

Abstract
Background:
Post-traumatic headache is the most common and long-lasting impairment observed following mild traumatic brain injury, and frequently has migraine-like characteristics. The mechanisms underlying progression from mild traumatic brain injury to post-traumatic headache are not fully understood. The aim of this study was to develop a mouse model of post-traumatic headache and identify mechanisms and novel targets associated with this disorder.
Methods:
We combined the closed head weight-drop method and the nitroglycerin chronic migraine model. To induce mild traumatic brain injury, a weight was dropped onto intact crania of mildly-anesthetized mice, and mechanical responses to chronic- intermittent administration of nitroglycerin, a human migraine trigger, were determined at multiple time-points post-injury.
Results:
Low dose nitroglycerin (0.1 mg/kg) evoked acute periorbital and hind paw allodynia in both mild traumatic brain injury and sham animals. However, only mild traumatic brain injury mice developed chronic hypersensitivity to low dose nitroglycerin. Migraine medications, sumatriptan and topiramate, inhibited post-traumatic headache-associated allodynia. In addition, the delta opioid receptor agonist, SNC80, also blocked post-traumatic headache-associated allodynia. Finally, we examined the expression of calcitonin gene-related peptide within this model and found that it was increased in trigeminal ganglia two weeks post-mild traumatic brain injury
Conclusions:
Overall, we have established a mouse model of post-traumatic headache and identified the delta opioid receptor as a novel therapeutic target for this disorder.
Keywords: Delta opioid receptor, hyperalgesia, migraine, nitroglycerin, mild traumatic brain injury
Introduction
Post-traumatic headache (PTH) is a debilitating secondary headache disorder that occurs after traumatic brain injury (TBI) (1–3). Within the United States, more than 1 million Americans experience mild TBIs (mTBIs) , and a follow-up study indicated that up to 58% of mTBI patients developed chronic PTH that persisted 1 year after injury (4–8). The most severe PTH has a migraine-like phenotype, develops within seven days to a year after injury, and typically progresses to a chronic condition (1). Chronic migraine associated with PTH is defined as 15 headache days or more per month, lasts for three or more months (1), and is not easily resolved.
To date, there are no PTH-specific pharmacotherapies. In general, post-traumatic migraine is clinically similar to atraumatic migraine, and many PTH patients are highly dependent on migraine therapies for acute and preventive treatment. However, these medications do not provide sufficient pain relief in all patients (9) and, similar to migraineurs, PTH patients continue to have unmet medical needs (10,11). Although migraine is commonly observed following TBI, the central mechanisms by which brain trauma leads to migraine are unclear. A predictive model of PTH, especially one highlighting the more severe migraine-like phenotype, would aid in understanding the mechanisms regulating this disorder, and would also provide a tool to screen novel pharmacotherapies.
PTH is typically induced by mTBI; however, many of the preclinical TBI models involve craniotomy and/ or penetrative brain injuries, such as controlled cortical impact (CCI) (12). The weight-drop model produces a non-invasive closed-head injury similar to a concussive injury observed in humans (13,14). This mouse model of mTBI does not induce substantial anatomical damage to the brain nor is there notable damage to the blood-brain barrier (13). In addition, this model has been used in rats to model cephalic pain associated with PTH (15). Considering that the most debilitating PTH has a chronic migraine phenotype (2,16), the aim of our study was to combine the closed head weight drop model with the nitroglycerin (NTG) model of chronic migraine-associate allodynia. NTG is a reliable human migraine trigger (17,18) and has been shown to evoke allodynia in mice (19–22), an effect that is amplified in a genetic model of familial migraine (23). In addition, NTG produces light-aversive behavior (24,25), and increased meningeal blood flow (26). We have shown previously that chronic intermittent NTG produces both acute allodynia and a basal hypersensitivity that acts as a model of migraine chronification (20,21). In this study we examined the effect of mTBI on NTG-induced acute and chronic allodynia, and validated this model using established migraine pharmacotherapies. We also tested an agonist for the delta opioid receptor (DOR), which we have previously identified as a novel target for migraine (19). Additionally, we examined the effect of mTBI on expression of the pro-migraine neuropeptide, calcitonin gene related peptide (CGRP), thus providing a link between head trauma and PTH.
Methods
Animals
All experiments used male C57BL/6J mice (Jackson Laboratories, Bar Harbor, ME, USA; Charles River Laboratories, Wilmington, MA, USA), weighing 25–30 g. Mice were group housed in a 12–12 light-dark cycle, where the lights were on from 07:00–19:00. Food and water were available ad libitum. All animals were randomly assigned to either sham or mTBI groups, and then randomly to the different treatment groups. All responses were collected in a blinded fashion by one to two experimenters. Weight was recorded at time of mTBI, and on each test day for all experiments. mTBI did not significantly affect weight gain and did not affect mortality. All experimental procedures were approved by the University of Illinois at Chicago Office of Animal Care and Institutional Biosafety Committee, in accordance with Association for Assessment and Accreditation of Laboratory Animal Care International (AAALAC) guidelines and the Animal Care Policies of the University of Illinois at Chicago. All results are reported according to Animal Research: Reporting of In Vivo Experiments (ARRIVE) guidelines.
Mild traumatic brain injury
Mild traumatic brain injury (mTBI) was induced using the closed head weight-drop method, as described previously (13). Briefly, mice were mildly anesthetized with 2.5% isoflurane with an oxygen flow rate of 0.6–0.8 liters per minute. Mice were placed chest down on a foam sponge (dimensions: 7.5 in × 5.5 in × 1.875 in) to support the head and body, which allowed for anterior-posterior motion without any rotational movement at the moment of impact. The mouse and sponge were placed directly underneath the weight-drop device. The weight-drop device consisted of a hollow cylindrical tube (inner diameter 2.54 cm, 80 cm height) placed approximately 1 cm vertically over the mouse’s head, in between the ear and eye. To induce mTBI, a 30g weight (13 mm diameter, 34 mm height) was dropped through the tube, striking the mouse and causing a closed head injury. Immediately after mTBI, mice were returned to their home cages for recovery for 3 days, 2, 4, or 12 weeks. All mice regained consciousness and were ambulatory within five minutes of mTBI. Sham animals were anesthetized but not subjected to the weight-drop. Sham animals regained consciousness and were ambulatory within a minute of the sham procedure.
Drug preparation and experimental outline
All drug injections were 10 ml/kg. Nitroglycerin (NTG) was purchased at a concentration of 5.0mg/mL, in 30% alcohol, 30% propylene glycol and water (American Reagent, NY, USA). NTG was freshly diluted on each test day in 0.9% saline to a concentration of 1 mg/mL for high dose (10 mg/kg), and 0.01 mg/mL (0.1 mg/kg) for a low dose. The vehicle used in these experiments was 0.9% saline. We previously found that there was no significant difference in mechanical thresholds between 0.9% saline and the solution in which high dose NTG was dissolved (6% propylene glycol, 6% alcohol, 0.9% saline) (20).
