

Abstract
Background
More and more recent studies have clearly shown that long non-coding RNA (lncRNA) should be considered as a fundamental part of the ceRNA network, mainly because lncRNA can act as miRNA sponges to regulate the protein-coding gene expression. Nevertheless, it is still not clear how lncRNA-mediated ceRNAs function in cervical squamous cell carcinoma (CESC). Moreover, information about the ceRNA regulatory mechanism is also remarkably limited; thus, prediction of CESC prognosis using ceRNA-related information remains challenging.
Material/Methods
We collected 306 RNA (lncRNA, miRNA, and mRNA) expression profile datasets obtained from cervical squamous cancer tissues plus 3 more from adjacent cervical tissues via the TCGA database. Subsequently, we constructed a lncRNAs-miRNAs-mRNAs CESC ceRNA network, and Gene Ontology (GO) analysis was carried out.
Results
We identified a total of 30 DElncRNAs, 70 DEmiRNAs, and 1089 DEmRNAs in CESC. Subsequently, to reveal the expression patterns of dysregulated genes, weighted gene co-expression network analysis was carried out, resulting in 3 co-expression modules with significantly related clinical properties. The constructed aberrant lncRNAs-miRNAs-mRNAs CESC ceRNA network was composed of 17 DEmiRNAs, 5 DElncRNAs, and 7 DEmRNAs. Moreover, the survival analysis was performed for DElncRNAs, DEmiRNAs, and DEmRNAs.
Conclusions
The present study shows the involvement of the lncRNA-related ceRNA network in the pathogenesis of CESC. We believe the newly generated ceRNA network will provide more insights into the lncRNA-mediated ceRNA regulatory mechanisms.
MeSH Keywords: Genes, Neoplasm; Genes, rRNA; Genes, vif; Uterine Cervical Neoplasms
Background
As one of the most common malignant tumors with incidence second only to breast cancer, cervical cancer is associated with high mortality and morbidity in women [1]. According to the World Health Organization, each year approximately 530 000 women are estimated to have developed cervical cancer, and 270 000 women die due to this disease [2]. Globally, 10–15% of cancer-associated deaths in women are caused by cervical squamous cell carcinoma (CESC) [3,4]. After being diagnosed with this disease, the status of most of cervical cancer patients (80%) has already deteriorated into the invasive stages. A slowly decreasing average age at diagnosis was documented. More seriously, distant metastasis of cervical cancer still contributes to the high morbidity and mortality [5–9]. Stage IVB cervical cancer has a 5-year survival rate of about 9% [10]. At present, due to multiple reasons, there is still no effective treatment for cervical cancer. Therefore, finding new treatments is urgent and necessary, and a better understanding of the specific CESC mechanisms may boost treatment efficiency and minimize the adverse effects typically observed when applying standard treatment.
The recent breakthrough of next-generation sequencing (NGS) technology has provided us with an excellent opportunity to illustrate the complexity of the human genome. In the human transcriptome, non-coding RNAs (ncRNAs), including micro-RNAs (miRNAs) and long non-coding RNAs (lncRNAs), represent about 98% of the total transcripts, and it has been well documented that they have important biological functions [11]. As a consequence, it has become one of the most intriguing challenges to explore the RNA for cancer biologists today. More importantly, the characteristics of lncRNAs can be utilized as potential new biomarkers and for designing targeted drugs. Over the past decade, the competing endogenous RNA (ceRNA) hypothesis introduced by Salmena et al. has attracted consideration attention. According to this hypothesis, in addition to miRNAs, multiple molecules also participated in post-transcriptional regulatory networks; other ncRNAs harboring miRNA response elements (MREs), such as lncRNAs, cirRNAs, as well as some pseudogenes, can likewise play ceRNA to suppress miRNA and interfere with mRNAs [12]. Following this hypothesis, an increasing amount of evidence has indicated that, many non-coding RNA (lncRNA) that were previously considered as transcriptional “desert island” actually are master regulators affecting the expression levels of numerous target genes [13,14]. Multiple studies coming after the discovery of ceRNA regulation role have reported that lncRNA, mRNA, and other RNA have the ability to suppress miRNA function by acting as natural miRNA sponges. LncRNA can mimic mRNA to compete for shared miRNA [12,15,16].
Over 20 years ago, National Cancer Institute (NCI) and the National Human Genome Research Institute (NHGRI) launched the Cancer Genome Atlas (TCGA) project. This gigantic project has so fat provided us with comprehensive cancer genomic information and greatly advanced our diagnostic methods; therefore, new therapy standards was set and more effective strategies of disease prevention were invented. Meanwhile, the latest progress of bioinformatics has also promoted the integrated multidimensional analyses which provide novel insights into the cancerous pathogenic mechanism [17]. Weighted gene co-expression network analysis (WGCNA), one of systems computational approaches, is an easy way to functional correlate genes with similar expression patterns [18]. It also can be extended to uncover highly correlated molecules and group separate modules, revealing the connection between hub nodes and external sample traits. WGCNA based algorithms are widely used in genomics of human diseases, animals and plants. In cancer research filed, the power of WGCNA is reflected by many important genes, biological processes, and candidate biomarkers identified by it. However, while WGCNA is relatively straightforward and TCGA database is publicly available, few researchers have performed WGCNA using TCGA data for understanding cervical cancer.
To our knowledge, this is the first WGCNA based study that utilizes the data collected from TCGA to study the cervical cancer specific lncRNA.
