

Abstract
Background and AIMS:
Terbinafine is an antifungal agent that has been associated with rare instances of hepatotoxicity. Study aims include a description of the presenting features and outcomes of patients with terbinafine hepatotoxicity as well as investigation of the role of HLA- A *33:01.
Methods:
Consecutive high causality cases of terbinafine hepatotoxicity enrolled into the Drug Induced Liver Injury Network were reviewed. DNA samples underwent high resolution confirmatory HLA sequencing using the Ilumina MiSeq platform.
Results:
All 15 patients with terbinafine hepatotoxicity were more than 40 years old (median = 57 years), 53% female and the median latency to onset was 38 days (range: 24 to 114 days). At DILI onset, 80% were jaundiced, median serum alanine aminotransferase was 448 U/L and alkaline phosphatase was 333 U/L. During follow-up, one subject required liver transplantation for acute liver failure, and 7 of the 13 (54%) remaining subjects had ongoing liver injury at 6 months with 4 demonstrating persistently abnormal liver biochemistries at month 24. High resolution HLA genotyping confirmed that 10 of the 11 (91%) European ancestry participants were carriers of the HLA- A* 33:01, B*14:02, C* 08:02 haplotype which has a carrier frequency of 1.6% in European Ancestry population controls. One African American patient was also a HLA- A * 33:01 carrier while 2 East Asian patients were carriers of a similar HLA type: A* 33:03. Molecular docking studies indicated that terbinafine may interact with HLA- A*33:01 and A*33:03.
CONCLUSIONS:
Patients with terbinafine hepatotoxicity most commonly present with a mixed or cholestatic liver injury profile and frequently have residual evidence of chronic cholestatic injury. A strong genetic association of HLA- A * 33:01 with terbinafine DILI was confirmed amongst Caucasians.
Keywords: Genetic polymorphisms, drug induced liver injury, hepatotoxicity
Graphical Abstract
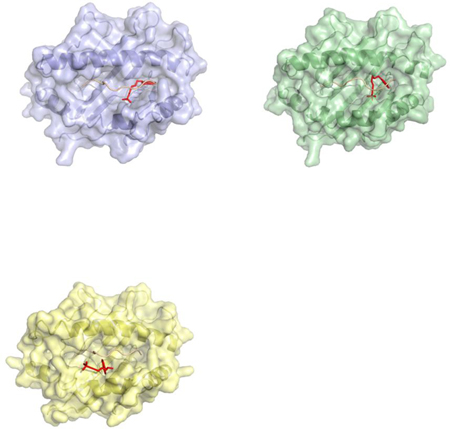
Example: A ribbon diagram demonstrating docking of a terbinafine molecule in the protein binding cleft of HLA-A * 33:01 (blue), HLA-B * 14:02 (green), and HLA-C * 08:02 locus.
Lay summary:
A locus in the human leukocyte antigen gene (HLA-A *33:01, B*14:02, C * 08:02) was significantly overrepresented in Caucasian and African American patients with liver injury attributed to terbinafine compared to population controls. These data along with the molecular docking studies demonstrate that this genetic polymorphism is a plausible risk factor for developing terbinafine hepatotoxicity and could be used in the future to help doctors make a diagnosis more rapidly and confidently.
Introduction
Terbinafine is an orally administered antifungal agent that is commonly used to treat superficial fungal infections of the skin and nails. It was approved for use in the United States in 1998 and is typically given in an oral dose of 250 mg daily for 6 to 12 weeks. Terbinafine use is associated with mild liver biochemistry abnormalities in < 1% of treated patients. However, the incidence of clinically significant liver injury attributed to terbinafine is not known due to lack of a sensitive and reliable means to detect hepatotoxicity in a prospective, population based cohort. The liver injury may be hepatocellular at initial presentation but usually evolves into a cholestatic profile over time (1). Hypersensitivity and autoimmune features are uncommon and some studies suggest that a reactive metabolite may be involved that can accumulate in the bile (2). In addition, rare instances of severe hepatitis, acute liver failure and fatalities have been described with terbinafine use (3,4).
The Drug Induced Liver Injury Network (DILIN) has been conducting studies of the presenting features, risk factors, and outcomes of patients with drug induced liver injury (DILI) (5,6). In addition, mechanistic studies exploring the potential genetic susceptibility in DILI patients have been undertaken using genome wide association (GWA) studies. A recent genetic analysis of 862 European ancestry DILI patients demonstrated a significant association between imputed HLA-A*33:01 and terbinafine liver injury in 14 European ancestry patients compared to population controls with an odds ratio (OR) of 40.5 (95% CI=12.5–288.9, p=6.7 × 10−10) (7). The replication stage of this same study included 8 additional terbinafine patients, 6 of whom were carriers of the single nucleotide polymorphism (SNP) rs114577328, the proxy marker of the HLA-A*33:01 allele. The overall carrier frequency of imputed HLA-A*33:01 positive alleles in the 22 European ethnicity terbinafine cases was 54% compared to an expected carrier frequency of 2.2% in European controls. Of note, 14 of these 22 terbinafine cases had a cholestatic or mixed liver injury pattern at presentation and 11 were enrolled into DILIN. The DILIN has now enrolled over 2,000 patients into the ongoing prospective registry study wherein patients undergo extensive phenotyping including assessment of laboratory, clinical and histological features through 6 months of follow-up (5 ,8, 9). The aim of the current study was to describe the clinical, laboratory, and histological features of 15 patients with liver injury attributed to terbinafine that were enrolled into DILIN. In addition, we set out to identify the HLA alleles associated with terbinafine hepatotoxicity in populations other than those of European ancestry and carry out molecular docking studies to define potential HLA terbinafine interactions.
METHODS
DILIN Prospective study-
All subjects were enrolled into the ongoing DILIN prospective study (5). Injury onset was defined as the first date after a subject taking terbinafine met the predefined laboratory criteria of either a serum aspartate aminotransferase (AST) or alanine aminotransferase (ALT) level that exceeded 5 times the upper limit of normal (ULN) (or 5 times pretreatment baseline if baseline abnormal), a serum alkaline phosphatase (ALP) that exceeded 2 times the ULN (or 2 times pretreatment baseline if baseline abnormal), a total bilirubin of 2.5 mg/dl or above, or an international normalized ratio (INR) greater than 1.5 on two consecutive blood draws. All study participants provided written informed consent and were enrolled within 6 months of DILI onset.
A detailed medical history was obtained at the baseline study visit and additional laboratory and radiological testing were performed to more fully characterize the DILI event and exclude competing etiologies. Specifically, testing for hepatitis A, B, C, E, HIV, anti-nuclear antibody (ANA) and smooth muscle antibody (SMA) titers, CMV, and EBV infection were obtained at the local laboratory. Cases without available hepatitis E virus testing or without HCV RNA results were tested centrally using stored serum samples. Enrolled patients were seen for a follow-up study visit at 6 months after initial enrollment and those with evidence of chronic injury were asked to return for additional 12 and 24 month study visits (5). Chronic DILI in the DILIN Prospective study was defined as any patient having a persistently elevated serum AST, ALT, or ALP level, histological evidence of liver injury, or clinical evidence of portal hypertension at 6 months after DILI onset (6).
The severity of the DILI episode was categorized on a 5-point scale from mild (1), moderate (2), moderate-hospitalized (3), severe (4), and fatal (5), where a fatal score was assigned only if the patient died or had liver transplant due to DILI within six months of onset (6). Of note, clinical case reports and serial laboratory results from all 15 cases are available on the LiverTox website (see http://livertox.nlm.nih.gov/terbinafine).
