

Abstract
A small molecule library of pyrido[2,3-d]pyrimidine-2,4-dione derivatives 6–16 was synthesized from 6-amino-1,3-disubstituted uracils 18, characterized, and screened for inhibitory activity against eukaryotic elongation factor-2 kinase (eEF-2K). To understand the binding pocket of eEF-2K, structural modifications of the pyrido[2,3-d]pyrimidine were made at three regions (R1, R2, and R3). A homology model of eEF-2K was created, and compound 6 (A-484954, Abbott laboratories) was docked in the catalytic domain of eEF-2K. Compounds 6 (IC50 = 420 nM) and 9 (IC50 = 930 nM) are found to be better molecules in this preliminary series of pyrido[2,3-d]pyrimidine analogs. eEF-2K activity in MDA-MB-231 breast cancer cells is significantly reduced by compound 6, to a lesser extent by compound 9, and is unaffected by compound 12. Similar inhibitory results are observed when eEF-2K activity is stimulated by 2-deoxy-D-glucose (2-DOG) treatment, suggesting that compounds 6 and 9 are able to inhibit AMPK-mediated activation of eEF-2K to a notable extent. The results of this work will shed light on the further design and optimization of novel pyrido[2,3-d]pyrimidine analogs as eEF-2K inhibitors.
Keywords: Pyrido[2,3-d]pyrimidine-24-dione; Uracil; eEF-2K inhibitor; Kinase inhibitor; Homology modeling
Graphical Abstract
1. Introduction
Eukaryotic elongation factor-2 kinase (eEF-2K, EC 2.7.11.20) is an atypical Ser/Thr-protein kinase, and is also known as a calcium/calmodulin-dependent protein kinase-III (CaM kinase-III). eEF-2K is not structurally similar to conventional protein kinases, and the structure of eEF-2K has not been reported. eEF-2K phosphorylates eEF2 at Thr-56 and Thr-58, and inactivates eEF2, and thus regulates protein synthesis at the elongation step of translation.1,2 Nutrients and growth factors inactivate eEF-2 kinase whereas starvation activates it.3 In 2013, Leprivier et al.4 demonstrated a crucial role for eEF-2K in protecting normal cells from nutrient deprivation through inhibition of eEF-2, and they suggested how cancer cells could exploit this pathway by activating eEF-2K to adapt to metabolic stress. eEF-2K has been reported to be upregulated in various malignant cells such as breast cancer cells,5 malignant glioma cells,6 HL60 leukemia cells,7 and pancreatic cancer (PaCa) cells,8 and has been identified as a potential cancer-therapeutic target.9
Several small molecule inhibitors of eEF-2K have been reported (Figure 1), including: Rottlerin 1, which is found to be a non specific and cytotoxic inhibitor,10 1,3-selenazine analog 2, which is suggested to block phosphorylation of eEF-2 in cells and to react with cysteine residues,11 the imidazolium derivative (NH125) 3,12 which does not inhibit eEF-2K in cells,13,16 and thieno[2,3-b]pyridine 4, which is an ATP-competitive inhibitor that lacks potency in cells.14 Recently, high-throughput screens for eEF-2K inhibitors by us,15 and by Abbott laboratories,16 revealed thiopyran-dicarbonitrile analog 5, and pyrido-pyrimidinedione derivative A-484954 (compound 6), respectively. A-484954, a small molecule discovered by Abbott, is an ATP-competitive and cell-permeable inhibitor of eEF-2K (IC50 = 0.28 μM).
Figure 1.

Structures of eEF-2K inhibitors.
We are interested in developing a more potent and selective eEF-2K inhibitor to probe the function of eEF-2K in the regulation of protein synthesis. In this paper, we report the synthesis of a preliminary series of pyrido[2,3-d]pyrimidine-2,4-dione derivatives 6–16 and their inhibitory activity against eEF-2K to explore structure-activity relationships (SAR) of A-484954. Moreover, a homology model has been generated and used to dock compound 6 to provide insight into its binding in the catalytic domain of eEF-2K.
2. Results and discussion
2.1. Chemistry
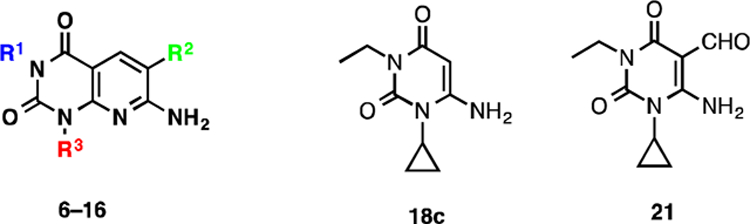
Pyrido[2,3-d]pyrimidine-2,4-dione derivatives 6–16 were prepared according to Schemes 1, 2, and 3. Direct alkylation of commercially available uracil derivatives 17 with either ethyl iodide or n-propyl iodide in the presence of 10–15% aqueous NaOH afforded the corresponding 1,3-disubstituted-6-aminouracils 18c–18e in 40–53% yield (Scheme 1).17 To construct the pyridine ring of pyrido[2,3-d]pyrimidine 6, uracil 18c was treated with the Vilsmeier reagent 19, which was generated in situ from phosphoryl chloride in dimethyl formamide, followed by solvent evaporation and treatment of the residue with triethylamine and cyanoacetamide in ethanol.18 However, addition of traces of water present in the reaction mixture to the hydrochloride salt 20c resulted in hydrolyzed product 21 in 66% yield, but not the required pyrido[2,3-d]pyrimidine 6 (Scheme 2).
Scheme 1.

Alkylation of uracil derivatives 17.