Material and Methods
Collection of Data
All RNA-seq data and clinical information used were download from TCGA database and filtered the following criteria: i) clinical data should contain enough details, including diagnosis age, race, clinical stage, neoplasm histologic grade, HPV status, menopause status, lymphovascular involvement, TNM stage; ii) clinical data included follow-up time. In total, 306 CESC tumor tissues and 3 adjacent normal tissues were analyzed in this study. Because this study strictly followed the ethics guidelines recommended by TCGA ethics committee, the approval of an ethics committee was not required.
Study design and data processing
Data preparation: i) gene expression matrix was merge by RNA and miRNA sequencing data; ii) converted ensemble ID to gene symbol in gene expression matrix; iii) extract clinical data of CESC patients; iv) outlier removal with hierarchical clustering.
Data analysis: i) used R package of edgeR to screen DElncRNAs, DEmiRNAs and DEmRNAs; ii) then WGCNA was utilized to detect module for DEmRNAs; iii) screen genes which interaction score greater than 0.9 in modules by string database; iv) utilized Cytoscape to identify hub genes of DEmRNAs and visualization; v) ceRNA network construction; vi) survival analysis. A detailed flow diagram is present to illustrate the design of this study (Figure 1).
Figure 1.

Workflow of the data preparation and analysis.
DElncRNAs, DEmiRNAs, and DEmRNAs Screening
We merged gene expression of tumor and normal tissues to generate gene expression matrix based on RNA and miRNA sequencing data from TCGA database. Then used the R package of “edgeR” to screen the DElncRNAs after removing low expression of genes (expression means <1) between CESC tumor tissues and adjacent normal tissues. The threshold of differential expression was set as adjusted p value <0.05 and |log2fold change (FC)| >2. Furthermore, the same condition was used to screen DEmiRNAs and DEmRNAs.
Identifying hub DEmRNAs
The construction of weighted gene co-expression network
R package of “WGCNA” was used to detect scale-free gene modules for DEmRNAs. First, to improve the accuracy and confidence of co-expression network construction, the outlier samples were identified and removed. Soft threshold power parameter of the network construction was set to 3 in order to make the weighted adjacency matrix scale-free for all pair-wise genes, so it only highlights significant correlations and weak correlations were ignored. In the second step, the obtained matrix was transformed, resulting in a topological overlap matrix (TOM), and then, by using dynamic tree cut, we calculated the dissimilarity of module eigengenes (1-TOM) to identify hierarchical clustering genes and modules. For this, the minimum size cut-off value of each module was set to 30, and modules which below 0.25 would be merged.
Construction of PPI network
To obtain protein-protein interaction (PPI) information, Search Tool for the Retrieval of Interacting Genes (STRING) (https://string-db.org/) was used. STRING is a widely used PPI network database to discover CESC-related DEGs distributing in different modules. In addition, STRING database was also used to evaluate the interrelationship of DEmRNAs. Data are visualized using Cytoscape software. We set the threshold for interactions score >0.9 as significant.
Selection of hub modules and their functional enrichment
For hub modules selection using our PPI network, a Cytoscape plug-in Molecular Complex Detection (MCODE) was installed and used here. Basic cut-off criteria are: degree=2, node score=0.2, k-score=2 and maximal.depth=100. Then, to perform functional analysis, selected genes were submitted to The Database for Annotation, Visualization and Integrated Discovery (DAVID) (https://david.ncifcrf.gov). Two Gene Ontology (GO) analysis were performed: Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways analysis and biological process (BP) analysis.
Clinical feature associated modules
Clinical features are usually correlated with gene expression modules. To explore their relationship, the Person’s correlation of all modules at 10 different conditions were calculated and filtered with relation cut-off and p value, and their association was displayed as heatmap. The conditions are diagnosis age, race, clinical stage, neoplasm histologic grade, HPV status, menopause status, lymphovascular involvement, pathology T/N/M stages.
ceRNA network construction
The construction of the ceRNA network, consisting of hub dysregulated genes associated in CESC, includes the following 5 steps. i) target lncRNAs prediction: to coordinate the DElncRNAs and DEmiRNAs, miRcode (http://www.mircode.org) database was used for predicting the lncRNAs-miRNAs interactions and 2 types of DE RNAs were mapped accordingly. ii) prediction of target miRNAs of mRNAs: miRDB (http://www.mirdb.org), miRTarBase (http://mirtarbase.mbc.nctu.edu.tw) and TargetScan (http://www.targetscan.org) database to predict to predict the target miRNAs of mRNAs; iii) prediction of mRNAs targeted by hub mRNAs: intersection target mRNAs and hub mRNAs; iv) hub DEmRNAs were further aligned with the predicted mRNAs/hub mRNAs interactions; v) ceRNA network construction: lncRNA-miRNA-mRNA interactions that are CESC-specific were used in the ceRNA network construction. The visualization of the final network was done using Cytoscape 3.6.1 software.
Survival analysis
To further characterize the constructed network and identify the network signatures, the clinical data were integrated, and we labelled each node of the ceRNA network with the life curve of those samples. R package “survival” was used to conduct the survival analysis using a standard Kaplan-Meier univariate curve. If a molecule had a p value <0.05, we considered had statistical significance.
Results
DElncRNAs, DEmiRNAs and DEmRNAs
A large gene expression dataset of tumor tissues (306) and adjacent normal tissues (3) was analyzed in this study. We used R package of “edgeR” to obtain DElncRNAs by selecting adjusted p value <0.05 and |log2FC| >2 as the cutoff criteria. A total of 30 DElncRNAs were effectively identified: 13 of them were upregulated and 17 were downregulated. Furthermore, the same condition was set to obtain DEmiRNAs and DEmRNAs by “edgeR” R package. Finally, we obtained 70 DEmiRNAs, of which 33 were upregulated and 37 were downregulated and 1089 DEmRNAs, of which 532 were upregulated and 557 were downregulated in CESC tumor tissues compared to the adjacent normal tissues. The top 10 DElncRNAs, DEmiRNAs, and DEmRNAs with |log2FC| >2 are listed in Table 1.