Liver histopathology-
Available liver biopsies were reviewed by a single expert liver histopathologist (DEK). All samples were scored for multiple histological features as well as an overall pattern of liver injury (10).
Causality assessment-
The causal relationship between the liver injury episode and terbinafine use was evaluated in a standardized fashion by the DILIN causality committee (5,8). A DILIN expert opinion causality score varying from 1 (Definite > 95% likelihood), 2 (Highly Likely 75%−95% likelihood), 3 (Probable 50%−74% likelihood), 4 (Possible 25%−49% likelihood) to 5 (unlikely < 25% likelihood) was assigned by consensus agreement of committee members for all of the retrospective and prospective DILIN cases. In subjects with 2 or more implicated drugs, an overall causality score was assigned to the case and then an individual causality score for each drug was given. For the current analysis, only cases with DILIN causality scores for terbinafine of definite, highly likely or probable were included.
Genome-wide genotyping and HLA sequencing
Genome-wide SNP genotypes were used to confirm the self-reported ethnicities of 14 of the cases. Genome-wide genotyping of DILI cases was performed by the Broad Institute in Boston by Illumina Infinium HumanCoreExome BeadChip (n=2), at the Duke Center for Human Genome Variation on the Illumina 1Mduo array (n=8), or at the Duke Molecular Genomics Shared Resource on the Illumina Multi-Ethnic Genotyping Array (MEGA; n=4) (11). Genetic ancestry was determined using EIGENSTRAT axes.
A proxy for HLA-A * 33:01, rs148631562, was typed with a TaqMan® SNP genotyping assay (ThermoFisher Scientific, Waltham, MA) in accordance with the manufacturer’s recommendations in 7 additional European ethnicity cases. The top associated HLA type was further validated by high resolution Class I HLA A B C and Class II DR DQ DP genotyping at Vanderbilt University and Institute for Immunology & Infectious Diseases, Western Australia. A 2.5 μg aliquot of DNA in EDTA buffer was PCR amplified using sample specific Multiplex Identified (MID) tagged primers that amplify polymorphic exons from class I (A, B, C exons 2 and 3) and Class II (DQ, Exons 2 and 3, DRB and DPB1, Exon 1) major histocompatibility genes. Amplified DNA products from unique MID-tagged products were then pooled and subjected to library preparation. Normalized libraries were sequenced on the Ilumina MiSeq platform using the MiSeq V3 600 cycle kit. Sequences were then separated by MID tags and alleles called using an in house accredited HLA allele software pipeline IIID HLA Analysis Suite (version 3.11), the reporting tool was used to perform all quality checks on the allele calls, condense and assign putative calls and ambiguous results and prepare the formatted report for release. Alleles were called using the latest IMGT HLA allele database (version 3270).
HLA type frequency data from population controls were obtained from allelefrequencies.net (12). In particular, the European ancestry population frequencies are taken from the USA NMDP European Caucasian population, with a sample size of 1,242,890 (13) ; the African ancestry frequencies are taken from the USA NMDP African American population, with a sample size of 416,581; the Hispanic ancestry population frequencies are taken from the USA NMDP Mexican or Chicano, Caribbean Hispanic, and Hispanic South or Central American populations, with respective sample sizes of 261,235, 115,374, and 146,714; the East Asian population frequencies are taken from the USA NMDP Chinese, Japanese, and Korean populations, with ample sizes of 99,672, 24,582, and 77,584, respectively; the Northern European population frequencies are taken from the Ireland Northern, Ireland South, England North West, Germany DKMS - United Kingdom minority, and Germany DKMS - France minority, and Germany pop 6 populations, with respective sample sizes of 1,000, 250, 298, 1,043, 1,406, and 8,862; and the Jewish ancestry population frequencies are taken from the Israel USA Jewish population, with a sample size of 6,058 (14–17).
Molecular Docking
Sequences of HLA-A*33:01, HLA-A * 33:03, HLA-B*14:02, and HLA-C*08:02 were obtained from the HLA/IGMT database (http://www.ebi.ac.uk/ipd/imgt/hla/allele.html). Atomic homology models for each were generated with SWISS-MODELLER (18). A model of HLA-C*08:02 was generated based on the most closely related crystal structure, PDB code 4NT6 (PMC4178881). For HLA-A*33:01, the model was based on 3RL1 (19) and for HLA-B*14:02 it was based on 3BVN (20). To generate peptide/HLA complex models, we used a polyglycine peptide extracted from the crystal structure of a peptide HLA complex (3UPR). The peptide was positioned into the antigen binding clefts of HLA-A*33:01, HLA-A * 33:03, HLA-B*14:02, and HLA-C*08:02 models using SSM in the COOT program package (21).
Terbinafine was docked on models of HLA-A*33:01, HLA-A * 33:03, HLA-B*14:02, and HLA-C*08:02 using AutoDock Vina (22). The structure of terbinafine was translated from SMILES string (23) to PDB file using the NCI/CADD translator (https://cactus.nci.nih.gov/translate/). The resulting PDB file was prepared for molecular docking using AutoDock tools (e.g., add H, generate charges for each atom to be used in scoring). Scoring grids were 20 × 20 × 20 Å, and centered on 3 sites within the antigen binding clefts of HLA-A*33:01, HLA-A * 33:03, HLA-B*14:02, and HLA-C*08:02: 1) a site corresponding to the Cα of the first peptide position (P1), 2) a site corresponding to the Cα of the fifth peptide position (P5), the middle of the antigen binding cleft, and 3) a site corresponding to Cα of the terminal peptide position (P9). Terbinafine was docked with exhaustiveness set to 10. The top 8 scoring orientations were output and compared. PyMol was used to generate molecular graphics (The PyMOL Molecular Graphics System, Version 1.8 Schrödinger, LLC.).
Results
Patient population-
Between January 2004 and February 2017, 1786 cases were enrolled into the ongoing DILIN prospective study. Amongst the 1549 adjudicated cases, there were 19 with terbinafine listed as an implicated agent but an alternative cause of liver injury was identified as more likely in the 4 cases with low causality scores of only possible or unlikely (Figure 1). The presenting clinical features and demographics of the 15 terbinafine cases with a causality score of definite (10), very likely (4), or probable (1) are shown in Table 1. The median age was 56.6 years (range: 40 to 74.8), 53% were female, and 73% of European ancestry. All patients were prescribed terbinafine at a dose of 250 mg/day for a fungal nail infection and the median duration of terbinafine use before DILI onset was 37 days (range: 20 to 105 days). Of note, none of the patients reported ever having taken terbinafine in the past.
Figure 1-. Overview of study cohort.