Scheme 2.

Hydrolysis of hydrochloride salt 20c.
Scheme 3.

Synthesis of pyrido[2,3-d]pyrimidine-2,4-dione derivatives 6–16.
Alternatively, when a catalytic amount of dimethyl formamide and oxalyl chloride were used to prepare the Vilsmeier reagent 22, and the resulting HCl salt 20 was treated with triethylamine and cyanoacetamide in ethanol, compound 6 was isolated in 78% yield over the two steps (Scheme 3). Other pyrido[2,3-d]pyrimidine-2,4-dione derivatives 7, 9, 12–16 were also synthesized using this procedure. Compound 8 was isolated in 47% yield by direct methylation of compound 7 in presence of KOtBu as a base. On treatment with sodium hydride and p-nitrobenzylbromide, compound 7 resulted in 61% of benzylated product 10 as an off-white solid, which was reduced with SnCl2 in an ethyl acetate and methanol mixture to afford amine 11 in 70% yield.
2.2. Computer homology modeling
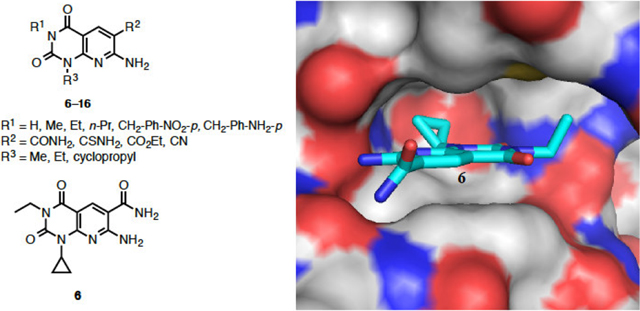
The structure of eEF-2K was generated using homology modeling with Dictyostelium myosin heavy chain kinase A (MHCK A) as the template and compound 6 was docked into the ATP-binding pocket of eEF-2K (Figure 2). The inhibitor 6 is shown in cyan. The protein is shown in cartoon representation, with the hinge region in purple and the Gly-rich loop in blue. The binding of inhibitor 6 involves hydrogen bonds to residues K170, I232 and G234. The hydrophobic cyclopropyl and ethyl groups are buried deep inside the adenine-binding pocket and underneath the Gly-rich-loop respectively. Docking was performed using Gold5.119 with ChemPLP fitness function.20 Twenty independent docking attempts were performed for the ligand using generic algorithm. The results of each attempt were ranked according to the ChemPLP fitness score and were virtually inspected. The highest ranked pose of the ligand also happened to be very similar to the most of the high-ranking poses that resulted from the twenty attempts (data not shown). This suggested that the docking attempts reach an optimal state, and thus supporting the likelihood of this pose in docking. Details on generating the homology model can be found in our recent work.21
Figure 2.

Compound 6 docked into the ATP-binding pocket of eEF-2K. A) eEF-2K is rendered using solvent accessible surface and compound 6 (sticks) is shown well fitted into the pocket. B) Docking determined the four key residues that are shown in sticks. Hydrogen bonds are shown as blue dashed lines.
2.3. Biological activity
2.3.1. In vitro activity against recombinant eEF-2K
It has been reported that compound 6 (A-484954, Abbott labs), inhibits eEF-2K activity in vitro with an IC50 value of 0.28 μM.16 In addition to compound 6, we examined a series of pyrido[2,3-d]pyrimidine-2,4-dione derivatives (7–16) to evaluate their potential in terms of modification of the initial compound, and to probe their structure-activity relationships. A radioactive assay utilizing [γ−32P]ATP was used to assess the inhibition of recombinant human eEF-2K by the compounds, where kinase activity was determined by measuring the rate of incorporation of 32P into a peptide substrate.22,23
The inhibition found for target compounds 6–16, 18c, and 21 against recombinant eEF-2K activity in vitro are summarized in Table 1. Compound 6 has an IC50 of 0.42 μM, which differs from the reported value of 0.28 μM (Figure 3).16 This can be accounted for by the difference in assay conditions, where our assay uses 10 μM ATP compared to 5 μM ATP by Chen et al. Because the compound is reported to be an ATP-competitive inhibitor, it is expected that an increase in ATP concentration in the assay would increase the IC50 value. When the R1 group on the scaffold is modified from hydrogen to benzyl derivatives, the potency increased from hydrogen (IC50 = 6.6 μM) to ethyl (IC50 = 0.42 μM) and was then decreased to benzyl derivatives (IC50 > 25 μM). Complete loss of inhibitory activity is observed on changing the CONH2 group (IC50 = 0.42 μM) to CSNH2, CN and CO2Et (compounds 12, 13, and 14, respectively) (IC50 > 25 μM). Moderate loss of activity is observed when the cyclopropyl group (IC50 = 0.42 μM) at R3 is changed to an ethyl group (IC50 = 1–2 μM). The inactivity of compounds 18c and 21 stresses the importance of a pyridine ring for this class of molecules. These results provide new insights, which may aid in the rational design of inhibitors for the kinase.
Table 1.
