic Acids Res. 38:W7–W13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschup S., Gish W., Miller W., Myers E., Lipman D.. 1990. Basic local alignment search. Tool. J. Mol. Biol. 215:403–410. [DOI] [PubMed] [Google Scholar]
- Armstrong M.R., Blok V.C., Phillips M.S.. 2000. A multipartite mitochondrial genome in the potato cyst nematode Globodera pallida. Genetics 154:181–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babbucci M., Basso A., Scupola A., Patarnello T., Negrisolo E.. 2014. Is it an ant or a butterfly? Convergent evolution in the mitochondrial gene order of Hymenoptera and Lepidoptera. Genome Biol. Evol. 6:3326–3343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker S.C., Whiting M., Johnson K.P., Murrell A.. 2003. Phylogeny of the lice (Insecta, Phthiraptera) inferred from small subunit rRNA. Zool. Scr. 32:407–414. [Google Scholar]
- Boore J.L., Brown W.M.. 1998. Big trees from little genomes: mitochondrial gene order as a phylogenetic tool. Curr. Opin. Genet. Dev. 8:668–674. [DOI] [PubMed] [Google Scholar]
- Boore J.L., Lavrov D.V., Brown W.M.. 1998. Gene translocation links insects and crustaceans. Nature 392:667. [DOI] [PubMed] [Google Scholar]
- Burger G., Forget L., Zhu Y., Gray M.W., Lang B.F.. 2003. Unique mitochondrial genome architecture in unicellular relatives of animals. Proc. Natl. Acad. Sci. U.S.A. 100:892–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron S.L. 2014. Insect mitochondrial genomics: implications for evolution and phylogeny. Annu. Rev. Entomol. 59:95–117. [DOI] [PubMed] [Google Scholar]
- Cameron S.L., Johnson K., Whiting M.. 2007. The mitochondrial genome of the screamer louse Bothriometopus (Phthiraptera: Ischnocera): effects of extensive gene rearrangements on the evolution of the genome. J. Mol. Evol. 65:589–604. [DOI] [PubMed] [Google Scholar]
- Cameron S.L., Yoshizawa K., Mizukoshi A., Whiting M.F., Johnson K.P.. 2011. Mitochondrial genome deletions and minicircles are common in lice (Insecta: Phthiraptera). BMC Genomics 12:394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clay T. 1970. The Amblycera (Phthiraptera: Insecta). Bull. Br. Mus. (Nat. Hist. Entomol.) 24:75–98. [Google Scholar]
- Covacin C., Shao R., Cameron S., Barker S.C.. 2006. Extraordinary number of gene rearrangements in the mitochondrial genomes of lice (Phthiraptera: Insecta). Insect Mol. Biol. 15:63–68. [DOI] [PubMed] [Google Scholar]
-
Cruickshank R.H., Johnson K.P., Smith V.S., Adams R.J., Clayton D.H., Page R.D..
2001.
Phylogenetic analysis of partial sequences of elongation factor 1
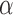 identifies major groups of lice (Insecta: Phthiraptera). Mol. Phylogenet. Evol. 19:202–215. [DOI] [PubMed] [Google Scholar]
identifies major groups of lice (Insecta: Phthiraptera). Mol. Phylogenet. Evol. 19:202–215. [DOI] [PubMed] [Google Scholar] - Dickey A.M., Kumar V., Morgan J.K., Jara-Cavieres A., Shatters R.G., McKenzie C.L., Osborne L.S.. 2015. A novel mitochondrial genome architecture in thrips (Insecta: Thysanoptera): extreme size asymmetry among chromosomes and possible recent control region duplication. BMC Genomics 16:439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong W.G., Dong W.G., Song S., Guo X.G., Jin D.C., Yang Q., Barker S.C., Shao R.. 2014a. Fragmented mitochondrial genomes are present in both major clades of the blood-sucking lice (suborder Anoplura): evidence from two Hoplopleura rodent lice (family Hoplopleuridae). BMC Genomics 15:751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong W.G., Song S., Jin D.C., Guo X.G., Shao R.. 2014b. Fragmented mitochondrial genomes of the rat lice, Polyplax asiatica and Polyplax spinulosa: intra-genus variation in fragmentation pattern and a possible link between the extent of fragmentation and the length of life cycle. BMC Genomics 15:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowton M., Cameron S.L., Dowavic J.I., Austin A.D., Whiting M.F.. 2009. Characterization of 67 mitochondrial tRNA gene rearrangements in the Hymenoptera suggests that mitochondrial tRNA gene position is selectively neutral. Mol. Biol. Evol. 26:1607–1617. [DOI] [PubMed] [Google Scholar]
- Ferris G.F. 1951. The sucking lice. Mem. Pacific Coast Entomol. Soc. 1:1–320. [Google Scholar]
- Gibson T., Blok V.C., Dowton M.. 2007. Sequence and characterization of six mitochondrial subgenomes from Globodera rostochiensis: multipartite structure is conserved among close nematode relatives. J. Mol. Evol. 65:308–315. [DOI] [PubMed] [Google Scholar]