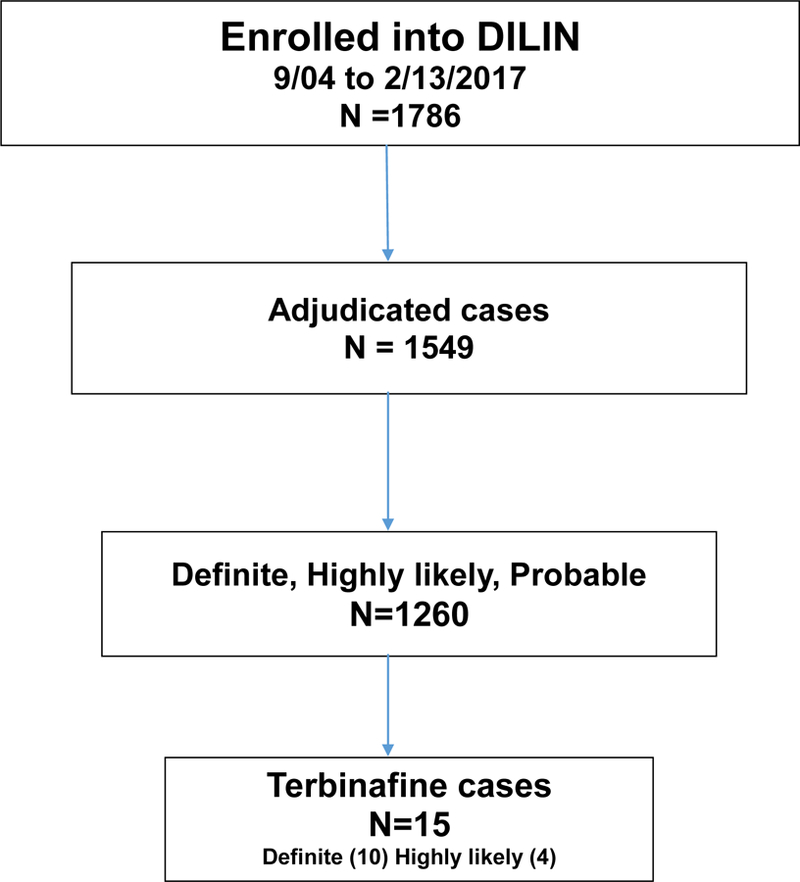
Among 1786 cases of suspected drug-induced liver injury enrolled in the DILIN between September 2004 and February 2017, 1549 underwent formal causality assessment by the time of formal analysis, of which 1260 cases were judged to be definite, highly likely or probable; 15 of these (1.2%) were attributed to terbinafine.
Table 1.
Presenting clinical Features of 15 patients with terbinafine hepatotoxicity stratified by HLA-A * 33:01 genotype.
| Overall all Cohort N=15 | HLA- A *33:01(+) N= 11 HLA |
HLA- A *33:01 (−) N=4 | p-value | |
|---|---|---|---|---|
| Median Age (years) | 56.6 (40– 74.8) | 57.5 (43.7– 4.8) | 46 (40– 68) | 0.30 |
| % Female | 53% | 55% | 50% | >0.99 |
| Race | ||||
| % European | 73% | 91% | 25% | 0.017 |
| % African | 7% | 9 | 0% | |
| % East Asian | 13% | 0 | 50% | |
| % Multiracial | 7% | 0 | 25% | |
| % Hispanic | 13% | 0 | 50% | 0.06 |
| Median BMI (kg/m2) | 24.2 (19.8– 37.6) | 23.8 (19.8– 29.6) | 28.0 (20.9– 37.6) | 0.30 |
| Medical history | ||||
| %Drug allergies | 33% | 18% | 75% | 0.08 |
| % Diabetes | 20% | 18% | 25% | >0.99 |
| % Alcohol use | 60% | 73% | 25% | 0.24 |
| Symptoms | ||||
| % Jaundice | 80% | 73% | 100% | 0.52 |
| % Pruritus | 73% | 82% | 50% | 0.52 |
| % Rash | 20% | 18% | 25% | >0.99 |
| % Fever | 7% | 0% | 25% | 0.27 |
| Duration of use prior to DILI onset (days) | 38 (24–114) | 38 (28– 114) | 39 (24–48) | 0.74 |
| Labs at DILI onset | ||||
| ALT (U/L) | 448 (131–1071) | 419 (131–1071) | 546 (293–778) | 0.79 |
| ALP (U/L) | 333 (143–452) | 333 (203– 433) | 303 (143–452) | 0.79 |
| Bilirubin (mg/dL) | 6.6 (0.7–23.7) | 6.6 (0.7–14.9) | 7.3 (2.2–23.7) | 0.60 |
| % ANA (+) | 21% | 20% | 25% | > 0.99 |
| % SmAb (+) | 8% | 0% | 25% | 0.38 |
| % Hepatocellular | 33% | 27% | 50% | 0.77 |
| % Mixed | 47% | 55% | 25% | |
| % Cholestatic | 20% | 18% | 25% | |
| Peak labs | ||||
| ALT (U/L) | 709 (131–1133) | 709 (131–1133) | 613 (372–912) | 0.70 |
| ALP (U/L) | 375 (143.0– 677.0) | 366 (311– 667) | 480 (143–677) | 0.43 |
| Bilirubin (mg/dL) | 12.9 (3.8– 29.5) | 11.0 (4.0– 20.8) | 17.3 (3.8–29.5) | 0.43 |
| Treatment | ||||
| % Corticosteroids | 40% | 36% | 50% | >0.99 |
| % Ursodiol | 27% | 36% | 0% | 0.52 |
| Chronic injury at 6 months | 7/13 (54%) | 6/10 (60%) | 1/3 (33%) | 0.56 |
Data presented as Median (range).
At presentation, only 6% of patients reported fever, 7% had eosinophilia and 20% reported having a rash. However, 73% reported symptoms of pruritus and 80% were visibly jaundiced. The median serum ALT at DILI onset was 448 U/L (range: 131 to 1071), ALP 333 U/L (range: 143 to 451), and bilirubin 6.6 mg/dL (range: 0.7 to 23.7). The median R ratio (ALT/ALP both expressed as multiple of ULN) at onset was 3.3 and ranged from 0.9 to 19 with 20% in the cholestatic (R <2), 47% in the mixed (R 2–5) and 33% in the hepatocellular (R>5) range. Detectable ANA and SMA were noted in only 21% and 8% of patients, respectively.
Two patients were hospitalized and one had a severe and progressive course. A 41-year-old Hispanic woman (Case #12) with a history of a Roux-en-Y gastric bypass for morbid obesity and a BMI of 38. 6 kg/m2 presented with a severe acute hepatocellular liver injury pattern with an initial R ratio of 10.5, a bilirubin of 23.7 mg/dL and INR of 2.68. Further testing revealed an ANA of 1: 1280, SMA 1: 126, elevated IgG of 2690 mg/dL, and a liver biopsy demonstrating severely active hepatitis with collapse and regenerative changes. She was treated with high doses of corticosteroids, but had progressive liver injury and required an urgent liver transplant 18 days after DILI onset. Of note, she did not possess any of the HLA-DRB1 alleles associated with autoimmune hepatitis and did not have HLA-A* 33:01 nor A* 33:03 (Patient # 12 in Supplementary Table 4). (24)
The median peak serum ALT in the cohort was 619 U/L (range 131 to 1133), median peak ALP was 375 U/L (range: 143 to 677), and median peak total bilirubin was 12.9 mg/dL (range: 3.8 to 29.5). The median R ratio at the time of peak bilirubin values was 2.4 and ranged from 0.4 to 5.1 with 47% in the cholestatic, 40% mixed and 13% hepatocellular range. The median time to bilirubin normalization was 34 days. During follow-up, 6 subjects were treated with corticosteroids while 4 subjects were treated with ursodeoxycholic acid.
Clinical outcomes 6 months after DILI onset-
Laboratory tests from a 6 month follow up visit were available in 13 patients, of whom 7 still had liver test abnormalities. The presenting clinical and laboratory features of the patients who met criteria for our protocol determined definition of chronic liver injury were similar to those with self-limited injury (Supplemental Table 1). At the month 12 study visit, the 6 evaluable patients with abnormalities at 6 months all had a persistent cholestatic liver injury profile with a median serum ALT of 101 U/L (range: 50 to 199), ALP of 245 U/L (range: 111 to 378) but normal total bilirubin levels. At 24 months, 7 patients were still being followed and 3 continued to have mild elevations in serum ALP (range: 76 to 244 U/L) but all were asymptomatic and total bilirubin levels were normal (Supplemental Table 2).