In vitro activity of the target compounds 6–16, 18c, and 21 against eEF-2K.
 | ||||
|---|---|---|---|---|
| compound | R1 | R2 | R3 | IC50 (MM) |
| 7 | H | CONH2 | cyclopropyl | 6.6 ± 0.2 |
| 8 | Me | CONH2 | cyclopropyl | 6.1 ± 0.2 |
| 6 | Et | CONH2 | cyclopropyl | 0.42 ± 0.01 |
| 9 | n-Pr | CONH2 | cyclopropyl | 0.93 ± 0.03 |
| 10 | CH2-Ph-NO2-p | CONH2 | cyclopropyl | >25 |
| 11 | CH2-Ph-NH2-p | CONH2 | cyclopropyl | >25 |
| 12 | Et | CSNH2 | cyclopropyl | >25 |
| 13 | Et | CO2Et | cyclopropyl | >25 |
| 14 | Et | CN | cyclopropyl | >25 |
| 15 | Et | CONH2 | Et | 1–2 |
| 16 | Me | CONH2 | Me | >25 |
| 18c | - | - | - | >25 |
| 21 | - | - | - | >25 |
Inhibitor dose response assays were performed with 5 nM recombinant eEF-2K enzyme at various concentrations of the inhibitor, in the presence of 50 μM free Ca2+, 200 nM CaM and 10 μM [γ−32P]ATP, against 150 μM peptide substrate as described under the experimental section. The inhibitor was incubated with eEF-2K for 30 min at 30 °C before initiating the assay with peptide and [γ−32P]ATP. Kinase activity in each case was determined by calculating the rate of phosphorylation of the peptide (μM/s), and dose-response curves for data conforming to inhibition were fit to equation 1.
Figure 3.

Inhibition of recombinant eEF-2K activity by compounds. Inhibitor dose response assays were performed with 5 nM eEF-2K enzyme at various concentrations of the inhibitor, in the presence of 50 μM free Ca2+, 200 nM CaM and 10 μM [γ−32P]ATP, against 150 μM peptide substrate as described under the experimental section. The inhibitor was incubated with eEF-2K for 30 min at 30 °C before initiating the assay with peptide and [γ−32P]ATP. Data are plotted as the percentage of kinase activity as a function of inhibitor concentration, and fit to equation 1. (A) Compound 6. (B) Compound 9.
2.3.2. Cellular activity against eEF-2K
Chen et al. have reported that treatment of various cell lines with compound 6 inhibits eEF-2K activity in a dose-dependent manner, as indicated by reduced levels of phosphorylated eEF-2 (Thr-56).16 This was observed in human prostate cancer (PC3), human cervical cancer (HeLa), human non-small cell lung cancer (H460 and H1299), and rat glial (C6) cells. Their results are most prominent under conditions of nutrient deprivation (Hanks’ Balanced Salt Solution, HBSS), which is known to activate eEF-2K presumably through inhibition of the nutrient-sensitive mTOR pathway.16,24 To determine if similar inhibition was observed in breast cancer cells, compounds 6 (IC50 = 0.42 μM), 9 (IC50 = 0.93 μM), and 12 (IC50 > 25 μM) were assayed in the MDA-MB-231 cell line. The level of phosphorylated eEF-2 (Thr-56) was used to estimate the activity of eEF-2K, and as indicated in Figure 4, results of cellular inhibition are similar to the in vitro kinase assay data. In low-glucose (5.6 mM) media, eEF-2K activity is significantly reduced by the addition of compound 6, to a lesser extent by compound 9, and is unaffected by compound 12 (Figure 4). To activate the AMPK-eEF-2K axis, 2-DOG was used. 2-DOG inhibits glycolysis and its production of ATP, thus activating AMPK, which in turn activates eEF-2K through phosphorylation at Ser-398.25 Similar inhibitory results are observed when eEF-2K activity is stimulated by 2-DOG treatment (Figure 4), suggesting that compounds 6 and 9 are able to inhibit AMPK-mediated activation of eEF-2K to a notable extent.
Figure 4.

Cellular analysis of compounds against eEF-2K activity. MDA-MB-231 tumorigenic breast cells were treated with 75 μM compound 6, 9, or 12 for 1 h in low-glucose media ± 2-DOG, along with the appropriate controls, as described under the experimental section. One set of samples was stimulated with 25 mM 2-DOG (30 min) to activate the AMPK-eEF-2K axis. Cell lysates were then analyzed by Western blotting using antibodies specific for phospho-eEF-2 (Thr-56), total eEF-2, total eEF-2K and pan-Actin as described under the experimental section.
3. Summary
In summary, a series of pyrido[2,3-d]pyrimidine-2,4-dione derivatives 6–16 was synthesized and screened against eEF-2K inhibitory activity. Ethyl group at R1, CONH2 at R2 and cyclopropyl at R3 are found to be optimal, and the pyridine ring and CONH2 group in 6 are essential for the activity of the studied compounds. Compounds 6 (IC50 = 420 nM) and 9 (IC50 = 930 nM) are found to be the best molecules in this preliminary series of pyrido[2,3-d]pyrimidine analogs. eEF-2K activity in MDA-MB-231 breast cancer cells is significantly reduced by compound 6, and to a lesser extent by compound 9. Because there is no eEF-2K crystal structure, the knowledge of the structural features from this preliminary series of analogs along with in-depth computer modeling should yield insights to develop a more potent eEF-2K inhibitor.
4. Experimental Section
4.1. Chemistry
Reagents and starting materials including 17a, 17b, and 18f were purchased from various commercial sources and used without further purification unless otherwise stated. All reactions were carried out in oven- or flame-dried glassware under argon. Thin-layer chromatography (TLC) was performed using pre-coated TLC plates with silica gel 60 F254 (EMD) or with aluminum oxide 60 F254 neutral. Flash column chromatography was performed using 40–63 μm (230–400 mesh ASTM) silica gel (EMD). Melting points were recorded on a Thomas Hoover capillary melting point apparatus. NMR spectra were recorded on a Varian MR spectrometer. High-resolution mass and liquid chromatography mass spectral data were obtained at the University of Texas at Austin. Compounds were characterized by NMR and HRMS or LCMS.
