

Abstract
Gle2/Rae1 is highly conserved from yeast to humans and has been described as an mRNA export factor. Additionally, it is implicated in the anaphase‐promoting complex‐mediated cell cycle regulation in higher eukaryotes. Here we identify an involvement for Saccharomyces cerevisiae Gle2 in septin organization, which is crucial for cell cycle progression and cell division. Gle2 genetically and physically interacts with components of the septin ring. Importantly, deletion of GLE2 leads to elongated buds, severe defects in septin‐assembly and their cellular mislocalization. Septin‐ring formation is triggered by the septin‐regulating GTPase Cdc42, which establishes and maintains cell polarity. Additionally, activity of the master cell cycle regulator Cdc28 (Cdk1) is needed, which is, besides other functions, also required for G2/M‐transition, and in yeast particularly responsible for initiating the apical–isotropic switch. We show genetic and physical interactions of Gle2 with both Cdc42 and Cdc28. Most importantly, we find that gle2∆ severely mislocalizes Cdc42, leading to defects in septin‐complex formation and cell division. Thus, our findings suggest that Gle2 participates in the efficient organization of the septin assembly network, where it might act as a scaffold protein. © 2017 The Authors. Yeast published by John Wiley & Sons, Ltd.
Keywords: cell cycle regulation, mRNA export, Gle2, Rae1, septins
Introduction
Septins are highly conserved eukaryotic proteins that also in human are increasingly recognized as novel components of the cytoskeleton (Mostowy and Cossart, 2012). Their dysfunction is linked to various diseases, including cancer, neurological disorders and infections. All septins are GTP‐binding proteins that form hetero‐oligomers and higher‐order structures resulting in filaments, bundles or rings (Mostowy and Cossart, 2012), which are necessary to control cellular processes that require localization, for instance at the division site (Joo et al., 2005; Kinoshita and Noda, 2001) or the plasma membrane (Hagiwara et al., 2011; Sellin et al., 2011). Septins control cellular processes by being scaffolds for protein recruitment and by establishment of structures that provide diffusion barriers important for cell division (Mostowy and Cossart, 2012). Their ability to form filaments was shown to be crucial for septin function and in case of errors activate the morphogenesis checkpoint to halt cell division (Kim et al., 2011; Lew, 2003; McMurray et al., 2011). Septins associate with cellular membranes, actin filaments and microtubules (Kinoshita et al., 2002; Sellin et al., 2011; Surka et al., 2002; Tanaka‐Takiguchi et al., 2009). However, the regulatory mechanisms for the directed and timely septin assembly are only partly understood.
Here we show that Gle2 (RAE1 in humans) is involved in proper septin organization. Gle2 was identified as a Nup116‐ and Nup100‐associated protein, which helps to sustain the structural integrity of the nuclear pore complex (NPC) (Ho et al., 1998). Deletion or mutation of GLE2 leads to NPC‐clustering (Bucci and Wente, 1997) and accumulation of poly(A)+ containing RNAs in the nucleus (Bailer et al., 1998; Murphy et al., 1996). The protein is highly conserved from Saccharomyces cerevisiae to metazoans and an involvement in mRNA‐export has also been documented for Saccharomyces pombe (Yoon et al., 2000) and human (Bharathi et al., 1997; Blevins et al., 2003).
Interestingly, in addition to its involvement in mRNA export, a mutation in S. pombe RAE1 (rae1–1) leads to an arrest in cell cycle at the G2/M boundary with perturbations of the cytoskeleton (Brown et al., 1995; Whalen et al., 1997). Crystal structure analysis revealed that the kinetochore checkpoint protein hBub3 and Gle2/Rae1 both are seven‐bladed WD40 repeat propeller proteins, which are typical scaffold proteins, and studies in human cells revealed that they are both involved in the progression through mitosis (Larsen and Harrison, 2004; Larsen et al., 2007; Reddy et al., 2008; Ren et al., 2010). There, a Rae1–Nup98 complex interacts with the early Cdh1 activated form of the anaphase promoting complex (APCCdh1) (Jeganathan et al., 2005). Ubiquitinylation of securin and mitotic cyclins by the APC with subsequent proteasomal degradation leads to chromosome segregation and entry into mitotic exit (Baker et al., 2007). Defects in this process cause chromosome missegegation and subsequent aneuploidy, leading to cancer and in particular leukaemia (Funasaka et al., 2011; Jeganathan et al., 2005). Another cell cycle‐related function of Rae1/Gle2 is the localization of an mRNA/Rae1 complex to microtubules (Kraemer et al., 2001; Sitterlin, 2005), where it is required for microtubule dynamics and spindle assembly (Blower et al., 2005).
All of these findings argue for a broad but in detail still undefined role of Gle2/Rae1 in the cell. Our study unravels an involvement for Gle2 in cell cycle regulation and in particular in septin‐ring formation, which is essential for cytokinesis.
Material and methods
Yeast strains, plasmids and oligonucleotides
All yeast strains used in this study are listed in the Supporting information Table 1 and plasmids in Table 2. Plasmids and yeast strains were generated by conventional methods. Unless stated differently all yeast strains derived from the BY4741 strain background.
Table 1.
Yeast strains used in this study.