Liver histology-
Nine subjects underwent a liver biopsy as part of their diagnostic evaluation of which 5 were available for central review. The median time from DILI onset to the biopsy (among all cases) was 27 days (range: 9 to 57) (Table 2). Four of the five cases reviewed showed a consistent pattern of injury with moderate to marked zone 3 canalicular and hepatocellular cholestasis combined with generally mild, predominantly portal inflammation (Figure 2) with little to no interface hepatitis. Neither plasma cells nor eosinophils were seen in significant numbers and no case had significant fibrosis. There was no ductal paucity in any of the cases and only two of the four showed duct injury. The remaining patient (Case #12) differed in that no canalicular cholestasis was identified, but the biopsy was small. This case showed moderate chronic inflammation without plasma cells or eosinophils, mainly in the portal areas, with at least bridging fibrosis. The hepatocytes showed a diffuse microvacuolar change consistent with microvesicular steatosis. None of the biopsies showed histological features suggestive of autoimmune hepatitis.
Table 2:
Histopathological features of 5 cases of terbinafine hepatotoxicity.
| Subject No | HLA-A * 33:01 status | Age/ sex | Time from onset to biopsy (days) | R value at or near time of biopsy | Histological findings | Outcome |
|---|---|---|---|---|---|---|
| Case 2 | (+) | 75 M | 52 | 2.6 | Cholestatic hepatitis | Resolved |
| Case 4 | (+) | 48 F | 57 | 9.7 | Cholestatic hepatitis | Corticosteroids-chronic |
| Case 11 | (+) | 48 M | 27 | 0.6 | Cholestatic hepatitis | Resolved |
| Case 12 | (−) | 41 F | 9 | 4.2 | Chronic hepatitis with microvesicular steatosis | Transplant at 18 days |
| Case 13 | (−) | 40 M | 22 | 4.2 | Acute Cholestasis | Corticosteroids-resolved |
Figure 2-. Characteristic histological changes of terbinafine liver injury.
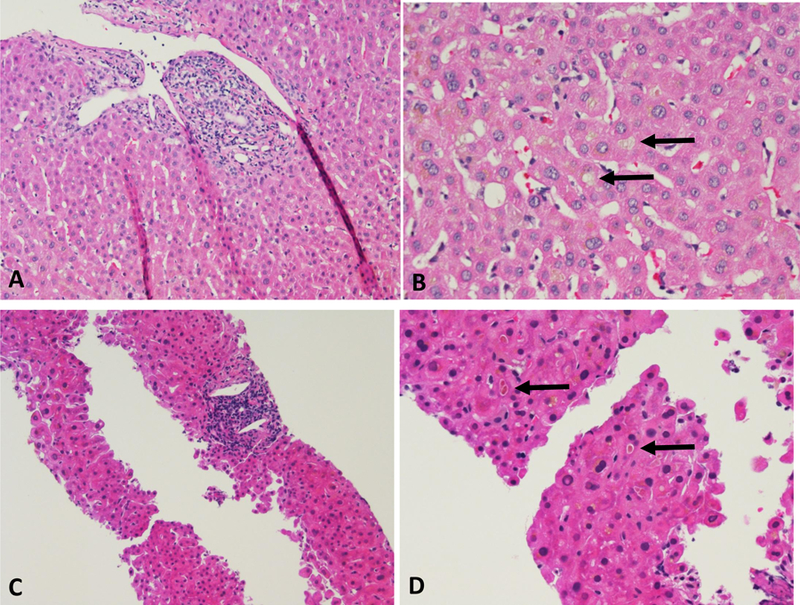
Portal inflammation (A, C, both H & E stains, 200x) and cholestasis (B, D, both H & E stains, 400x) from two cases: Case #11 (106–0007) (A, B) and case #2 (109–0102) (C, D). Arrows indicate bile plugs in canaliculi
Genetic analyses:
Previous studies using GWA with HLA imputation identified a highly significant association between terbinafine hepatotoxicity and HLA-A *33:01 (8). In the current study, the patients imputed as HLA-A*33:01 carriers were confirmed as having this rare HLA allele using high resolution HLA genotyping. In addition, one African American patient was genotyped for the first time and found to a carrier of HLA-A * 33:01. Furthermore, all 11 carriers were shown to have the extended haplotype: HLA A*33:01, HLA-B*14:02 and HLA-C*08:02. In addition, testing of further cases of terbinafine hepatotoxicity in patients from different racial groups showed that this association was present in the European ancestry subjects as well as an African American patient (Table 3, Supplementary Table 4). Ten of the 11 Caucasian but none of the Asian or mixed race subjects were HLA-A *33:01 carriers and none had HLA-B*14:02 or HLA-C*08:02. None of the 11 HLA A*33:01 positive patients were homozygous for this allele. Comparison of subjects with and without this allele showed no demographic, clinical or biochemical differences other than ancestral background (Table 1).
Table 3:
Frequency of HLA-A *33:01 and A* 33:03 in Terbinafine Hepatotoxicity Cases vs Population Controls
| HLA-A allele | HLA-A*33:01/−03 Carrier Frequency | |
|---|---|---|
| Cases (n=14) | Controls (n= see text) | |
| HLA-A *33:01 | ||
| European (n=11) | 91 % | 1.6% |
| Northern European (n=3) | 66% | 1.6% |
| Jewish (n=6) | 100% | 6.8% |
| African American (n=1) | 100% | 4.1% |
| HLA-A *33:03 | ||
| East Asian (n=2) | 100% | 23.4% |
The carrier frequency of the HLA-A *33:01 allele in the 11 European ancestry terbinafine patients is strikingly higher than that seen in population controls of European ancestry (91% vs 1.6%). The genome-wide SNP data available from participants was used to define ancestry more precisely to ensure that this was not a population-specific effect. Six of the 11 European ancestry cases were of Jewish ancestry: all 6 were HLA-A*33:01 carriers, though the carrier frequency of this HLA type is just 6.8% in Jewish population controls. Among 3 cases identified as having Northern European ancestry, 2 were HLA-A*33:01 carriers, despite a carrier frequency of just 1.6% in Northern European population controls (1.2% in England and Ireland). The single European ancestry case who was not heterozygous for HLA-A*33:01 was of Northern European ancestry and had the genotype HLA-A*24:02/HLA-A*32:01, which have carrier frequencies of 17% and 7% in European ancestry population controls, respectively.
The carrier frequency of HLA-A*33:01 is much lower in East Asian populations (0.08%) and was not found in either of the 2 East Asian cases. Although HLA-A*33:01 is more common in Hispanic ancestry population controls (carrier frequency 4.4%), it was still not seen in the single Hispanic case. Instead, the Hispanic case was homozygous for HLA-A*24:02, which has a carrier frequency of 24.4% in Hispanic population controls, and the two East Asian cases were of the type HLA-A*24:02/HLA-A*33:03, which have carrier frequencies of 40.6% and 23.4%, respectively, in East Asian population controls. HLA-A*33:03 shows sequence homology and shared peptide binding specificities with HLA-A*33:01 with differing amino acids at residues 131, 171 and 186 (Supplemental Table 3). We also had one case of African American ancestry and this patient was found to have HLA-A * 33:01. The carrier frequency of this allele in African Americans is 4.6%.