Table 1.
The top 10 DElncRNAs, DEmiRNAs, and DEmRNAs.
| Gene symbol | |log2FC| | P value | FDR | Dysregulated stage |
|---|---|---|---|---|
| Top 10 DElncRNA | ||||
| PGM5-AS1 | 8.6326 | 2.41E-93 | 2.28E-90 | Down |
| EMX2OS | 4.25477 | 1.05E-09 | 1.25E-07 | Down |
| MIR100HG | 3.42444 | 2.24E-10 | 3.55E-08 | Down |
| AC093010.3 | 3.11185 | 1.57E-19 | 7.47E-17 | Down |
| LINC01139 | 2.68524 | 0.00067 | 0.015134 | Down |
| MIR205HG | 6.509706 | 0.001037 | 0.020499 | Up |
| AL049555.1 | 5.540556 | 3.75E-05 | 0.001482 | Up |
| AL354766.2 | 5.362799 | 0.002883 | 0.0456 | Up |
| MIR9-3HG | 5.004024 | 0.000879 | 0.017754 | Up |
| U62317.2 | 4.734125 | 0.002055 | 0.034215 | Up |
| Top 10 DEmiRNA | ||||
| hsa-mir-133a-2 | 4.50689 | 2.19E-21 | 2.87E-19 | Down |
| hsa-mir-133a-1 | 4.49975 | 3.40E-22 | 5.57E-20 | Down |
| hsa-mir-145 | 4.28937 | 3.90E-32 | 1.28E-29 | Down |
| hsa-mir-1-1 | 4.11872 | 9.70E-17 | 1.06E-14 | Down |
| hsa-mir-1-2 | 4.10623 | 1.87E-16 | 1.76E-14 | Down |
| hsa-mir-944 | 8.032189 | 0.000672 | 0.005372 | Up |
| hsa-mir-205 | 7.807328 | 1.69E-05 | 0.000231 | Up |
| hsa-mir-203a | 5.991917 | 0.000272 | 0.002586 | Up |
| hsa-mir-31 | 5.640633 | 9.80E-05 | 0.001071 | Up |
| hsa-mir-96 | 5.595909 | 1.07E-07 | 3.19E-06 | Up |
| Top 10 DEmRNA | ||||
| DES | 7.61144 | 3.51E-56 | 3.16E-53 | Down |
| CNN1 | 7.54789 | 2.85E-77 | 1.19E-73 | Down |
| ACTG2 | 7.37523 | 2.16E-54 | 1.60E-51 | Down |
| PI16 | 6.9519 | 1.09E-34 | 3.05E-32 | Down |
| MYH11 | 6.9064 | 6.97E-55 | 5.48E-52 | Down |
| CALML3 | 8.505356 | 0.006408 | 0.040904 | Up |
| PITX1 | 7.588409 | 3.30E-06 | 6.28E-05 | Up |
| KRT15 | 7.476535 | 0.005863 | 0.038285 | Up |
| KRT6A | 7.469412 | 0.002422 | 0.019027 | Up |
| KRT5 | 7.393729 | 0.001477 | 0.012715 | Up |
Identifying hub module DEmRNAs
Weighted gene co-expression network construction
We used R package of “WGCNA” to build the correlation network based on DEmRNAs, selecting the soft threshold power parameter as 3 to ensure a scale-free network. Following the step of using average linkage to group the DEmRNAs whose expression patterns were similar into modules whose different colors representing different modules, we identified a total of 3 modules colored as turquoise, blue, and brown modules. Genes that were not grouped into any modules were marked grey (Figure 2).
Figure 2.

Weighted gene co-expression network construction.
PPI network analysis
PPI networks obtained from STRING database was used to evaluate the interrelationship of DEmRNAs in modules. In this analysis, interactions score >0.9 was set as the threshold for significant PPI; therefore, only protein interactions with highest confidence were included in our network. Eventually, 1066 nodes and 2510 edges were contained by the PPI network of DEmRNAs, in which 884 nodes and 2312 edges were included in the turquoise module, 138 nodes and 128 edges in the blue module, and 44 nodes and 70 edges in the brown module.
Hub modules selection and functional enrichment analysis
Hub modules were identified and visualized by the Cytoscape plug-in MCODE with the settings: node score cut-off=0.2, k-core=2, Degree cut-off=2, and max.depth=100. For the KEGG pathways/Biological process analysis of each module, data were uploaded to the DAVID database. Finally, a total of 3 hub modules were identified. As is shown in Figure 3, the 3 hub module networks consist of 120 nodes and 816 edges. There are 36 nodes and 628 edges exhibited in the turquoise hub module, while the blue module contained 66 nodes and 119 edges. In the brown module, we found 18 nodes and 69 edges.
Figure 3.

Module analysis of PPI network. (A) Turquoise hub module. (B) Blue hub module. (C) Brown hub module.