| Number | Name | Genotype | Source |
|---|---|---|---|
| HKY124 | — | MATα ura3–52 leu2∆1 his3∆200 rat7–1 | Gorsch et al. (1995) |
| HKY381 | — | MATα ura3∆0 leu2∆0 his3∆1 lys2∆0 | Euroscarf |
| HKY1154 | — |
MATα ura3∆0 leu2∆0 his3∆1 met15∆0
can1::STE2pr‐SP_his5 lyp1::STE3pr‐LEU2 gle2::NatR |
SGA screen |
| HKY1159 | — |
MATα ura3∆0 leu2∆0 his3∆1 met15∆0 lyp1
LYS2 can1::STE2pr‐SP_his5 ura3::NatR |
SGA screen |
| HKY1163 | Y7092 |
MATα can1::STE2pr‐SP_his5 lyp1 ura3∆0 leu2∆0
his3∆1 met15∆0 |
Tong and Boone (2007) |
| HKY1282 | — |
MATa ura3∆0 leu2∆0 his3∆1 met15∆0
CDC10‐GFP:HIS3MX6 |
Invitrogen |
| HKY1450 | — |
MATa ura3–52 leu2_3 trp1–289 his3∆1 MAL2‐8 cc
SUC2 (CEN.PK2‐1Ca) |
Entian et al. (1999) |
| HKY1451 | — |
MATa ura3–52 leu2_3 trp1–289 his3∆1 MAL2‐8 cc
SUC2 (CEN.PK2‐1Ca) gle2::kanMX4 |
This study |
| HKY1501 | — |
MATa ura3∆0 leu2∆0 his3∆1 met15∆0
CDC11‐GFP:HIS3MX6 |
Invitrogen |
| HKY1524 | — | MATa ura3∆0 leu2∆0 his3∆1 met15∆0 cdc10–1:kanR | SGA screen |
| HKY1525 | — | MATa ura3∆0 leu2∆0 his3∆1 met15∆0 cdc14–8:kanR | SGA screen |
| HKY1526 | — | MATa ura3∆0 leu2∆0 his3∆1 met15∆0 cdc15–2:kanR | SGA screen |
| HKY1527 | — | MATa ura3∆0 leu2∆0 his3∆1 met15∆0 cdc16–1:kanR | SGA screen |
| HKY1528 | — | MATa ura3∆0 leu2∆0 his3∆1 met15∆0 cdc20–2:kanR | SGA screen |
| HKY1529 | — | MATa ura3∆0 leu2∆0 his3∆1 met15∆0 cks1–38:kanR | SGA screen |
| HKY1531 | — | MATα ura3∆0 cdc14–8:kanR gle2::NatR | This study |
| HKY1532 | — | MATa ura3∆0 leu2∆0 his3∆1 met15∆0 cdc15–2:kanR gle2::NatR | This study |
| HKY1533 | — | MATα ura3∆0 his3∆1 cdc16–1:kanR gle2::NatR | This study |
| HKY1534 | — | MATα ura3∆0 his3∆1 lyp1::STE3pr‐LEU2 cdc20–2:kanR gle2::NatR + p CEN URA3 GLE2 | This study |
| HKY1535 | — | MATa ura3∆0 leu2∆0 cks1–38:kanR gle2::NatR | This study |
| HKY1538 | — | MATa ura3∆0 leu2∆0 his3∆1 cdc28–13:kanR gle2::NatR | This study |
| HKY1539 | — | MATa ura3∆0 LEU2 CDC10‐GFP:HIS3MX6 + p CEN URA3 GLE2‐myc | This study |
| HKY1540 | — | MATa ura3∆0 LEU2 CDC10‐GFP:HIS3MX6 gle2::NatR + p CEN URA3 GLE2‐myc | This study |
| HKY1541 | — | MATa ura3∆0 LEU2 CDC14‐GFP:HIS3MX6 gle2::NatR | This study |
| HKY1542 | — | MATα ura3∆0 LEU2 CDC15‐GFP:HIS3MX6 gle2::NatR | This study |
| HKY1543 | — | MATα ura3∆0 leu2∆ CDC16‐GFP:HIS3MX6 gle2::NatR | This study |
| HKY1544 | — | MATα ura3∆0 LEU2 CDC20‐GFP:HIS3MX6 gle2::NatR | This study |
| HKY1545 | — | MATα ura3∆0 LEU2 CDC28‐GFP:HIS3MX6 gle2::NatR | This study |
| HKY1546 | — | MATα ura3∆0 LEU2 CKS1‐GFP:HIS3MX6 gle2::NatR | This study |
| HKY1564 | — | MATa ura3∆0 LEU2 CDC11‐GFP:HIS3MX6 gle2::NatR | This study |
| HKY1600 | RLY8492 |
MATa ura3–52, leu2_3, MFA1‐3xGFP::HIS5
+ minichromosome CEN3.L..YFS5.1 MATalpha‐LEU2 |
Zhu et al. (2015) |
| HKY1602 | RLY8496 | MATa ura3–52, leu2_3, MFA1‐3xGFP::HIS5 mad1∆::natMX + minichromosome CEN3.L..YFS5.1 MATalpha‐LEU2 | Zhu et al. (2015) |
| HKY1610 | — | MATa ura3∆0 leu2∆0 his3∆1 met15∆0 cdc28–13:kanR | SGA screen |
| HKY1614 | — |
MATa ura3–52 leu2∆1 his3∆200 trp1∆63
GFP‐linker‐CDC42:URA3 |
This study |
| HKY1615 | — |
MATa ura3–52 leu2∆1 his3∆200 TRP
GFP‐linker‐CDC42:URA3 gle2::NatR |
This study |
| HKY1618 | — |
MATa ura3–52 leu2_3 MFA1‐3xGFP:HIS5 gle2::kanMX4
+ minichromosome CEN3.L..YFS5.1 MATalpha‐LEU2 |
This study |
| HKY1625 | — |
MATa ura3–52 leu2_3 MFA1‐3xGFP:HIS5 gle2::kanMX4
+ minichromosome CEN3.L..YFS5.1 MATalpha‐LEU2 |
This study |
| HKY1627 | — | MATa ura3∆0 leu2∆0 cdc10–1:kanR gle2::NatR | This study |
| HKY1755 | — | MATa ura3∆0 leu2∆0 his3∆1 met15∆0 cdc3–3:kanR | SGA screen |
| HKY1758 | — | MATa ura3∆0 leu2∆0 his3∆1 met15∆0 cdc11–3:kanR | SGA screen |
| HKY1761 | — | MATa ura3∆0 leu2∆0 his3∆1 met15∆0 cdc12–1:kanR | SGA screen |
| HKY1763 | — | MATa ura3∆0 leu2∆0 his3∆1 met15∆0 cdc42–1:kanR | SGA screen |
| HKY1769 | — | MATα ura3∆0 leu2∆0 his3∆1 cdc3–3:kanR gle2::NatR | This study |
| HKY1770 | — | MATα ura3∆0 leu2∆0 his3∆1 cdc11–3:kanR gle2::NatR | This study |
| HKY1771 | — | MATa ura3∆0 leu2∆0 cdc12–1:kanR gle2::NatR | This study |
| HKY1772 | — | MATa ura3∆0 leu2∆0 his3∆1 cdc42–1:kanR gle2::NatR | This study |
SGA, Synthetic genetic array.
Table 2.
Plasmids used in this study.
| Number | Features | Source |
|---|---|---|
| pHK87 | CEN LEU2 | Sikorski and Hieter (1989) |
| pHK88 | CEN URA3 | Sikorski and Hieter (1989) |
| pHK101 | 2 μ HIS3 | Sikorski and Hieter (1989) |
| pHK1384 | CEN URA3 GLE2 | This study |
| pHK1385 | 2 μ HIS3 GLE | This study |
| pHK1386 | CEN URA3 P ADH1 :GLE2‐3xmyc | This study |
| pHK1387 | CEN URA3 P ADH1 :3xmyc‐GLE2 | This study |
| pHK1427 | CEN URA3 P ADH1 :CDC10‐3xmyc | This study |
| pHK1507 | CEN LEU2 P ADH1 : 6xmyc‐GLE2 | This study |
Drop dilution tests
Cells were spotted in serial dilution (107 to 103 cells/10 μL per drop) onto rich medium (Figures 1b, 3a and 4a) or selective medium (Figure S1b). Plates were incubated for 3 days at the indicated temperatures.
Figure 1.