Molecular Docking Studies
To determine if terbinafine has the potential to interact with HLA-A*33:01, HLA-A * 33:03, HLA-B*14:02 and HLA-C*08:02, models of each antigen binding cleft (residues 1–180) were generated based on the most closely related crystal structures. Since class I HLA molecules are unstable in the absence of peptide, the models of terbinafine-associated HLA molecules were complexed with peptide. As sequences of peptides complexed with terbinafine and HLA-A*33:01, HLA-B*14:02 and HLA-C*08:02 are not known, potential bias that the peptide sequence may impose was reduced by removing each side chain in the peptide used for docking by altering each position to glycine. Molecular docking, using AutoDock Vina, suggested that terbinafine has the potential to interact with HLA-A*33:01, HLA-A * 33:03, HLA-B*14:02 and HLA-C*08:02 with moderate affinities (>−6.5 kcal per mole, low affinity, −6.5 to −9.0 moderate affinity, <−10 high affinity): molecular docking scores were estimated to be ΔG values of −7.2, −7.6, −7.1 and −7.3 kcal per mole, respectively (Figure 3). By comparison, abacavir, shown to bind a structural pocket of HLA-B*57:01 accommodating the side chain of the terminal peptide position, P9, and is predicted to bind the HLA-B*57:01 with a score of −7.2 in the absence of peptide and −9.1 in presence of peptide. These data indicated that the drug abacavir was predicted (and validated), to bind with higher affinity in the presence of a specific peptide. To determine if the peptide may alter the binding of terbinafine to HLA-A*33:01, HLA-A * 33:03, HLA-B*14:02 and HLA-C*08:02, terbinafine was docked in the absence of peptide, resulting in AutoDock Vina scores of −7.1, − 5.0, −7.7, and −7.4 kcal per mole, respectively. These data suggest peptides have the potential to influence terbinafine binding to HLA.
Figure 3-. Potential terbinafine binding to HLA molecules.
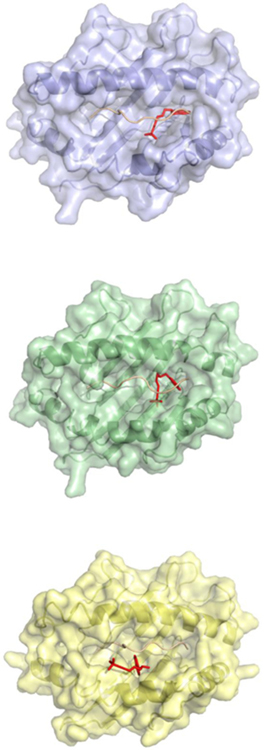
Terbinafine has the potential to interact with HLA molecules that have been linked to terbinafine induced liver injury. Molecular docking of terbinafine (red) on models of the antigen binding clefts of HLA-A*33:01 (violet), HLA-B*14:02 (green), and HLA-C*08:02 (yellow) predicts binding in sites comprised of peptide (salmon or pink) and HLA.
The predicted site of interaction between terbinafine and HLA-A*33:01 and HLA-B*14:02 was similar to the abacavir/HLA-B*57:01 interaction in which the drug was bound to the P9 pocket forming contact with peptide and HLA (Figure 3, top panels). Terbinafine was predicted to bind a distinct site on HLA-C*08:02 in which the drug was solvent accessible, potentially serving as an antigenic structure. Terbinafine was predicted to interact with the central portion of the antigen binding cleft (Figure 3, bottom panel). These molecular docking studies suggest terbinafine may interact with complexes comprised of peptides bound to HLA-A*33:01, HLA- A * 33:03, HLA-B*14:02 and HLA-C*08:02.
Discussion
Fifteen well characterized patients with liver injury attributed to terbinafine who were enrolled into the ongoing DILIN Prospective registry study are presented. Although the majority of patients were symptomatic with pruritus and jaundice, only 2 were hospitalized and 1 required eventual liver transplantation. Consistent with prior reports, hypersensitivity and autoimmune features were uncommon and most patients had either a cholestatic or mixed liver injury pattern at onset and a generally favorable prognosis (8, 25–29). The only potential fatal case in this series had a distinctly severe hepatocellular injury at onset and underwent liver transplantation within 3 weeks of presentation. During follow-up, 6 patients were treated with corticosteroids in an effort to hasten the resolution of liver injury. Nevertheless, the majority of patients (54%) followed had evidence of protocol determined chronic liver injury at a rate far higher than that observed in the overall DILIN cohort (18%) (7). In comparison to prior studies, the age and serum ALP levels at onset of those who did versus did not go on to develop chronic injury were not substantially higher but the total number of patients included was limited (Supplemental Table 1) (30). Of note, none of the patients with chronic injury had known underlying chronic liver disease. In addition, the median BMI of the terbinafine patients was substantially lower than that seen in overall DILIN cohort (24.2 vs 27 kg/m2) (8).
A recent review of the literature of 15 published cases of terbinafine hepatotoxicity noted that the majority of afflicted patients were over the age of 40 as observed in the current series but none of those cases developed chronic liver injury (31). However, the duration of follow-up was variable and many of the cases were identified retrospectively. A review of the available liver biopsies in our 5 patients obtained at a median of 27 days after DILI onset demonstrated a consistent pattern of cholestasis with or without mild hepatitis in 4 of 5 cases available for central review (Table 2). In these four cases the inflammation was mainly in the portal areas and lacked features of autoimmune hepatitis. The absence of bile duct loss in the presence of prominent zone 3 cholestasis suggests functional inhibition of bile transport at the level of the canalicular membrane. Interestingly, none of the patients who underwent an early biopsy for diagnostic purposes had neocholangiogenesis or other findings suggestive of early ductopenia despite prior case reports of progressive ductopenia in some patients with terbinafine hepatotoxicity (3,32). Although one patient (case #12) had detectable autoantibodies, the HLA alleles associated with autoimmune hepatitis were not present and her liver explant did not show histological features suggestive of autoimmune hepatitis. Although she would meet clinical criteria for a diagnosis of probable autoimmune hepatitis, other patients with suspected DILI and autoimmune features also score high on this scale. For example, the majority of patients with hepatotoxicity associated with nitrofurantoin and minocycline have prominent autoimmune features at presentation (33).
In a recent analysis of the International FDA Adverse Event Reporting System (FAERS) database there were 1964 total DILI cases associated with anti-mycotic agents (25). Interestingly, the oral antimycotics accounted for nearly 3% of the total liver injury events reported despite the fact that there are many other drugs more commonly used in the general population. Terbinafine was implicated in the largest number of cases (422) compared to the other orally administered antifungal agents. These data may reflect the fact that terbinafine prescriptions represented > 50% of the total systemic antifungal use in many European countries in 2007 (26). However, causality assessment was not possible in these cases due to incomplete reporting of clinical data and lack of systematic follow-up. In contrast, a population based study from Taiwan conducted between 2002 and 2008 demonstrated that terbinafine only accounted for 2 of 51 cases of antimycotic hepatotoxicity reported and was associated with the lowest incidence of hepatotoxicity amongst the agents studied (27). Similarly, in a United Kingdom study of over 69,000 patients treated with an oral antifungal agent, terbinafine was associated with the lowest overall rate of hepatotoxicity (28,29). In both of these studies, a longer duration of antifungal agent use was associated with a greater risk of hepatotoxicity. Of note, the FDA removed the need for liver biochemistry monitoring in patients treated with terbinafine in 2001 due to its favorable safety profile in post-marketing surveillance.