4.1.1. General procedure for the synthesis of 6-amino-1-alkyl-3-alkylpyrimidine-2,4(1H,3H)-dione (Compounds 18c–18e)
A mixture of 6-amino-1-alkyl uracil 17 (1 mmol), sodium hydroxide 10–15% (0.3 mL) and 95% EtOH (0.9 mL) was heated under reflux for 15 min and cooled to room temperature. To the reaction mixture was added the corresponding alkylating agent (R1-I) (2 mmol) dropwise. The resulting solution was heated under reflux for 3–8 h. The volatiles were removed under reduced pressure and the residue was partitioned between chloroform/water (2:1 ratio v/v, 9 mL). The organic layer was washed with water, brine, dried over Na2SO4 and evaporated to dryness to give the corresponding 3-substituted 6-amino-1-alkyl-uracils 18c–18e. Unreacted starting material was recovered from the aqueous phase by evaporation to dryness. The obtained crude from organic layer was purified by flash chromatography using 0–10% methanol in dichloromethane.
4.1.1.1. 6-Amino-1-cyclopropyl-3-ethylpyrimidine-2,4(1H,3H)-dione (Compound 18c)
Prepared from 6-amino-1-cyclopropylpyrimidine-2,4(1H,3H)-dione (Compound 17a) and ethyl iodide in 40% yield as a white solid. Mp 186−187 °C. 1H NMR (400 MHz, CDCl3): δ 4.91 (s, 1H), 4.90 (brs, 2H), 3.93 (q, J = 7.0 Hz, 2H), 2.66 (m, 1H), 1.24 (m, 2H), 1.19 (t, J = 7.0 Hz, 3H), 0.99 (m, 2H). LCMS: m/z [M+H]+ 196.2.
4.1.1.2. 6-Amino-1-cyclopropyl-3-propylpyrimidine-2,4(1H,3H)-dione (Compound 18d)
Prepared from 6-amino-1-cyclopropylpyrimidine-2,4(1H,3H)-dione (Compound 17a) and propyl iodide in 53% yield as a white solid. Mp 154–155 °C. 1H NMR (400 MHz, CDCl3): δ 4.90 (s, 1H), 4.87 (brs, 2H), 3.88–3.70 (m, 2H), 2.72–2.60 (m, 1H), 1.61 (m, 2H), 1.24 (m, 2H), 1.01–0.95 (m, 2H), 0.92 (t, J = 7.5 Hz, 3H). LCMS: m/z [M+H]+ 210.2.
4.1.1.3. 6-Amino-1,3-diethylpyrimidine-2,4(1H,3H)-dione (Compound 18e)
Prepared from 6-amino-1-ethylpyrimidine-2,4(1H,3H)-dione (Compound 17b) and ethyl iodide in 42% yield as a white solid. Mp 192−193 °C. 1H NMR (400 MHz, DMSO-d6): δ 6.79 (brs, 2H), 4.64 (s, 1H), 3.82 (q, J = 7.0 Hz, 2H), 3.73 (q, J = 7.0 Hz, 2H), 1.09 (t, J = 7.0 Hz, 3H), 1.02 (t, J = 7.0 Hz, 3H). LCMS: m/z [M+H]+ 184.4.
4.1.2. Synthesis of 6-amino-1-cyclopropyl-3-ethyl-2,4-dioxo-1,2,3,4-tetrahydropyrimidine-5-carbaldehyde (Compound 21)
To a suspension of 18c (20 mg, 0.102 mmol) in DMF (0.6 mL) was added phosphoryl chloride (10 μL, 0.113 mmol) dropwise at below 20 °C. The reaction mixture was stirred for 30 min at room temperature. After evaporation of volatiles, the crude was taken to the next step. To a mixture of the crude in ethanol (1 mL) was added cyanoacetamide (9 mg, 0.102 mmol) and then triethylamine (17 μL, 0.123 mmol) dropwise at 0 °C. The suspension was stirred for 2 hr at room temperature. Solvent was removed and the obtained crude was purified by flash chromatography using 0–5% methanol in dichloromethane to afford 21 as a white solid in 66% yield. Mp 149−150 °C. 1H NMR (400 MHz, CDCl3): δ 10.63 (brs, 1H), 9.93 (s, 1H), 6.19 (brs, 1H), 3.97 (q, J = 7.2 Hz, 2H), 2.67 (m, 1H), 1.33 (m, 2H), 1.21 (t, J = 7.2 Hz, 3H), 1.00 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 188.8, 162.1, 157.9, 150.2, 91.3, 36.2, 23.5, 13.1, 9.1. LCMS: m/z [M+H]+ 224.2.
4.1.3. General procedure for the synthesis and characterization of pyrido[2,3-d]pyrimidine-2,4-dione derivatives (Compounds 6, 7, 9, 12–16).
To a solution of dimethylformamide (0.11 mmol) in dichloromethane (1.5 mL) was added oxalyl chloride (0.11 mmol) dropwise at ca. 10 °C. The reaction mixture was stirred for 30 min at room temperature. Compound 18 (0.10 mmol) was added to the resulting white suspension and stirred for another 30 min. Volatiles were removed and the crude was taken to the next step without further purification. To the mixture of the crude in dry ethanol (1.0 mL) was added an active methylene compound such as cyanoacetamide, cyanothioacetamide, ethyl cyanoacetate, or malononitrile (0.10 mmol) and then triethylamine (0.12 mmol) dropwise at 0 °C. The suspension was allowed to room temperature and stirred for 2 hr. Filtered the resulting precipitate and washed with ethanol. In the case of no precipitation, solvent was removed and the obtained crude was purified by flash chromatography (either neutral Al2O3 or silica gel) using 0–10% methanol in dichloromethane.