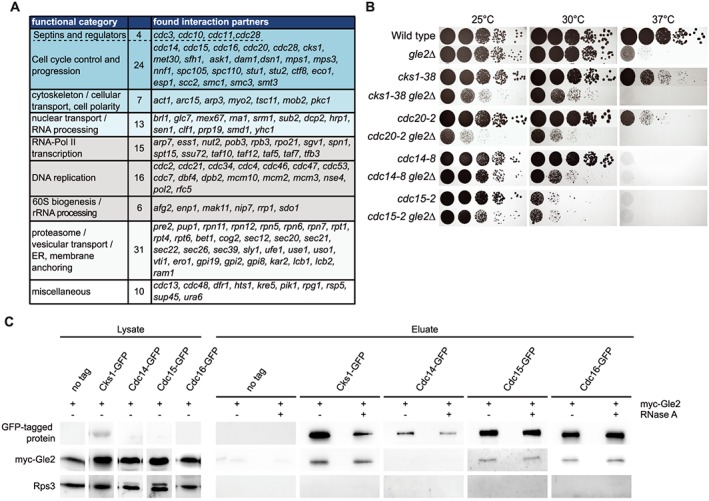
Gle2 interacts with cell cycle regulators. (a) Synthetic genetic array (SGA) screen with essential temperature sensitive alleles reveals interactions of GLE2 with several groups functioning in cell cycle progression. A gle2∆ strain was crossed in an automated setup with each of the SGA strains and synthetic sickness or lethality was analysed. (b) Combination of gle2∆ with cell cycle mutants aggravates their growth defects, as visualized on agar plates in serial dilutions. (c) Gle2 interacts physically with several proteins involved in cell cycle regulation. Western blots showing co‐immunoprecipitations of myc‐Gle2 with GFP‐tagged versions of proteins involved in cell cycle progression. Rps3 served as a negative control.
Figure 3.
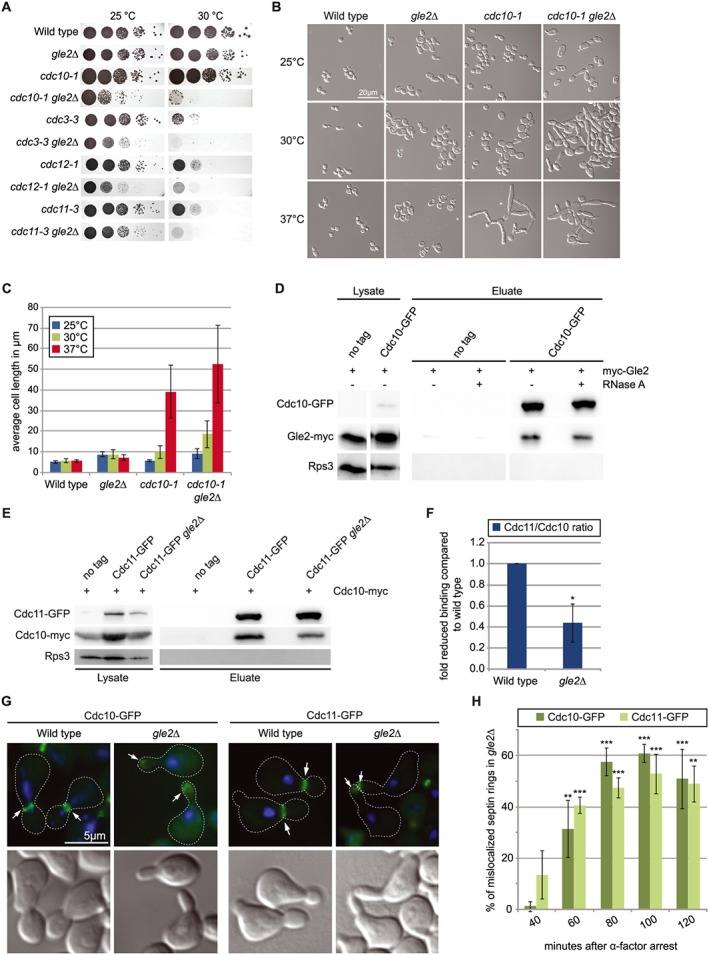
Gle2 is needed for correct formation of the septin ring. (a) Drop dilution test shows genetic interactions of gle2∆ with all septin mutants. (b) The temperature sensitive phenotype of the cdc10–1 mutant, regarding cell size and shape, is drastically enhanced when combined with a deletion of GLE2. (c) Quantification of the average cell length of the strains shown in (b). (d) Western blots of co‐immunoprecipitations (co‐IPs) show interactions of Cdc10 with Gle2. Rps3 served as a negative control. (e) Interaction of the septin ring components Cdc10 and Cdc11 is disturbed in gle2∆ cells as shown by western blots of co‐IPs between the septins. (f) Quantification of three different experiments shown in (e). (g) Cdc10‐GFP and Cdc11‐GFP are drastically mislocalized from the bud neck to the bud tip in strains deleted for GLE2. (h) Quantification of three different experiments shown in (g).
Figure 4.
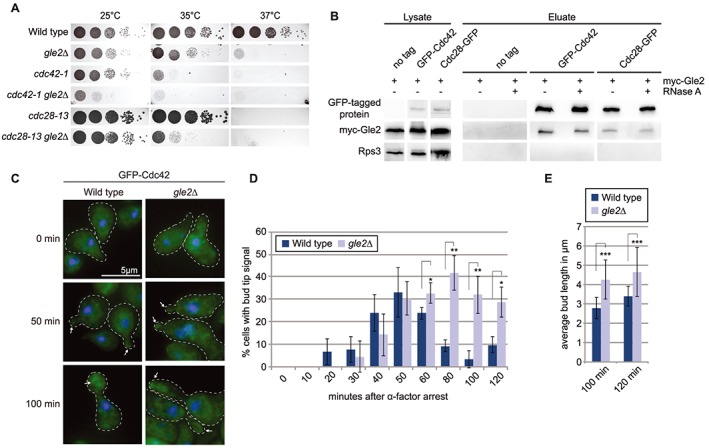
The cell cycle regulating GTPase Cdc42 requires Gle2 for correctly timed localization. (a) Drop dilution tests uncover genetic interaction of gle2∆ with mutant alleles of CDC42 and the major cell cycle kinase CDC28 (CDK1). (b) Co‐immunoprecipitation and western blot experiments reveal physical interaction of Cdc42 and Cdc28 with Gle2. (c) GFP‐microscopy during a time course experiment with synchronized cells show a prolonged presence of Cdc42 at the bud tip in gle2∆ cells. (d) Quantification of three different experiments shown in (c). A minimum of 100 cells was counted for each time point. (e) Average bud length of cells shown in (c) was determined and reveals significant elongation for cells lacking GLE2.