An experimental outline is depicted in Figure 1. After mTBI, mice were returned to their home cage. After recovery, sham and mTBI mice were randomly assigned to different treatment groups. To induce chronic migraine-associated pain, mice were treated with NTG or vehicle every other day for nine days (5 treatment days total). On a test day, basal responses were determined, NTG/vehicle injected, and post-treatment responses determined 2h later. For experiments in Figures 2 and 3, animals were tested with vehicle, a low/subthreshold dose of NTG (0.1 mg/kg, ip), or a high dose of NTG (10 mg/kg, ip), and after the final treatment day, they were tested every other day until basal thresholds recovered to post-treatment levels. For experiments in Figures 3–7, animals were tested with NTG 2 weeks post-mTBI/sham. On test days, basal responses were determined and animals were given a low dose of NTG (0.1 mg/kg, ip) or vehicle. One hour and 15 minutes post-NTG, mice were injected with vehicle, sumatriptan (0.6 mg/kg, ip), topiramate (30 mg/kg, ip), or SNC80 (10 mg/kg, ip), and were tested 45 min later (2h post-NTG). For topiramate experiments, mice were also pretreated with topiramate for 2 days before NTG treatment, and also on the days in between test days. To determine CGRP expression (Figure 8), mice underwent mTBI or sham, and 2 weeks post-injury tissue was collected for immunohistochemical analysis.
Figure 1.
Schematic of experimental outline.
Figure 2.
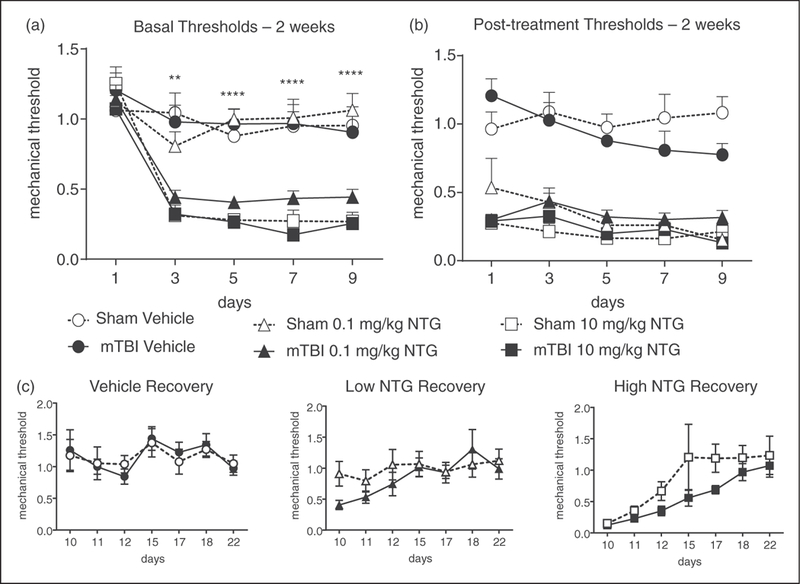
mTBI increases mechanical hypersensitivity to a low-dose of NTG 2 weeks after closed head injury. Post-sham or injury, C57BL/6J mice received vehicle, low (0.1 mg/kg, ip), or high (10 mg/kg, ip) dose NTG every other day for 9 days (five test days total). (a) Basal mechanical thresholds, assessed prior to vehicle or NTG administration, revealed that mTBI animals treated with low dose NTG had significantly lower basal thresholds compared to corresponding sham controls. p<0.01 treatment, time, and interaction; two-way RM ANOVA and Holm-Sidak post hoc analysis. **p<0.01, ***p<0.001, n=8–12/group. (b) In the same mice tested 2 h post-NTG/vehicle, both low- and high-dose NTG evoked hyperalgesia which did not differ between sham and mTBI groups. (c) Recovery from NTG did not differ between mTBI and sham animals for any of the groups. mTBI animals are more susceptible to developing NTG-induced chronic pain.
Figure 3.
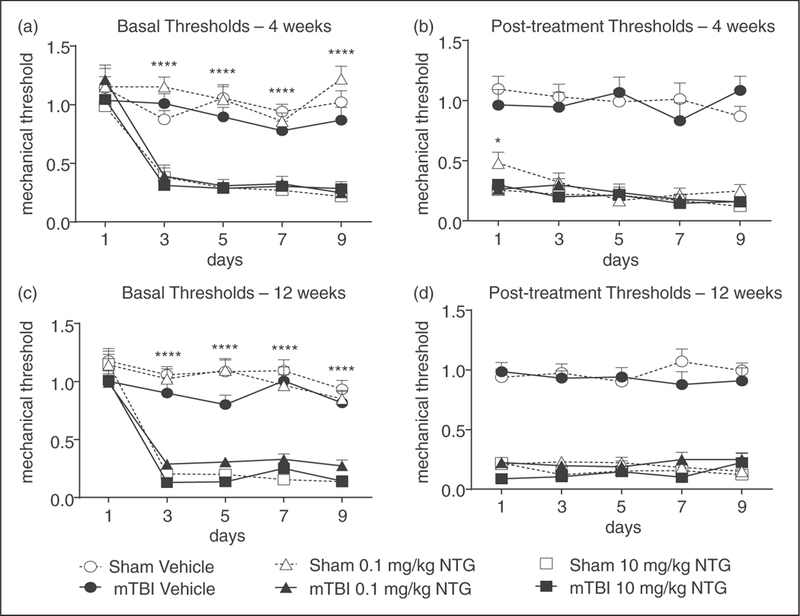
mTBI increases mechanical hypersensitivity to a low dose of NTG 4 and 12 weeks after closed head injury. Post-sham or injury, C57BL/6J mice received vehicle, low (0.1 mg/kg, ip), or high (10 mg/kg, ip) dose NTG every other day for 9 days (five test days total). (a) and (c) In both 4 and 12 week groups, assessment prior to vehicle or NTG administration revealed that mTBI animals treated with low dose NTG had significantly lower basal thresholds compared to corresponding shams. p < 0.01 treatment, time, and interaction; two-way RM ANOVA and Holm-Sidak post hoc analysis. **p < 0.01, ***p < 0.001, n = 11–18/group. (b) and (d) In the same animals tested 2 h post-NTG/vehicle, both low-and high-dose NTG evoked hyperalgesia that did not differ between sham and mTBI groups. Even after 12 weeks post-injury, mTBI animals are more susceptible to developing NTG-induced chronic pain.
Sumatriptan was purchased at a concentration of 12mg/mL and was diluted to 0.06mg/mL in 0.9% saline (Sandoz, NC, USA). Topiramate (Johnson & Johnson) and SNC80 (Tocris Bioscience) were made fresh on each test day in saline, or 0.33% 1N HCl/ 0.9% saline, respectively.
Sensory sensitivity testing
For all behavioral experiments, mice were counterbalanced into groups following the first basal test for mechanical sensitivity. The experimenter was blinded to the injury condition of the animal and the drug condition being tested. No adverse events were observed in any of the experiments. All mice were tested in a separate behavior room with low-light (~35–50 lux) and low-noise conditions, between 09:00 and 16:00. For hind paw sensitivity, the threshold for responses to punctate mechanical stimuli (mechanical allodynia) was tested according to the up-and-down method (27,28). Briefly, the plantar surface of the mouse hind paw was stimulated with a series of eight von Frey hair filaments (bending force ranging from 0.008 g to 2g). A response was defined as a lifting, shaking, or licking of the paw upon stimulation. The first filament tested was 0.4 g. In the absence of a response, a heavier filament (up) was tried, and in the presence of a response, a lighter filament (down) was tested. This pattern was followed for a maximum of four filaments following the first response. Mice were tested as follows: 20 minutes’ habituation on a testing rack, measurement of basal mechanical responses to von Frey hair filaments, administration of vehicle/NTG, home cage for 1 hour and 40 minutes, 20 minutes’ habituation on the testing rack, measurement of post-treatment mechanical responses to von Frey hair filaments. For cephalic sensitivity, mice were tested in 4 oz paper cups, to which they had been previously habituated for 1 h/day for 2 days. The periorbital region caudal to the eyes and near the midline was tested, similar to the up-down method described above, and herein (29).