Except for the grey module, Gene Ontology and KEGG pathway analyses were carried out for 3 modules to explore the involved biological process and pathways with connections to CESC (Figure 4). We found that hub module DEmRNAs in the turquoise module were enriched in biological processes of cell communication and signal transduction and were related to mitotic prometaphase, mitotic prophase, mitotic M-M/G1 phases, M Phase, and DNA Replication pathways. In the blue module, hub module DEmRNAs were enriched in biological processes of cell communication and signal transduction, and were related to integrin family cell surface interactions pathway. In the brown module, hub module DEmRNAs were enriched in biological processes of cell communication, signal transduction, and regulation of nucleobase, nucleoside, nucleotide, and nucleic acid metabolism pathways. For all the hub module DEmRNAs in the 3 modules, biological process results showed that hub module DEmRNAs most enriched in cell communication and signal transduction. KEGG pathways analysis indicated that the hub module DEmRNAs enriched in “cell cycle, mitotic”, “DNS replication”, “mitotic M-M/G1 phases”, “M phase”, and “mitotic prometaphase” pathways. All enrichment pathways had statistical significance (Figure 4).
Figure 4.

Gene Ontology and KEGG pathway analysis.
Module clinical feature associations
Clinical and pathological information were obtained and are listed in Table 2. Using this information, a feature heatmap was constructed for examining the relationship between modules and clinical characteristics. As shown in Figure 5, we noticed that HPV status (r=0.3, p=2e-06) in blue module and HPV status (r=−0.29, p=8e-06) in turquoise module displayed the highest correlation coefficient. However, the remaining clinical features had no significant associations with these 3 modules. Based on our data, we speculate that the DEmRNAs located in these modules are likely to be the critical regulators controlling CESC development.
Table 2.
Clinical and pathological information of CESC patients.
| Clinical feature | Variable | Patients (%) |
|---|---|---|
| Age at diagnosis (years) | >60 | 59 (19.4) |
| ≤60 | 245 (80.6) | |
| Race | White | 209 (68.8) |
| Black or African American | 30 (9.87) | |
| Native Hawaiian or other Pacific islander | 2 (0.61) | |
| Asian | 20 (6.58) | |
| American Indian or Alaska native | 7 (2.3) | |
| Other | 36 (11.84) | |
| Clinical stage | Stage I | 162 (53.29) |
| Stage II | 69 (22.7) | |
| Stage III | 45 (14.8) | |
| Stage IV | 21 (6.91) | |
| Other | 7 (2.3) | |
| Neoplasm histologic grade | g1 | 18 (5.92) |
| g2 | 135 (44.41) | |
| g3 | 118 (38.82) | |
| g4 | 1 (0.33) | |
| gx | 24 (7.89) | |
| Other | 8 (2.63) | |
| HPV status | Indeterminate | 1 (0.33) |
| Negative | 22 (7.24) | |
| Positive | 281 (92.43) | |
| Menopause status | Pre (<6 months since lmp and no prior bilateral ovariectomy and not on estrogen replacement) | 124 (40.79) |
| Post (prior bilateral ovariectomy or >12 mo since lmp with no prior hysterectomy) | 82 (26.97) | |
| Peri (6–12 months since last menstrual period) | 25 (8.22) | |
| Indeterminate (neither pre or postmenopausal) | 3 (0.99) | |
| Other | 70 (23.03) | |
| Lymphovascular involvement | Absent | 71 (23.36) |
| Present | 79 (25.99) | |
| Other | 154 (50.65) | |
| Pathology T stage | T1 | 140 (46.05) |
| T2 | 71 (23.36) | |
| T3 | 20 (6.58) | |
| T4 | 10 (3.29) | |
| Tis | 1 (0.33) | |
| Tx | 17 (5.59) | |
| Other | 45 (14.8) | |
| Pathology N stage | N0 | 133 (43.75) |
| N1 | 60 (19.74) | |
| Nx | 66 (21.71) | |
| Other | 45 (14.8) | |
| Pathology M stage | M0 | 116 (38.16) |
| M1 | 10 (3.29) | |
| Mx | 128 (42.1) | |
| Other | 50 (16.45) |
Figure 5.

Module-trait relationships.
Construction of CESC ceRNA network
We utilized the results of “edgeR” and “WGCNA”, which included DElncRNAs, DEmiRNAs, and hub module DEmRNAs, to construct a competing endogenous RNAs network to further study the regulatory role of lncRNA and miRNA in the pathogenesis of cervical cancer. We constructed a ceRNA network by following these steps:
Constructing DElncRNAs-DEmiRNAs interaction pairs
The relationship between CESC-related lncRNA and miRNA was further investigated here to reveal their potential role. The target miRNAs of DElncRNAs were predicted by mirCode database and matched with DEmiRNAs to construct the DElncRNAs-DEmiRNAs interactions.
As a result, 17 DEmiRNAs related to 5 DElncRNAs and 37 interactions were identified, and all of them are previously reported cancer-associated miRNAs.
Constructing DEmiRNAs-hub DEmRNAs interaction pairs
miRNAs-mRNAs interactions were predicted by miRDB, miRTarBase, and TargetScan database. We obtained hub DEmRNAs by taking the intersection of the target mRNAs of DEmiRNAs and the hub module DEmRNAs. Then, hub DEmRNAs were matched with predicted target mRNAs of DEmiRNAs to construct DEmiRNAs-hub DEmRNAs interactions. Eventually, 7 DEmiRNAs related to 7 DEmRNAs and 7 interactions were identified, and most hub DEmRNAs were associated with cancer.