Synthetic genetic array screen
Synthetic genetic array (SGA) analyses were carried out as described using a Singer RoToR HDA (Tong and Boone, 2006). The query strain was a gle2Δ::natMX4 derivative of Y7092 (HKY1163), which was kindly provided by C. Boone, University of Toronto. The library was a collection of temperature sensitive mutants, also kindly provided by C. Boone. Growth defects were detected by comparing the growth of double mutants with the combined growth of single and double mutants. As a measure for growth, colony areas were taken, which were quantitated from plate scans using ‘Balony’ (Young and Loewen, 2013).
Cell cycle arrest and flow cytometric analysis
Overnight cultures were diluted in rich medium to a density of 0.5 × 107 cells/mL and incubated at 25°C for 2 h. Cells were arrested in their cell cycle by addition of α‐factor to a final concentration of 30 μg/mL and incubated for 2 h at 25°C. After addition of another 10 μg/mL α‐factor per milliliter culture and 1 h incubation, the 0 min time point was taken and cells were fixed with 70% ethanol. The rest of the culture was washed five times with fresh medium to remove the α‐factor and brought into the same volume of fresh medium as before. Samples were taken at time points indicated in the experiments and fixed as described above. For flow cytometry, fixed cells were washed with 50 mm sodium citrate pH 7.0 and treated with 0.25 mg/mL RNase A at 50°C for 1 h. After removal of RNase by washing with sodium citrate, samples were sonicated (15 s, 30% output level, Branson Sonifier 250) with a micro‐tip to separate cells from each other. Samples were washed twice with sodium citrate and incubated with 0.2 μL Sytox‐Green® (Thermo Fisher) per milliliter suspension at room temperature in the dark for 30 min. Analysis of the cells was performed using the BD FACS Canto Cytometer.
Determination of chromosome loss rates
Chromosome loss rates were determined according to Zhu et al. (2015). A gle2Δ::kanMX4 deletion was introduced into RLY8492 (HKY1600) and confirmed via PCR analysis. Two independently isolated clones were analysed. RLY8492 served as wild type and RLY8496 (HKY1602) (mad1∆) as positive control. Overnight cultures of each strain were grown in SC medium lacking leucine at 25°C. These cultures served to determine the GFP−/GFP+ ratio at starting time. The cultures were diluted in YPD to a cell density of 2 × 106 cells/mL and grown at 25°C to cell densities of 0.75–1.5 × 108 cells/mL. The theoretical number of doublings was calculated for each culture. Cells were fixed with 4% formaldehyde and analysed by flow cytometry. Chromosome loss rates were calculated as described previously (Zhu et al., 2015).
Microscopic studies
For the analysis of live cells as depicted in Figure 1(c) and Figure S2, cells were grown in rich medium at the indicated temperatures overnight, harvested and examined directly. For green fluorescent protein (GFP) microscopy cells were arrested in cell cycle as described above and fixed with 4% formaldehyde for a maximum of 5 min. Samples were washed twice with P‐solution (0.1 m potassium phosphate buffer pH 6.5, 1.2 m sorbitol), permeabilized with 0.5% Triton® X‐100 in P‐solution on a polylysine coated slide and DNA was stained with Hoechst 33 342 (Sigma). Fluorescent in situ hybridization experiments were used for visualization of poly(A)+ RNAs (Figure 2c and Figure S2) as described before (Zander et al., 2016). Cells were grown to log phase and shifted to 37°C for 1 h before they were fixed with 4% formaldehyde for 1 h. Zymolyase (Amsbio) treatment resulted in spheroblasts that were further permeabilized with 0.5% Triton® X‐100 in P‐solution on a polylysine coated slide. Samples were pre‐hybridized with Hybmix (50% deionized formamide, 5× SSC, 1× Denhardts, 500 μg/mL tRNA, 500 μg/mL salmon sperm DNA, 50 μg/mL heparin, 2.5 mm EDTA pH 8.0, 0.1% Tween® 20, 10% dextran sulphate) for 1 h at 37°C and hybridized with a Cy3‐labelled oligo d(T)50 probe (0.5 μm) in Hybmix at 37°C overnight. DNA was stained as described above. For microscopic studies a Leica AF6000 microscope was used and pictures were obtained by using the LEICA DFC360FX camera and processed with the LAS AF 2.7.3.9 software (Leica).
Figure 2.
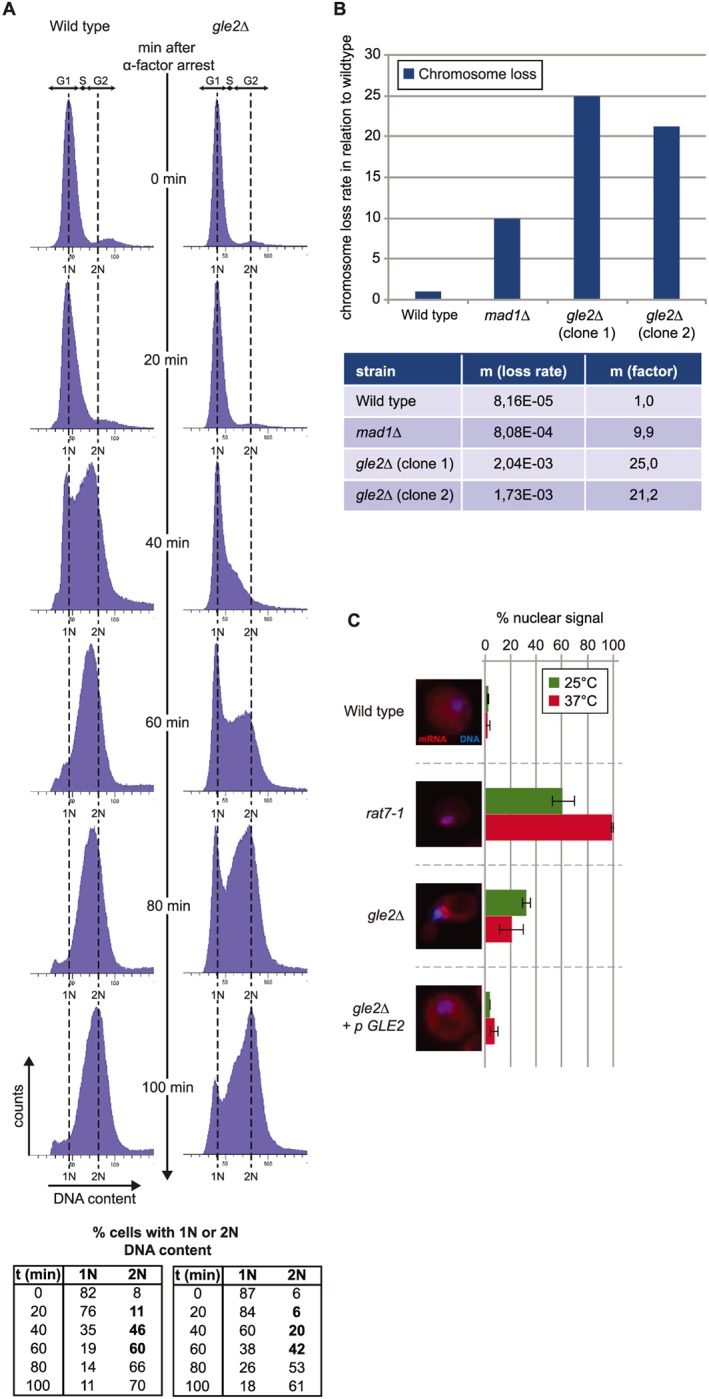
Gle2 has a role in cell cycle regulation. (a) Deletion of GLE2 delays cell cycle progression. Flow cytometric analysis of wild type and gle2∆ cells after arrest with α‐factor (top). The percentage of cells with a haploid (1 N) or diploid (2 N) genome was calculated (bottom). (b) Deletion of GLE2 causes chromosome missegregation. Loss rates relative to wild type (top) and loss rates per cell division are depicted (bottom). mad1∆, defective in the spindle attachment checkpoint served as a positive control. (c) Nuclear mRNA export defects in gle2∆ are weak, when compared to the mRNA export mutant rat7–1 (nup159). Poly(A)+‐containing RNA was stained with a Cy3‐labelled oligo d(T)50 probe (red); DNA was stained with Hoechst (blue) in fluorescence in situ hybridization experiments.