4.1.3.2. Synthesis of 7-amino-1-cyclopropyl-3-ethyl-2,4-dioxo-1,2,3,4-tetrahydropyrido[2,3-d]pyrimidine-6-carboxamide (Compound 6).
Isolated in 78% yield as a white solid. Mp >250 °C. 1H NMR (400 MHz, DMSO-d6): δ 8.55 (s, 1H), 8.22 (s, 2H), 7.36 (s, 2H), 3.89 (q, J = 8.0 Hz, 2H), 2.76 (m, 1H), 1.11 (t, J = 8.0 Hz, 3H), 1.08 (m, 2H), 0.86–0.71 (m, 2H). 13C NMR (100 MHz, DMSO-d6): δ 168.8, 161.1, 160.0, 153.6, 151.1, 138.3, 105.4, 99.5, 35.7, 25.8, 13.1, 9.7. HRMS: m/z calcd. for C13H15N5O3 [M]+ 289.1175, found 289.1182.
4.1.3.1. Synthesis of 7-amino-1-cyclopropyl-2,4-dioxo-1,2,3,4-tetrahydropyrido[2,3-d]pyrimidine-6-carboxamide (Compound 7).
Isolated in 81% yield as a yellow solid. Mp >250 °C. 1H NMR (400 MHz, DMSO-d6): δ 11.17 (s, 1H), 8.51 (s, 1H), 8.20 (s, 1H), 7.33 (s, 1H), 2.70 (m, 1H), 1.14–0.98 (m, 2H), 0.85–0.73 (m, 2H). 13C NMR (150 MHz, DMSO-d6): δ 168.8, 161.2, 160.9, 154.9, 151.1, 137.9, 105.0, 100.0, 25.0, 9.4. LCMS: m/z [M+H]+ 262.1.
4.1.3.3. Synthesis of 7-amino-1-cyclopropyl-2,4-dioxo-3-propyl-1,2,3,4-tetrahydropyrido[2,3-d]pyrimidine-6-carboxamide (Compound 9).
Isolated in 66% yield as a white solid. Mp 234−235 °C. 1H NMR (400 MHz, DMSO-d6): δ 8.54 (s, 1H), 8.23 (s, 1H), 7.36 (s, 1H), 3.91–3.72 (m, 2H), 2.77 (m, 1H), 1.64–1.46 (m, 2H), 1.17–1.02 (m, 2H), 0.86 (t, J = 7.4 Hz, 3H), 0.81–0.71 (m, 2H). 13C NMR (100 MHz, DMSO-d6): δ 168.9, 161.1, 160.2, 153.6, 151.3, 138.4, 105.4, 99.4, 42.1, 25.8, 20.8, 11.3, 9.9. HRMS: m/z calcd. for C14H17N5O3 [M]+ 303.1331, found 303.1331.
4.1.3.4. Synthesis of 7-amino-1-cyclopropyl-3-ethyl-2,4-dioxo-1,2,3,4-tetrahydropyrido[2,3-d]pyrimidine-6-carbothioamide (Compound 12)
Isolated in 73% yield as a yellow solid. Mp 222−223 °C. 1H NMR (400 MHz, DMSO-d6): δ 9.74 (d, J = 69.6 Hz, 2H), 8.11 (d, J = 1.9 Hz, 1H), 8.02 (brs, 1H), 3.89 (dd, J = 7.1, 2.0 Hz, 1H), 2.77 (m, 1H), 1.11 (m, 2H), 0.79 (m, 2H). 13C NMR (100 MHz, DMSO-d6): δ 196.5, 160.1, 159.3, 152.7, 151.1, 135.6, 114.0, 99.3, 35.8, 25.8, 13.1, 9.6. LCMS: m/z [M+H]+ 306.0.
4.1.3.5. Synthesis of ethyl 7-amino-1-cyclopropyl-3-ethyl-2,4-dioxo-1,2,3,4-tetrahydropyrido[2,3-d]pyrimidine-6-carboxylate (Compound 13).
Isolated in 65% yield as a white solid. Mp 220−221 °C. 1H NMR (400 MHz, CDCl3): δ 8.85 (s, 1H), 8.23 (s, 1H), 5.73 (s, 1H), 4.35 (q, J = 7.1 Hz, 2H), 4.06 (q, J = 7.1 Hz, 2H), 2.82 (m, 1H), 1.39 (t, J = 7.1 Hz, 3H), 1.27–1.23 (m, 3H), 1.23–1.19 (m, 2H), 0.90–0.83 (m, 2H). 13C NMR (100 MHz, CDCl3): δ 166.3, 161.1, 160.5, 154.8, 151.7, 142.4, 103.1, 101.8, 61.2, 36.8, 26.1, 14.3, 13.2, 9.9. LCMS: m/z [M+H]+ 319.2.
4.1.3.6. Synthesis of 7-amino-1-cyclopropyl-3-ethyl-2,4-dioxo-1,2,3,4-tetrahydropyrido[2,3-d]pyrimidine-6-carbonitrile (Compound 14).
Isolated in 79% yield as an off-white solid. Mp 189−190 °C. 1H NMR (400 MHz, DMSO-d6): δ 8.32 (s, 1H), 7.80 (brs, 2H), 3.86 (m, 2H), 2.74 (m, 1H), 1.37–0.90 (m, 5H), 0.89–0.57 (m, 2H). LCMS: m/z [M+H]+ 272.2.