Polymorphisms in human drug metabolizing enzymes, drug transporters, and immune response genes have been implicated in the pathogenesis of idiosyncratic DILI. In a prior study that used an unbiased GWA approach in 862 well phenotyped DILI cases of European ancestry, the imputed HLA-A *33:01 allele was associated with hepatotoxicity due to several drugs and particularly mixed or cholestatic DILI (7). The strongest associations were seen for individual drugs including terbinafine (OR = 40.5 (12.5, 131.14), p = 6.7 × 10−10), fenofibrate (OR: 58.7 (12.3, 279.8), p=3.2 × 10−7), ticlodipine (OR: 163.1 (16.2 1642), p=0.00002), and sertraline (OR = 29 (4, 207), p=0.0008). However, the number of cases of hepatotoxicity with each agent was small and limited to those of European ancestry only. This prior study also suggested that the haplotype of HLA-A*33:01, B*14:02 and C*08:02 may confer risk for DILI as opposed to HLA-A*33:01 alone. In the current study, HLA genotyping confirmed that 10 of the 11 European ancestry terbinafine cases were carriers of HLA-A*33:01. The HLA genotypes in 3 newly enrolled high causality cases of terbinafine hepatotoxicity in non-European patients were also identified. Two of these patients were East Asian, and both were carriers of HLA-A*33:03, which has a high prevalence in East Asian control populations (carrier frequency 23.4%) and is very similar to HLA-A*33:01 in sequence, 99.5 %, identical (Supplemental Table 3). In the previous publication a single East Asian terbinafine DILI case was investigated and also found to be a carrier of HLA-A*33:03. Given the relatively high prevalence of this allele in East Asian populations, we would expect a higher rate of DILI due to terbinafine in East Asians compared to the rate in other groups. However, the small total sample size of terbinafine cases here precludes us from assessing the risk of toxicity based upon patient ethnicity. The other new terbinafine case in the current series was of Hispanic ancestry and homozygous for HLA-A*24:02, which has a carrier frequency of 24.6% in Hispanic populations and is not overtly similar to HLA-A*33:01 in structure and function. HLA-A*24:02 is 89.3 % identical to HLA-A*33:01 with differences in the antigen binding cleft. Of note, no significant differences in the clinical presentation or outcome based upon HLA-A *33:01 genotype was observed in our series (Table 1). A larger number of terbinafine DILI cases could help determine if the HLA- A *33:01 carriers have any unique clinical or laboratory features compared to non-carriers with hepatotoxicity.
The increased frequency of the HLA-A*33:03 risk allele in those of East Asian ancestry appears to correspond to a higher incidence of terbinafine DILI than occurs in those of European ancestry. We found that 0.5% of our European ancestry DILI cases had terbinafine as their causal drug, compared to 7.7% of those of East Asian ancestry. Likewise, the risk allele HLA-A*33:01 is at a higher frequency in those of Jewish ancestry than in other populations, again appearing to correspond to a higher incidence of terbinafine DILI, with 6.7% of the Jewish or Middle Eastern ancestry DILI cases having terbinafine as the causal drug. Although those of Jewish or Middle Eastern ancestry only made up 5.1% of our cohort overall, and 6.9% of those with European ancestry, they made up 33% of our terbinafine DILI cases. Those of Jewish ancestry make up approximately 2.2% of the American population, and HLA-A*33:01 has a carrier frequency of 6.8% in this group; likewise, those of East Asian ancestry make up approximately 2.6% of the American population, and HLA-A*33:03 has a carrier frequency of 22% in this group (34, 35). More studies are required to quantify baseline terbinafine use in different populations, but our results imply that those of East Asian or Jewish ancestry might benefit from screening for these highly penetrant HLA risk alleles before terbinafine use. Furthermore, testing for HLA-A*33:01 and 33:03 might be helpful in diagnosis of terbinafine induced liver injury in patients exposed to several potential injurious agents at least among persons of European or East Asian ancestory. In support of this, none of our 4 cases with a low causality score for terbinafine DILI (3 Caucasians and 1 Asian) were HLA-A * 33:01 or 33:03 carriers.
Molecular docking studies suggest mechanism(s) by which one or several HLA molecules may present immunogenic ligands in terbinafine DILI patients. Although these studies demonstrated that terbinafine has the potential to directly interact with HLA-A*33:01, HLA- A * 33:03, HLA-B*14:02 and HLA-C*08:02 (Figure 3), metabolites of terbinafine may also contribute to the formation of immunogenic peptide/HLA complexes (3). Terbinafine was predicted to bind HLA-A*33:01 and HLA-B*14:02 in the same site where abacavir bound HLA-B*57:01 (P9 or F pocket) in crystal structures, suggesting possible similarities in terms of the mechanism (35). Abacavir binding at this site altered the preference of HLA-B*57:01 for specific peptides, resulting in presentation of peptides for which the host was not tolerant. Terbinafine may stimulate T cell responses through multiple mechanisms involving one or more class I HLA molecules, the drug and/or its metabolites, and peptides that bind HLA preferentially in the presence of drug or its metabolites. Additional studies that may prove informative include the testing of human hepatocytes and macrophages in an in vitro test system from individuals that are HLA-A *33:01 carriers and non-carriers with and without terbinafine or its metabolites. Measurement of peptide binding affinities in the presence and absence of terbinafine or its metabolites would also help determine if direct intermolecular interactions occur with the HLA-A *33:01 antigen binding cleft.
Strengths of the current study include the prospective identification and enrollment of terbinafine DILI patients of varying ethnicities from multiple medical centers throughout the United States which improves the generalizability of our results. In addition, all patients were followed for at least 6 months, a thorough investigation was undertaken to exclude competing causes of liver injury, and a standardized causality assessment method was used. As has been reported in a recent systematic review, all of the patients in our cohort were older than 40 further strengthening the generalizability of our observations (31). Limitations of our study include the small total number of cases and lack of an external validation cohort. In addition, liver histology and other objective measures of liver disease severity such as liver elastography were not available in patients who met our protocol determined definition of chronic DILI to better define their long-term outcomes. Lastly, although all of the patients had normal total bilirubin levels at month 24 of follow-up, we previously showed in a larger cohort of DILIN patients that quality of life was impaired in patients with persistent biochemical abnormalities and that some patients may develop advanced fibrosis or evidence of portal hypertension during follow-up (30). To address these limitations, the DILIN prospective study protocol was recently amended to extend follow-up out to 4 years and include non-invasive measures of liver disease severity including liver elastography.
The consistent mixed or cholestatic liver injury profile with jaundice and pruritus suggests that terbinafine may be associated with a fairly characteristic and recognizable clinical phenotype. The use of a high resolution genotyping platform confirmed our prior imputed GWA results implicating the HLA- A * 33:01, B* 14:02, C*08:02 haplotype in terbinafine hepatotoxicity. The magnitude of this HLA association is quite strong (i.e. OR =40) having only been exceeded by the studies of HLA- B*57:01 and flucoxacillin hepatotoxicity. Finally, the molecular docking studies that we completed suggest that terbinafine itself with or without a peptide may interact with any of the haplotypic HLA alleles (HLA-A*33:01, B*14:02 and C*08:02). Further studies exploring the mechanism of this HLA association are planned.
Supplementary Material
Highlights:
Patients with terbinafine hepatotoxicity most commonly present with a mixed or cholestatic liver injury profile and up to 50% have residual evidence of chronic cholestasis during follow-up
High resolution HLA genotyping demonstrated that Caucasian patients were significantly more likely to be carriers of HLA-A * 33:01, B *14:02, C* 08:02 compared to population controls (91% vs 1.6%) and a similar HLA haplotype of HLA-A *33:03 was also over-represented in Asian American terbinafine hepatotoxicity cases compared to population controls (100% vs 23%).