Co‐immunoprecipitation (IP) experiments
IPs were essentially done as described previously (Zander et al., 2016). Briefly, late log phase cells (2–3 × 107 cells/mL) were harvested and lysed in IP buffer (1 × PBS, 3 mm KCl, 2.5 mm MgCl2, 0.5% Triton X‐100, vanadyl phosphatase inhibitors and protease inhibitors from Roche). The resulting lysate was incubated with GFP‐Trap®_A beads (Chromotek) and if applicable 200 μg/mL RNase A for 3–4 h at 4°C. Afterwards beads were washed five times with IP buffer and proteins were detected by Western blot analyses with the indicated antibodies [GFP (Pierce) 1:5000; c‐myc (9E10, Santa Cruz) 1:1000; Rps3 (rabbit, own serum) 1:700]. Signals were detected with the Fusion SL system (PeqLab). Intensities were quantified using the Bio1D software.
Quantification
All experiments shown in this work were performed at least three times independently with the exception of the SGA screen and the chromosome misseggregation. Error bars represent the standard deviation. p‐Values shown in Figure 3(f) were calculated using a two‐tailed, two‐sample unequal variance t‐test. p‐Values shown in Figures 3(h) and 4(d, e) and Figure S3(a) were calculated using a two‐tailed, two‐sample equal variance t‐test. p‐Values are indicated as follows: *** p < 0.001, ** p < 0.01, * p < 0.05. For quantification of cells with displayed phenotypes (Figures 2c, 3h and 4d and Figure S3) for each experiment a minimum of 100 cells were counted. For Figures 2(d) and 3(c) three times 20 cells were measured.
Results and discussion
Gle2 interacts with cell cycle regulators
In order to characterize cellular functions of Gle2 we performed an sSGA analysis with temperature‐sensitive (ts) alleles of over 600 essential genes (kindly provided by C. Boone). We prepared a gle2∆ strain, crossed it with the library and analysed haploid segregants, a method described earlier (Tong and Boone, 2006).
More than 100 mutant alleles show genetic interactions with the deletion of GLE2 (Figure 1a). Surprisingly, the amount of interacting genes involved in nuclear transport or RNA processing was quite small (13 alleles). However, we found many genes involved in cell cycle progression and regulation, such as genes encoding proteins of the APC, the kinetochore, the spindle and the cytoskeleton (Figure 1a). To confirm these interactions we generated new double mutants of the APC (cdc20–2), Cks1 (cks1–38), important for G1/S and G2/M transition, and members of the mitotic exit network (cdc14–8, cdc15–2) with gle2∆ via tetrad dissection. Detailed analysis of these mutants showed enhanced growth defects (Figure 1b), increased cell size and defects in morphology, which reflected mostly malfunction at different stages of cell division, when combined with gle2∆ (Figure S1a). Interestingly, the abnormalities in growth and morphology of cks1–38 were suppressed by high copy (2 μ) GLE2 (Figure S1b–d), suggesting a direct interaction of these proteins. Indeed, physical interactions of Gle2 specifically with the cell cycle regulators Cks1, Cdc15 and Cdc16, but not Cdc14, are shown in co‐immunoprecipitation (co‐IP) analyses (Figure 1c). Interactions with the RNA‐binding protein Gle2 are insensitive to RNase treatment, suggesting that they are not mediated and dependent on RNA. These findings support an involvement of Gle2 in regulation of the cell cycle.
To address if Gle2 alone affects cell cycle regulation, we performed flow cytometry experiments and found significant cell cycle delay in cells deleted for GLE2 (Figure 2a). Wild‐type and gle2∆ cells were arrested in the G1 phase of the cell cycle using the mating pheromone α‐factor. Washing away this factor re‐starts the cell cycle and SYTOX®‐green staining of the DNA allowed monitoring the synchronous population going through replication and cytokinesis. About 40 min after release, the portion of cells with a diploid (2 N) genome is ~46% in the wild‐type strain. In contrast, less than half (~20%) of the cells in gle2∆ have a 2 N content (Figure 2a, bottom). At 60 min most of the wild‐type cells have reached the 2 N stadium, while in gle2∆ this does not happen until 100 min, suggesting that cells lacking GLE2 face trouble entering S‐phase and progress from there. These data argue for an already early function of Gle2 in cell cycle progression, although its exact role remains to be studied in more detail.
Our findings that Gle2 genetically and physically interacts with components involved in the regulation of the APC support research in higher eukaryotes that also linked Rae1 with the APC (Jeganathan et al., 2005). As the APC is a major regulator of the correct timing for chromosome segregation and we found a physical interaction of Gle2 with the APC‐component Cdc16 (Figure 1c), we addressed whether Gle2 is required for proper chromosome segregation, using a GFP‐based quantitative chromosome transmission fidelity assay that allows sensitive and quantitative detection of chromosome loss (Zhu et al., 2015). We found that the deletion of GLE2 causes massive chromosome missegregation (Figure 2b). Interestingly, the effect is much stronger than that of the spindle assembly checkpoint regulator Mad1, which controls proper attachment of the microtubules to the chromosomes and else delays division of the sister chromatids. Deletion of MAD1 leads to a ~ 10‐fold higher loss of the mini‐chromosome compared with wild type, an increase that is more than doubled in gle2∆ (~21‐ and ~25‐fold higher than wild type). This defect in maintaining chromosomal stability in gle2∆ might result from misorientation of the mitotic spindle or from a general perturbation of cell cycle controlling complexes. Nevertheless, this striking effect underlines the general importance of Gle2 in cell cycle regulation.