Immunohistochemistry
Trigeminal ganglia (TG) were collected 2 weeks after mTBI or sham. Mice were anesthetized with Somnasol (100 μL/mouse; 390mg/mL pentobarbital sodium; Henry Schein, SKU#024352), and perfused intracardially with 15 mL of ice-cold phosphate buffer (0.1M PB, pH 7.2) and subsequently 50mL of ice-cold 4% paraformaldehyde (PFA)/0.1M PB (pH 7.4). TG was harvested from the mice and post-fixed overnight in 4% PFA/0.1M PB at 4°C. Tissue was cryoprotected in 30% sucrose/0.1M PB for 24–36 hours, or until it sank. TG was flash frozen in 2-methyl butane on dry ice, and sections of the TG were sliced at 16 μM. Upon slicing, TG sections were immediately mounted onto slides. Slides were blocked with 5% normal donkey serum in 0.1M phosphate-buffered saline with 0.3% Triton X-100 (NDST) for 1 hour at room temperature. Slides were incubated overnight at room temperature with primary sheep anti-CGRP antibody (RRID AB_725809; ab22560; Abcam; 1:1000 dilution) made in 1% NDST. Slides were washed with 1% NDST before incubating with a secondary antibody solution (Alexa Fluor 555 Donkey anti-Sheep; Life Technologies; 1:1000) made in 1% NDST for 2 hours at room temperature. Slides were washed with 0.1M phosphate buffer, and cover slipped with Mowiol-DAPI mounting solution. Images for quantification were taken by two observers in a blinded manner using an EVOS FL Auto Cell Imaging System, using a 20X objective. All images collected were used for analysis. Expression of CGRP was quantified by observers blinded to treatment groups. All CGRP- positive cells from all sections containing both right and left ganglia per mouse were analyzed (n = 8/ group). Confocal images were taken by a Zeiss Laser Scanning Microscope (LSM) 710 using a 25X objective.
Statistical Analysis
Data are expressed as mean ± s.e.m. All mice tested were included in the analysis. All statistical analyses were performed with SigmaStat software, and graphs were generated using GraphPad Prism. For all behavioral experiments, a two-way repeated-measures analysis of variance (ANOVA) was performed, with injury (sham/mTBI) and time (days) as factors. For experiments with sumatriptan, topiramate, or SNC80, a 2-way repeated-measures ANOVA was performed, with drug and time as factors. When a significant interaction occurred, subsequent Holm-Sidak post-hoc analysis was performed. In this case, all groups were compared to thresholds for sham mice on day 1, and to sham-vehicle groups. For CGRP experiments, a Student’s t-test was performed. A significance level of p < 0.05 was used throughout this study. For the proposed experiments, we performed the following power analysis: Minimal detectable differences in means = 0.3, expected standard deviation of residuals = 0.4, desired power = 0.8, alpha = 0.05, n = 15/group. Based on experience, we decreased this number accordingly.
Results
A detailed description of the experiments performed in this study is outlined in Figure 1.
We initially tested the effect of mTBI alone on basal allodynia, and observed that 3 days post-injury mTBI caused a significant decrease in hindpaw (mean ± SEM, sham vs. mTBI; 1.28 ± 0.16 vs. 0.78 ± 0.14), and cephalic (sham vs. mBTI; 0.58 ± 0.04 vs. 0.27 ± 0.05) responses. As post-traumatic migraine can develop and persist long after initial injury, we wanted to test at a time when animals had recovered from the pain induced by injury alone. We therefore tested at least 2 weeks post-injury, a time at which mTBI alone no longer affected basal thresholds in hindpaw or cephalic regions (see below Figures 2 and 7, day 1).
mTBI increases sensitivity to low-dose NTG
To determine the effect of mTBI on susceptibility to develop migraine-associated pain, mice were tested 2 weeks post-injury in the chronic NTG model. Varying doses of NTG (1–10 mg/kg, ip) have been shown previously to produce acute allodynia, and only higher doses of NTG (3–10 mg/kg, ip) produced chronic basal hypersensitivity (20,30). To determine whether mTBI increased the susceptibility to developing migraine-associated pain 2 weeks’ post-injury, we tested a sub-threshold dose of NTG (0.1 mg/kg, ip) to evoke acute but not chronic allodynia, as well as the standard high dose NTG (10 mg/kg, ip). Vehicle, low, or high dose NTG was administered every other day for 9 days (five total test days). Mechanical thresholds were tested before (basal threshold) and 2 hours after (post-treatment threshold) vehicle/NTG administration on each test day. At this 2-week post-injury time point, mTBI alone did not produce a significant decrease in basal hind paw mechanical thresholds (Figure 2(a), day 1). In both sham and mTBI groups, a high-dose of NTG evoked both a progressive and sustained basal hypersensitivity (Figure 2(a)), and acute allodynia 2 h post-injection (Figure 2(b)). Interestingly, a low-dose of NTG only produced a significant decrease in basal responses in the mTBI group, an effect not observed in the sham controls (Figure 2(a)); while both groups showed a significant acute allodynia to this low dose (Figure 2(b)). Following the final treatment day (day 9), recovery from NTG-induced basal hypersensitivity was determined, and animals were followed until their mechanical responses returned to pre-NTG thresholds as determined on day 1. There was no significant effect of mTBI on recovery time after NTG administration (Figure 2(c)). These results indicate that mTBI increases sensitivity to chronic migraine-associated pain induced by repeated administration of NTG.
mTBI has a long-lasting effect on sensitivity to NTG-induced chronic pain
To determine whether the sensitivity induced by mTBI persists beyond 2 weeks, sham and mTBI groups were tested 4 and 12 weeks post-mTBI. As in Figure 2, animals were treated with vehicle, low-, or high-dose NTG every second day for 9 days (five total test days). Again, mTBI alone did not alter basal responses on day 1 (Figure 3(a) and (c)). Similar to 2 weeks post-injury, both sham and mTBI groups developed basal hypersensitivity and acute allodynia to a high dose of NTG, and an acute response to low-dose NTG (Figure 3(b) and (d)). However, only the mTBI group developed a basal hypersensitivity to the low dose of NTG, an effect not observed in shams (Figure 3(a) and (c)). We tested mice following the final injection of NTG/vehicle to determine when their baselines returned to post-NTG levels. There was no difference in recovery time between sham and mTBI groups at 4 or 12 weeks post-injury (data not shown). The effects of mTBI are long-lasting, and even 12 weeks following injury mice were more susceptible to develop chronic NTG-induced pain.