4.1.3.7. Synthesis of 7-amino-1,3-diethyl-2,4-dioxo-1,2,3,4-tetrahydropyrido[2,3-d]pyrimidine-6-carboxamide (Compound 15).
Isolated in 61% yield as a light yellow solid. Mp 249−250 °C. 1H NMR (400 MHz, DMSO-d6): δ 8.58 (s, 1H), 8.24 (brs, 2H), 7.37 (brs, 2H), 4.17 (q, J = 6.9 Hz, 2H), 3.92 (q, J = 7.0 Hz, 2H), 1.19 (t, J = 6.9 Hz, 3H), 1.13 (t, J = 6.9 Hz, 3H). 13C NMR (100 MHz, DMSO-d6): δ 168.8, 161.5, 159.7, 151.8, 150.3, 138.6, 105.5, 99.1, 36.7, 35.8, 13.1. HRMS: m/z calcd. for C12H15N5O3 [M]+ 277.1175, found 277.1177.
4.1.3.8. Synthesis of 7-amino-1,3-dimethyl-2,4-dioxo-1,2,3,4-tetrahydropyrido[2,3-d]pyrimidine-6-carboxamide (Compound 16).
Isolated in 72% yield as a light yellow solid. Mp >250 °C. 1H NMR (400 MHz, DMSO-d6): δ 8.59 (s, 1H), 8.25 (brs, 2H), 7.37 (brs, 2H), 3.46 (s, 3H), 3.25 (s, 3H). LCMS: m/z [M+H]+ 250.2.
4.1.3.2. Synthesis of 7-amino-1-cyclopropyl-3-methyl-2,4-dioxo-1,2,3,4-tetrahydropyrido[2,3-d]pyrimidine-6-carboxamide (Compound 8).
To a mixture of 7 (30 mg, 0.191 mmol) in DMF (2 mL) was added KOtBu (19.3 mg, 0.172 mmol) and methyl iodide (11 μL, 0.172 mmol) at room temperature. After stirring for 12 h, volatiles were removed under reduced pressure and the residue was purified by flash chromatography (neutral Al2O3) using 0–5% methanol in dichloromethane to obtain 8 in 47% yield as an off-white solid. Mp >250 °C. 1H NMR (400 MHz, DMSO-d6): δ 8.56 (s, 1H), 8.24 (s, 2H), 7.35 (s, 2H), 3.22 (s, 3H), 2.77 (m, 1H), 1.17–1.01 (m, 2H), 0.79 (m, 2H). 13C NMR (150 MHz, DMSO-d6): δ 168.8, 161.1, 160.4, 153.5, 151.6, 138.2, 105.4, 99.5, 27.6, 25.9, 9.6. LCMS: m/z [M+H]+ 276.0.
4.1.3.5. Synthesis of 7-amino-1-cyclopropyl-3-(4-nitrobenzyl)-2,4-dioxo-1,2,3,4-tetrahydropyrido[2,3-d]pyrimidine-6-carboxamide (Compound 10).
To a suspension of sodium hydride (60% dispersion in mineral oil) (15.3 mg, 0.383 mmol) in DMF (2 mL) was slowly added compound 7 (50 mg, 0.191 mmol) at 0 °C. After stirring for 30 min, p-nitrobenzylbromide (83 mg, 0.383 mmol) was added at 0 °C and the reaction mixture was allowed to room temperature and stirred for 12 h. Volatiles were removed under reduced pressure and the residue was purified by flash chromatography (neutral Al2O3) using 0–3% methanol in dichloromethane to yield 61% of 10 as an off-white solid. Mp >250 °C. 1H NMR (400 MHz, DMSO-d6): δ 8.57 (s, 1H), 8.23 (brs, 2H), 8.17 (d, J = 8.7 Hz, 2H), 7.56 (d, J = 8.8 Hz, 2H), 7.38 (brs, 2H), 5.16 (s, 2H), 2.85–2.71 (m, 1H), 1.12–1.06 (m, 2H), 0.90–0.73 (m, 2H). LCMS: m/z [M+H]+ 397.2.
4.1.3.6. Synthesis of 7-amino-3-(4-aminobenzyl)-1-cyclopropyl-2,4-dioxo-1,2,3,4-tetrahydropyrido[2,3-d]pyrimidine-6-carboxamide (Compound 11).
A mixture of 10 (27 mg, 0.068 mmol), SnCl2 (52 mg, 0.273 mmol) in ethyl acetate:methanol (6 ml, 5:6 ratio v/v) was heated at reflux for 12 h. After cooling to room temperature, the solvent was evaporated and the residue was purified by flash chromatography (neutral Al2O3) using 0–5% methanol in dichloromethane to isolate 70% of 11 as an off-white solid. Mp >250 °C. 1H NMR (400 MHz, DMSO-d6): δ 8.55 (s, 1H), 8.21 (brs, 2H), 7.36 (brs, 2H), 7.02 (d, J = 8.3 Hz, 2H), 6.45 (d, J = 8.4 Hz, 2H), 4.98 (s, 2H), 4.86 (s, 2H), 2.77 (m, 1H), 1.09 (m, 2H), 0.77 (m, 2H). LCMS: m/z [M+H]+ 367.1.