Merged DElncRNAs-DEmiRNAs-hub DEmRNAs and ceRNA network analysis
Finally, we obtained 5 DElncRNAs, 17 DEmiRNAs, and 7 DEmRNAs, which are likely to be the critical nodes underpinning the whole ceRNA network. All of the above data were integrated to construct the lncRNA-miRNA-mRNA integrated CESC ceRNA network, and network visualization was realized with Cytoscape (Figure 6). A summary of all node information is shown in Table 3. The present network contains many nodes that have been described as molecules associated with cancer, such as FGFR3, CCNB1, E2F1, KLF5, PMAIP1, and ITGA2 (except for HCAR2). A key lncRNA, EPB41L4A-AS1, was identified in our analysis and it was connected with 8 key miRNAs: hsa-mir-141, hsa-mir-383, hsa-mir-183, hsa-mir-205, hsa-mir-106a, hsa-mir-195, hsa-mir-200a, and hsa-mir-429, although no research had demonstrated its function to date. The network hub feature of EPB41L4A-AS1 suggests that it can act as a ceRNA, thus inhibiting target miRNAs and regulating the translation of associated hub genes such as PMAIP1, CCNB1, E2F1, and TGA2. Another lncRNA of interest is MIR205HG, which can bind hsa-mir-183, hsa-mir-143, hsa-mir-145, hsa-mir-31, hsa-mir-205, hsa-mir-383, and hsa-mir-204; therefore, it is likely endogenous and indirectly associated with many targeted transcripts (e.g., CCNB1, KLF5, and HCAR2). In addition, AC132872.1 is a lncRNA on which no research has been reported. We found that AC132872.1 may impact on CESC through its interactions with hsa-mir-195, hsa-mir-106a, hsa-mir-204, hsa-mir-143, hsa-mir-205, hsa-mir-210, and hsa-mir-100, which then regulate the target hub mRNAs E2F1, ITGA2, and FGFR3. There has been abundant research on lncRNAs of CRNDE, most of which are related to the pathological process of cancer. CRNDE is also a key lncRNA in our study, which acts by binding 7 miRNAs (hsa-mir183, hsa-mir-145, hsa-mir-140, hsa-mir-32, hsa-mir-31, hsa-mir-205, and hsa-mir-143), which can indirectly regulate the target hub mRNAs of KLF5 and CCNB1. Moreover, EMX2OS also serves as the central molecule of 6 miRNAs: hsa-mir-183, hsa-mir-143, hsa-mir-182, hsa-mir-31, hsa-mir-210, and hsa-mir-205. Hence, we speculate that EPB41L4A-AS1, EMX2OS, MIR205HG, AC132872.1, and CRNDE are intimately related to the pathogenesis of CESC.
Figure 6.
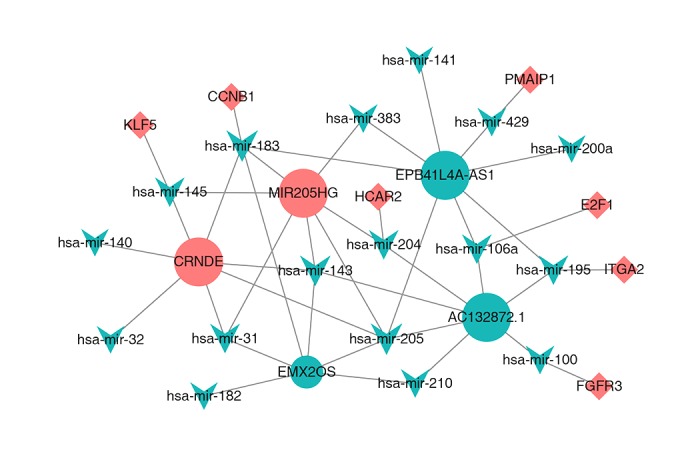
The dysregulated lncRNA-mRNA-miRNA ceRNA network. Rounds denotes lncRNA, and inverted triangles represents miRNA and diamonds represents mRNA. All shapes in red and green stand for up-regulation and down-regulation, respectively.
Table 3.
All node information of ceRNA network.
| Nodes | |log2FC| | FDR | Dysregulated stage |
|---|---|---|---|
| Hub lncRNAs | |||
| MIR205HG | 6.509705742 | 0.020499198 | Up |
| EMX2OS | 4.254768887 | 1.25034E-07 | Down |
| CRNDE | 2.557103416 | 0.014930541 | Up |
| EPB41L4A-AS1 | 2.414220907 | 6.16839E-12 | Down |
| AC132872.1 | 2.191392699 | 5.06E-12 | Down |
| Hub miRNAs | |||
| hsa-mir-205 | 7.807328074 | 0.000231499 | Up |
| hsa-mir-31 | 5.6406334 | 0.001071158 | Up |
| hsa-mir-183 | 5.533759671 | 0.0000159 | Up |
| hsa-mir-141 | 5.082110951 | 0.000000023 | Up |
| hsa-mir-210 | 4.930713592 | 0.000333648 | Up |
| hsa-mir-182 | 4.515097043 | 0.0000834 | Up |
| hsa-mir-145 | 4.28937124 | 1.28E-29 | Down |
| hsa-mir-429 | 4.147416534 | 0.000191854 | Up |
| hsa-mir-204 | 4.077599564 | 0.00000123 | Down |
| hsa-mir-200a | 3.994337067 | 0.000091 | Up |
| hsa-mir-383 | 3.747450302 | 0.004375965 | Down |
| hsa-mir-106a | 3.616312461 | 0.008870532 | Up |
| hsa-mir-100 | 3.219979103 | 4.12E-09 | Down |
| hsa-mir-143 | 3.20692291 | 6.1E-13 | Down |
| hsa-mir-140 | 2.778828132 | 1.68E-23 | Down |
| hsa-mir-32 | 2.246555022 | 0.000218652 | Up |
| hsa-mir-195 | 2.151037526 | 0.0000451 | Down |
| Hub mRNAs | |||
| HCAR2 | 4.881356753 | 0.043430907 | Up |
| FGFR3 | 4.159902463 | 0.026845557 | Up |
| CCNB1 | 4.009891379 | 3.62E-10 | Up |
| E2F1 | 4.006738743 | 0.00000522 | Up |
| KLF5 | 3.575849722 | 0.0000849 | Up |
| PMAIP1 | 3.520103023 | 0.002564182 | Up |
| ITGA2 | 2.965511951 | 0.037274851 | Up |
Survival analysis for genes in the ceRNA network
To further investigate the relationship between the expression pattern and clinical information of the CESC samples, Kaplan-Meier curves were calculated to identify survival-associated key nodes (with p<0.05). We selected 5 hub DElncRNAs, 17 hub DEmiRNAs, and 7 hub DEmRNAs from our ceRNA network to perform survival analysis. The results revealed that the expression levels of E2F1 and hsa-mir-204 were positively correlated with overall survival time, as patients with higher expression of them also had long survival time. All results of the survival analyses are shown in Figure 7.