Sumatriptan alleviates acute, but not chronic, allodynia within the PTH model
To pharmacologically validate this model, we investigated the effects of the migraine abortive sumatriptan on PTH-associated pain. At 2 weeks post-mTBI or sham, all animals were tested every other day for 9 days with a low-dose of NTG (0.1 mg/kg, ip), followed by vehicle or sumatriptan (0.6 mg/kg, ip; SUMA). In vehicle controls, low dose NTG induced basal allodynia in mTBI animals, but not sham controls (Figure 4(a)); and produced acute allodynia in both groups (Figure 4(b)). Sumatriptan significantly inhibited the post-treatment allodynia induced by NTG in both sham and mTBI mice (Figure 4(b)). Consistent with our previous findings using chronic high-dose NTG (21), sumatriptan did not affect the development of basal hypersensitivity to chronic low-dose NTG treatment in mTBI animals. However, we also observed that in sham animals, sumatriptan administration progressively lowered the basal mechanical thresholds (Figure 4(a)). These results indicate that while sumatriptan can reverse the acute effects of NTG after mTBI, it does not affect the progression of basal hypersensitivity that occurs with repeated NTG exposure. Furthermore, chronic treatment with sumatriptan alone could potentially synergize with NTG to worsen chronic migraine-associated pain.
Figure 4.
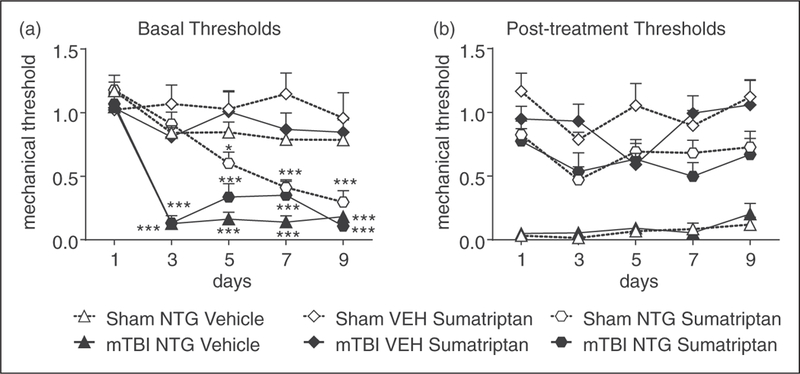
Sumatriptan inhibits acute PTH-associated allodynia. Two weeks post-injury, C57BL/6J mice were injected every second day with low-dose NTG (0.1 mg/kg, ip), and 1 h 15 min later with vehicle or sumatriptan (SUMA, 0.6 mg/kg, ip). (a) Basal mechanical thresholds, assessed prior to drug administration, were significantly decreased in mTBI animals regardless of drug treatment. p<0.001 drug, time, and interaction, 2-way RM ANOVA as compared to sham-NTG-vehicle on day 1. There was also a time-dependent effect of sumatriptan on sham animals, and sumatriptan decreased the basal threshold by day 5 when compared to sham-NTG-vehicle controls; p<0.01 effect of drug, time, and interaction two-way RM ANOVA, Holm-Sidak post hoc analysis. n¼8/group, *p<0.05, ***p<0.001 as compared to sham-NTG-vehicle on day 1. (b) Regardless of injury, low dose NTG produced acute hyperalgesia 2 hours post-NTG, which was significantly attenuated by sumatriptan. 2-way RM ANOVA, p<0.001 for drug only.
Topiramate attenuates acute and chronic allodynia within the PTH model
To further validate our model, we investigated the effects of the migraine preventive topiramate on PTH-associated pain. At 2 weeks post-mTBI or sham, mice were injected with either vehicle or topiramate (TOPI, 30 mg/kg, ip) every day for 11 days. On days 3, 5, 7, 9 and 11 all animals were tested with vehicle or low-dose NTG (0.1 mg/kg, ip). As above, low-dose NTG induced basal hypersensitivity in mTBI-vehicle treated animals, but not sham-vehicle treated controls (Figure 5(a)). Low-dose NTG produced acute allodynia in all vehicle controls (Figure 5(b)). Topiramate significantly attenuated the basal hypersensitivity to chronic low-dose NTG treatment in mTBI animals (Figure 5(a)). Furthermore, topiramate significantly inhibited post-NTG evoked allodynia in both sham and mTBI mice (Figure 5(b)). These results suggest that topiramate can reverse the acute effects of NTG after mTBI, and partially reduce the progression of basal hypersensitivity that occurs with repeated NTG exposure.
Figure 5.
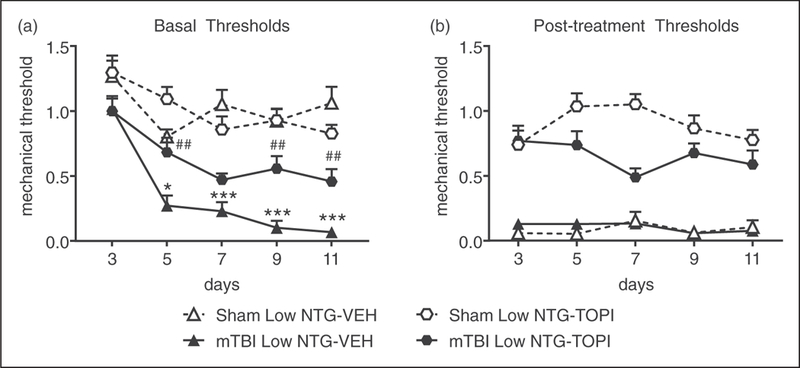
Topiramate inhibits both acute and chronic hyperalgesia induced by NTG. Two weeks post-injury, C57BL/6J mice were injected every day with vehicle or topiramate (TOPI, 30 mg/kg, ip). On days 3, 5, 7, 9 and 11, mice were treated with low-dose NTG (0.1 mg/kg, ip), and 1 h 15 min later with vehicle or topiramate. (a) Basal mechanical thresholds, assessed prior to drug administration, were significantly decreased in the mTBI group treated with vehicle compared to their sham counterparts, and that effect was attenuated by topiramate. p < 0.001, effect of injury, time and interaction two-way RM ANOVA as compared to sham-vehicle, Holm-Sidak post hoc analysis, *p < 0.05, ***p < 0.001 as compared to sham-vehicle day 1; mTBI-vehicle vs. mTBI-topiramate p < 0.05 drug, time, interaction, two-way RM ANOVA, ##p < 0.01 as compared to mTBI-vehicle day 1. n = 8/group. (b) Regardless of injury, low-dose NTG produced acute allodynia 2 hours post-NTG, which was significantly inhibited by topiramate. p < 0.001 effect of drug only, 2-way RM ANOVA.
SNC80 inhibits acute and chronic allodynia within the PTH model
We next tested the delta opioid receptor (DOR) agonist, SNC80, within this model of PTH-associated pain. As above, at 2 weeks post-injury all animals were treated every other day for 9 days with low-dose NTG (0.1 mg/kg, ip), and subsequently with vehicle or SNC80 (10 mg/kg, ip). Again, low-dose NTG evoked basal hypersensitivity only in mTBI animals (Figure 6(a)); and acute post-treatment allodynia in both sham and mTBI mice (Figure 6(b)). Treatment with SNC80 significantly attenuated chronic basal hypersensitivity induced by low-dose NTG in the mTBI group (Figure 6(a)). Furthermore, SNC80 reversed NTG- induced acute allodynia in both sham and mTBI groups on each test day (Figure 6(b)). These data indicate that SNC80 may not only inhibit acute PTH-associated pain but may also restrict the development of chronic PTH-associated pain.
Figure 6.