4.2. In vitro recombinant kinase inhibition assays
Recombinant human eEF-2K (expressed in bacteria in the absence of λ-phosphatase) was purified as described earlier.22,23 Inhibitor dose response assays were performed in Buffer A [5 mM HEPES (pH 7.5), 2 mM DTT, 0.15 μM BSA, 50 mM KAcO, 100 mM EGTA, 150 μM CaCl2, 200 nM CaM and 10 mM MgCl2], against 150 μM peptide substrate using 5 nM eEF-2K enzyme, 10 μM [γ−32P]ATP (100–1000 cpm/pmol) and various concentrations of the inhibitor, in a final reaction volume of 100 μL. The reaction mixture containing the inhibitor and eEF-2K was incubated at 30 °C for 30 min before initiating the assay with peptide and [γ−32P]ATP. At set time points, 10 μL aliquots were taken and spotted onto P81 cellulose filters (Whatman, 2 × 2 cm). The filter papers were then washed thrice with 50 mM phosphoric acid (15 min each wash), once with acetone (15 min), and dried. The amount of labeled peptide associated with each paper was determined by measuring the cpm on a Packard 1500 scintillation counter. Kinase activity in each case was determined by calculating the rate of phosphorylation of the peptide (μM/s), and dose-response curves for data conforming to inhibition were fit to equation 1.
| (Equation 1) |
The parameters are defined as follows: , apparent rate; , apparent rate in the absence of the inhibitor (maximum rate); , apparent rate at saturating concentration of the inhibitor; [i], concentration of the inhibitor; IC50, inhibitor concentration required to achieve half maximal apparent rate. When is converted to percentage maximal activity, is taken as 100.
4.3. Cellular kinase inhibition assays
4.3.1. Cell lines and culture conditions
The human breast cancer cell line MDA-MB-231 was obtained from American Type Culture Collection (Manassas, VA). Cells were cultured in DMEM/F12 supplemented with 10% FBS, 50 units/mL penicillin and 50 μg/mL streptomycin. Cell cultures were maintained at 37 °C in a humidified incubator containing 5% CO2. All cell culture reagents were from Invitrogen or Sigma-Aldrich.
4.3.2. Treatment of cells with stimuli and inhibitors
MDA-MB-231 breast cancer cells were seeded in 6-well plates, and treated when ~ 80% confluent. To activate the AMPK-eEF-2K axis, cells were treated with 2-DOG as follows. Cells were washed with phosphate buffered saline (PBS, pH 7.4), and then pre-incubated in low-glucose (5.6 mM) DMEM supplemented with 10% FBS for 2 h. Then in the same low-glucose media, cells were initially treated with 75 μM compound 6, 9, or 12 for 30 min, followed by incubation ±25 mM 2-DOG in the presence of the compounds for an additional 30 min. Treatments with the appropriate DMSO controls were also performed.
4.3.3. Cell lysis and western blot analysis
Following treatments, cells were washed twice in ice-cold PBS (pH 7.4), and lysed in ice-cold Buffer B [50 mM HEPES (pH 7.4), 150 mM NaCl, 1.5 mM MgCl2, 1% Triton X-100, 1 mM EGTA, 100 mM NaF, 10 mM Na pyrophosphate, 1 mM Na3VO4 and 10% glycerol], supplemented with PhosSTOP Phosphatase Inhibitor Cocktail (Roche Diagnostics, Indianapolis, IN), complete EDTA-free Protease Inhibitor Cocktail (Roche Diagnostics) and Halt Protease and Phosphatase Inhibitor Cocktail (Thermo Fisher Scientific). Lysates were subjected to one freeze-thaw cycle, and then clarified by centrifugation at 15,000 × g for 15 min. Total protein concentration for each sample was determined by Bradford assay (Bio-Rad Laboratories, Hercules, CA). Western blotting was carried out with 5–10 μg protein. Equal amounts of protein from cell lysate samples were resolved by 10% SDS-PAGE and then transferred to Amersham Hybond-P PVDF membranes (GE Healthcare, Piscataway, NJ). Membranes were blocked with 5% BSA or non-fat dry milk in Tris-buffered saline/Tween 20 (TBST) for 1 h, and then incubated with primary antibodies at 4 °C overnight, according to the manufacturer’s protocol. The membranes were washed with TBST and incubated with the appropriate HRP-conjugated secondary antibody at room temperature for 1 h. After washing with TBST, chemiluminescence detection was performed with Amersham ECL Plus™ Western Blotting Detection Reagents (GE Healthcare).
4.3.4. Commercial antibodies
eEF-2K (C-term) (EEF-2K) antibody RabMAb® (#1754–1, 1:3000) was purchased from Epitomics (Burlingame, CA). Phospho-eEF2 (Thr56) Antibody (#2331, 1:3000) was obtained from Cell Signaling Technology (Danvers, MA). Anti-eEF2 (C-term) (#07–1382, 1:25000) and Anti-Actin Antibody, clone C4 (#MAB1501, 1:40000) were from Millipore (Billerica, MA). Bio-Rad Laboratories supplied Goat Anti-Rabbit IgG (H+L)-HRP Conjugate (#172–1019, 1:2000) and Goat Anti-Mouse IgG (H+L)-HRP Conjugate (#172–1011, 1:2000) secondary antibodies.
Supplementary Material
Acknowledgements
This research work is supported in part by the grants from the National Institutes of Health (GM59802), and the Welch Foundation (F-1390). RE acknowledges Cancer Prevention and Research Institute of Texas (CPRIT) postdoctoral training award (RP101501).