Figure 7.

Kaplan-Meier survival curves of ceRNA network for the overall survival in CESC.
Discussion
Long non-coding RNAs (long ncRNAs, lncRNA) are defined as transcripts longer than 200 nucleotides that are not translated into protein. Nevertheless, much research in recently years has shown that some lncRNAs are involved in the regulation of multiple processes in the cell.
In this study, a total of 5 lncRNAs (MIR205HG, EMX2OS, CRNDE, EPB41L4A-AS1, and AC132872.1) were characterized as hub lncRNAs. According to the results of our integrated analysis, these lncRNAs were differentially enriched and are located in the constructed ceRNA network. A more careful inspection of the network pinpointed EPB41L4A-AS1 as the network center, which probably can indirectly regulate the target genes of 8 competing miRNAs (hsa-mir-383, hsa-mir-183, hsa-mir-205, hsa-mir-106a, hsa-mir-141, hsa-mir-195, hsa-mir-200a, and hsa-mir-429). Although EPB41L4A-AS1 plays a quite important role in the ceRNA network, no research has been reported yet. We found that CRNDE is also a key lncRNA, acting by binding 7 miRNAs (hsa-mir183, hsa-mir-145, hsa-mir-140, hsa-mir-32, hsa-mir-31, hsa-mir-205, and hsa-mir-143). Some previous reports have shown that CRNDE can deregulate gene expression in different types of tumors, and thus it has the potential to be used as diagnostic and prognostic biomarkers [19]. Cheng et al. had found that CRNDE was significantly increased in bladder cancer, and overexpression of CRNDE was positively correlated with advanced TNM stage of bladder cancer patients [20]. Sun et al. found that CRNDE negatively regulates the expression of miR-384 and is positively correlated with pleiotrophin in papillary thyroid cancer tissues. Moreover, the CRNDE/miR-384/pleiotrophin axis may play an important role in the regulation of papillary thyroid cancer progression [21].
Our WGCNA analysis demonstrated that highly expressed CRNDE acts as miRNA competitors inhibiting the expression of targets genes categorized into the turquoise and blue module (e.g., CCNB1 and KLF5), which are 2 genes involved in our ceRNA network. Taking clinical characteristics into consideration, the WGCNA turquoise and blue modules further demonstrated that they had the high correlation coefficient in the CESA of HPV status (r=−0.29, p=8e-06; r=0.3, p=2e-06). Thus, we hypothesized that knocking down the expression of CRNDE can transform HPV status into negative in CESC. Nevertheless, given that the survival analysis for CRNDE showed no significant p values, these results need to be confirmed by further research to elucidate its effect on cervical cancer.
In addition, MIR205HG has the key lncRNA interrelated with 7 key miRNAs in the ceRNA network. Di et al. showed that downregulation of MIR205HG expression can lead to significantly reduced clonogenic activity of head and neck cancer cells, thus lowering the rate of cell proliferation and cell migration. MIR205HG can compete with endogenous miR-590-3p in vivo to increase the expression of cyclin B, cdk1, and YAP, and these genes are upregulated in head and neck of squamous cell carcinoma patients, with poor prognosis [22]. We found that highly expressed MIR205HG can compete with 7 DEmiRNAs (hsa-mir-183, hsa-mir-145, hsa-mir-31, hsa-mir-143, hsa-mir-205, hsa-mir-383, and hsa-mir-204) to regulate target (CCNB1, KLF5, HCAR2) gene expression. Nonetheless, the survival analysis for MIR205HG showed no significant p values, and these findings require further research to explore its cervical cancer-related functions. In the construction of the ceRNA network, another 3 key DElncRNAs – EMX2OS, EPB41L4A-AS1, and AC132872.1 – were not relate to clinical outcome of CESC patients.