- Gissi C., Iannelli F., Pesole G.. 2008. Evolution of the mitochondrial genome of Metazoa as exemplified by comparison of congeneric species. Heredity 101:301–320. [DOI] [PubMed] [Google Scholar]
- Harrison L. 1928. Host and parasite. Proc. Linn. Soc. NSW. 53:9–31. [Google Scholar]
- Herd K., Barker S.C., Shao R.. 2015. The mitochondrial genome of the chimpanzee louse, Pediculus schaeffi: insights into the process of mitochondrial genome fragmentation in the blood-sucking lice of great apes. BMC Genomics 16:661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkins G.H.E. 1949. The host-associations of the lice of mammals. J. Zool. 119:387–604. [Google Scholar]
- Howe C.J., Nisbet R.E.R., Barbrook A.C.. 2008. The remarkable chloroplast genome of dinoflagellates. J. Exp. Bot. 59:1035–1045. [DOI] [PubMed] [Google Scholar]
- Jiang H.W., Barker S.C., Shao R.. 2013. Substantial variation in the extent of mitochondrial genome fragmentation among blood-sucking lice of mammals. Genome Biol. Evol. 5:1298–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson K.P., Whiting M.F.. 2002. Multiple genes and the monophyly of Ischnocera (Insecta: Phthiraptera). Mol. Phylogenet. Evol. 22:101–110. [DOI] [PubMed] [Google Scholar]
- Johnson K.P., Nguyen N-P., Sweet A.D., Boyd B.M., Warnow T., Allen J.M.. 2018. Simultaneous radiation of bird and mammal lice following the K-Pg boundary. Biol. Lett. 14:20180141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K., Standley D.M.. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30:772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearse M., Moir R., Wilson A., Stones-Havas S., Cheung M., Sturrock S., Thierer T.. 2012. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28:1647–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeling P.J., Archibald J.M.. 2008. Organelle evolution: what’s in a name? Curr. Biol. 18:345–347. [DOI] [PubMed] [Google Scholar]
- Kim K.C. 1988. Evolutionary parallelism in Anoplura and eutherian mammals In: Service M.W., editor. Biosystematics of haematophagous insects. Oxford: Claredon Press; p. 91–114. [Google Scholar]
- Kim K.C., Ludwig H.W.. 1982. Parallel evolution, cladistics, and classification of parasitic Psocodea. Ann. Entomol. Soc. Am. 75:537–548. [Google Scholar]
- Koumandou V.L., Nisbet R.E.R., Barbrook A.C., Howe C.J.. 2004. Dinoflagellate chloroplasts—where have all the genes gone? Trends Genet. 20:261–267. [DOI] [PubMed] [Google Scholar]
- Lartillot N., Rodrigue N., Stubbs D., Richer J.. 2013. PhyloBayes MPI: phylogenetic reconstruction with infinite mixtures of profiles in a parallel environment. Syst. Biol. 62:611–615. [DOI] [PubMed] [Google Scholar]
- Laslett D., Canback B.. 2008. ARWEN: a program to detect tRNA genes in metazoan mitochondrial nucleotide sequences. Bioinformatics 24:172–175. [DOI] [PubMed] [Google Scholar]
- Lavrov D.V., Pett W., Voigt O., Wörheide G., Forget L., Lang B.F., Kayal E.. 2012. Mitochondrial DNA of Clathrina clathrus (Calcarea, Calcinea): six linear chromosomes, fragmented rRNAs, tRNA editing, and a novel genetic code. Mol. Biol. Evol. 30:865–880. [DOI] [PubMed] [Google Scholar]
- Li H., Shao R., Song N., Song F., Jiang P., Li Z., Cai W.. 2015. Higher-level phylogeny of paraneopteran insects inferred from mitochondrial genome sequences. Sci. Rep. 5:8527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe T.M., Eddy S.R.. 1997. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25:955–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyal C.H.C. 1985. Phylogeny and classification of the Psocodea, with particular reference to the lice (Psocodea: Phthiraptera). Syst. Entomol. 10:145–165. [Google Scholar]
- Marande W., Burger G.. 2007. Mitochondrial DNA as a genomic jigsaw puzzle. Science 318:415–415. [DOI] [PubMed] [Google Scholar]
- Marshall A.G. 1981. The ecology of ectoparasitic insects London: Academic Press. [Google Scholar]
- Miller M., Pfeiffer W., Schwartz T.. 2010. Creating the CIPRES science gateway for inference of large phylogenetic trees. Proceedings of the Gateway Computing Environments Workshop (GCE), 14November 2010, New Orleans, LA. p. 1–8. [Google Scholar]
- Peng Y., Leung H.C.M., Yiu S.M., Chin F.Y.L.. 2012. IBDA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 28:1420–1428. [DOI] [PubMed] [Google Scholar]
- Price R.D., Hellenthal R.A., Palma R.L.. 2003. World checklist of chewing lice with host associations and keys to families and genera In: Price R.D., Hellenthal R.A., Palma R.L., Johnson K.P., Clayton D.H., editors. The chewing lice: world checklist and biological overview. Illinois: Illinois Natural History Survey Special Publication 24. p. 1–448. [Google Scholar]