Abbreviations:
- eEF-2K
eukaryotic elongation factor-2 kinase
- 2-DOG
2-deoxy-D-glucose
- DMF
dimethylformamide
- DMSO
dimethylsulfoxide
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supporting Information
The NMR spectra and High Resolution Mass Spectrometry (HRMS) data for some of the compounds 18c–18e, 21, and 6–16 are available at http://
References and notes
- 1.Ryazanov AG; Shestakova EA; Natapov PG Nature, 1988, 334, 170. [DOI] [PubMed] [Google Scholar]
- 2.Redpath NT; Price NT; Severinov KV; Proud CG Eur. J. Biochem 1993, 213, 689. [DOI] [PubMed] [Google Scholar]
- 3.Proud CG Biochem. J 2007, 403, 217. [DOI] [PubMed] [Google Scholar]
- 4.Leprivier G; Remke M; Rotblat B; Dubuc A; Mateo AR; Kool M; Agnihotri S; El-Naggar A; Yu B; Somasekharan SP; Faubert B; Bridon G; Tognon CE; Mathers J; Thomas R; Li A; Barokas A; Kwok B; Bowden M; Smith S; Wu X; Korshunov A; Hielscher T; Northcott PA; Galpin JD; Ahern CA; Wang Y; McCabe MG; Collins VP; Jones RG; Pollak M; Delattre O; Gleave ME; Jan E; Pfister SM; Proud CG; Derry WB; Taylor MD; Sorensen PH Cell 2013, 153, 1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Parmer TG; Ward MD; Yurkow EJ; Vyas VH; Kearney TJ; Hait WN Br. J. Cancer 1999, 79, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bagaglio DM; Hait WN Cell Growth Differ. 1994, 5, 1403. [PubMed] [Google Scholar]
- 7.Nilsson A; Nygard O Biochim. Biophys. Acta 1995, 1268, 263. [DOI] [PubMed] [Google Scholar]
- 8.Ashour AA; Abdel-Aziz AA; Mansour AM; Alpay SN; Huo L; Ozpolat B Apoptosis 2014, 19, 241. [DOI] [PubMed] [Google Scholar]
- 9.Hait WN; Wu H; Jin S; Yang JM Autophagy, 2006, 2, 294. [DOI] [PubMed] [Google Scholar]
- 10.(a) Gschwendt M; Kittstein W; Marks F FEBS Lett 1994, 338, 85. [DOI] [PubMed] [Google Scholar]; (b) Soltoff SP Trends Pharmacol. Sci 2007, 28, 453. [DOI] [PubMed] [Google Scholar]; (c) Parmer TG; Ward MD; Hait WN Cell Growth Differ. 1997, 8, 327. [PubMed] [Google Scholar]
- 11.Cho SI; Koketsu M; Ishihara H; Matsushita M; Nairn AC; Fukazawa H; Uehara Y Biochim. Biophys. Acta 2000, 1475, 207. [DOI] [PubMed] [Google Scholar]
- 12.Arora S; Yang JM; Kinzy TG; Utsumi R; Okamoto T; Kitayama T; Ortiz PA; Hait WN Cancer Res. 2003, 63, 6894. [PubMed] [Google Scholar]
- 13.Devkota AK; Tavares CD; Warthaka M; Abramczyk O; Marshall KD; Kaoud TS; Gorgulu K; Ozpolat B; Dalby KN Biochemistry 2012, 51, 2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lockman JW; Reeder MD; Suzuki K; Ostanin K; Hoff R; Bhoite L; Austin H; Baichwal V; Adam Willardsen J Bioorg. Med. Chem. Lett 2010, 20, 2283. [DOI] [PubMed] [Google Scholar]
- 15.Devkota AK; Warthaka M; Edupuganti R; Tavares CD; Johnson WH; Ozpolat B; Cho EJ; Dalby KN J. Biomol. Screen 2014, 19, 445. [DOI] [PubMed] [Google Scholar]
- 16.Chen Z; Gopalakrishnan SM; Bui MH; Soni NB; Warrior U; Johnson EF; Donnelly JB; Glaser KB J. Biol. Chem 2011, 286, 43951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nieto MI; Balo MC; Brea J; Caamaño O; Cadavid MI; Fernández F; Mera XG; López C; Rodríguez-Borges JE Bioorg. Med. Chem 2009, 17, 3426. [DOI] [PubMed] [Google Scholar]
- 18.Hirota K; Kitade Y; Senda SJ Heterocyclic. Chem 1985, 22, 345. [Google Scholar]
- 19.Hartshorn MJ; Verdonk ML; Chessari G; Brewerton SC; Mooij WT; Mortenson PN; Murray CW J. Med. Chem 2007, 50, 726. [DOI] [PubMed] [Google Scholar]
- 20.Korb O; Stützle T; Exner TE J. Chem. Inf. Model 2009, 49, 84. [DOI] [PubMed] [Google Scholar]
- 21.Devkota AK; Edupuganti R; Yan C; Shi Y; Jose J; Wang Q; Kaoud TS; Cho EJ; Ren P; Dalby KN manuscript in prepartion. [DOI] [PMC free article] [PubMed]
- 22.Tavares CD; O’Brien JP; Abramczyk O; Devkota AK; Shores KS; Ferguson SB; Kaoud TS; Warthaka M; Marshall KD; Keller KM; Zhang Y; Brodbelt JS; Ozpolat B; Dalby KN Biochemistry 2012, 51, 2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Abramczyk O; Tavares CD; Devkota AK; Ryazanov AG; Turk BE; Riggs AF; Ozpolat B; Dalby KN Protein Expr. Purif 2011, 79, 237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Proud CG; Wang X; Patel JV; Campbell LE; Kleijn M; Li W; Browne GJ Biochem. Soc. Trans 2001, 29, 541. [DOI] [PubMed] [Google Scholar]
- 25.Browne GJ; Finn SG; Proud CG J. Biol. Chem 2004, 279, 12220. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.