Given the involvement of Gle2 in mRNA export, one might speculate that the cell cycle perturbations seen in gle2∆ might be due to a shortage of proteins evoked by insufficient nuclear export of the respective mRNAs. However, analysis of mRNA export shows only very minor defects (Figure 2c and Figure S2) and mutants that have stronger mRNA export defects like rat7–1 show none of the morphological phenotypes that can be observed for a deletion of GLE2 (Figure S2).
This involvement of Gle2 in cell cycle regulation is a new finding for S. cerevisiae and in accordance with data from higher eukaryotes that identified a role for Gle2/Rae1 in the microtubule organization, cell cycle regulation and prevention of aneuploidy (Nakano et al., 2011), showing once more that basic principles are conserved in all eukaryotes.
Gle2 is involved in septin organization
Besides the interactions of Gle2 with cell cycle regulators, we found a novel interesting group of genes that are important for cell division that belong to the septin family and its regulatory network (Figure 1a). Drop dilution tests with mutants of all septins revealed that their combination with gle2∆ leads to significantly reduced growth compared with the single mutants (Figure 3a). Strikingly, cdc10–1 gle2∆ double mutants show a drastic increase in the defects in morphology with about 10‐fold elongated cells compared with wild type (Figure 3b and c), clearly indicating defects in entering isotropic bud growth and separation of mother and daughter cells. These data suggest that Gle2 might be important for septin‐ring formation. To support our findings, we investigated physical interactions of Gle2 with the septin Cdc10 by co‐IP analyses and found strong physical interactions (Figure 3d).
Because the interaction of GLE2 with CDC10 is quite strong on a genetic level and the two proteins show a very stable physical interaction, we analysed this aspect in more detail. Cdc10 together with Cdc3, Cdc11 and Cdc12 is one of the four main septins in yeast. Their ordered interaction leads to formation of hetero‐octameric filaments that localize to the incipient bud site (McMurray et al., 2011). Over the course of budding the single filaments interact with each other and build a highly structured meshwork called the septin ring. This ring is necessary for correct bud formation and cell division and represents a barrier between mother and daughter cell (Bi and Park, 2012).
To investigate if the formation of the septin ring would be affected by missing Gle2, we analysed the interaction between two septins in gle2∆. Strikingly, co‐IPs clearly showed a reduced interaction of Cdc10 and Cdc11 in gle2∆ cells (Figure 3e). In fact, quantification of several of these co‐IPs revealed a ~ 60% reduced interaction of the septins when GLE2 was deleted (Figure 3f). These findings could suggest a direct function of Gle2 in assisting septin assembly.
The reduced septin interaction in gle2∆ could be a result of incorrect hetero‐octamer formation itself or hindered multimerization of the filaments, or it might be that already assembled filaments are rather unstable in gle2∆ cells. Another possibility could be that the correct cue that triggers localized formation is missing. Therefore, we first investigated possible disturbance of the septin‐ring localization by using GFP‐tagged versions of septin proteins that allowed monitoring of the formation and localization of the septin ring during cell division with GFP microscopy. After synchronization with α‐factor we took samples of wild‐type and gle2∆ cells every 20 min. Septin rings become visible about 40 min after release (Figure S3a and S3b) and reach their maximum after around 80–100 min. While in the wild type nearly all cells form a visible septin ring, <80% of the gle2∆ cells show this structure (Figure S3a). In addition to the reduced amount in gle2∆, the most striking difference from wild‐type cells is the change in localization of septin rings. At 80 min after release, cells are in the middle of the budding event with a clearly distinguishable bud and the mother‐bud‐neck visible. In wild type the septin ring is located at the mother–daughter border, while this signal can be found prominently at the bud tip and not the bud neck in gle2∆ cells (Figure 3g). Not only for Cdc10, but also for Cdc11, this mislocalization is observed, indicating that the GFP‐signal really represents the septin ring and not a defect in a single septin protein alone. This wrong localization of the septin ring to the bud tip in gle2∆ is not a rare event. Quantification of mislocalized septin rings in gle2∆ indeed revealed that around 50% of the cells show this defect (Figure 3h). This is highly significant compared with wild type in which <1% of the cells have a mislocalized septin ring. So when Gle2 is missing, the septin ring can either not assemble properly or cannot be maintained at its natural position at the mother‐bud‐neck.
Gle2 is involved in the Cdc42‐mediated apical–isotropic switch
Gle2 is a WD40- -propeller protein, a typical protein structure that recruits regulators such as kinases or phosphatases (Reddy et al., 2008). The organization of the septin filaments into a ring is tightly regulated and coupled with other checkpoints of the cell cycle (Bi and Park, 2012) and the lack of the correct placement of the septin ring in gle2∆ could be a reason for defects in regulation, which in turn might argue for a role of Gle2 as a scaffold for septin‐regulating proteins.
The GTPase Cdc42 acts as a major regulator in cell cycle progression and morphology in all eukaryotes. In yeast, Cdc42 controls bud emergence, septin recruitment and the switch between apical and isotropic bud growth (Johnson, 1999). While its localization to the bud tip during bud emergence is required for bud growth, it is distributed along the daughter cell membrane in G2/M‐phase (Bi and Park, 2012). This re‐localization is triggered by the kinase Cdc28 (Johnson, 1999). Strikingly, both proteins show strong genetic interactions with gle2∆ (Figure 4a). Moreover, Cdc42 and Cdc28 physically interact with Gle2 as shown by co‐IPs (Figure 4b). Most importantly, we show that Gle2 is required for correct Cdc42 localization, as shown by synchronized cells. In wild type, Cdc42 is located at the bud tip with a peak at 50 min after release from the cell cycle arrest (Figure 4c and d). In contrast, in gle2∆ cells Cdc42 remains localized to the bud tips after 50 min and even after 120 min upon release (Figure 4c and d). Another apparent difference is the shape and length of the newly formed bud (Figure 4c). Quantification shows that cells lacking GLE2 form buds that are significantly elongated compared with wild type (Figure 4e). This phenotype could be a result of the prolonged stay of Cdc42 at the bud tip, which delays the apical–isotropic switch.
Together, our data have identified an involvement of Gle2 in cell cycle regulation and the APC‐mediated chromosome separation, underlining that this function is highly conserved from S. cerevisiae to humans. Additionally, we found a novel function for Gle2 in septin organization that is important for cell cycle progression. As this novel function presumably occurs before its APC‐mediated function, it will be interesting to see how far they are connected. One could speculate that Gle2 as a WD40 repeat propeller protein might be a scaffold for septin‐complex formation. In this function it could provide a platform for proteins and complexes that regulate the bud emergence, growth and cell cycle in general. Gle2 interacts not only with the GTPase Cdc42, but also with the kinase Cdc28/Cdk1 (Figure 4b), which as the Clb1–2/Cdc28 complex coordinates re‐localization of Cdc42, important for the apical–isotropic switch in the daughter cell and entering of the G2/M‐phase (Johnson, 1999). A localization of Clb2/Cdc28 to the bud neck has been shown previously (Eluere et al., 2012; Hood et al., 2001) and it is tempting to suggest that Gle2 supports this as a scaffold and allows a coordinated regulation of these processes. Whether Gle2/Rae1 impacts septin‐complex formation in humans remains to be shown; however, owing to the fact that the septins are increasingly recognized as important components of the cytoskeleton and as such are involved in the organization of cytokinesis (Mostowy and Cossart, 2012), a function of Gle2/Rae1 in this process is most appealing.