Molecular docking studies suggest that terbinafine may interact with these HLA alleles.
Testing for HLA-A * 33:01 and HLA * 33:03 in Caucasian and Asian Americans with suspected terbinafine hepatotoxicity, respectively, may prove worthwhile as a confirmatory diagnostic biomarker
Acknowledgements
DILIN Clinical Sites:
Indiana University-Purdue: Naga Chalasani, MD, PI; Marwan S. Ghabril, MD, Sub-I; Marwan Ghabril, MD, Sub-I; Raj Vuppalanchi, MD, Sub-I; Study Coord; Wendy Morlan, RN, Study Coord;
University of Michigan-Ann Arbor: Robert J. Fontana, MD, PI; Hari Conjeevaram, MD, Sub-I; Frank DiPaola, MD, Sub-I; [Dora Siettas, Study Coord; Ben Johnson, Study Coord];
University of North Carolina-Chapel Hill: Paul Watkins, MD, PI; Jama Darling, MD, Sub-I; Michael Fried, Sub-I; Paul H. Hayashi, MD, Sub-I; Steven Lichtman, MD, Sub-I; Steven Zacks, MD, MPH, Sub-I; [Tracy Russell, CCRP, Study Coord];
Satellite Sites:
Asheville: William Harlan, MD, PI; [Tracy Russell, CCRP, Study Coord];
Wake Forest Baptist Medical Center: Herbert Bonkovsky, MD, PI; Pradeep Yarra, MD, Sub-I; [Denise Faust, Study Coord];
University of Southern California: Andrew Stolz, MD, PI; Neil Kaplowitz, MD, Sub-I; John Donovan, MD, Sub-I; [Susan Milstein, RN, BSN, Study Coord];
Satellite Sites:
University of California-Los Angeles (Pfleger Liver Institute): Francisco A. Durazo, MD, PI; [Yolanda Melgoza, Study Coord; Val Peacock, RN, BSN, Co-Coord]
Albert Einstein Medical Center: Victor J. Navarro, MD, PI; Simona Rossi, MD, Sub-I; [Maricruz Vega, MPH, Study Coord; Manisha Verma, MD, MPH, Study Coord]
Icahn School of Medicine at Mount Sinai: Joseph Odin, MD, PhD, PI; Jawad Ahmad, MD, PI; Nancy Bach, Sub-I; Meena Bansal, MD, Sub-I; Charissa Chang, MD, Sub-I; Douglas Dieterich, MD, Sub-I; Priya Grewal, MD, Sub-I; Lawrence Liu, MD, Sub-I; Thomas Schiano, MD, Sub-I; [Monica Taveras, Study Coord]
National Institutes of Health Clinical Center: Christopher Koh, MD, PI; [Beverly Niles, Study Coord]
DILIN Data Coordinating Center at Duke Clinical Research Institute: Huiman X. Barnhart, PhD, PI; Katherine Galan, RN, Project Lead; Theresa O’Reilly, Lead CRA; Elizabeth Mocka, CRA; Olivia Pearce, CTA; Michelle McClanahan-Crowder, Data Management; Coleen Crespo-Elliott, Data Management; Hoss Rostami, Data Management; Qinghong Yang, Programmer-Statistics; Jiezhun (Sherry) Gu, PhD, Statistician; Tuan Chau, Lead Safety Associate; Liz Cirulli-Rogers, PhD, Pharmacogenetics statistician
National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK): José Serrano, MD, Project Scientist; Sherry R. Hall, MS, Clinical Trials Specialist; Jay Hoofnagle, MD, Scientific Advisor; Averell H. Sherker, MD, FRCP(C), Program Official.
National Cancer Institute: David E. Kleiner, MD, PhD, Hepatopathologist
Financial support: The DILIN Network is structured as a U01 cooperative agreement with funds provided by the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) under grants: 2U01-DK065176–06 (Duke), 2U01-DK065201–06 (UNC), 2U01-DK065184–06 (Michigan), 2U01-DK065211–06 (Indiana), 5U01DK065193–04 (UConn), 5U01-DK065238–08 (UCSF/CPMC), 1U01-DK083023–01 (UTSW), 1U01-DK083027–01 (TJH/UPenn), 1U01-DK082992–01 (Mayo), 1U01-DK083020–01 (USC). Additional funding is provided by CTSA grants: UL1 RR025761 (Indiana), UL1TR000083 (UNC), UL1 RR024134 (UPenn), UL1 RR024986 (Michigan), UL1 RR024982 (UTSW), UL1 RR024150 (Mayo) and in part by the Intramural Research Program of the NIH, National Cancer Institute. This study was also supported in part by 1P50GM115305–01, 1P30AI110527–01A1, 1 R13AR71267–01, National Health and Medical Research Council (Australia) (E.J.P), and RO1 AI103348 (DAO).
Abbreviations
- ALP
Alkaline phosphatase
- ALT
Alanine aminotransferase
- ANA
Anti-nuclear antibody
- AST
Aspartate aminotransferase
- BMI
Body mass index
- DILI
Drug induced liver injury
- CI
Confidence interval
- DILIN
Drug induced liver injury network
- FAERS
FDA Adverse Event Reporting system
- GWA
Genome wide association
- HLA
Human leukocyte antigen
- iSAEC
International Serious Adverse Events Consortium
- MAF
Minor allele frequency
- MID
Multiplex Identified OR Odds ratio
- RUCAM
Rousell- Uclaf Causality Assessment Method
- SMA
Smooth muscle antibody
- SNP
Single nucleotide polymorphisms
- ULN
Upper limit of normal
- VBDS
Vanishing bile duct syndrome
Footnotes
Potential conflicts of interest
Dr. Fontana has received research support from BMS and Gilead and has consulted for Alnylam
Dr. Phillips has consulted for Biocryst Pharmaceuticals Inc. and Xcovery Pharmaceuticals
Drs Hoofnagle, Kleiner, Barnhart, Ostrov, Schutte and Gu have no potential conflicts
Dr. Cirulli is currently an employee of Human Longevity but was at Duke University when studies were completed.
Dr. Watkins consults for Novartis which is a producer of terbinafine, but has not consulted with the company regarding any issues related to terbinafine.
Dr. Chalasani consults for several pharmaceutical companies but has no reported conflicts for this manuscript.