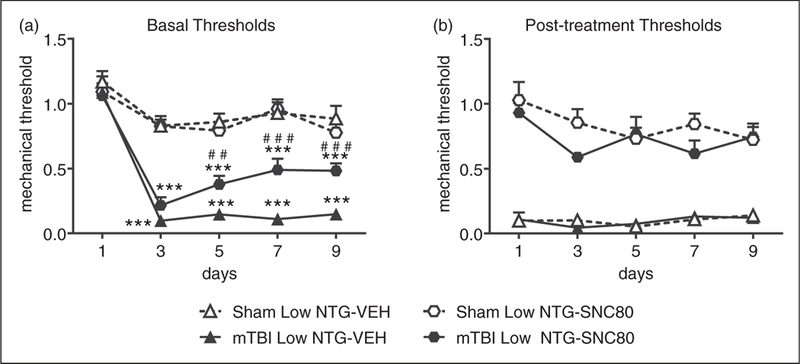
SNC80 inhibits both acute and chronic allodynia induced by NTG. Two weeks post-injury, C57BL/6J mice were injected every second day with low-dose NTG (0.1 mg/kg, ip), and 1 h 15 min later with vehicle or SNC80 (10 mg/kg, ip). (a) Basal mechanical thresholds, assessed prior to drug administration, were significantly decreased in mTBI groups treated with vehicle compared to their sham counterparts, an effect that was attenuated by SNC80. p < 0.05, effect of injury, time and interaction two-way RM ANOVA, Holm-Sidak post hoc analysis, ***p < 0.001 as compared to sham-vehicle. p < 0.001 drug, time, interaction, two-way RM ANOVA mTBI-vehicle vs. mTBI-SNC80, ##p < 0.01, ###p < 0.001 as compared to vehicle day 1. n = 8–14/group. (b) Regardless of injury, low-dose NTG produced acute hyperalgesia as determined 2 hours post-NTG, which was significantly inhibited by SNC80. p < 0.001 effect of drug only, 2-way RM ANOVA.
mTBI increases cephalic hypersensitivity to low-dose NTG
To determine whether cephalic responses differed from hind paw responses, we tested the effect of low dose NTG on cephalic allodynia. At 2 weeks post-injury, mTBI and sham mice had similar cephalic mechanical thresholds on day 1 (Figure 7(a), day 1). On each test day, low-dose NTG produced acute periorbital allodynia 2h post-administration regardless of injury (Figure 7(b)). In addition, mTBI mice treated with low-dose NTG also developed a profound basal hypersensitivity, an effect that was not seen in sham mice (Figure 7(a)). These results indicate that similar to the hind paw, mTBI results in heightened sensitivity to the development of chronic migraine-associated pain.
Figure 7.
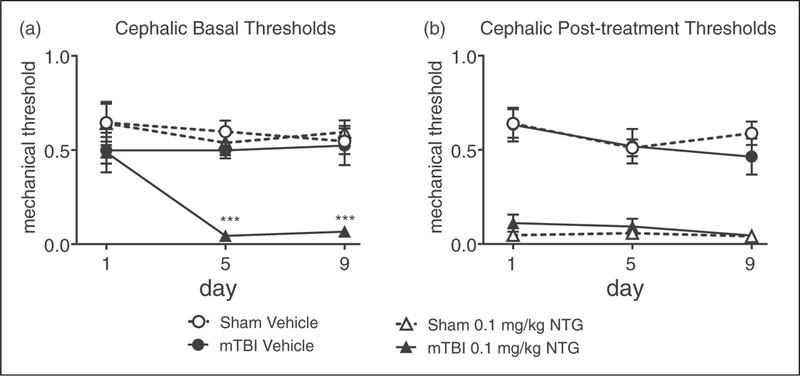
mTBI increases cephalic mechanical hypersensitivity to a low-dose of NTG 2 weeks after closed head injury. Post-sham or injury, C57BL/6J mice received either a vehicle or low dose NTG (0.1 mg/kg, ip) every day over 9 days, and tested every 4th day (days 1.5, 9). (a) Basal thresholds, assessed prior to vehicle or NTG administration, revealed that mTBI animals treated with low dose NTG had significantly lower basal cephalic thresholds than their sham counterparts. p < 0.001 treatment, time and interaction; two-way RM ANOVA, Holm-Sidak post hoc analysis, ***p < 0.001, n = 8/group. (b) In the same mice tested 2 h post NTG/vehicle, NTG evoked hyperalgesia that did not differ between sham and mTBI groups. mTBI increases the development of cephalic hypersensitivity to a low dose of NTG.
mTBI increases expression of CGRP within the trigeminal ganglia 2 weeks post-injury
CGRP is considered to be an endogenous migraine generator and plays a critical role in the regulation of migraine pain (31). Trigeminal ganglia (TG) are first order cells that regulate head-specific pain, and we determined immunohistochemically if mTBI affected the amount and number of CGRP expressing (CGRP+) cells within this region. We observed that,2 weeks post-injury, mTBI produced a significant increase in the overall expression of CGRP in each cell (Figure 8(a)), as well as an increase in the total number of CGRP + cells (Figure 8(b)) relative to sham controls. Our results indicate that this mTBI procedure dynamically alters the expression of CGRP within the TG.
Figure 8.
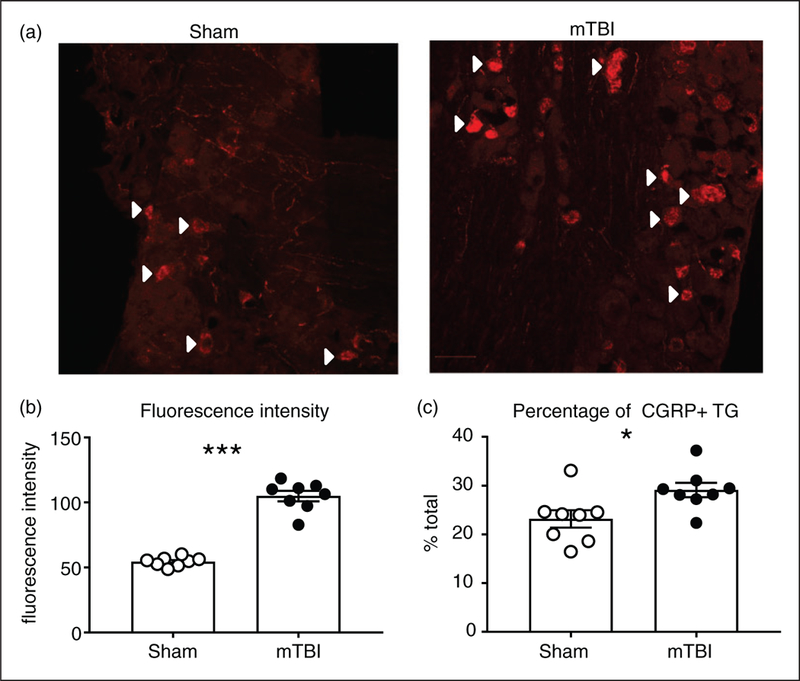
mTBI causes an increase in the expression of the pro-migraine neuropeptide, CGRP, in the trigeminal ganglia. C57BL/6J mice underwent a sham/mTBI procedure, and trigeminal ganglia was analyzed for CGRP quantification at 2 weeks post-injury. (a) Representative images of trigeminal ganglia from sham and mTBI mice. White arrow heads indicate some, but not all CGRP + ganglia.Quantification of the fluorescent intensity of CGRP positive cells shows that mTBI significantly increases the amount of CGRP in the TG. ***p < 0.001, t-test, n = 8 mice/group. (c) Quantification of the percentage of CGRP positive cells show that mTBI significantly increased the overall number of TGs expressing CGRP. p < 0.05, t-test, n = 8 mice/group.