In comparison with miRNAs, a category of small ncRNA molecules whose role in various human disease have been well documented, but far fewer lncRNAs have been comprehensively studied in terms of their disease-associated function. In the present study, by constructing a novel ceRNA network, we were able to clearly define the relationship of lncRNAs and miRNAs. A total of 17 miRNAs were identified and will be subject to further investigation: hsa-mir-141, hsa-mir-383, hsa-mir-183, hsa-mir-205, hsa-mir-106a, hsa-mir-195, hsa-mir-200a, hsa-mir-429, hsa-mir-140, hsa-mir-32, hsa-mir-31, hsa-mir-143, hsa-mir-145, hsa-mir-204, hsa-mir-182, hsa-mir-210, and hsa-mir-100. The mir-143, mir-183, and mir-205 are of particular importance in the ceRNA network core. Zhao et al. reported that microRNA-143/145 cluster is downregulated in hepatitis B virus-associated hepatocellular carcinoma, thus serving as a potential biomarker for tumorigenesis in patients with chronic hepatitis B [23]. Supic et al. reported that mir-183 can be a potential progression marker and biomarker for tongue carcinoma [24]. Yang proved that downregulating miR-205 can suppress the tumorigenesis of cervical cancer [25]. Pang et al. showed that miR-205 serves as a prognostic factor and suppresses proliferation in human cervical cancer by targeting insulin-like growth factor [26]. Moreover, Yue et al. confirmed that miR-205 mediates the inhibition of cervical cancer cell proliferation [27]. However, the result of survival analysis showed that clinical information of CESC patients is not significantly related with mir-143, mir-183, and mir-205. More work is needed to verify the significance or specific role of these aberrant miRNAs in CESC tumorigenesis. Notably, in the new ceRNA network, several protein coding genes (e.g., CCNB1 [28], KLF5 [29], E2F1 [30,31], and FGFR3 [32]) have been recently reported to be key cervical cancer oncogenes affecting disease development and progression. Furthermore, our survival analysis results suggest that low expression of E2F1 can result in a poor CESC outcome.
The KEGG pathways analysis enabled us to identify 5 enriched pathways: Mitotic Prometaphase, M Phase, Cell Cycle, Mitotic M-M/G1 phases, and DNA Replication.
Currently, although an increasingly effort has been made to study the lncRNAs-miRNAs-mRNAs network, there have been few investigations focusing on ncRNA-mediated human diseases. Our study is the first to use WGCNA to establish a co-expression network of lncRNAs-miRNAs-mRNAs in CESC. However, some limitations also exist in our analysis. Because only limited samples were used and the sample information was not complete, we were not able to conduct any subtypes analysis. Therefore, further experimental work is very much needed to validate our results and elucidate the functions of ceRNA in CESC.
In conclusion, an advanced integrative network-based systems biology approach was used in this study to characterize both non-coding and coding RNA expression profiles generated from cervical cancer samples and controls. Some hub genes of potential importance were selected using the WGCNA algorithm, and the newly constructed lncRNA-miRNA-mRNA ceRNA interactive network will likely provide a basis for further investigation of the regulatory mechanism of CESC.
Conclusions
In summary, we provided insights into the lncRNA-mediated ceRNA regulatory mechanisms by constructing a lncRNA-miRNA-mRNA interactive network. This may contribute to the discovery of molecular mechanisms underlying the initiation and development of cervical cancer.
Footnotes
Source of support: This work was supported by the National Natural Sciences Foundation of China (no. 81673848 and 81603520), the Natural Sciences Foundation of Guangdong Province (no. 2016A030310084 and 2017A030313658), the Science and Technical Plan of Guangzhou, Guangdong, China (no. 201804010213 and 201707010100), the Administration of Traditional Medicine of Guangdong Province (no. 20161063 and 20181068), the Fundamental Research Funds for the Central Universities (no. 21616315), and the Medical Scientific Research Foundation of Guangdong Province (no. 2016111221315850)
References
- 1.Munksgaard PS, Blaakaer J. The association between endometriosis and gynecological cancers and breast cancer: A review of epidemiological data. Gynecol Oncol. 2011;123(1):157–63. doi: 10.1016/j.ygyno.2011.06.017. [DOI] [PubMed] [Google Scholar]
- 2.World Health Organization. Comprehensive cervical cancer prevention and control: A healthier future for girls and women (2013) 2015. Tersedia di: http://appswhoint/iris/bitstream/10665/78128/3/9789241505147_eng pdf.
- 3.Ojesina AI, Lichtenstein L, Freeman SS, et al. Landscape of genomic alterations in cervical carcinomas. Nature. 2014;506(7488):371–75. doi: 10.1038/nature12881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.zur Hausen H. Papillomaviruses in the causation of human cancers – a brief historical account. Virology. 2009;384(2):260–65. doi: 10.1016/j.virol.2008.11.046. [DOI] [PubMed] [Google Scholar]
- 5.Bresadola F, Terrosu G, Uzzau A, Bresadola V. Distant metastases from cervical esophagus cancer. J Otorhinolaryngol Relat Spec. 2001;63(4):229–32. doi: 10.1159/000055747. [DOI] [PubMed] [Google Scholar]
- 6.Kulkarni PR, Rani H, Vimalambike MG, Ravishankar S. Opportunistic screening for cervical cancer in a tertiary hospital in Karnataka, India. Asian Pac J Cancer Prev. 2013;14(9):5101–5. doi: 10.7314/apjcp.2013.14.9.5101. [DOI] [PubMed] [Google Scholar]
- 7.Wang IT, Chou SC, Lin YC. Zoledronic acid induces apoptosis and autophagy in cervical cancer cells. Tumour Biol. 2014;35(12):11913–20. doi: 10.1007/s13277-014-2460-5. [DOI] [PubMed] [Google Scholar]