References
- 1.Fernandes NF, Geller SA, Fong TL. Terbinafine hepatotoxicity: case report and review of the literature. Am J Gastroenterol 1998. March;93(3):459–60. [DOI] [PubMed] [Google Scholar]
- 2.Iverson SL, Uetrecht JP. Identification of a reactive metabolite of terbinafine: insights into terbinafine-induced hepatotoxicity. Chem Res Toxicol 2001. February;14(2):175–81. [DOI] [PubMed] [Google Scholar]
- 3.Anania FA, Rabin L. Terbinafine hepatotoxicity resulting in chronic biliary ductopenia and portal fibrosis. Am J Med 2002; 112:741–2. [DOI] [PubMed] [Google Scholar]
- 4.Perveze Z, Johnson MW, Rubin RA, Sellers M, Zayas C, Jones JL, Cross R, et al. Terbinafine-induced hepatic failure requiring liver transplantation. Liver Transpl 2007; 13:162–4. [DOI] [PubMed] [Google Scholar]
- 5.Fontana RJ, Watkins PB, Bonkovsky HL, Chalasani N, Davern T, Serrano J, Rochon J. Drug-Induced Liver Injury Network (DILIN) prospective study: rationale, design and conduct. Drug Saf 2009; 32:55–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lucena MI, Molokhia M, Shen Y, Urban TJ, Aithal GP, Andrade RJ, et al. Susceptibility to Amoxicillin-Clavulanate-Induced Liver Injury is Influenced by Multiple HLA Class I and II Alleles. Gastroenterology 2011. July;141(1)338–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nicoletti P, Aithal GP, Bjornsson ES, Andrade RJ, Sawle A, Arrese M, et al. Association of Liver Injury from Specific Drugs, or Groups of Drugs, with Polymorphisms in HLA and Other Genes in a Genome-wide Association Study. Gastroenterology 2017. April;152(5):1078–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chalasani N, Bonkovsky HL, Fontana R, Lee W, Stolz A, Talwalkar J, et al. Features and Outcomes of 899 Patients with Drug-Induced Liver Injury: The DILIN Prospective Study. Gastroenterology 2015. June;148(7):1340–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fontana RJ, Hayashi PH, Gu J, Reddy KR, Barnhart H, Watkins PH, et al. Idiosyncratic drug-induced liver injury is associated with substantial morbidity and mortality within 6 months from onset. Gastroenterology 2014. July;147(1):96–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kleiner DE, Chalasani NP, Lee WM, Fontana RJ, Bonkovsky HL, Watkins PB, et al. Hepatic histological findings in suspected drug-induced liver injury: systematic evaluation and clinical associations. Hepatology 2014. February;59(2):661–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet 2006. August;38(8):904–9. [DOI] [PubMed] [Google Scholar]
- 12.Gonzalez-Galarza FF, Takeshita LY, Santos EJ, Kempson F, Maia JH, da Silva AL, et al. Allele frequency net 2015 update: new features for HLA epitopes, KIR and disease and HLA adverse drug reaction associations. Nucleic Acids Res 2015. January;43: D784–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gragert L, Madbouly A, Freeman J, Maiers M. Six-locus high resolution HLA haplotype frequencies derived from mixed-resolution DNA typing for the entire US donor registry. Hum Immunol 2016. October;74(10):1313–20. [DOI] [PubMed] [Google Scholar]
- 14.Schmidt AH, Baier D, Solloch UV, Stahr A, Cereb N, Wassmuth R, Ehninger G, Rutt C. Estimation of high-resolution HLA-A, -B, -C, -DRB1 allele and haplotype frequencies based on 8862 German stem cell donors and implications for strategic donor registry planning. Hum Immunol 2009. November;70(11):892–902. [DOI] [PubMed] [Google Scholar]
- 15.Pingel J, Solloch UV, Hofmann JA, Lange V, Ehninger G, Schmidt AH. High-resolution HLA haplotype frequencies of stem cell donors in Germany with foreign parentage: how can they be used to improve unrelated donor searches? Hum Immunol 2013. March;74(3):330–40. [DOI] [PubMed] [Google Scholar]
- 16.Williams Fionnuala, Meenagh Ashley, Single Rich, Mark McNally Philip Kelly, Nelson Mark P, et al. High resolution HLA-DRB1 identification of a Caucasian population. Hum Immunol 2004. January;65(1):66–77. [DOI] [PubMed] [Google Scholar]
- 17.Manor S, Halagan M, Shriki N, Yaniv I, Zisser B, Maiers M, et al. High-resolution HLA A~B~DRB1 haplotype frequencies from the Ezer Mizion Bone Marrow Donor Registry in Israel. Hum Immunol 2016. December;77(12):1114–1119. [DOI] [PubMed] [Google Scholar]
- 18.Bordolo L, Kiefer F, Arnold K, Benkert P, Battey J, Schwede T. Protein structure homology modeling using SWISS-MODEL workspace. Nat Protoc 2009;4(1):1–13. [DOI] [PubMed] [Google Scholar]
- 19.Zhang S, Liu J, Cheng H, Tan S, Qi J, Yan J, Gao GF. Structural basis of cross-allele presentation by HLA-A*0301 and HLA-A*1101 revealed by two HIV-derived peptide complexes. Mol Immunol 2011. October;49(1–2):395–401. [DOI] [PubMed] [Google Scholar]
- 20.Kumar P, Vahedi-Faridi A, Saenger W, Merino E, Lopez de Castro JA, Uchanska-Ziegler B, Ziegler A. Structural basis for T cell alloreactivity among three HLA-B14 and HLA-B27 antigens. J Biol Chem 2009. October 23;284(43):29784–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr 2010. April;66(Pt 4):486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J Comput Chem 2009. December;30(16):2785–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weininger D SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J Chem Inf Comput Sci 1988. 28(1):31–36. [Google Scholar]
- 24.de Boer YS, van Gerven NM, Zwiers A, et al. Genome wide association study identifies variants associated with autoimmune hepatitis type 1. Gastroenterology 2014; 147: 443–452. [DOI] [PubMed] [Google Scholar]
- 25.Raschi Emanuel, Poluzzi Elisabetta, Koci Ariola, Caraceni Paolo, Fabrizio De Ponti. Assessing liver injury associated with antimycotics: Concise literature review and clues from data mining of the FAERS database. World J Hepatol 2014. August 27;6(8):601–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Adriaenssens N, Coenen S, Versporten A, Goossens H. Outpatient systemic antimycotic and antifungal use in Europe: new outcome measure provides new insight. Int J Antimicrob Agents 2013. November;42(5):466–470. [DOI] [PubMed] [Google Scholar]
- 27.Kao WY, Su CW, Huang YS, Chou YC, Chen YC, Chung WH, et al. Risk of oral antifungal agent- induced liver injury in Taiwanese. Br J Clin Pharmacol 2014. January;77(1):180–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Elewski B, Tavakkol A. Safety and tolerability of oral antifungal agents in the treatment of fungal nail disease: a proven reality. Ther Clin Risk Manag 2005. December;1(4):299–306. [PMC free article] [PubMed] [Google Scholar]
- 29.Garcia Rodriguez LA, Duque A, Castellsaque J, Perez-Gutthann S, Stricker BH. A cohort study on the risk of acute liver injury among users of ketoconazole and other antifungal drugs. Br J Clin Pharmacol 1999. December;48(6):847–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fontana RJ, Hayashi PH, Barnhart H, Kleiner DE, Reddy KR, Chalasani N, et al. Persistent liver biochemistry abnormalities are more common in older patients and those with cholestatic drug induced liver injury. Am J Gastroenterol 2015. October;110(10):1450–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang J, Wang X, Chen S. Systematic review of severe acute liver injury caused by terbinafine. Int J Clin Pharm 2014. August;36(4):679–83. [DOI] [PubMed] [Google Scholar]
- 32.Mallat A, Zafrani ES, Metreau JM, Dhumeaux D. Terbinafine-induced prolonged cholestasis with reduction of interlobular bile ducts. Dig Dis Sci 1997. July;42(7):1486–8. [DOI] [PubMed] [Google Scholar]
- 33.DeBoer YS, Kosinski AS, Urban TJ, Zhao Z, Long N, Chalasani N, et al. Features of autoimmune hepatitis in patients with Drug-induced liver injury. Clin Gastro & Hepatol 2017; 15: 103–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.US Census 2010; http://www.census.gov/prod/cen2010/briefs/c2010br-11.pdf
- 35.Ostrov DA, Grant BJ, Pompeu TA, Sidney J, Harndahl M, Southwood S, et al. Drug hypersensitivity caused by alteration of the MHC-presented self-peptide repertoire. PNAS 2012;109(25):9959–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.