Discussion
Despite the high prevalence of PTH, the mechanisms underlying the progression from head trauma to PTH remain unclear. A primary goal of this study was to characterize a mouse model of PTH which combined published models of closed head injury and chronic migraine (13,20). We demonstrate that this mTBI procedure alone produced mechanical allodynia at 3 days post-injury, but that hypersensitivity was resolved by 2 weeks post-injury. However, mTBI mice were more sensitive to the development of chronic migraine-associated pain as induced by low dose NTG, an effect observed in both cephalic and somatic regions. Acute allodynia within this model was blocked by the migraine abortive sumatriptan, and acute and chronic PTH-associated pain was inhibited by the migraine preventive topiramate. We also found that the selective delta opioid receptor agonist, SNC80, inhibited acute and chronic allodynia in this model, identifying this receptor as a novel therapeutic target for PTH. Additionally, 2 weeks following closed head injury we observed an increase in the expression of the migraine-associated neuropeptide, CGRP, in the trigeminal ganglia, which provides a potential mechanism for the heighted sensitivity to the development of chronic migraine associated with mTBI.
We have previously shown that chronic intermittent treatment with higher doses of NTG (3–10 mg/kg) can produce a progressive basal hypersensitivity in mice (20). In this study, we tested a lower dose of NTG that did not cause basal hypersensitivity in sham controls but significantly reduced mechanical thresholds in mTBI animals. This effect was long lasting, as sensitivity to low-dose NTG was still seen 12 weeks post-injury. Our findings are in keeping with the original characterization of the closed head weight-drop model in which long-term cognitive deficits were observed in the absence of structural damage to the brain (13). However, this relatively mild TBI can still cause adaptations, especially at the level of inflammatory responses. Increased gene expression of the cytokine CCL13 was observed up to 7 days post-injury in this model (32), and increased dural mast cell degranulation was also found up to 30 days post-injury (33). Furthermore, closed head injury models using a heavier weight (50 g, as compared to 30 g used herein) have resulted in elevated levels of tumor necrosis factor-alpha (TNF-α) post-TBI (34). One possibility is that neuroinflammation induced by mTBI can ultimately trigger sensitization of the trigeminovascular complex, resulting in PTH (3). In patients, PTH can develop 1 week to 1 year after injury and may even manifest outside of this time frame (1,3,4). The mild nature of the injury used in this model may reflect sensitization to sub-concussive head trauma and may contribute to the major inflammatory changes shown in previous studies. Future studies will focus on characterizing the effect of anti-inflammatory agents within our model of post-traumatic migraine. Our results reflect the finding that a single mTBI can have long-term effects on the susceptibility to developing chronic PTH. It should be noted that we only used C57BL/6J mice, as the NTG dosing regimen has been well characterized in this mouse strain (20). Other mouse strains may respond differently to mTBI, and/or have a different dose response to NTG.
To determine the predictive validity of this model of post-traumatic migraine, we tested the migraine abortive, sumatriptan, and the preventive, topiramate. A clinical study examining the treatment of PTH in soldiers found that triptans significantly alleviated PTH, and topiramate could act as an effective preventive (35). In our study, sumatriptan significantly inhibited the acute allodynia induced by low dose NTG in mTBI animals, which is consistent with previous work using high doses of NTG (19,20,22). We were surprised to find that in sham animals treated with low dose NTG and sumatriptan there was a decrease in basal responses. Chronic daily treatment with sumatriptan can be used to model medication overuse headache (21,36), although in our study sumatriptan was only administered every other day. Chronic treatment with sumatriptan alone may act with low-dose NTG to exacerbate migraine-associated pain through a yet undetermined mechanism. Chronic daily administration of the migraine preventive, topiramate, alleviated both the acute allodynia and chronic basal hypersensitivity induced by low-dose NTG in the mTBI animals. These results are consistent with clinical reports that show that topiramate can be effective in the treatment of chronic PTH (35,37), and it has been used extensively as a migraine preventive (38). These experiments were performed in the periphery. We have previously shown that sumatriptan and topiramate (20), along with the migraine preventive propranolol (21), can block migraine-associated pain induced by high dose NTG also assessed in the periphery. In addition, in the dural inflammation model, application of inflammatory mediators to the dura produced mechanical sensitivity in both cephalic and hind paw regions (39,40), similar to the effects observed in our study; these results likely reflect the development of central sensitization, which may be mediated through neurons within the thalamus (41). Together, our pharmacological results support the use of this mouse PTH model as a pharmacological screening tool.
There are limited therapeutic options for the treatment of PTH, and many patients use established migraine therapies that do not provide sufficient pain relief in all patients (9). We have previously shown in preclinical models that DOR activation can inhibit multiple migraine-associated symptoms, including allodynia, negative affect, and aura (19). In addition, anatomical studies have shown that DOR can be co-expressed with CGRP in the TG (42), thus further supporting the role of DOR as a potential therapy for migraine-associated pain. In our study, we found that SNC80 could block PTH-related acute allodynia and that it had a protective effect on the mTBI-NTG induced basal hypersensitivity. DOR may be a particularly promising target for TBI-associated pathologies. For example, DOR agonists are effective in models of peripheral hyperalgesia (43,44), and chronic pain conditions, including headache, are a major source of disability following TBI (45). Importantly, DOR agonists produce anxiolytic and antidepressant effects (43,46,47). Emotional dysregulation is often comorbid with chronic pain and migraine and contributes to a feed forward cycle of disability. Post-traumatic stress disorder is especially comorbid with PTH, and its presence is associated with increased severity of PTH (2,48–50). The ability of DOR agonists to alleviate negative emotional states would be beneficial in these more complicated clinical situations. The delta opioid receptor may be uniquely positioned to alleviate multiple aspects of mTBI-related pathologies, including PTH.
We observed that CGRP expression was significantly increased in TG following mTBI, and we postulate that this augmentation likely promotes the development of PTH from traumatic brain injury (3). This increase was observed 2 weeks post-injury, a time at which allodynia induced by mTBI alone was already resolved. CGRP is an endogenous migraine generator, and this neuropeptide plays a critical role in migraine pathophysiology. CGRP infusion can induce headache (51), and levels of CGRP in the circulation are upregulated during acute migraine attacks (52). Additionally, CGRP receptor antagonists are effective in aborting migraine (53), and antibodies targeting CGRP and its receptor are currently in drug development with promising results in late stage clinical trials (54,55). In terms of mTBI-related pain, experiments performed in rats found that both TBI by controlled cortical impact (2,12,56) and repeated mild head injury (57) resulted in increased CGRP expression in the trigeminal nucleus caudalis as compared to controls. The TG is a major source of CGRP to the trigeminal nucleus caudalis (58,59), and together these structures form part of the trigeminovascular complex, which regulates head-specific pain. The CGRP antagonist MK8825 was also found to attenuate both periorbital allodynia and photosensitivity evoked by controlled cortical impact injury (56). Furthermore, in a rat model, CGRP inhibition blocked increased sensitivity to NTG (15). In this study, weight-drop increased acute periorbital allodynia evoked by NTG up to 30 days post-injury, and this allodynia was blocked by a CGRP antibody (15). NTG also increased conditioned place aversion in mTBI rats, an effect that was blocked by CGRP antibody treatment (15). Our study further supports the role of CGRP as a link for the development of PTH following mTBI and expands the role of this neuropeptide for PTH with a chronic migraine-like phenotype. Taken together, these results suggest that our model reflects the role of CGRP in the development to PTH and support the notion that upcoming CGRP- targeted therapies will be promising for the treatment of this disorder.