- 8.International Agency for Research on Cancer. IARC Monographs on the evaluation of carcinogenic risks to humans. International Agency for Research on Cancer; Lyon: 1995. [PMC free article] [PubMed] [Google Scholar]
- 9.International Agency for Research on Cancer. Review of human carcinogens: Biological agents. World health organization; 2012. [Google Scholar]
- 10.Hopkins MP, Morley GW. Prognostic factors in advanced stage squamous cell cancer of the cervix. Cancer. 1993;72(8):2389–93. doi: 10.1002/1097-0142(19931015)72:8<2389::aid-cncr2820720816>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 11.Baltimore D. Our genome unveiled. Nature. 2001;409(6822):814–16. doi: 10.1038/35057267. [DOI] [PubMed] [Google Scholar]
- 12.Salmena L, Poliseno L, Tay Y, et al. A ceRNA hypothesis: The Rosetta Stone of a hidden RNA language? Cell. 2011;146(3):353–58. doi: 10.1016/j.cell.2011.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Caley DP, Pink RC, Trujillano D, Carter DR. Long noncoding RNAs, chromatin, and development. ScientificWorldJournal. 2010;10:90–102. doi: 10.1100/tsw.2010.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang L, Duff MO, Graveley BR, et al. Genomewide characterization of non-polyadenylated RNAs. Genome Biol. 2011;12(2):R16. doi: 10.1186/gb-2011-12-2-r16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ergun S, Oztuzcu S. Oncocers: ceRNA-mediated cross-talk by sponging miRNAs in oncogenic pathways. Tumour Biol. 2015;36(5):3129–36. doi: 10.1007/s13277-015-3346-x. [DOI] [PubMed] [Google Scholar]
- 16.Qi X, Zhang DH, Wu N, et al. ceRNA in cancer: Possible functions and clinical implications. J Med Genet. 2015;52(10):710–18. doi: 10.1136/jmedgenet-2015-103334. [DOI] [PubMed] [Google Scholar]
- 17.Tomczak K, Czerwinska P, Wiznerowicz M. The Cancer Genome Atlas (TCGA): An immeasurable source of knowledge. Contemp Oncol (Pozn) 2015;19(1A):A68–77. doi: 10.5114/wo.2014.47136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Langfelder P, Horvath S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liang C, Zhang B, Ge H, et al. Long non-coding RNA CRNDE as a potential prognostic biomarker in solid tumors: A meta-analysis. Clin Chim Acta. 2018;481:99–107. doi: 10.1016/j.cca.2018.02.039. [DOI] [PubMed] [Google Scholar]
- 20.Cheng J, Chen J, Zhang X, et al. Overexpression of CRNDE promotes the progression of bladder cancer. Biomed Pharmacother. 2018;99:638–44. doi: 10.1016/j.biopha.2017.12.055. [DOI] [PubMed] [Google Scholar]
- 21.Sun H, He L, Ma L, et al. LncRNA CRNDE promotes cell proliferation, invasion and migration by competitively binding miR-384 in papillary thyroid cancer. Oncotarget. 2017;8(66):110552–65. doi: 10.18632/oncotarget.22819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Di Agostino S, Valenti F, Sacconi A, et al. Long non-coding MIR205HG depletes hsa-miR-590-3p leading to unrestrained proliferation in head and neck squamous cell carcinoma. Theranostics. 2018;8(7):1850–68. doi: 10.7150/thno.22167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhao Q, Sun X, Liu C, et al. Expression of the microRNA-143/145 cluster is decreased in hepatitis B virus-associated hepatocellular carcinoma and may serve as a biomarker for tumorigenesis in patients with chronic hepatitis B. Oncol Lett. 2018;15(5):6115–22. doi: 10.3892/ol.2018.8117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Supic G, Zeljic K, Rankov AD, et al. miR-183 and miR-21 expression as biomarkers of progression and survival in tongue carcinoma patients. Clin Oral Investig. 2018;22(1):401–9. doi: 10.1007/s00784-017-2126-y. [DOI] [PubMed] [Google Scholar]
- 25.Yang W, Hong L, Xu X, et al. LncRNA GAS5 suppresses the tumorigenesis of cervical cancer by downregulating miR-196a and miR-205. Tumour Biol. 2017;39(7) doi: 10.1177/1010428317711315. 1010428317711315. [DOI] [PubMed] [Google Scholar]
- 26.Pang H, Yue X. MiR-205 serves as a prognostic factor and suppresses proliferation and invasion by targeting insulin-like growth factor receptor 1 in human cervical cancer. Tumour Biol. 2017;39(6) doi: 10.1177/1010428317701308. 1010428317701308. [DOI] [PubMed] [Google Scholar]
- 27.Yue Z, Yun-Shan Z, Feng-Xia X. miR-205 mediates the inhibition of cervical cancer cell proliferation using olmesartan. J Renin Angiotensin Aldosterone Syst. 2016;17(3) doi: 10.1177/1470320316663327. 1470320316663327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rajkumar T, Sabitha K, Vijayalakshmi N, et al. Identification and validation of genes involved in cervical tumourigenesis. BMC Cancer. 2011;11:80. doi: 10.1186/1471-2407-11-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ma D, Chang LY, Zhao S, et al. KLF5 promotes cervical cancer proliferation, migration and invasion in a manner partly dependent on TNFRSF11a expression. Sci Rep. 2017;7(1):15683. doi: 10.1038/s41598-017-15979-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Singh S, Gupta M, Seam RK, Changotra H. E2F1 genetic variants and risk of cervical cancer in Indian women. Int J Biol Markers. 2018 doi: 10.1177/1724600818768459. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 31.Wang X, Gao P, Wang M, et al. Feedback between E2F1 and CIP2A regulated by human papillomavirus E7 in cervical cancer: Implications for prognosis. Am J Transl Res. 2017;9(5):2327–39. [PMC free article] [PubMed] [Google Scholar]
- 32.Tamura R, Yoshihara K, Saito T, et al. Novel therapeutic strategy for cervical cancer harboring FGFR3-TACC3 fusions. Oncogenesis. 2018;7(1):4. doi: 10.1038/s41389-017-0018-2. [DOI] [PMC free article] [PubMed] [Google Scholar]