- Rokas A., Holland P.W.H.. 2000. Rare genomic changes as a tool for phylogenetics. Trends Ecol. Evol. 15:454–459. [DOI] [PubMed] [Google Scholar]
- Shao R., Barker S.C.. 2011. Chimeric mitochondrial minichromosomes of the human body louse, Pediculus humanus: evidence for homologous and non-homologous recombination. Gene 473:36–43. [DOI] [PubMed] [Google Scholar]
- Shao R., Barker S.C, Li H., Su Y.. 2015. Fragmented mitochondrial genomes in two suborders of parasitic lice of eutherian mammals (Anoplura and Rhynchophthirina, Insecta). Sci. Rep. 5:17389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao R., Campbell N.J.H., Barker S.C.. 2001. Numerous gene rearrangements in the mitochondrial genome of the wallaby louse, Heterodoxus macropus (Phthiraptera). Mol. Biol. Evol. 18: 858–865. [DOI] [PubMed] [Google Scholar]
- Shao R., Kirkness E.F., Barker S.C.. 2009. The single mitochondrial chromosome typical of animals has evolved into 18 minichromosomes in the human body louse, Pediculus humanus. Genome Res. 19:904–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao R., Li H., Barker S.C., Song S.. 2017. The mitochondrial genome of the guanaco louse, Microthoracius praelongiceps: insights into the ancestral mitochondrial karyotype of sucking lice (Anoplura, Insecta). Genome Biol. Evol. 9:431–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao R., Zhu X.Q., Barker S.C., Herd K.. 2012. Evolution of extensively fragmented mitochondrial genomes in the lice of humans. Genome Biol. Evol. 4:1088–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith D.R., Kayal E., Yanagihara A.A., Collins A.G., Pirro S., Keeling P.J.. 2011a. First complete mitochondrial genome sequence from a box jellyfish reveals a highly fragmented linear architecture and insights into telomere evolution. Genome Biol. Evol. 4:52–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith V.S., Ford T., Johnson K.P., Johnson P.C., Yoshizawa K., Light J.E.. 2011b. Multiple lineages of lice pass through the K-Pg boundary. Biol. Lett. 7:782–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song F., Li H., Jiang P., Zhou X., Liu J., Sun C., Vogler A.P., Cai W.. 2016. Capturing the phylogeny of Holometabola with mitochondrial genome data and Bayesian site-heterogeneous mixture models. Genome Biol. Evol. 8:1411–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song S., Barker S.C., Shao R.. 2014. Variation in mitochondrial minichromosome composition between blood-sucking lice of the genus Haematopinus that infest horses and pigs. Parasit. Vectors 7:144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talavera G., Castresana J.. 2007. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 56:564–577. [DOI] [PubMed] [Google Scholar]
- Trifinopoulos J., Nguyen L.T., von Haeseler A., Minh B.Q.. 2016. W-IQ-TREE: a fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 44:W232–W235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voigt O., Erpenbeck D., Wörheide G.. 2008. A fragmented metazoan organellar genome: the two mitochondrial chromosomes of Hydra magnipapillata. BMC Genomics 9:350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace D.C. 1982. Structure and evolution of organelle genomes. Microbiol. Rev. 46:208–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe K.I., Bessho Y., Kawasaki M., Hori H.. 1999. Mitochondrial genes are found on minicircle DNA molecules in the mesozoan animal Dicyema. J. Mol. Biol. 286:645–650. [DOI] [PubMed] [Google Scholar]
- Wei D.D., Shao R., Yuan M.L., Dou W., Barker S.C., Wang J.J.. 2012. The multipartite mitochondrial genome of Liposcelis bostrychophila: insights into the evolution of mitochondrial genomes in bilateral animals. PLoS One 7:e33973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshizawa K., Johnson K.P.. 2003. Phylogenetic position of Phthiraptera (Insecta: Paraneoptera) and elevated rate of evolution in mitochondrial 12S and 16S rDNA. Mol. Phylogenet. Evol. 29:102–114. [DOI] [PubMed] [Google Scholar]
- Yoshizawa K., Johnson K.P.. 2010. How stable is the “polyphyly of lice” hypothesis (Insecta: Psocodea)?: a comparison of phylogenetic signal in multiple genes. Mol. Phylogenet. Evol. 55:939–951. [DOI] [PubMed] [Google Scholar]
- Zhang Z., Green B.R., Cavalier-Smith T.. 1999. Single gene circles in dinoflagellate chloroplast genomes. Nature 400:155–159. [DOI] [PubMed] [Google Scholar]