PTH is a debilitating disorder that can result in chronic disability and decreased quality of life. A better understanding of the mechanisms that regulate PTH would allow for the discovery of more targeted approaches to treat this disorder. Here, we have characterized a novel mouse model of PTH, one which specifically reflects the more severe post-traumatic chronic migraine phenotype. The development of this model opens up the possibility for investigators to easily use genetic, opto-and chemogenetic approaches that have been optimized for use in mice. In addition, this model can be used to screen novel therapies for PTH, and we have used it to identify the delta opioid receptor as a promising target. We also recapitulate findings that CGRP is an important facilitator between mTBI and the development of PTH. Future studies will use this model to further identify the molecular mechanisms regulating PTH.
Article highlights.
We developed a mouse model of post-traumatic migraine by combining the closed-head weight drop model and the nitroglycerin chronic migraine model.
mTBI induces an increased sensitivity to nitroglycerin in cephalic and peripheral regions.
We identified the delta opioid receptor as a novel therapeutic target for post-traumatic headache.
CGRP expression is upregulated in trigeminal ganglia weeks following mTBI and could serve as a link between injury and headache chronification.
Acknowledgements
We would like to acknowledge the Confocal Microscopy Facility of the Research Resources Center at the University of Illinois at Chicago for their expertise and use of their confocal microscope. We would also like to thank Dr Amul Sakharkar for helpful discussion about mTBI models.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Research was supported by DOD grant PR141746 (AAP), NIH Grant DA040688 (AAP), and the NIAAA funded Center for Alcohol Research in Epigenetics - AA022538 (SCP). LM is a member of the UIC Graduate Program for Neuroscience.
Footnotes
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
References
- 1.The International Classification of Headache Disorders, 3rd edition (beta version). Cephalalgia 2013; 33: 629–808. [DOI] [PubMed] [Google Scholar]
- 2.Theeler B, Lucas S, Riechers RG 2nd, et al. Post-traumatic headaches in civilians and military personnel: A comparative, clinical review. Headache 2013; 53: 881–900. [DOI] [PubMed] [Google Scholar]
- 3.Moye LS and Pradhan AA. From blast to bench: A translational mini-review of posttraumatic headache. J Neurosci Res 2017; 95: 1347–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.National Center for Injury Prevention and Control. Report to Congress on mild traumatic brain injury in the United States: Steps to prevent a serious public health problem. Atlanta, GA: Centers for Disease Control and Prevention, 2003. [Google Scholar]
- 5.VA/DoD clinical practice guideline for management of concussion/mild traumatic brain injury. J Rehabil Res Dev 2009; 46: Cp1–Cp68. [PubMed] [Google Scholar]
- 6.Couch JR and Bearss C. Chronic daily headache in the posttrauma syndrome: Relation to extent of head injury. Headache 2001; 41: 559–564. [DOI] [PubMed] [Google Scholar]
- 7.Vargas BB and Dodick DW. Posttraumatic headache. Curr Opin Neurol 2012; 25: 284–289. [DOI] [PubMed] [Google Scholar]
- 8.Lucas S, Hoffman JM, Bell KR, et al. A prospective study of prevalence and characterization of headache following mild traumatic brain injury. Cephalalgia 2014; 34: 93–102. [DOI] [PubMed] [Google Scholar]
- 9.Visser WH, de Vriend RH, Jaspers NH, et al. Sumatriptan-nonresponders: A survey in 366 migraine patients. Headache 1996; 36: 471–475. [DOI] [PubMed] [Google Scholar]
- 10.Bigal ME, Serrano D, Reed M, et al. Chronic migraine in the population: Burden, diagnosis, and satisfaction with treatment. Neurology 2008; 71: 559–566. [DOI] [PubMed] [Google Scholar]
- 11.Lipton RB, Buse DC, Serrano D, et al. Examination of unmet treatment needs among persons with episodic migraine: Results of the American Migraine Prevalence and Prevention (AMPP) Study. Headache 2013; 53: 1300–1311. [DOI] [PubMed] [Google Scholar]
- 12.Elliott MB, Oshinsky ML, Amenta PS, et al. Nociceptive neuropeptide increases and periorbital allodynia in a model of traumatic brain injury. Headache 2012; 52: 966–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zohar O, Schreiber S, Getslev V, et al. Closed-head minimal traumatic brain injury produces long-term cognitive deficits in mice. Neuroscience 2003; 118: 949–955. [DOI] [PubMed] [Google Scholar]
- 14.Zohar O, Rubovitch V, Milman A, et al. Behavioral consequences of minimal traumatic brain injury in mice. Acta Neurobiol Exp (Wars) 2011; 71: 36–45. [DOI] [PubMed] [Google Scholar]
- 15.Bree D and Levy D. Development of CGRP-dependent pain and headache related behaviours in a rat model of concussion: Implications for mechanisms of post-traumatic headache. Cephalalgia 2016; 38: 246–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hoffman JM, Lucas S, Dikmen S, et al. Natural history of headache after traumatic brain injury. J Neurotrauma 2011; 28: 1719–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Iversen HK, Olesen J and Tfelt-Hansen P. Intravenous nitroglycerin as an experimental model of vascular headache. Basic characteristics. Pain 1989; 38: 17–24. [DOI] [PubMed] [Google Scholar]
- 18.Christiansen I, Thomsen LL, Daugaard D, et al. Glyceryl trinitrate induces attacks of migraine without aura in sufferers of migraine with aura. Cephalalgia 1999; 19: 660–667. [DOI] [PubMed] [Google Scholar]
- 19.Pradhan AA, Smith ML, Zyuzin J, et al. Delta-opioid receptor agonists inhibit migraine-related hyperalgesia, aversive state and cortical spreading depression in mice. Br J Pharmacol 2014; 171: 2375–2384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pradhan AA, Smith ML, McGuire B, et al. Characterization of a novel model of chronic migraine. Pain 2014; 155: 269–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tipton AF, Tarash I, McGuire B, et al. The effects of acute and preventive migraine therapies in a mouse model of chronic migraine. Cephalalgia 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bates EA, Nikai T, Brennan KC, et al. Sumatriptan alleviates nitroglycerin-induced mechanical and thermal allodynia in mice. Cephalalgia 2010; 30: 170–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brennan KC, Bates EA, Shapiro RE, et al. Casein kinase idelta mutations in familial migraine and advanced sleep phase. Sci Transl Med 2013; 5: 183ra–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sufka KJ, Staszko SM, Johnson AP, et al. Clinically relevant behavioral endpoints in a recurrent nitroglycerin migraine model in rats. J Headache Pain 2016; 17: 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Farajdokht F, Babri S, Karimi P, et al. Chronic ghrelin treatment reduced photophobia and anxiety-like behaviors in nitroglycerin-induced migraine: Role of pituitary adenylate cyclase-activating polypeptide. Eur J Neurosci 2017; 45: 763–772. [DOI] [PubMed] [Google Scholar]