Author contributions
Experiments were designed and data interpreted by G.Z, W.K. and H.K.; experiments were performed by G.Z. (Figures 1b, c; 2a, c; 3a–h, 4a–e), W.K. (Figures 1a, 2b) and A.S. (Figure 2a). The manuscript was written by H.K. and G.Z.; all authors discussed the results and commented on the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Supporting info item
Fig. S1. Deletion of GLE2 affects cell morphology and size. (A) Abnormal cell morphology of cell cycle mutants increases with rising temperature and is detectable already at lower temperatures when combined with gle2∆. Strains that are lethal at the respective temperature (see Figure 1B) are marked with a black frame and show sometimes weaker phenotypes, as they die quickly. A quantification of the average cell length is shown for each strain and temperature (bottom) (B) Overexpression of GLE2 can partially rescue the growth and temperature sensitivity of cks1‐38 as shown in drop dilution experiments. (C) Overexpression of GLE2 alleviates the cks1‐38 phenotype in cell size and morphology. (D) Quantification of the average cell length of the strains shown in (C) reveals a slight reduction of cell length when GLE2 is overexpressed.
Fig. S2. Fluorescence in situ hybridization experiments presented in Fig 2C showing several cells and single channels. The enlarged cells in Fig 2C are indicated by the boxes.
Fig. S3. (A) Quantification of the amount of septin‐rings detectable in wild type and gle2∆ cells of the experiment shown in Fig 3G. Three different experiments were analyzed, in which for each a minimum of 100 cells was counted at every time point. p‐values were calculated to the corresponding wild type time point (***p < 0.001, **p < 0.01, *p < 0.05). (B) Microscopic images showing Cdc10‐GFP in wild type and gle2∆ cells at every time point after α‐factor arrest analysed for (A).
Fig. S4. Uncropped western blots are depicted. Areas shown in the main figures are marked with a green box. (A) Western blot shown in Figure 1C. (B) Western blot shown in Figure 3D. (C) Western blot shown in Figure 3E. (D) Western blot shown in Figure 4B.
Acknowledgements
We thank C. Boone for providing the genetic library. We are grateful to G. Braus and D.J. Lew and for providing plasmids, strains or antibodies. This work was funded by grants from the Deutsche Forschungsgemeinschaft and the SFB860 awarded to H.K.
Zander, G. , Kramer, W. , Seel, A. , and Krebber, H. (2017) Saccharomyces cerevisiae Gle2/Rae1 is involved in septin organization, essential for cell cycle progression. Yeast, 34: 459–470. doi: 10.1002/yea.3249.
References
- Bailer SM, Siniossoglou S, Podtelejnikov A, Hellwig A, Mann M, Hurt E. 1998. Nup116p and Nup100p are interchangeable through a conserved motif which constitutes a docking site for the mRNA transport factor Gle2p. EMBO J 17: 1107–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker DJ, Dawlaty MM, Galardy P, Van Deursen JM. 2007. Mitotic regulation of the anaphase‐promoting complex. Cell Mol Life Sci 64: 589–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bharathi A, Ghosh A, Whalen WA, et al 1997. The human Rae1 gene is a functional homologue of Schizosaccharomyces Pombe Rae1 gene involved in nuclear export of poly(A)+ RNA. Gene 198: 251–258. [DOI] [PubMed] [Google Scholar]
- Bi E, Park HO. 2012. Cell Polarization and cytokinesis in budding yeast. Genetics 191: 347–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blevins MB, Smith AM, Phillips EM, Powers MA. 2003. Complex formation among the rna export proteins Nup98, Rae1/Gle2, and Tap. J Biol Chem 278: 20979–20988. [DOI] [PubMed] [Google Scholar]
- Blower MD, Nachury M, Heald R, Weis K. 2005. A Rae1‐containing ribonucleoprotein complex is required for mitotic spindle assembly. Cell 121: 223–234. [DOI] [PubMed] [Google Scholar]
- Brown JA, Bharathi A, Ghosh A, Whalen W, Fitzgerald E, Dhar R. 1995. A mutation in the Schizosaccharomyces Pombe Rae1 gene causes defects in poly(A)+ RNA export and in the cytoskeleton. J Biol Chem 270: 7411–7419. [DOI] [PubMed] [Google Scholar]
- Bucci M, Wente SR. 1997. In vivo dynamics of nuclear pore complexes in yeast. J Cell Biol 136: 1185–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eluere R, Varlet I, Bernadac A, Simon MN. 2012. Cdk And the anillin homolog Bud4 define a new pathway regulating septin organization in yeast. Cell Cycle 11: 151–158. [DOI] [PubMed] [Google Scholar]
- Entian KD, Schuster T, Hegemann JH, et al 1999. Functional analysis of 150 deletion mutants in Saccharomyces cerevisiae by a systematic approach. Mol Gen Genet 262: 683–702. [DOI] [PubMed] [Google Scholar]
- Funasaka T, Nakano H, Wu Y, et al 2011. RNA export factor Rae1 contributes to Nup98‐Hoxa9‐mediated leukemogenesis. Cell Cycle 10: 1456–1467. [DOI] [PubMed] [Google Scholar]
- Gorsch L, Dockendorff TC, Cole CN. 1995. A conditional allele of the novel repeat‐containing yeast nucleoporin. Jcb 129: 939–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagiwara A, Tanaka Y, Hikawa R, et al 2011. Submembranous septins as relatively stable components of actin‐based membrane skeleton. Cytoskeleton (Hoboken) 68: 512–525. [DOI] [PubMed] [Google Scholar]
- Ho AK, Raczniak GA, Ives EB, Wente SR. 1998. The integral membrane protein Snl1p is genetically linked to yeast nuclear pore complex function. Mol Biol Cell 9: 355–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hood JK, Hwang WW, Silver PA. 2001. The Saccharomyces cerevisiae cyclin Clb2p is targeted to multiple subcellular locations by cis‐ and trans‐acting determinants. J Cell Sci 114: 589–597. [DOI] [PubMed] [Google Scholar]
- Jeganathan KB, Malureanu L, Van Deursen JM. 2005. The Rae1–Nup98 complex prevents aneuploidy by inhibiting securin degradation. Nature 438: 1036–1039. [DOI] [PubMed] [Google Scholar]
- Johnson DI. 1999. Cdc42: An essential rho‐type gtpase controlling eukaryotic cell polarity. Microbiol Mol Biol Rev 63: 54–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joo E, Tsang CW, Trimble WS. 2005. Septins: Traffic control at the cytokinesis intersection. Traffic 6: 626–634. [DOI] [PubMed] [Google Scholar]
- Kim MS, Froese CD, Estey MP, Trimble WS. 2011. Sept9 occupies the terminal positions in septin octamers and mediates polymerization‐dependent functions in abscission. J Cell Biol 195: 815–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita M, Noda M. 2001. Roles of septins in the mammalian cytokinesis machinery. Cell Struct Funct 26: 667–670. [DOI] [PubMed] [Google Scholar]