- 26.Greco R, Meazza C, Mangione AS, et al. Temporal profile of vascular changes induced by systemic nitroglycerin in the meningeal and cortical districts. Cephalalgia 2011; 31: 190–198. [DOI] [PubMed] [Google Scholar]
- 27.Chaplan SR, Bach FW, Pogrel JW, et al. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods 1994; 53: 55–63. [DOI] [PubMed] [Google Scholar]
- 28.Moye LS and Pradhan AAA. Animal model of chronic migraine-associated pain. Curr Protoc Neurosci 2017; 809 60 1–9 9. ). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ben Aissa M, Tipton AF, Bertels Z, et al. Soluble guanylyl cyclase is a critical regulator of migraine-associated pain. Cephalalgia 2017; 333102417737778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bates EA, Nikai T, Brennan KC, et al. Sumatriptan alleviates nitroglycerin-induced mechanical and thermal allodynia in mice. Cephalalgia 2010; 30: 170–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bigal ME, Walter S and Rapoport AM. Calcitonin generelated peptide (CGRP) and migraine current understanding and state of development. Headache 2013; 53: 1230–1244. [DOI] [PubMed] [Google Scholar]
- 32.Israelsson C, Wang Y, Kylberg A, et al. Closed head injury in a mouse model results in molecular changes indicating inflammatory responses. J Neurotrauma 2009; 26: 1307–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Levy D, Edut S, Baraz-Goldstein R, et al. Responses of dural mast cells in concussive and blast models of mild traumatic brain injury in mice: Potential implications for post-traumatic headache. Cephalalgia 2016; 36: 915–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Baratz R, Tweedie D, Wang JY, et al. Transiently lowering tumor necrosis factor-alpha synthesis ameliorates neuronal cell loss and cognitive impairments induced by minimal traumatic brain injury in mice. J Neuroinflammation 2015; 12: 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Erickson JC. Treatment outcomes of chronic post-traumatic headaches after mild head trauma in US soldiers: An observational study. Headache 2011; 51: 932–944. [DOI] [PubMed] [Google Scholar]
- 36.De Felice M, Ossipov MH, Wang R, et al. Triptan-induced enhancement of neuronal nitric oxide synthase in trigeminal ganglion dural afferents underlies increased responsiveness to potential migraine triggers. Brain 2010; 133: 2475–2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Minen MT, Boubour A, Walia H, et al. Post-concussive syndrome: A focus on post-traumatic headache and related cognitive, psychiatric, and sleep issues. Curr Neurol Neurosci Rep 2016; 16: 100. [DOI] [PubMed] [Google Scholar]
- 38.Diener HC, Bussone G, Van Oene JC, et al. Topiramate reduces headache days in chronic migraine: A randomized, double-blind, placebo-controlled study. Cephalalgia 2007; 27: 814–823. [DOI] [PubMed] [Google Scholar]
- 39.Edelmayer RM, Ossipov MH and Porreca F. An experimental model of headache-related pain. Methods Mol Biol 2012; 851: 109–120. [DOI] [PubMed] [Google Scholar]
- 40.Edelmayer RM, Le LN, Yan J, et al. Activation of TRPA1 on dural afferents: A potential mechanism of headache pain. Pain 2012; 153: 1949–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Burstein R, Jakubowski M, Garcia-Nicas E, et al. Thalamic sensitization transforms localized pain into wide-spread allodynia. Ann Neurol 2010; 68: 81–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rice FL, Xie JY, Albrecht PJ, et al. Anatomy and immunochemical characterization of the non-arterial peptidergic diffuse dural innervation of the rat and Rhesus monkey: Implications for functional regulation and treatment in migraine. Cephalalgia 2016; 37: 1350–1372. [DOI] [PubMed] [Google Scholar]
- 43.Pradhan AA, Befort K, Nozaki C, et al. The delta opioid receptor: An evolving target for the treatment of brain disorders. Trends Pharmacol Sci 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Charles A and Pradhan AA. Delta-opioid receptors as targets for migraine therapy. Curr Opin Neurol 2016; 29: 314–319. [DOI] [PubMed] [Google Scholar]
- 45.Nampiaparampil DE. Prevalence of chronic pain after traumatic brain injury: A systematic review. Jama 2008; 300: 711–719. [DOI] [PubMed] [Google Scholar]
- 46.Filliol D, Ghozland S, Chluba J, et al. Mice deficient for delta- and muopioid receptors exhibit opposing alterations of emotional responses. Nat Genet 2000; 25: 195–200. [DOI] [PubMed] [Google Scholar]
- 47.Lutz PE and Kieffer BL. Opioid receptors: Distinct roles in mood disorders. Trends Neurosci 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Theeler BJ, Mercer R and Erickson JC. Prevalence and impact of migraine among US Army soldiers deployed in support of Operation Iraqi Freedom. Headache 2008; 48: 876–882. [DOI] [PubMed] [Google Scholar]
- 49.Scofield DE, Proctor SP, Kardouni JR, et al. Risk factors for mild traumatic brain injury and subsequent post-traumatic stress disorder and mental health disorders among US army soldiers. J Neurotrauma 2017. [DOI] [PubMed] [Google Scholar]
- 50.O’Neil ME, Carlson K, Storzbach D, et al. Complications of mild traumatic brain injury in veterans and military personnel: A systematic review. Washington, DC:22, 2013. [PubMed]
- 51.Lassen LH, Haderslev PA, Jacobsen VB, et al. CGRP may play a causative role in migraine. Cephalalgia 2002; 22: 54–61. [DOI] [PubMed] [Google Scholar]
- 52.Goadsby PJ, Edvinsson L and Ekman R. Vasoactive peptide release in the extracerebral circulation of humans during migraine headache. Ann Neurol 1990; 28: 183–187. [DOI] [PubMed] [Google Scholar]
- 53.Olesen J, Diener HC, Husstedt IW, et al. Calcitonin generelated peptide receptor antagonist BIBN 4096 BS for the acute treatment of migraine. N Engl J Med 2004; 350: 1104–1110. [DOI] [PubMed] [Google Scholar]
- 54.Hou M, Xing H, Cai Y, et al. The effect and safety of monoclonal antibodies to calcitonin gene-related peptide and its receptor on migraine: A systematic review and meta-analysis. J Headache Pain 2017; 18: 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tso AR and Goadsby PJ. Anti-CGRP monoclonal antibodies: The next era of migraine prevention? Curr Treat Options Neurol 2017; 19: 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Daiutolo BV, Tyburski A, Clark SW, et al. Trigeminal pain molecules, allodynia, and photosensitivity are pharmacologically and genetically modulated in a model of traumatic brain injury. J Neurotrauma 2016; 33: 748–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tyburski AL, Cheng L, Assari S, et al. Frequent mild head injury promotes trigeminal sensitivity concomitant with microglial proliferation, astrocytosis, and increased neuropeptide levels in the trigeminal pain system. J Headache Pain 2017; 18: 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Edvinsson L The trigeminovascular pathway: Role of CGRP and CGRP receptors in migraine. Headache 2017; 57: S47–S55. [DOI] [PubMed] [Google Scholar]
- 59.Goadsby PJ, Holland PR, Martins-Oliveira M, et al. Pathophysiology of migraine: A disorder of sensory processing. Physiol Rev 2017; 97: 553–622. [DOI] [PMC free article] [PubMed] [Google Scholar]