- Kinoshita M, Field CM, Coughlin ML, Straight AF, Mitchison TJ. 2002. Self‐ and actin‐templated assembly of mammalian septins. Dev Cell 3: 791–802. [DOI] [PubMed] [Google Scholar]
- Kraemer D, Dresbach T, Drenckhahn D. 2001. Mrnp41 (Rae 1p) Associates with microtubules in hela cells and in neurons. Eur J Cell Biol 80: 733–740. [DOI] [PubMed] [Google Scholar]
- Larsen NA, Harrison SC. 2004. Crystal structure of the spindle assembly checkpoint protein Bub3. J Mol Biol 344: 885–892. [DOI] [PubMed] [Google Scholar]
- Larsen NA, Al‐Bassam J, Wei RR, Harrison SC. 2007. Structural analysis of Bub3 interactions in the mitotic spindle checkpoint. Proc Natl Acad Sci U S A 104: 1201–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lew DJ. 2003. The morphogenesis checkpoint: How yeast cells watch their figures. Curr Opin Cell Biol 15: 648–653. [DOI] [PubMed] [Google Scholar]
- Mcmurray MA, Bertin A, Garcia G, 3rd , Lam L, Nogales E, Thorner J. 2011. Septin filament formation is essential in budding yeast. Dev Cell 20: 540–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mostowy S, Cossart P. 2012. Septins: The fourth component of the cytoskeleton. Nat Rev Mol Cell Biol 13: 183–194. [DOI] [PubMed] [Google Scholar]
- Murphy R, Watkins JL, Wente SR. 1996. Gle2, a Saccharomyces cerevisiae homologue of the Schizosaccharomyces Pombe export factor Rae1, is required for nuclear pore complex structure and function. Mol Biol Cell 7: 1921–1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano H, Wang W, Hashizume C, Funasaka T, Sato H, Wong RW. 2011. Unexpected role of nucleoporins in coordination of cell cycle progression. Cell Cycle 10: 425–433. [DOI] [PubMed] [Google Scholar]
- Reddy DM, Aspatwar A, Dholakia BB, Gupta VS. 2008. Evolutionary analysis of Wd40 super family proteins involved in spindle checkpoint and rna export: molecular evolution of spindle checkpoint. Bioinformation 2: 461–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren Y, Seo HS, Blobel G, Hoelz A. 2010. Structural and functional analysis of the interaction between the nucleoporin Nup98 and the mRNA export factor Rae1. Proc Natl Acad Sci U S A 107: 10406–10411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellin ME, Holmfeldt P, Stenmark S, Gullberg M. 2011. Microtubules support a disk‐like septin arrangement at the plasma membrane of mammalian cells. Mol Biol Cell 22: 4588–4601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikorski RS, Hieter P. 1989. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae . Genetics 122: 19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sitterlin D. 2005. Aster lights on RNA. Nat Struct Mol Biol 12: 479–480. [DOI] [PubMed] [Google Scholar]
- Surka MC, Tsang CW, Trimble WS. 2002. The mammalian septin Msf localizes with microtubules and is required for completion of cytokinesis. Mol Biol Cell 13: 3532–3545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka‐Takiguchi Y, Kinoshita M, Takiguchi K. 2009. Septin‐mediated uniform bracing of phospholipid membranes. Curr Biol 19: 140–145. [DOI] [PubMed] [Google Scholar]
- Tong AH, Boone C. 2006. Synthetic genetic array analysis in Saccharomyces cerevisiae . Meth Mol Biol 313: 171–192. [DOI] [PubMed] [Google Scholar]
- Tong AHY, Boone C. 2007. High‐throughput strain construction and systematic synthetic lethal screening in Saccharomyces cerevisiae . Yeast Gene Anal 36: 369. [Google Scholar]
- Whalen WA, Bharathi A, Danielewicz D, Dhar R. 1997. Advancement through mitosis requires Rae1 gene function in fission yeast. Yeast 13: 1167–1179. [DOI] [PubMed] [Google Scholar]
- Yoon JH, Love DC, Guhathakurta A, Hanover JA, Dhar R. 2000. Mex67p Of Schizosaccharomyces Pombe interacts with Rae1p in mediating mRNA Export. Mol Cell Biol 20: 8767–8782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young BP, Loewen CJ. 2013. Balony: A software package for analysis of data generated by synthetic genetic array experiments. BMC Bioinformat 14: 354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zander G, Hackmann A, Bender L, et al 2016. mRNA quality control is bypassed for immediate export of stress‐responsive transcripts. Nature 540: 593–596. [DOI] [PubMed] [Google Scholar]
- Zhu J, Heinecke D, Mulla WA, et al 2015. Single‐cell based quantitative assay of chromosome transmission fidelity. G3 (Bethesda) 5: 1043–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting info item
Fig. S1. Deletion of GLE2 affects cell morphology and size. (A) Abnormal cell morphology of cell cycle mutants increases with rising temperature and is detectable already at lower temperatures when combined with gle2∆. Strains that are lethal at the respective temperature (see Figure 1B) are marked with a black frame and show sometimes weaker phenotypes, as they die quickly. A quantification of the average cell length is shown for each strain and temperature (bottom) (B) Overexpression of GLE2 can partially rescue the growth and temperature sensitivity of cks1‐38 as shown in drop dilution experiments. (C) Overexpression of GLE2 alleviates the cks1‐38 phenotype in cell size and morphology. (D) Quantification of the average cell length of the strains shown in (C) reveals a slight reduction of cell length when GLE2 is overexpressed.
Fig. S2. Fluorescence in situ hybridization experiments presented in Fig 2C showing several cells and single channels. The enlarged cells in Fig 2C are indicated by the boxes.
Fig. S3. (A) Quantification of the amount of septin‐rings detectable in wild type and gle2∆ cells of the experiment shown in Fig 3G. Three different experiments were analyzed, in which for each a minimum of 100 cells was counted at every time point. p‐values were calculated to the corresponding wild type time point (***p < 0.001, **p < 0.01, *p < 0.05). (B) Microscopic images showing Cdc10‐GFP in wild type and gle2∆ cells at every time point after α‐factor arrest analysed for (A).
Fig. S4. Uncropped western blots are depicted. Areas shown in the main figures are marked with a green box. (A) Western blot shown in Figure 1C. (B) Western blot shown in Figure 3D. (C) Western blot shown in Figure 3E. (D) Western blot shown in Figure 4B.